Back to Journals » International Journal of Nanomedicine » Volume 12
Supersaturable solid self-microemulsifying drug delivery system: precipitation inhibition and bioavailability enhancement
Authors Quan G ![]() , Niu B, Singh V
, Niu B, Singh V ![]() , Zhou Y, Wu C, Pan X, Wu C
, Zhou Y, Wu C, Pan X, Wu C
Received 22 August 2017
Accepted for publication 24 October 2017
Published 13 December 2017 Volume 2017:12 Pages 8801—8811
DOI https://doi.org/10.2147/IJN.S149717
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Linlin Sun
Guilan Quan,1 Boyi Niu,1 Vikramjeet Singh,1 Yixian Zhou,1 Chuan-Yu Wu,2 Xin Pan,1 Chuanbin Wu1
1School of Pharmaceutical Sciences, Sun Yat-sen University, Guangzhou, China; 2Department of Chemical and Process Engineering, University of Surrey, Guildford, UK
Abstract: Solid self-emulsifying drug delivery system (SSEDDS), which incorporates liquid SEDDS into a solid dosage form, has been recently introduced to improve the oral bioavailability of poorly water-soluble drugs. However, supersaturated drug generated by SSEDDS is thermodynamically unstable and tends to precipitate rapidly prior to absorption, resulting in compromised bioavailability. The aim of this study was to construct a novel supersaturated SSEDDS (super-SSEDDS) by combining SSEDDS with appropriate precipitation inhibitor. Fenofibrate (FNB), a sparingly soluble drug, was selected as a model drug in this study. An optimized SSEDDS was prepared by solvent evaporation by using mesoporous silica Santa Barbara Amorphous-15 as the inert carrier. Supersaturation assay was conducted to evaluate the precipitation inhibition capacity of different polymers, and the results showed that Soluplus® could retard the FNB precipitation more effectively and sustain a higher apparent concentration for ~120 min. This effect was also clearly observed in the dissolution profiles of FNB from SSEDDS under supersaturated condition. The study of the mechanism suggested that the inhibition effect might be achieved both thermodynamically and kinetically. The area under the concentration–time curve of the super-SSEDDS was 1.4-fold greater than that of SSEDDS in the absence of Soluplus, based on an in vivo pharmacokinetic study conducted in beagle dogs. This study has demonstrated that the approach of combining SSEDDS with Soluplus as a supersaturation stabilizer constitutes a potential tool to improve the absorption of poorly water-soluble drugs.
Keywords: Soluplus, supersaturation, fenofibrate, mesoporous silica, poorly water-soluble drugs
Introduction
In drug discovery, almost 70% of new drug candidates exhibit low aqueous solubility which ultimately results in poor absorption.1,2 A number of alternative techniques including cyclodextrin complexation,3 particle size reduction,4 solid dispersions,5 and lipid-based formulations6 have been developed to overcome this problem.
Among all the formulations, lipid-based formulations, especially self-emulsifying drug delivery systems (SEDDS) have received much attention.7 Recently, solid SEDDS (SSEDDS), prepared by incorporating the conventional liquid SEDDS preconcentrate into powder or particles, has been introduced to enhance the oral bioavailability of sparingly soluble drugs. It combines the advantages of liquid SEDDS (improved solubility and bioavailability) with those of solid dosage forms (storage stability and patient compliance).8,9 SSEDDS are supposed to maintain the self-emulsifying ability and capable of forming fine oil-in-water emulsions under gentle agitation provided by the gastrointestinal (GI) motion.10 The drug presented in a dissolved form with a huge interfacial area for absorption provided by emulsion droplets results in an enhanced bioavailability.11,12 In our previous study, solid self-emulsifying matrix combined with mesoporous silica was successfully prepared. When introduced to aqueous media under gentle agitation, the solid matrix exhibited excellent self-emulsifying properties of forming a uniform microemulsion.13
Nevertheless, the lipid-based formulation is limited by loss of solubilization capacity due to the digestion in GI tract. A supersaturated solution exceeding the equilibrium solubility will be generated after administration. Theoretically, drugs at high concentration have an increased driving force for flux across the GI membrane, and enhanced absorption could be achieved with sufficient time period.14 However, supersaturated drug with higher chemical potential tends to precipitate rapidly into an energetically favorable crystalline form prior to the absorption, resulting in compromised bioavailability.15 Therefore, ideal formulations should be able to maintain the supersaturated state for appropriate time period to allow better absorption.
