Back to Journals » International Journal of Nanomedicine » Volume 12
Synthesis, construction, and evaluation of self-assembled nano-bacitracin A as an efficient antibacterial agent in vitro and in vivo
Authors Hong W, Gao X, Qiu P, Yang J, Qiao M, Shi H, Zhang D, Tian C, Niu S, Liu M
Received 15 March 2017
Accepted for publication 7 June 2017
Published 30 June 2017 Volume 2017:12 Pages 4691—4708
DOI https://doi.org/10.2147/IJN.S136998
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Linlin Sun
Wei Hong, 1 Xiang Gao, 1 Peng Qiu, 1 Jie Yang, 1 Mingxi Qiao, 2 Hong Shi, 3 Dexian Zhang, 1 Chunlian Tian, 1 Shengli Niu, 1 Mingchun Liu 1
1Key Laboratory of Zoonosis of Liaoning Province, College of Animal Science and Veterinary Medicine, Shenyang Agricultural University, Shenhe, Shenyang, Liaoning, People’s Republic of China; 2Department of Pharmaceutics, School of Pharmacy, China Pharmaceutical University, Jiangning, Nanjing, 3Department of Pharmaceutics, School of Pharmacy, Shenyang Pharmaceutical University, Shenyang, Liaoning, People’s Republic of China
Abstract: Bacitracin A (BA) is an excellent polypeptide antibiotic that is active against gram-positive bacteria without triggering multidrug resistance. However, BA is inactive against gram-negative bacteria because of its inability to cross the outer membrane of these cells, and it has strong nephrotoxicity, thus limiting its clinical applications. Nanoantibiotics can effectively localize antibiotics to the periplasmic space of bacteria while decreasing the adverse effects of antibiotics. In this study, biodegradable hydrophobic copolymers of poly (D,L-lactide-co-glycolide) (PLGA) were attached to the N-termini of BA to design a novel class of self-assembled nano-bacitracin A (nano-BAs), and their potential as antibacterial agents was evaluated in vitro and in vivo. Nano-BAs had a core-shell structure with a mean diameter < 150 nm. Impressively, nano-BAs had strong antibacterial properties against both gram-positive and gram-negative bacteria, and the distribution of antibacterial activity as a function of PLGA block length was skewed toward longer PLGA chains. No cytotoxicity against HK-2 cells or human red blood cells (hRBCs) was observed in vitro, suggesting good biocompatibility. A high local density of BA mass on the surface promoted endocytotic cellular uptake, and hydrophobic interactions between the PLGA block and lipopolysaccharide (LPS) facilitated the uptake of nano-BAs, thereby leading to greater antibacterial activities. In addition, Nano-BA 5K was found to be effective in vivo, and it served as an anti-infective agent for wound healing. Collectively, this study provides a cost-effective means of developing self-assembling nano-polypeptide antibiotic candidates with a broader antibacterial spectrum and a lower toxicity than commercially available peptide antibiotics, owing to their modification with biodegradable copolymers.
Keywords: bacitracin A, poly (D,L-lactide-co-glycolide), nano-BAs, broader antibacterial spectrum, wound healing
Corrigendum for this paper has been published
Further Corrigendum for this paper has been published
Introduction
Bacitracin is a widely used polypeptide antibiotic produced by Bacillus subtilis and B. licheniformis.1,2 It is a polypeptide antibiotic with a narrow spectrum of activity that is primarily directed against gram-positive bacteria.3 Bacitracin is a mixture of several similar polypeptide components that differ in some amino acids, and the components designated as bacitracin A (BA) are known to be the most microbiologically active.4 BA is targeted to its site of action by binding to undecaprenyl pyrophosphate, thus preventing the recycling of the sugar carrier and inhibiting bacterial cell wall synthesis.5 In addition, BA disintegrates the bacterial cytoplasmic membrane, thus leading to the loss of various ions and amino acids and resulting in the death of bacteria.6–8 However, BA has a complex chemical structure and high molecular weight, and it is unable to pass through porins in the outer membrane of gram-negative bacteria to reach the cell wall area, which is their site of action. Therefore, gram-negative bacteria are intrinsically resistant to BA. Thus, efforts to broaden the spectrum of action of BAs against gram-negative bacteria have been dismissed.
Hydrophobic modification of polypeptide antibiotics can promote their membrane adsorption, insertion, permeabilization, and disruption potency, and these modifications usually translate into a broad spectrum of antimicrobial activities and low toxicity.9 Avrahami and Shai have attached lipophilic acids of different lengths (heptanoic, undecanoic, and palmitic acid) to the N-terminus of magainin, and the resulting peptidic micelles have been found to display antifungal activity.10 Malina and Shai have indicated that the attachment of different aliphatic acids (decanoic acid, dodecanoic acid, myristic acid, and palmitic acid) to the N-terminus of peptides increases their antibacterial activity and lipopolysaccharide (LPS)-binding activities.11 Furthermore, it has recently been shown that the attachment of a palmitoyl group, but not shorter fatty acids, to a membrane-inactive cationic peptide makes it membrane-active.12
Encapsulation into, or the association of antibiotics with, nanosized carriers called “nanoantibiotics” can effectively improve antibiotic interactions with pathogenic microorganisms.13 The capability of nanoantibiotics to control infections in vitro and in vivo has been explored and demonstrated. Unlike many antimicrobial agents currently in use in the clinical setting, nanoantibiotics may not have direct and acute adverse effects, although their potential toxicity after long-term exposure is unknown. Most importantly, nanoantibiotics act on multiple biological pathways found in broad species of microbes, and many concurrent mutations would need to occur in order for an organism to develop resistance against the antimicrobial activities of nanoantibiotics. The preparation of nanoantibiotics can be cost-effective, as compared with that of antibiotic synthesis, and they are sufficiently stable for long-term storage and have a prolonged shelf-life. In addition, some nanoantibiotics can withstand harsh conditions, such as high-temperature sterilization, under which conventional antibiotics are inactivated. Nanoantibiotics have multiple advantages over antibiotics: 1) they are controllable and have a relatively uniform distribution in the target tissue, 2) they have improved solubility, 3) they offer sustained and controlled release, 4) they provide improved patient compliance, 5) they have minimized side effects, and 6) they offer enhanced cellular internalization.14–16 Hydrophobic modification can also confer on the hydrophilic polypeptide antibiotics an amphiphilic structure that can spontaneously form nanoparticles with a core-shell structure in water. Wang et al have designed a short amphiphilic peptide (CG3R6TAT) containing a hydrophilic block of cell-penetrating peptide TAT, six arginine residues (R6), and a hydrophobic block of cholesterol (C). This peptide can easily self-assemble into core-shell nanoparticles, and it possesses a broad spectrum of strong antimicrobial activities that are much stronger than those of hydrophilic peptides, which are incapable of forming nanoparticles.17,18
In this study, we examined the possibility of using biodegradable polymers of poly (lactic-co-glycolic acid) (PLGA) to modify BA (BA–PLGA–BA) to form nano-BAs. Because PLGA has a well-documented ability to drive the self-assembly of PLGA-containing materials,19 amphipathic BA–PLGA–BA can easily form core-shell structured micelles (nano-BAs) with a hydrophobic PLGA core and a hydrophilic BA shell arranged toward the surrounding environment. In addition, controlled enzymatic degradation is another advantage of PLGA, because compounds such as fatty acids – which are fully protected from enzymatic degradation – are less suitable for therapy. The physicochemical characterization of BA–PLGA–BA and nano-BAs was first determined by 1H nuclear magnetic resonance (NMR) spectra, gel permeation chromatography (GPC), fluorescent probe technique, and particle-size measurements. Minimum inhibitory concentrations (MICs) of nano-BAs were then analyzed against both gram-positive and gram-negative bacteria in the absence or presence of physiological salts or human serum, respectively. In addition, harmful effects on the HK-2 cells and human red blood cells (hRBCs) were evaluated. Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) were used to investigate the potential mechanisms of Nano-BA5K against gram-negative bacteria. Furthermore, a preliminary evaluation of Nano-BA5K treatment on wound healing was undertaken.
Materials and methods
Materials
Reagents
DL-lactide and glycolide were obtained from Beijing CONAN Polymer R&D Center (Beijing, People’s Republic of China). BA, stannous 2-ethylhexanoate, t-butyldimethylsilanol, triphenylsilanol, N, N′-carbonyldiimidazole (CDI), and Triton X-100 were purchased from Sigma-Aldrich (Shanghai, People’s Republic of China). Mueller–Hinton broth (MHB) powder, Mueller–Hinton (MH) agar, Salmonella Shigella agar, MacConkey agar, and Edwards Medium (Modified) were purchased from AoBoX (Beijing, People’s Republic of China) and were used to prepare bacterial broths according to the manufacturer’s instructions. All the other regents and chemicals were of analytical or chromatographic grade and were purchased from Concord Technology (Tianjing, People’s Republic of China).
Bacteria
Escherichia coli (E. coli) American Type Culture Collection (ATCC) 25922, Salmonella typhimurium (S. typhimurium) ATCC 13311, Pseudomonas aeruginosa (P. aeruginosa) ATCC 27853, Staphylococcus aureus (S. aureus) ATCC 29213, Streptococcus pneumoniae (S. pneumoniae) ATCC 49619, and Trueperella pyogenes (T. pyogenes) ATCC 19411 were purchased from American Type Culture Collection (Manassas, VA, USA).
Cells
The human tubular epithelial cell line HK-2 was purchased from American Type Cell Culture (ATCC, USA). Dulbecco’s Modified Eagle’s Medium (DMEM) and fetal bovine serum (FBS) were purchased from Gibco BRL (Gaithersberg, MD, USA). Culture plates and dishes were purchased from Corning Inc. (NY, USA). HK-2 cells were cultured in DMEM with 15% FBS, 100 IU/mL penicillin, and 100 μg/mL streptomycin sulfate. Cells were cultured in a CO2 incubator with 5% CO2 at 37°C. All experiments were performed on cells in the logarithmic phase of growth. The hRBCs used in the experiment were kindly provided by Shengjing Hospital of China Medical University (Shenyang, People’s Republic of China). All blood donors provided informed consent for the use of their blood in research, and all procedures were approved by the Institutional Ethical Committee of Shenyang Agricultural University (Shenyang, People’s Republic of China).
Animals
Equal numbers of adult male and female Kunming mice (KM mice; weight 20–25 g) were used in the experiment. KM mice were supplied by the Department of Experimental Animals, Shenyang Pharmaceutical University (Shenyang, People’s Republic of China). Animal care and experiments were performed with the approval of the animal ethical committee of Shenyang Agricultural University (Shenyang, People’s Republic of China), according to the requirements of the National Act on the Use of Experimental Animals (People’s Republic of China). All animals were kept in a favorable environment and were acclimated at 25°C and 55% of humidity under natural light/dark conditions, with free access to a rodent diet and water. They were acclimated for 1 week before the beginning of the study. All procedures followed the guidelines of the institutional animal ethical committee of Shenyang Agricultural University (Shenyang, People’s Republic of China).
Synthesis and characterization of BA–b–PLGA–b–BA
BA–b–PLGA–b–BA was obtained by grafting PLGA onto BA through the N-terminus. Briefly, a copolymer of PLGA was first synthesized and then activated by CDI to yield CDI–b–PLGA–b–CDI. A coupling reaction of CDI–b–PLGA–b–CDI with the N-terminus of BA was then conducted to obtain the copolymer of BA–b–PLGA–b–BA. The overall scheme is presented in Figure 1.
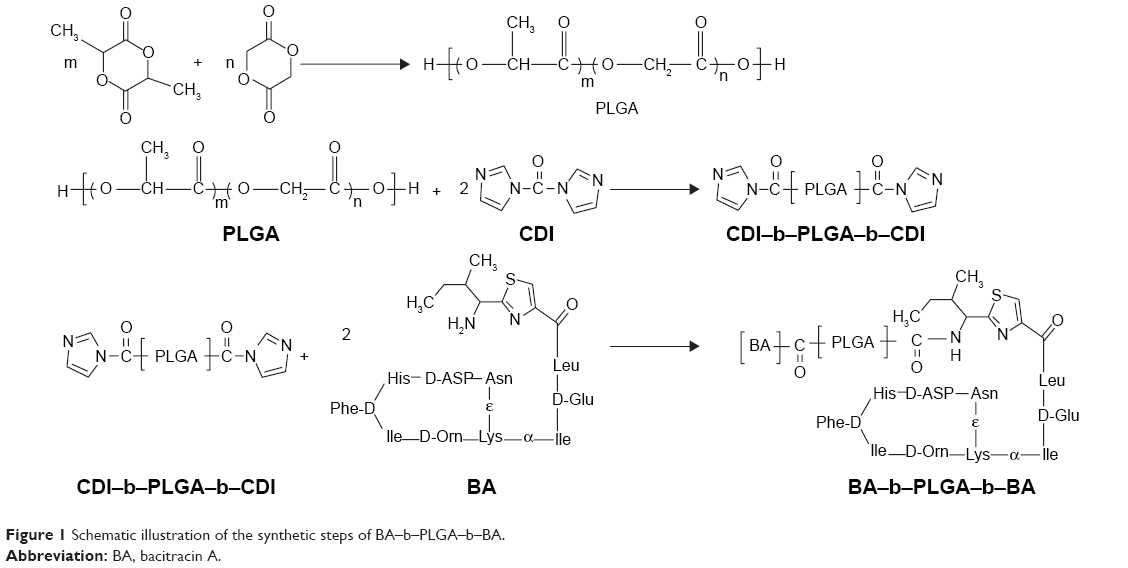 | Figure 1 Schematic illustration of the synthetic steps of BA–b–PLGA–b–BA. |
Synthesis of PLGA copolymer
The starting copolymer of PLGA was synthesized through a ring-opening melting polymerization of DL-lactide and glycolide with stannous octoate [Sn(Oct)2] as a catalyst and t-butyldimethylsilanol/triphenylsilanol as a molecular weight regulator. Briefly, precise amounts of DL-lactide and glycolide were mixed and introduced into a polymerization tube with the catalyst and molecular weight regulator. The mixtures were degassed with a vacuum pump for at least 12 h in order to remove moisture and oxygen. Then, the tube was sealed under dynamic vacuum at 10−3 M bar and the polymerization was allowed to proceed at 140°C for 10 h. Then, the product was purified by dialysis against deionized water for 3 days and lyophilized. The yield of the copolymers [weight of the product (PLGA) to total feed amount of DL-lactide and glycolide] was >85%.
Synthesis of CDI-activated PLGA
Excessive amounts of CDI dissolved in 10 mL of dry acetonitrile was added dropwise to the acetonitrile solution of PLGA (15 mL) at room temperature under a nitrogen atmosphere. After 24 h of reaction, the mixture solution was concentrated in a rotary evaporator and poured into an excess amount of ethyl ether. This process was repeated thrice to remove unreacted CDI. Then, the CDI-activated PLGA (CDI-b-PLGA-b-CDI) was dried for 3 days under vacuum (yield ~85%).
Synthesis of BA–b–PLGA–b–PLGA
The CDI-activated PLGA dissolved in 15 mL of DMSO was added dropwise to a DMSO solution of BA at room temperature for over 2 h under nitrogen atmosphere. The mixture was allowed to react for 3 days at room temperature. The crude product was further purified by dialysis against deionized water for 3 days at room temperature and then subjected to lyophilization (yield ~70%).
Characterization of intermediates and BA–b–PLGA–b–BA
1H-NMR spectra were recorded on a Bruker DRX-600 NMR instrument operated at 600 MHz and were used to determine the molecular structure and composition of the intermediates and BA–b–PLGA–b–BA copolymer. DMSO-d6 and CDCl3 were used as the solvent, including 0.03 vol.% tetramethylsilane (TMS). The composition and block length of PLGA were calculated by comparing the proton-peak integration of LA and GA from 1H-NMR spectra. The molecular weight and molecular weight distribution of the PLGA and BA–b–PLGA–b–BA were measured with a GPC system equipped with a Waters 515 HPLC Pump, a Waters StyragelTM HT3 column (300×7.8 mm), and a Waters 2414 refractive index detector. Tetrahydrofuran (THF) was used as the eluent, with a flow rate of 1 mL/min at 35°C. The concentration of the samples was 10 mg/mL. The molecular weight of the copolymer was determined relative to polystyrene standards.
The critical micelle concentration (CMC) of the BA–b–PLGA–b–BA was determined with a fluorescence technique using pyrene as a probe.20 Aliquots of pyrene solution (2.4×10−5 M, 50 μL) in acetone were poured into phosphate-buffered saline (PBS) solution, and the acetone was eliminated by stirring at 40°C for 6 h. After the solvent was evaporated, 10 mL of the aqueous copolymer solutions, with concentration ranging from 1×10−4 mg/L to 1×10−1 mg/L, were added. The final pyrene concentration in the copolymer solution was 1.2×10−7 mol/L. All solutions were stored in the dark for 24 h to reach solubilization equilibrium prior to measurements. The fluorescence spectra of pyrene were recorded on a RF-5301 fluorescence spectrophotometer (Shimadzu Spectrofluorophotometer, Japan). The intensity ratio (I385/I374) of the pyrene emission spectrum was plotted as a function of polymer concentration at the excitation wavelength of 350 nm.
Preparation and characterization of nano-BAs
Nano-BAs were prepared by a thin-film hydration method. Briefly, the copolymer (BA–b–PLGA–b–BA, 100 mg) was dissolved in acetonitrile (25 mL) in a round-bottomed flask. The solvent was evaporated under decreased pressure by rotary evaporation at 35°C for 1 h to obtain a thin film. Residual acetonitrile in the film was removed under vacuum at room temperature for another 12 h. The resultant thin film was hydrated with 20 mL of PBS (pH 7.4) at 35°C for 30 min to obtain a nano-BAs solution. This nano-BA solution was sonicated for 30 s with a KQ3200DB Ultrasonic Instrument, three times at 400 W, and then the resulting nano-BAs were freeze dried.
The hydrodynamic diameters, particle-size distributions, and zeta potential of the nano-BAs were determined with a NICOMP™ 380 ZLS (Santa Barbara, USA). Each sample was filtered through a 0.45-μM disposable filter prior to measurements. Each measurement was repeated thrice, and an average value was calculated. The morphology of nano-BAs was observed using a Hitachi HT-7700 instrument operating at an acceleration voltage of 80 kV without staining. Samples were prepared by dipping a copper grid into the nano-BAs solution, and additional solution was blotted away with a strip of filter paper. The solution was evaporated at room temperature for 2 h before TEM observation.
In vitro antibacterial activity assays
MICs of nano-BAs (Nano-BA3K and Nano-BA5K) were determined using a modified standard microtiter dilution method, as described previously.21,22 Briefly, bacterial cells were cultured overnight at 37°C to log-phase growth in MHB and were then diluted to a final concentration of 1×105 CFU/mL. Bacterial suspension (100 μL) was incubated in 96-well microtiter plates, and 100 μL of twofold dilutions of the tested formulations (nano-BAs and BA) was added to each well with a final volume of 200 μL. Final concentrations of the nano-BAs ranged from 0.25 to ~128 μM. MICs were defined as the concentration at which no visible growth of organisms was observed after incubation for 18–24 h at 37°C. Inhibition of growth was determined by measuring the absorbance at 600 nm with a multifunctional microplate reader (Tecan, Austria) after incubation for 18 h at 37°C. The broth with bacteria was used as the negative control. Tests were repeated at least three times.
To further evaluate the antibacterial potency of nano-BAs, the minimum bactericidal concentration (MBC) was also determined, as described previously. Briefly, bacterial cells were washed three times with PBS buffer and resuspended in the same buffer. The bacterial suspensions (~105 CFU/mL) in PBS were incubated with twofold dilutions of nano-BAs for 2 h at 37°C. After the treatment, cell samples were incubated on MH agar to determine the number of viable bacteria. The surviving colonies were counted the following day, and the lowest concentration of nano-BAs at which there was complete killing was taken as the MBC. The results given are the mean values of three independent determinations.