A variety of pharmaceutical polymers have been widely explored to generate and maintain a supersaturated state of drugs for an extended period.16 These polymers have been confirmed to kinetically or thermodynamically inhibit the precipitation by retardation in drug nucleation and crystal growth or changing the properties of aqueous medium.17 Studies of several poorly water-soluble drugs, such as silybin,18 celecoxib,19 AMG 517,20 and halofantrine hydrochloride,21 clearly demonstrated that the SEDDS formulations with polymeric precipitation inhibitors can result in better oral bioavailability when compared to the conventional SEDDS systems.
Soluplus (polyvinyl caprolactam–polyvinyl acetate–polyethylene glycol graft copolymer) is a graft amphiphilic polymer introduced by Badische Anilin-und-Soda-Fabrik (BASF) to produce solid dispersions by hot-melt extrusion method.22 It shows excellent solubilization properties for poorly water-soluble drugs and subsequently increases the dissolution and oral absorption.23 In this regard, Soluplus could be a promising supersaturation stabilizer for SSEDDS. Therefore, the aim of this study was to construct a supersaturated SSEDDS (super-SSEDDS) to improve the oral absorption of fenofibrate (FNB). Based on our previous report,13 SEDDS composed of ethyl oleate (EO) as oil, Cremophor RH40 as surfactant, and Transcutol® HP as cosurfactant was selected to solubilize FNB. Mesoporous silica was used as the solid reservoir due to its unique properties including huge surface area and well-ordered porous interior. Herein, a super-SSEDDS system of FNB was formulated by screening various kinds of hydrophilic polymers and amphiphilic copolymers including Soluplus as a precipitation inhibitor. The mechanism of precipitation inhibition was thermodynamically and kinetically investigated. In vitro dissolution profile under non-sink condition was applied to characterize the performance of the super-SSEDDS. The in vivo oral bioavailability study was also performed in beagle dogs to determine the advantage of super-SSEDDS in oral absorption enhancement of FNB.
Materials and methods
Materials
FNB was obtained from Kaifeng Pharmaceutical Factory (Henan, China). Indomethacin was used as the internal standard and purchased from Wuhan DKY Technology Co., Ltd (Wuhan, China). Transcutol HP was provided by Gattefosse (Saint-Priest Cedex, France). Cremophor RH40, polyvinylpyrrolidone Kollidon 30 (PVP K30), polyvinylpyrrolidone Kollidon 90 (PVP K90), vinylprrolidone–vinylacetate copolymer Kollidon VA64 (PVP VA64), and Soluplus were kindly donated by BASF (Ludwigshafen, Germany). Hydroxypropyl methyl cellulose E3 (HPMC E3), HPMC E5, and HPMC E15 were supplied by Dow Chemical Company (Midland, TX, USA). EO was purchased from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). All other chemicals and solvents were of reagent grade and used without further purification.
Preparation of SSEDDS matrix
The solid self-emulsifying formulation was prepared according to our previous report. Briefly, the formulation amount of FNB (15% w/w) was dissolved in the optimized liquid SEDDS consisting of 30% EO, 30% Cremophor RH40, and 40% Transcutol HP. The resulting mixture was vortexed until a clear solution was obtained. Subsequently, liquid SEDDS was dissolved in 10 mL of ethanol followed by addition of solid carrier, and then the mixture was continuously stirred at room temperature for 24 h to reach the absorption equilibrium. Finally, ethanol was evaporated at 45°C for 48 h and the remaining matrix powder was collected. Santa Barbara Amorphous-15 (SBA-15) mesoporous silica and Aerosil® 200 were used as solid absorbents. The fixed amounts of absorbent to liquid SEDDS in ratios of 1:1, 1:2, 1:3, and 1:4 (wt %) were adopted to evaluate the absorption capacity.
Characterization of SSEDDS matrix
The outer macroscopic structures of FNB powder and SSEDDS matrix were observed by scanning electron microscopy (JSM-6330F; JEOL, Tokyo, Japan). The physical state of samples was examined by X-ray diffraction (Bruker/D2 PHASER; Bruker, Karlsruhe, Germany) using Cu Kα radiation with the scanning rate of 5°/min in the range of 5°–40°. The nitrogen absorption–desorption isotherms were obtained at −196°C by using a surface area and pore size analyzer (ASAP 2020C; Micromeritics, Atlanta, GA, USA), and the surface area, pore volume, and pore diameter distribution of samples were calculated according to the isotherms.
Reconstitution test
The SSEDDS system should be able to redisperse quickly into emulsion when introduced to aqueous media under mild agitation. Samples of SSEDDS (0.4 g) and preconcentrated liquid SEDDS (0.3 g) were dispersed in 200 mL water with magnetic stirring (50 rpm). In order to minimize the interference of insoluble mesoporous silica particles, the resulting mixture was allowed to stand at ambient temperature for 12 h. Then the appearance of the supernatant was observed in the presence of a red laser light passageway. The size distribution and polydispersity index (PDI) of the reconstituted emulsions were measured at 25°C using Zetasizer Nano ZS90 (Malvern Instruments, Worcestershire, UK).