In vitro killing curve testing
Briefly, bacterial cells were cultured overnight at 37°C to log-phase growth in MHB and were then diluted to a final concentration of 5×106 CFU/mL, as measured with a spectrophotometer at 600 nm. Nano-BAs (Nano-BA3K and Nano-BA5K) were added at concentrations corresponding to 2× MIC. Samples were drawn at 0, 30, 60, 70, 120, 150, 180, 210, 270, and 300 min and were then diluted to various concentrations. The diluted samples were plated on MH Agar medium for S. aureus ATCC 29213 and MacConkey agar medium for E. coli ATCC 25922 and incubated at 37°C for 72 h. The number of CFUs was then counted. The experiments were performed in triplicate, and the results are expressed as the mean ± SD.
Salt and serum sensitivity assays
To determine whether the antibacterial activity was affected by the presence of salts or serum, the MICs in MHB supplemented with different salts at their physiological concentrations (150 mM NaCl, 4.5 mM KCl, 2 mM CaCl2, 6 μM NH4Cl, 8 μM ZnCl2, 1 mM MgCl2, and 4 μM FeCl3) or 20% human heat-inactivated serum were determined as described earlier. The assays were performed at least three times.
Scanning electron microscopy characterization
E. coli ATCC 25922 bacterial cells were grown to an exponential phase in MHB at 37°C under constant shaking at 220 rpm. After centrifugation at 1,000 g for 10 min, the cell pellets were harvested, washed twice with 10 mM PBS, and re-suspended to an optical density (OD600) of 0.2. Cells were incubated at 37°C for 2 h with Nano-BA5K at their 1× MICs. The negative control was run without Nano-BA5K or BA solution, whereas polymyxin B was used as a positive control. After incubation, the cells were centrifuged at 5,000 g for 5 min. The cell pellets were harvested, washed thrice with PBS, and subjected to fixation with 2.5% glutaraldehyde at 4°C overnight, and then washed twice with PBS. The cells were then dehydrated in a graded ethanol series (50%, 70%, 90%, and 100%) for 10 min and in 100% ethanol – a mixture (1:1) of 100% ethanol and tertiary butanol and absolute tertiary butanol – for 15 min. Finally, the specimens were dehydrated, dried, coated with gold, and examined using a Hitachi S-4800 SEM.
Transmission electron microscopy observations
Pretreatment with bacterial samples was conducted in the same manner as for SEM treatment. After pre-fixation with 2.5% glutaraldehyde at 4°C overnight, the bacteria cells were then post-fixed with 2% osmium tetroxide for 70 min. After dehydration with a graded ethanol series (50%, 70%, 90%, and 100%) for 8 min each, the bacteria samples were transferred to 100% ethanol – a mixture (1:1) of 100% ethanol and acetone and absolute acetone – for 10 min. Then, the specimens were transferred to 1:1 mixtures of absolute acetone and epoxy resin for another 30 min and to pure epoxy resin and were incubated overnight at a constant temperature. Finally, the specimens were sectioned with an ultramicrotome, stained with uranyl acetate and lead citrate, and examined using a Hitachi H-7650 TEM.
In vitro cell cytotoxicity
A thiazolyl blue tetrazolium bromide (MTT) assay was used to assess the in vitro cytotoxicity of nano-BAs against HK-2 cells. Briefly, HK-2 cells were seeded at a density of 5×103 cells per well in 96-well plates and then incubated for 24 h. Then, the growth medium was replaced with fresh medium containing an indicated concentration of the tested formulations (Nano-BA3K, Nano-BA5K, and BA). Control wells were treated with an equivalent volume of BA-free medium. The cells were incubated at 37°C for 48 h. After incubation, the wells were rinsed with PBS, and MTT solution (5 mg/mL) was added to each well, and the plate was incubated for 4 h, thus allowing the viable cells to decrease the yellow MTT into purple formazan crystals. Finally, the medium was completely removed, and 150 μL of dimethyl sulfoxide (DMSO) was added to each well to dissolve the purple formazan crystals. The absorbance was measured at 570 nm using a multifunctional microplate reader (Tecan, Austria). The IC50 values were calculated using nonlinear regression analysis, and cell cytotoxicity was assessed by quantifying the IC50 values of the tested formulations.
Measurement of hemolytic activity
Fresh hRBCs were washed with PBS (pH 7.4) three times, centrifuged at 1,000 g for 5 min, and resuspended in PBS to attain a dilution of ~1% (v/v) of the erythrocyte. Then, 100 μL hRBCs solution was placed in each well of 96-well plates, and 100 μL of serial dilution of nano-BAs was added to each well and incubated for 2 h at 37°C. Then, the mixture was centrifuged at 1,000 g for 5 min at 4°C, aliquots (100 μL) of the supernatant were transferred to 96-well plates, and the release of hemoglobin was measured by monitoring the OD at 576 nm using a multifunctional microplate reader (Tecan, Austria). The hRBCs in PBS and 0.5% Triton X-100 were used as negative and positive controls, respectively. The tests were repeated six times, and the data are expressed as the mean and standard deviation of six replicates. The percentage of hemolysis was calculated using the following equation:
|
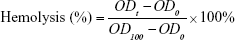
where ODt is the observed fluorescence at a given nano-BA concentration, OD0 is the observed fluorescence in PBS, and OD100 is the observed fluorescence in 0.5% Triton X-100.
Experimental wound infection and treatment
Female Kunming mice (weight 20–25 g) were used in the experiment. Mice were anesthetized with an intraperitoneal injection of sodium pentobarbital (30 mg/kg). After the selected dorsal region with a diameter of 3.0 cm was treated with depilatory cream and the surgical area was disinfected with 75% alcohol, a full-thickness 0.8-cm diameter wound was created by excising the skin and infecting by direct seeding with 20 μL of mixtures of S. aureus (5×107 CFU/mL) and E. coli (5×107 CFU/mL) on the wound. The wound was subsequently covered with sterile gauze to prevent cross contamination. Treatment was initiated at 48 h after bacterial inoculation. Physiological saline was used as a negative control; Polysporin® ointment was used as a positive control; and 2 μM Nano-BA5K was used with the treated group. The procedures were repeated twice daily starting from the day of infection, and the entire treatment lasted 6 days. A total of 3 or 6 days after treatment with Nano-BA5K, six mice from each group were sacrificed, and the infected sections of skin were aseptically contained, weighed, and homogenized in distilled water. The homogenate was then serially diluted, bacterial CFU counts were obtained, and statistical analysis was performed.
Measurement of hydroxyproline content
Full-thickness skin samples taken from the incisions were rinsed in ice-cold PBS (10 mM, pH 7.4) to remove excess blood and were then weighed. The samples were minced into small pieces, then homogenized in 1 mL of PBS with a glass homogenizer on ice, and maintained at −20°C overnight. The resulting suspensions were subjected to two freeze–thaw cycles to further disrupt the cell membrane. The homogenates were then centrifuged for 5 min at 5,000 g. Hydroxyproline levels in the supernatant were measured immediately according to the protocol supplied with the Mouse Hydroxyproline ELISA Kit (CUSABIO, Wuhan, People’s Republic of China).
Statistical analysis
All experiments were performed at least three times. Quantitative data are presented as the mean ± standard deviation (SD). Statistical comparisons were determined by analysis of variance (ANOVA) among ≥3 groups or Student’s t-test between two groups. P-values <0.05 and <0.01, respectively, were considered statistically significant.
Results
Characterization of BA–PLGA–BA copolymer
The 1H NMR spectra of the intermediates (BA, PLGA5K, and CDI-PLGA5K-CDI) and the final triblock copolymer (BA–PLGA5K–BA) are shown in Figure 2A–D. All the chemical shifts are expressed in parts per million (δ) relative to the solvent signal. The 1H NMR spectrum (DMSO-d6) of BA showed peaks at δa 0.82 ppm (-CH2CH3), δb 6.63 ppm (the proton “b” on the thiazole moiety), and δc 7.54 ppm (-NH2) (Figure 2A) – a result consistent with findings from a previous study.5 The 1H NMR spectrum (CDCl3) of PLGA5K showed peaks at δa 1.58 ppm (–COCH(CH3)O–), δb 5.21 ppm (–COCH(CH3)O–), and δc 4.82 ppm (–COCH2O–) (Figure 2B). The 1H NMR spectrum (CDCl3) of CDI-PLGA5K-CDI showed peaks at δd 8.34 ppm (the proton “d” on the imidazole moiety), δe 7.42 ppm (the proton “e” on the imidazole moiety), δf 7.11 ppm (the proton “f” on the imidazole moiety), as well as the characteristic peaks of PLGA (Figure 2C). The 1H NMR spectrum (DMSO-d6) of the final copolymer BA–PLGA5K–BA showed the characteristic peaks of BA at δa 0.82 ppm (–CH2CH3), δb 7.24 ppm (the proton “b” on the thiazole moiety), and δc 8.52 ppm (-NH2) as well as the characteristic peaks of PLGA at δd 1.58 ppm (-COCH(CH3) O-), δe 5.21 ppm (-COCH(CH3)O-), and δf 4.82 ppm (-COCH2O-) (Figure 2D), thus indicating that the final triblock copolymer of BA–PLGA–BA had been obtained.
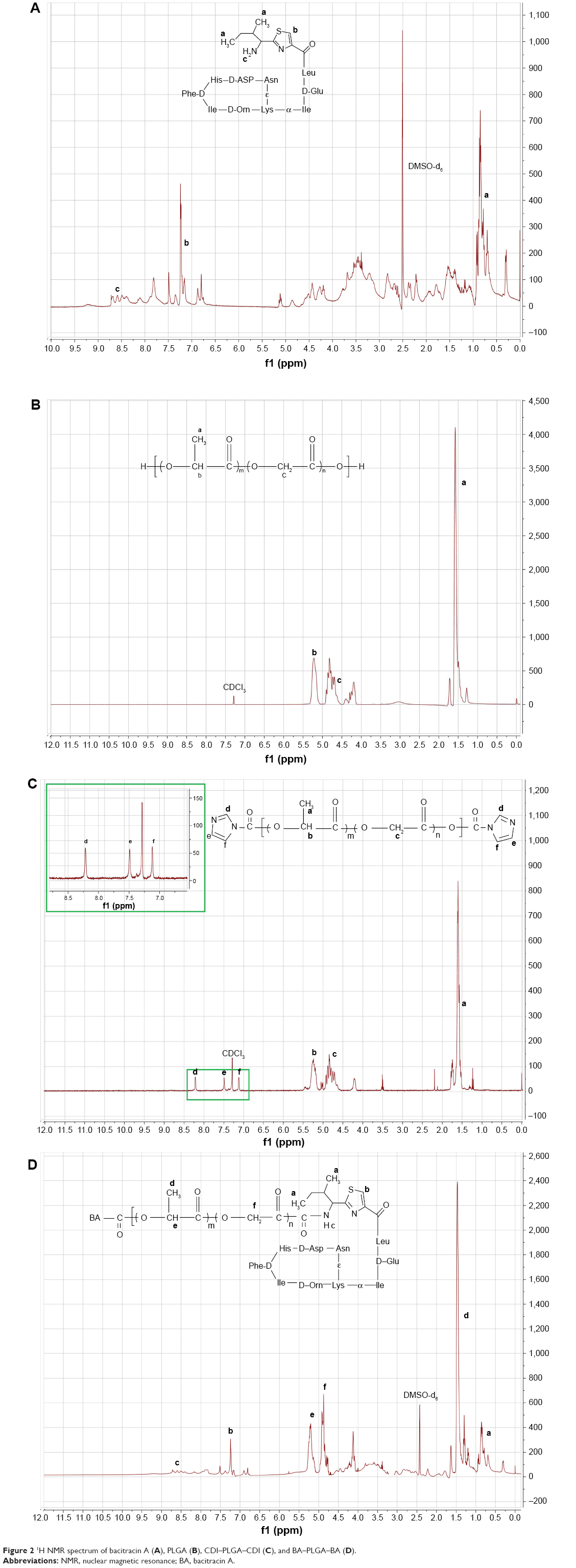 | Figure 2 1H NMR spectrum of bacitracin A (A), PLGA (B), CDI–PLGA–CDI (C), and BA–PLGA–BA (D). |
The GPC chromatograms of the synthetic copolymers are shown in Figure 3. All copolymers showed unimodal distribution with a polydispersity of <1.2, whereas the shorter elution time of copolymers indicated an increase in molecular weight compared with those of the intermediates. In addition, the composition and characterization of the PLGA and BA–PLGA–BA copolymers are presented in Table 1.
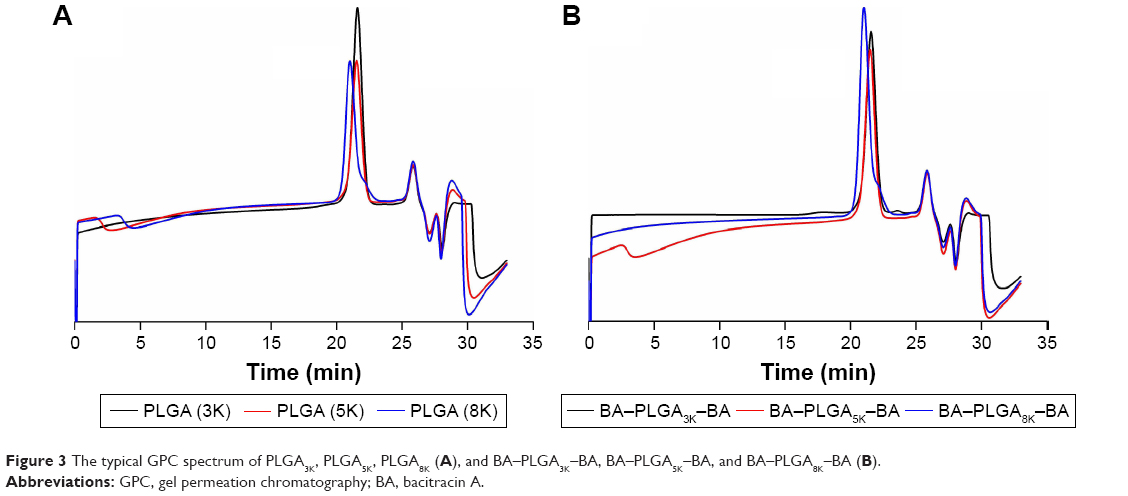 | Figure 3 The typical GPC spectrum of PLGA3K, PLGA5K, PLGA8K (A), and BA–PLGA3K–BA, BA–PLGA5K–BA, and BA–PLGA8K–BA (B). |
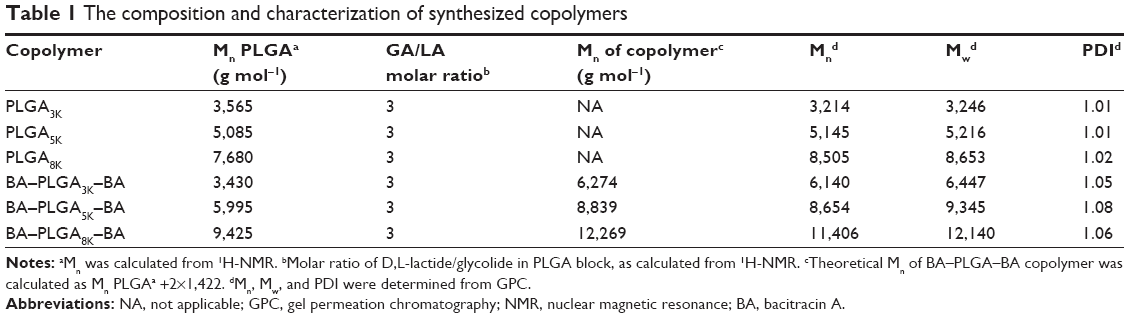 | Table 1 The composition and characterization of synthesized copolymers |
Micelle formation from BA–PLGA–BA
To investigate the effect of the molecular weight of PLGA on micellar stability, the CMC of BA–PLGA3K–BA, BA–PLGA5K–BA, and BA–PLGA8K–BA at biological pH (pH 7.4) was investigated using the pyrene-based fluorescent probe method. As shown in Table 2, the CMC values of BA–PLGA–BA copolymers decreased (BA–PLGA3K–BA: 1.82×10−2 g/L; BA–PLGA5K–BA: 1.36×10−2 g/L; and BA–PLGA8K–BA: 1.14×10−2 g/L) with an increase in the molecular weight of the PLGA block from 3K to 8K at pH 7.4. This result indicated that the copolymer with a longer PLGA block showed better micellar stability, owing to the relatively higher hydrophobicity of its PLGA (8K) block. However, with an increase in the molecular weight of PLGA, the solubility of BA–PLGA–BA sharply decreased (data not shown). The BA–PLGA–BA with modification by PLGA (5K) showed better stability and solubility compared with those of other BA–PLGA–BA.
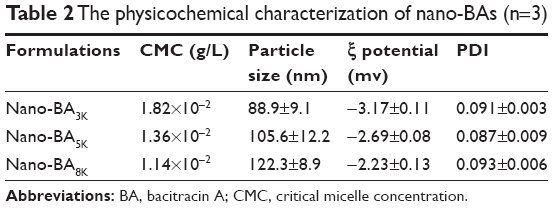 | Table 2 The physicochemical characterization of nano-BAs (n=3) |
The physical characterizations of the nano-BAs are summarized in Table 2. The mean diameters of the nano-BAs determined by dynamic light scattering technique (DLS) were between 80 and 130 nm with good polydispersity indexes (PDIs) <0.1. All nano-BAs exhibited similar slightly negative surface charges at pH 7.4. TEM images revealed that all nano-BAs were spherical and that the particle size correlated well with the results obtained by DLS (Figure 4).
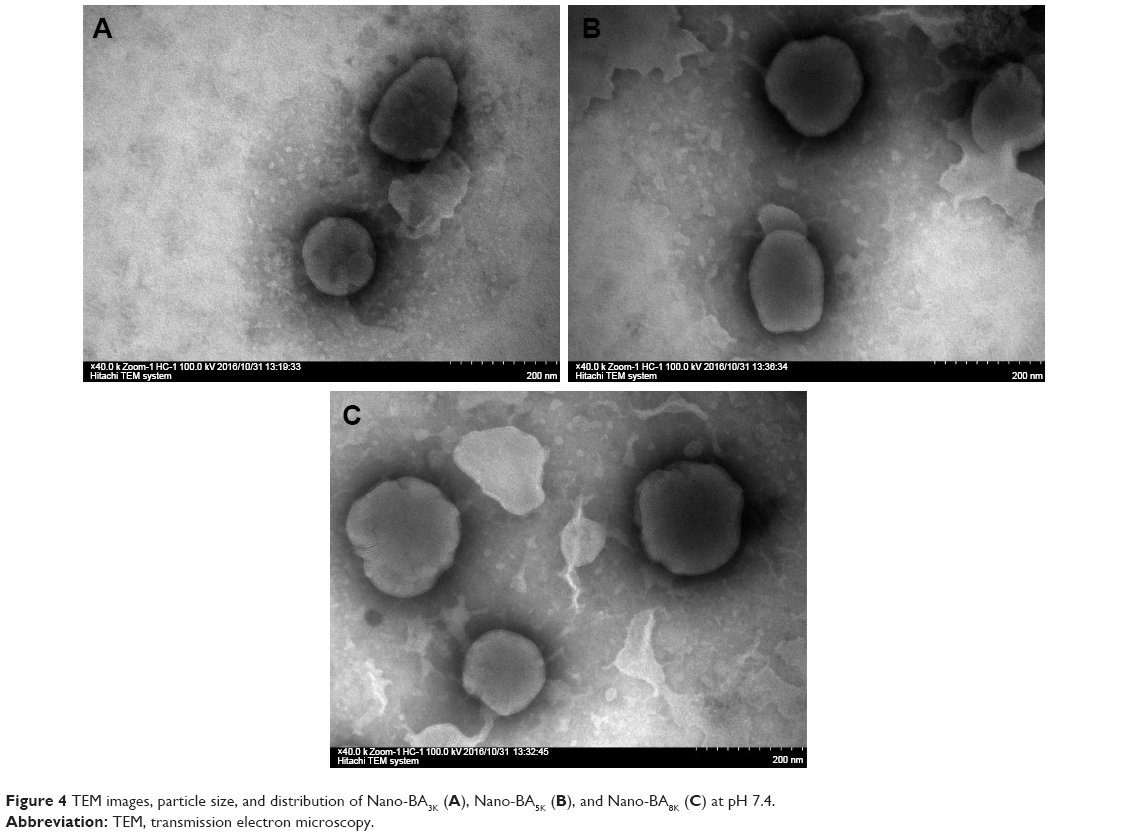 | Figure 4 TEM images, particle size, and distribution of Nano-BA3K (A), Nano-BA5K (B), and Nano-BA8K (C) at pH 7.4. |
Antibacterial activities of the nano-BAs
The MICs of the nano-BAs were determined against both gram-negative and gram-positive bacteria. Owing to the poor solubility of Nano-BA8K in the broth, its antimicrobial activity was not determined. As shown in Table 3, BA solution showed antibacterial activities against only gram-positive bacteria – a result consistent with previous findings.6,23–26 Nano-BAs (Nano-BA3K and Nano-BA5K) had high effectiveness against both gram-positive and gram-negative bacteria. Their MICs were cell-type dependent and were close to or even below the CMC of the triblock copolymers. In addition, the MICs of nano-BAs were much lower than those of BA solution, and the distribution of antibacterial activity as a function of PLGA block length was skewed toward longer PLGA chains.