Supersaturation assay
A solvent-shift method was employed to generate FNB supersaturation.24 The apparent drug concentration–time profile and the duration of the supersaturated state were determined using a USP II paddle method with a ZRS-8G dissolution tester (TDTF, Tianjing, China). A total medium volume of 200 mL with or without 0.05% (w/w) predissolved polymer was maintained at 37°C±0.5°C and the stirring speed was fixed at 100 rpm. The polymers used to stabilize the supersaturation were HPMC E3, HPMC E5, HPMC E15, PVP K30, PVP K90, PVP VA 64, and Soluplus. After adding 200 μL of FNB stock solution (100 mg/mL) in dimethylsulfoxide to the test medium, 2 mL of samples were withdrawn without volume replacement at 10, 30, 60, 90, and 120 min and filtered through a 0.22 μm filter. All filters were prerinsed with 5 mL of a saturated solution of FNB in order to prevent adsorption onto the filter. The resulting filtrate was diluted appropriately in absolute ethanol and assayed by high-performance liquid chromatography (HPLC LC-20AT; Shimadzu, Kyoto, Japan) at a wavelength of 286 nm. The area under the concentration–time curve in supersaturated state (AUCss) was computed according to the linear trapezoidal rule. Each dissolution test was performed in triplicate.
In vitro dissolution test under supersaturation condition for the SSEDDS
The apparent drug concentration–time profiles of SSEDDS formulations were determined using a USP II paddle method (100 rpm, 37°C±0.5°C, 200 mL dissolution medium) with a ZRS-8G dissolution tester. The powdered Soluplus was mixed with solid self-emulsifying matrix at various polymer/drug ratios, ranging from 1/0.2 to 1/10 (w/w). The mixed samples containing equivalent amounts of FNB (100 mg) were added to each vessel. Two milliliters of aliquot samples were taken at predetermined time intervals (10, 30, 60, 90, and 120 min). The samples were filtered through a 0.22 μm polytetrafluoroethylene syringe filter, and the first 1 mL of filtrate was discarded. Then 0.5 mL of filtrate was immediately diluted in 10 mL of absolute ethanol to prevent the precipitation, and the concentration of FNB in samples was measured by HPLC. Each dissolution test was performed in triplicate.
Solubility study
The solubility of FNB in deionized water containing various Soluplus concentrations (ranging from 0.00% to 0.50%) was measured to evaluate the mechanism of precipitation inhibition. Briefly, an excess amount of FNB was added to each capped vial containing 2 mL of Soluplus solution and mixed by vortexing. Then the mixture was kept at 37°C for 72 h in a shaking water bath to facilitate the equilibrium. The equilibrated sample was centrifuged at 10,000 rpm for 5 min to remove the undissolved drug. The supernatant was taken and diluted with methanol for quantification of FNB by HPLC system (Daojing, Japan) at 286 nm.
Cytotoxicity on Caco-2 cells
Human colorectal carcinoma Caco-2 cell line was obtained from American Type Culture Collection. The cells were cultured in DMEM supplemented with 10% fetal bovine serum, 1% L-glutamine, 1% nonessential amino acids, and 100 U/mL of penicillin–streptomycin. The culture was maintained in a humidified incubator containing 5% CO2 at 37°C. The growth medium was changed every 2 days prior to experimental use.
The cytotoxicity of liquid SEDDS formulation, mesoporous silica SBA-15, and Soluplus was evaluated in Caco-2 cells following the standard MTT viability assay. Cells were seeded in 96-well plates at a density of 5×103 cells/well and incubated for 48 h in 5% CO2 at 37°C till adherent growth. Subsequently, the medium was replaced with 200 μL of fresh medium containing different concentrations of test samples. After incubation with the samples for 48 h, the medium was replaced with 180 μL of fresh medium and 20 μL of MTT (5 mg/mL). After another 4 h of incubation at 37°C, the medium was rejected and 150 μL of dimethylsulfoxide was added in each cell to dissolve the purple formazan crystals. Finally, the optical density value of each well was measured at 490 nm by an ELX 800 micro-plate reader. Cells treated with pure medium were used as the blank control. The cell viability for each test group vs blank control group was calculated based on six independent wells.