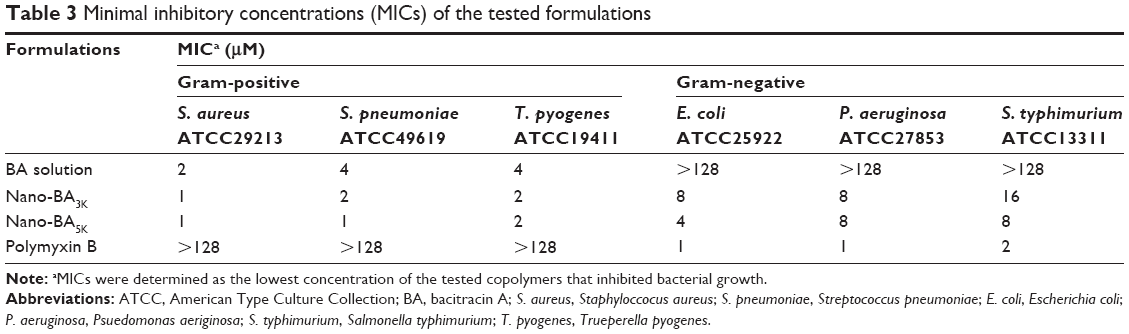 | Table 3 Minimal inhibitory concentrations (MICs) of the tested formulations |
To further test the ability of nano-BAs to kill bacteria, MBCs of the nano-BAs were also determined (Table 4). The results indicated that nano-BAs killed bacteria in a concentration-dependent manner, and at concentrations of 2 and 8 μM, Nano-BA5K effectively killed S. aureus or E. coli at 2 h of incubation, respectively. In summary, the activity of Nano-BA5K against gram-positive bacteria was much stronger than that of its unassembled BA counterpart, and it was effective against gram-negative bacteria – as is characteristic of broad-spectrum nanoparticles. However, the activity of Nano-BA5K against gram-negative bacteria was weaker than that of currently available antibiotics, such as polymyxin B.
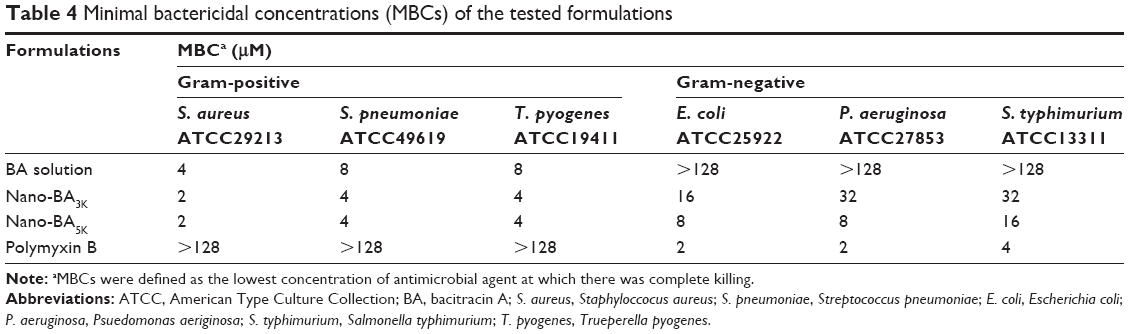 | Table 4 Minimal bactericidal concentrations (MBCs) of the tested formulations |
The time courses of antibacterial efficacy of nano-BAs against E. coli ATCC 25922 and S. aureus ATCC 29213 were also determined. For E. coli (Figure 5A), Nano-BA3K and Nano-BA5K were all bactericidal, showing a decrease in viable cell count after 2 h of treatment, whereas E. coli cells were completely killed after 4.5 and 4.0 h, respectively. Instead, BA solution showed no visible antibacterial efficacy against E. coli during the tested period. For S. aureus, nano-BAs were more efficient at killing bacteria than the BA solution – a finding consistent with the results of the MBC assay (Figure 5B).
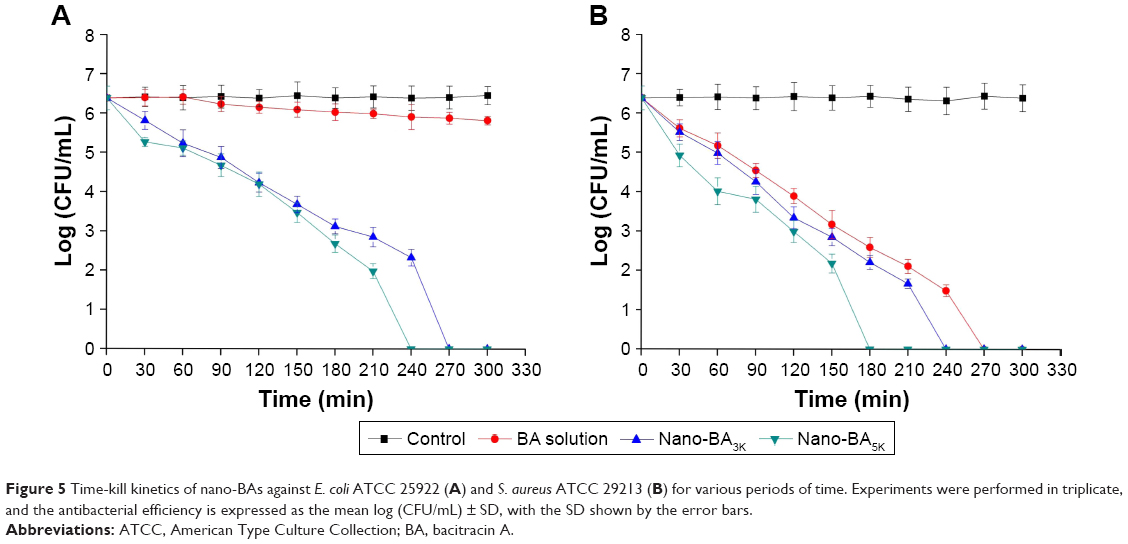 | Figure 5 Time-kill kinetics of nano-BAs against E. coli ATCC 25922 (A) and S. aureus ATCC 29213 (B) for various periods of time. Experiments were performed in triplicate, and the antibacterial efficiency is expressed as the mean log (CFU/mL) ± SD, with the SD shown by the error bars. |
Salt and serum sensitivity
To examine the influence of salt on their antibacterial activity, nano-BAs were also tested after the addition of physiological concentrations of different salts, because early studies have indicated that BA exhibits an interesting cation-dependent inhibition against bacteria.27 As indicated in Table 5, nano-BAs were highly potent against bacteria in the presence of divalent-(Mg2+ and Zn2+) cations. The presence of the mono-(Na+, K+, and NH4+) and trivalent (Fe3+) cations showed a limited effect on the anti-bacterial activity of nano-BAs. Furthermore, we treated the bacteria with increasing nano-BAs concentrations in the presence of human serum (20%). As shown in Table 5, the activity of nano-BAs remained unchanged or slightly decreased under serum conditions, thus indicating that all of the nano-BAs resisted serum challenges.
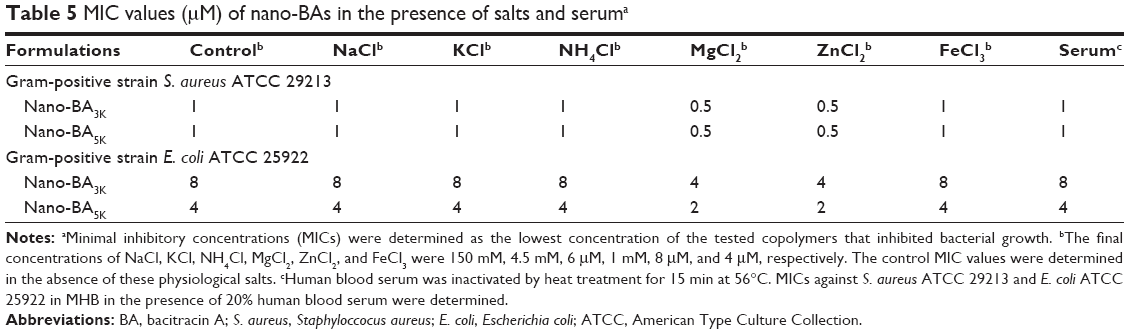 | Table 5 MIC values (μM) of nano-BAs in the presence of salts and seruma |
SEM and TEM observation
SEM observation
On the basis of the strongest antibacterial activity, the desirable stability and solubility, and the salt tolerance of the tested nano-BAs, Nano-BA5K was selected for SEM observation. We first investigated the morphological changes of E. coli before and after incubation with the Nano-BA5K at 2× MICs for 2-h incubation through SEM observations. Polymyxin B was selected as a positive control. SEM images of untreated E. coli exhibited a smooth surface (Figure 6A). E. coli treated with BA solution also showed a bright, smooth surface with no significant cell membrane-damaging effects observed during the incubation period (Figure 6B). In sharp contrast, treatment with Nano-BA5K induced a significant membrane-damaging effect. After 2 h of treatment with Nano-BA5K, significant rupture of the cell membrane was observed, and the protoplasm from the bacteria exhibited viscidity, which promoted aggregation (Figure 6C). Under the same treatment, the cell membrane of E. coli treated with polymyxin B was efficiently disrupted, with the appearance of membrane distortion, blisters, and breakage (Figure 6D).
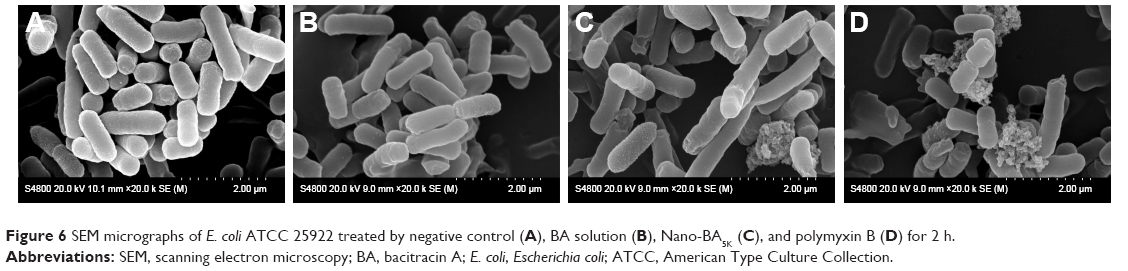 | Figure 6 SEM micrographs of E. coli ATCC 25922 treated by negative control (A), BA solution (B), Nano-BA5K (C), and polymyxin B (D) for 2 h. |
TEM observation
TEM was further used to study the morphological character and intracellular alternation of bacterial cells treated with Nano-BA5K. The results indicated that, in comparison to the negative control (Figure 7A) and BA solution (Figure 7B) – which resulted in bacteria with a smooth surface and dense internal structure – Nano-BA5K induced a significant decrease in the integrity of the E. coli membrane. After 2 h of treatment, the cytoplasmic membrane began to rupture and the cellular contents began to be released, but the cell morphology was still recognizable (Figure 7C). For polymyxin B, after 2-h incubation, some cells became ghost cells with a large amount of leakage of intracellular contents (Figure 7D).
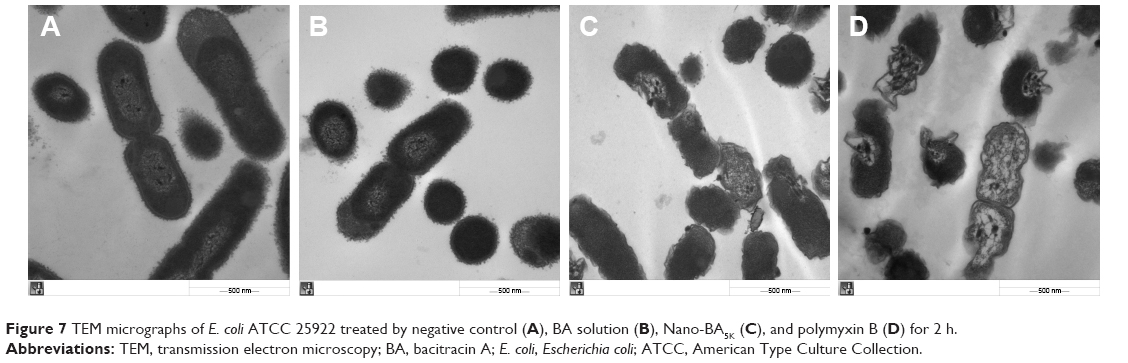 | Figure 7 TEM micrographs of E. coli ATCC 25922 treated by negative control (A), BA solution (B), Nano-BA5K (C), and polymyxin B (D) for 2 h. |
In vitro cytotoxicity assay against HK-2 cells and hemolytic activity
BA is not administered systemically because it is nephrotoxic, and it is used only as a last resort. Thus, the in vitro cytotoxicity of nano-BAs (Nano-BA3K and Nano-BA5K) were evaluated on human HK-2 tubular epithelial cells (HK-2 cells) by using the MTT assay (Figure 8A). Compared with BA solution (IC50: 4.22±0.68 μM), all nano-BAs exhibited higher IC50 values (Nano-BA3K: 26.13±1.22 μM, Nano-BA5K: 27.24±1.56 μM) against HK-2 cells. Furthermore, all nano-BAs exhibited low hemolytic activities (<20%), even at the highest concentration of 256 μM, that was much higher than their MICs. However, polymyxin B mediated more than 90% hemolysis at its MIC. These results indicated that all nano-BAs have greater potential for applications in the host than the currently available antibiotics, such as polymyxin B (Figure 8B).
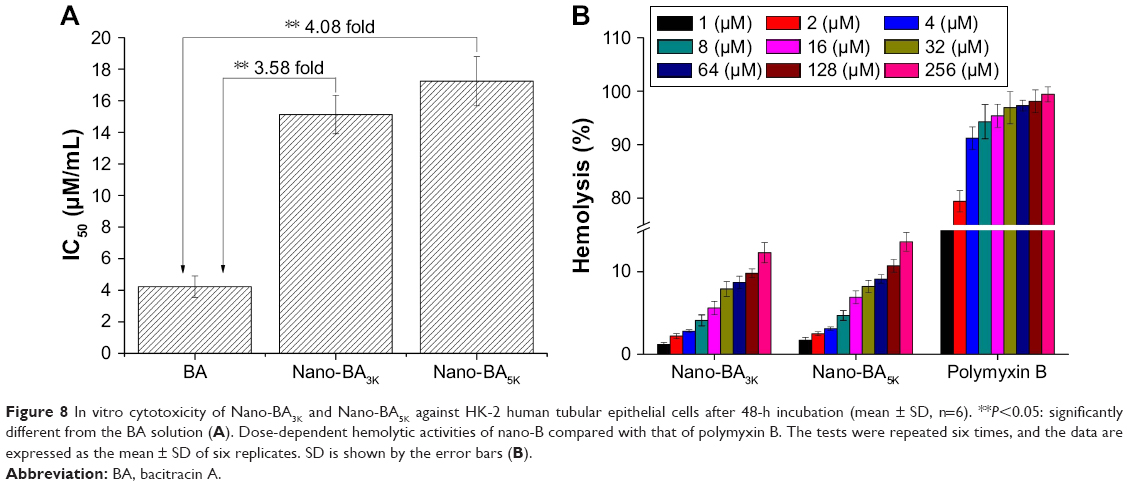 | Figure 8 In vitro cytotoxicity of Nano-BA3K and Nano-BA5K against HK-2 human tubular epithelial cells after 48-h incubation (mean ± SD, n=6). **P<0.05: significantly different from the BA solution (A). Dose-dependent hemolytic activities of nano-B compared with that of polymyxin B. The tests were repeated six times, and the data are expressed as the mean ± SD of six replicates. SD is shown by the error bars (B). |
Wound closure enhanced by nano-BA5K
The percentage of wound closure was approximately 39%–48% on day 3 (Figure 9A) and 92%–100% on day 6 (Figure 9B) in the groups treated with 1× MIC Nano-BA5K. Epithelialization was almost complete by the sixth postoperative day. By comparison, the group treated with Polysporin® ointment exhibited only ~30% wound closure on day 3 (Figure 9C) and 65% on day 6 (Figure 9D). No wound contraction was observed when the wound was treated with saline on day 3 (Figure 9E), although scab formation occurred on the surface of the wound on day 6 and the wound size decreased (Figure 9F).
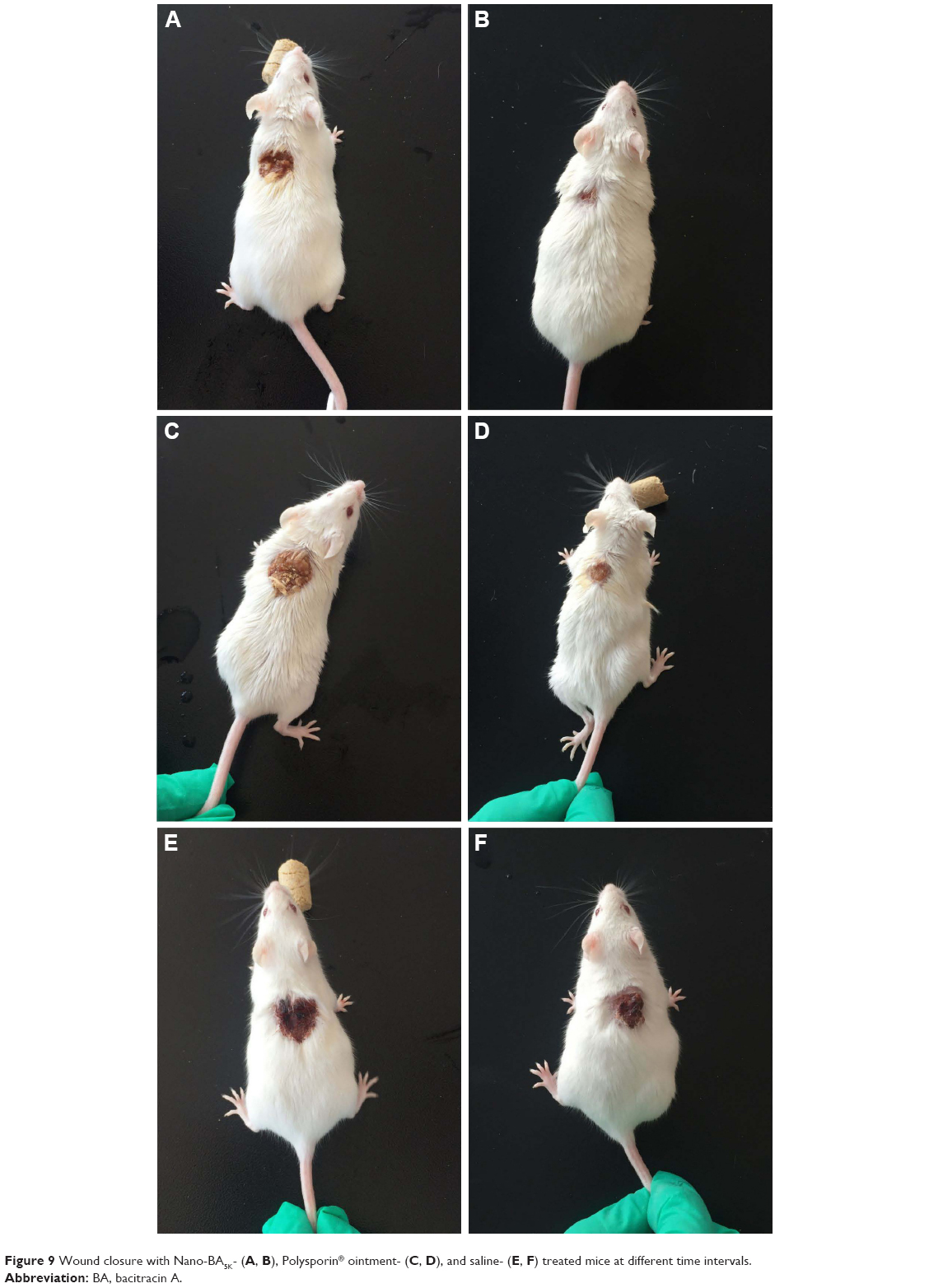 | Figure 9 Wound closure with Nano-BA5K- (A, B), Polysporin® ointment- (C, D), and saline- (E, F) treated mice at different time intervals. |
Furthermore, surface-wound bacterial counts were obtained to understand the dynamics of bacterial growth on the surface of the wound.28 As shown in Figure 10A, after treatment with 1× MIC Nano-BA5K, the bacterial count diminished by 69% on day 3 and by more than 96% on day 6. Similarly, bacterial counts diminished by 67% and 93% after treatment with Polysporin® ointment on days 3 and 6, respectively.
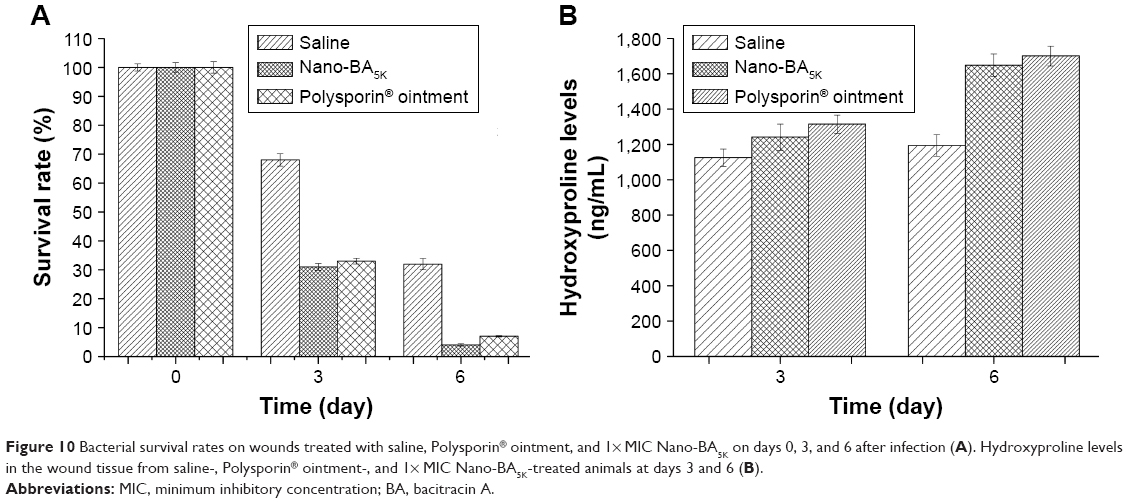 | Figure 10 Bacterial survival rates on wounds treated with saline, Polysporin® ointment, and 1× MIC Nano-BA5K on days 0, 3, and 6 after infection (A). Hydroxyproline levels in the wound tissue from saline-, Polysporin® ointment-, and 1× MIC Nano-BA5K-treated animals at days 3 and 6 (B). |
Collagen deposition is another important event in the development of granulation tissue. The formation of new collagen was confirmed by the detection of increased hydroxyproline levels in lesions at days 3 and 6 after wounding. The results indicated that hydroxyproline levels in the 1× MIC Nano-BA5K-treated group were higher than those in the Polysporin® ointment-treated group, and from days 3 to 6, the level increased by only 29% (Figure 10B). This result may suggest that the wound almost healed on day 3, in agreement with the previous wound closure observation.
Discussion
BA is an excellent polypeptide antibiotic, which is active against gram-positive bacterial strains and does not trigger multidrug resistance. However, BA is inactive against gram-negative bacteria because of its inability to cross the outer membrane of these cells. Previous studies have shown that self-assembling nanoantibiotics effectively localize antibiotics to the periplasmic space, thus allowing them to exert their bacterial activities against gram-negative bacteria.9,10,29 In an attempt to augment and hone this strategy, biodegradable hydrophobic copolymers of PLGA have been attached to the N-termini of BA to design a novel class of self-assembling nano-BAs. Compared with other compounds, such as fatty acids, PLGA has attracted considerable attention for its attractive characteristics: 1) biodegradability and biocompatibility, 2) US Food and Drug Administration and European Medicine Agency approval in drug delivery systems for parenteral administration, 3) well-described formulations and methods of production adapted to various types of drugs (eg, hydrophilic or hydrophobic small molecules or macromolecules), 4) ability to modify surface properties to provide stealth and/or better interaction with biological materials, and 5) ability to target nanoparticles to specific organs or cells. The starting copolymer of PLGA was synthesized in a ring-opening melting polymerization, and the final copolymer of BA–b–PLGA–b–BA was obtained in a coupling reaction. From the results of GPC, the increase in the average molecular weight from PLGA to BA–PLGA–BA was smaller than expected (Table 1). This discrepancy might have been because BA–PLGA–BA could be a physical mixture of BA–PLGA–BA, PLGA with one end coupled to BA, or PLGA. This result was also indicated by the small increase in the PDI of the triblock copolymers, as compared with the PLGA. During the synthesis of BA–PLGA–BA, an excess of CDI (the molar weight of CDI to PLGA was 10/1) was added to obtain a PLGA copolymer with both ends activated. Owing to high steric hindrance, the opportunity for the hydroxyl end groups of the D,L-lactide terminus to react with CDI was smaller than that for the hydroxyl end groups of the glycolide terminus. Moreover, because BA–PLGA–BA had a narrow molecular weight distribution, it was reasonable to assume that most of the copolymer had a triblock structure.
Nano-BAs were prepared by a thin-film hydration method. As shown in the TEM image, the dark region of the micellar image was probably attributable to the outer shell formed by the hydrophilic block of BA, whereas the bright region corresponded to the inner core formed by the hydrophobic block of PLGA.30 All nano-BAs exhibited similar slightly negative surface charges at pH 7.4, whereas the nearly net surface charge of the micelles was able to prevent the recognition of opsonin, thus leading to prolonged in vivo circulation.31 In addition, from the negative zeta-potential data, we can roughly identify the dominant component on the particle surface. Because PLGA is non-ionic, this negative surface charge indicated the presence of a BA layer on the surface of nano-BAs.
The antibacterial activities assay comprehensively revealed that nano-BAs were more effective against bacteria compared to BA, especially in the presence of divalent-(Mg2+ and Zn2+) cations. BA has been known to bind several divalent metal cations and to form 1:1 complexes. Thus, the physical concentrations of divalent-cations induce the high affinity of nano-BAs for the bacterial cell wall and lead to greater antibacterial activities. The results corroborate a proposed mechanism of action for BA in which the binding efficiency of metallobacitracin A to undecaprenyl pyrophosphate and the cell membrane can be enhanced by divalent-cations.
Impressively, it was worth noting that nano-BAs also showed antibacterial potency against gram-negative bacteria, whereas BA did not appear to have these activities. The electron microscopy observations revealed that gram-positive bacteria treated with Nano-BA5K were killed by disruption of cell membrane structures, which caused leakage of the intracellular contents, thereby leading to cell death. Thus, we presumed that the cell membrane might be the main target of Nano-BA5K against gram-negative bacteria. Previous studies have indicated that nanoantibiotics can efficiently travel across the outer membrane of gram-negative bacteria through endocytosis to cause steric hindrance.32 Therefore, Nano-BA5K might cross the outer membrane by endocytosis and enter into the periplasmic space of gram-negative bacteria. Moreover, Hancock has proposed a mechanism of peptide interaction with membranes, termed self-promoted uptake.33 According to this model, the initial action of the peptide involves a competitive displacement of LPS through binding to lipid A,34,35 which allows antibacterial drugs to gain access to outer membranes. It is possible that Nano-BA5K may also act in a similar way, because there may be hydrogen binding between the hydrophobic blocks (PLGA) of Nano-BA5K and the lipid A core of the LPSs of the cell membrane. These two factors may together facilitate Nano-BA5K transport across the outer cell membrane of gram-negative bacteria. After Nano-BA5K enters into the periplasmic space of gram-negative bacteria, it can damage the cytoplasmic membrane of the bacteria, owing to high local density of BA mass on the surface and a relatively large volume of micelles, thereby resulting in osmotic lysis of the cells. Collectively, nano-BA may predominantly bind LPS and consequently traverse through the lipid core of LPS and enter into the periplasmic space of bacteria cells. In addition, Nano-BA5K may then insert into the target cytoplasmic membrane, owing to the high local density of BA mass on the surface, thus eventually resulting in cell lysis. Thus, we presumed that the enhanced antibacterial activities of Nano-BA5K against gram-negative bacteria may stem from two aspects: 1) high outer membrane permeability and 2) high local density of BA mass on the surface.
Moreover, nano-BAs showed lower cytotoxicity than BA against HK-2 cells, probably because of the different uptake mechanisms between micelles and the drug solution. The quick, energy-independent passive diffusion of BA solution resulted in a high drug concentration in HK-2 cells, which may lead to high cytotoxicity. In contrast, the lower inhibition performance of nano-BAs against HK-2 cells might be due to the saturated and limited micelle cellular uptake capability of HK-2 cells. Such characteristics of cellular uptake of micelles have also been observed by others.31 Nano-BAs exhibited strong antibacterial activities against gram-negative bacteria by acting on the target cell membrane, which might decrease their biocompatibility in normal mammalian cells, especially hRBCs. Thus, the toxicity of nano-BAs against eukaryotic cells was measured as a function of their concentrations in the range of 1–256 μM. All of the tested nano-BAs showed lower cytotoxicity against hRBCs.
In addition to in vitro experiments, we conducted in vivo experiments to evaluate the efficacy of Nano-BA5K. We developed an infection model for both gram-positive and gram-negative bacteria that are encountered in wound infections. The selection of these test bacteria is appropriate because they are causative agents of open wound infections in humans. The surface wound bacterial counts increased after the wound was infected by the bacteria; however, after treatment with the Nano-BA5K, the overall bacterial counts decreased to the levels present before surgery and, essentially, no obvious changes were observed in the physiology of the saline-treated wound. The results indicated that the wound-healing effect of Nano-BA5K is significantly greater than that of Polysporin® ointment, according to bacterial counts and hydroproline content. However, the actual reason for the increased rate of wound infection closure in Nano-BA5K-treated wounds is not fully understood and requires further study.
Conclusion
In summary, a newly designed series of self-assembled, core-shell structured nano-BAs were constructed by modification of BA with PLGA. We demonstrated that the formation of nano-BAs strongly enhances antibacterial activities compared to unassembled BA. These nano-BAs possess a broad spectrum of antibacterial activities that efficiently inhibit the growth of various types of gram-positive and gram-negative bacteria with low MIC and MBC values, yet induce low hemolysis and cytotoxicity. Nano-BAs might cross the outer membrane of E. coli through binding to LPS and/or endocytosis, and then disrupt the cytoplasmic membrane, owing to the high local density of BA mass on the surface, thus ultimately leading to cell death. The effects of Nano-BA5K on wound healing in mice are more significant than those of the commonly used commercial product Polysporin® ointment. In this study, all of the data we presented should aid in the design of a new group of self-assembling nano-polypeptide antibiotic candidates, and Nano-BA5K could be a potent topical anti-infective agent for wound treatment.
Acknowledgments
The authors are grateful for the financial support from the National Science Foundation for Young Scientists of China (Grant No 31602108), the PhD Start-up Fund of Natural Science Foundation of Liaoning Province (Grant No 201601101), and the Key Laboratory of Zoonosis of Liaoning Province.
Disclosure
The authors report no conflicts of interest in this work.
References
Meleney FL, Johnson BA. Bacitracin. Am J Med. 1949;7(6):794–806. | ||
Johnson BA, Anker H, Meleney FL. Bacitracin: a new antibiotic produced by a member of the B. Subtilis Group. Science. 1945;102(2650):376–377. | ||
Chapnick EK, Gradon JD, Kreiswirth B, et al. Comparative killing kinetics of methicillin-resistant Staphylococcus aureus by bacitracin or mupirocin. Infect Control Hosp Epidemiol. 1996;17(3):178–180. | ||
Tsuji K, Robertson JH. Improved high-performance liquid chromatographic method for polypeptide antibiotics and its application to study the effects of treatments to reduce microbial levels in bacitracin powder. J Chromatogr. 1975;112:663–672. | ||
Ming LJ, Epperson JD. Metal binding and structure-activity relationship of the metalloantibiotic peptide bacitracin. J Inorg Biochem. 2002;91(1):46–58. | ||
Ciesiołka J, Jeżowska-Bojczuk M, Wrzesiński J, et al. Antibiotic bacitracin induces hydrolytic degradation of nucleic acids. Biochim Biophys Acta. 2014;1840(6):1782–1789. | ||
Joy SR, Li X, Snow DD, Gilley JE, Woodbury B, Bartelt-Hunt SL. Fate of antimicrobials and antimicrobial resistance genes in simulated swine manure storage. Sci Total Environ. 2014;481:69–74. | ||
Thevenard B, Besset C, Choinard S, et al. Response of S. thermophilus LMD-9 to bacitracin: involvement of a BceRS/AB-like module and of the rhamnose-glucose polysaccharide synthesis pathway. Int J Food Microbiol. 2014;177:89–97. | ||
Tamaki M, Fujinuma K, Harada T, et al. Fatty acyl-gramicidin S derivatives with both high antibiotic activity and low hemolytic activity. Bioorg Med Chem Lett. 2012;22(1):106–109. | ||
Avrahami D, Shai Y. Conjugation of a magainin analogue with lipophilic acids controls hydrophobicity, solution assembly, and cell selectivity. Biochemistry. 2002;41(7):2254–2263. | ||
Malina A, Shai Y. Conjugation of fatty acids with different lengths modulates the antibacterial and antifungal activity of a cationic biologically inactive peptide. Biochem J. 2005;390(Pt 3):695–702. | ||
Avrahami D, Shai Y. A new group of antifungal and antibacterial lipopeptides derived from non-membrane active peptides conjugated to palmitic acid. J Biol Chem. 2004;279(13):12277–12285. | ||
Li Q, Mahendra S, Lyon DY, et al. Antimicrobial nanomaterials for water disinfection and microbial control: potential applications and implications. Water Res. 2008;42(18):4591–4602. | ||
Mansour HM, Rhee YS, Wu X. Nanomedicine in pulmonary delivery. Int J Nanomedicine. 2009;4:299–319. | ||
Santos-Magalhães NS, Mosqueira VC. Nanotechnology applied to the treatment of malaria. Adv Drug Deliv Rev. 2010;62(4–5):560–575. | ||
Sosnik A, Carcaboso AM, Glisoni RJ, Moretton MA, Chiappetta DA. New old challenges in tuberculosis: potentially effective nanotechnologies in drug delivery. Adv Drug Deliv Rev. 2010;62(4–5):547–559. | ||
Liu L, Xu K, Wang H, et al. Self-assembled cationic peptide nanoparticles as an efficient antimicrobial agent. Nat Nanotechnol. 2009;4(7):457–463. | ||
Wang H, Xu K, Liu L, et al. The efficacy of self-assembled cationic antimicrobial peptide nanoparticles against Cryptococcus neoformans for the treatment of meningitis. Biomaterials. 2010;31(10): 2874–2881. | ||
Danhier F, Ansorena E, Silva JM, Coco R, Le Breton A, Préat V. PLGA-based nanoparticles: an overview of biomedical applications. J Control Release. 2012;161(2):505–522. | ||
Maiti S, Chatterji PR, Nisha CK, Manorama SV, Aswal VK, Goyal PS. Aggregation and polymerization of PEG-based macromonomers with methacryloyl group as the only hydrophobic segment. J Colloid Interface Sci. 2001;240(2):630–635. | ||
Ma QQ, Dong N, Shan AS, et al. Biochemical property and membrane-peptide interactions of de novo antimicrobial peptides designed by helix-forming units. Amino Acids. 2012;43(6):2527–2536. | ||
Dong N, Ma Q, Shan A, et al. Strand length-dependent antimicrobial activity and membrane-active mechanism of arginine- and valine-rich β-hairpin-like antimicrobial peptides. Antimicrob Agents Chemother. 2012;56(6):2994–3003. | ||
Mandal SM, Roy A, Mahata D, Migliolo L, Nolasco DO, Franco OL. Functional and structural insights on self-assembled nanofiber-based novel antibacterial ointment from antimicrobial peptides, bacitracin and gramicidin S. J Antibiot (Tokyo). 2014;67(11):771–775. | ||
Ikai Y, Oka H, Hayakawa J, et al. Total structures and antimicrobial activity of bacitracin minor components. J Antibiot (Tokyo). 1995;48(3):233–242. | ||
Yilmaz ER, Gurer B, Kertmen H, et al. The histopathological and ultrastructural effects of the topical application of bacitracin on the cerebral cortex in rats. Turk Neurosurg. 2015;25(1):78–84. | ||
Nie B, Ao H, Zhou J, Tang T, Yue B. Biofunctionalization of titanium with bacitracin immobilization shows potential for anti-bacteria, osteogenesis and reduction of macrophage inflammation. Colloids Surf B Biointerfaces. 2016;145:728–739. | ||
Toscano WA Jr, Storm DR. Bacitracin. Pharmacol Ther. 1982;16(2):199–210. | ||
Huang HN, Rajanbabu V, Pan CY, Chan YL, Wu CJ, Chen JY. Use of the antimicrobial peptide Epinecidin-1 to protect against MRSA infection in mice with skin injuries. Biomaterials. 2013;34(38):10319–10327. | ||
Tamaki M, Harada T, Fujinuma K, et al. Polycationic gramicidin S analogues with both high antibiotic activity and very low hemolytic activity. Chem Pharm Bull (Tokyo). 2012;60(9):1134–1138. | ||
Lo CL, Huang CK, Lin KM, Hsiue GH. Mixed micelles formed from graft and diblock copolymers for application in intracellular drug delivery. Biomaterials. 2007;28(6):1225–1235. | ||
Liu D, Liu F, Song YK. Recognition and clearance of liposomes containing phosphatidylserine are mediated by serum opsonin. Biochim Biophys Acta. 1995;1235(1):140–146. | ||
Düzgüne N, Nir S. Mechanisms and kinetics of liposome-cell interactions. Adv Drug Deliv Rev. 1999;40(1–2):3–18. | ||
Hancock RE. Peptide antibiotics. Lancet. 1997;349(9049):418–422. | ||
Meincken M, Holroyd DL, Rautenbach M. Atomic force microscopy study of the effect of antimicrobial peptides on the cell envelope of Escherichia coli. Antimicrob Agents Chemother. 2005;49(10):4085–4092. | ||
Glukhov E, Stark M, Burrows LL, Deber CM. Basis for selectivity of cationic antimicrobial peptides for bacterial versus mammalian membranes. J Biol Chem. 2005;280(40):33960–33967. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.