In vivo pharmacokinetic study
The in vivo pharmacokinetic study of SSEDDS (solid self-emulsifying matrix) and optimized super-SSEDDS was conducted in beagle dogs. All the experimental procedures were approved by the Institutional Animal Care and Use Committee of Sun Yat-sen University in accordance with the National Institute of Health and Nutrition Guidelines for the care and use of laboratory animals. The study was conducted in six healthy beagle dogs (10–12 kg) in a nonrandomized cross-over manner, with a washout period of 2 weeks between each treatment. The animals were fasted for 12 h prior to the experiment, but allowed free access to water. Hard capsules containing the test samples equivalent to a dose of 100 mg FNB were administered orally, together with 20 mL of water. Water and food were returned at 2 and 4 h postdosing, respectively. Blood samples (5 mL) were withdrawn from the cephalic vein of the hind leg into heparinized tubes before administration and at 0.25, 0.50, 0.75, 1, 1.5, 2, 3, 4, 6, 8, and 24 h postdosing. The plasma was harvested immediately by centrifugation of the blood samples at 5,000 rpm for 10 min and then kept frozen at −20°C until further analysis was conducted by the validated HPLC method.
Quantitative analysis of FNB in plasma
A Daojin series HPLC system (LC-20AT pump, DGU-20A5R system controller, and SPD-20A ultraviolet–visible detector; Daojin, Japan) and Phenomenex C18 column (4.6×250 mm, 5 μm) with guard column were used. The mobile phase was composed of acetonitrile and 0.1% acetic acid (65:35, v/v) with a flow rate of 1.0 mL/min, and the run time was 30 min. The column working temperature was set to 40°C and the ultraviolet detection was performed at 286 nm. Indomethacin was used as the internal standard.
The samples for HPLC analysis were prepared by liquid–liquid extraction. Briefly, 50 μL of methanol solution of the internal standard (50 μg/mL) was added to 500 μL of plasma, followed by 200 μL of HCl solution (1 mol/L). After vortex-mixing for 1 min, 4 mL of diethyl ether was added and the mixture was vortexed for another 5 min. After centrifuging at 8,000 rpm for 5 min, the organic layer was transferred to a microtube and evaporated under a stream of nitrogen at 40°C. Subsequently, the organic layer residue was reconstituted in 200 μL of mobile phase and vortexed for 5 min. Finally, the resulting mixture was centrifuged at 15,000 rpm for 5 min. Then 50 μL of the supernatant was injected into HPLC for analysis.
Pharmacokinetic analysis and statistical analysis
The pharmacokinetic parameters were obtained by noncompartmental analysis using WinNonlin V. 3.3 software (Pharsight, Sunnyvale, CA, USA). The maximum plasma concentration (Cmax) and the time to reach Cmax (Tmax) were directly obtained from the plasma concentration–time curve. The AUC was estimated according to the linear trapezoidal rule.
All the data were expressed as the mean ± SD. The statistical analysis of the mean pharmacokinetic parameters was performed using the Student’s t-test following normality and equal variance tests (SPSS 13.0). Also, the difference was considered to be statistically significant if the probability value was <0.05 (P<0.05).
Results and discussion
Preparation of SSEDDS matrix
Lipid SEDDS formulations could be transformed into solid dosage forms by using suitable absorbents. However, the reduced drug loading capacity in the final products is the main disadvantage of this formulation technique. The dilution of the liquid SEDDS during mixing with the solid absorbents and further dilution by addition of excipients to obtain different solid dosage forms is mainly responsible for reduced drug loading capacity.25 Therefore, the ideal solid absorbents which possess the ability to hold considerable amount of liquid formulation are essential in the development of a desirable SSEDDS.
In our previous study,13 a novel SSEDDS matrix of FNB was originally designed by using well-ordered 2-D hexagonal mesoporous silica SBA-15 as the inert solid absorbent. In this study, the absorption capacity of Aerosil 200, a colloidal silica, which has been adopted as one of the most commonly used solid carriers in previous studies,8,26–28 was also measured. When the ratio of Aerosil 200 to liquid SEDDS increased from 1:1 to 1:2 (w/w), the powdered solid samples were changed to sticky aggregates. However, the powder properties of the matrix were still obtained at a higher ratio of 1:3 (SBA-15:liquid SEDDS), which confirmed the ability of SBA-15 to hold a larger amount of liquid SEDDS than conventional silica Aerosil 200. These results were consistent with the nitrogen adsorption–desorption measurements. As shown in Figure 1A, SBA-15 exhibited typical type-IV isotherms containing H1-type hysteresis, suggesting the highly regular mesoporous structure of SBA-15. Whereas, the nitrogen adsorption–desorption isotherms of Aerosil 200 were quite different (type-III) from those of SBA-15, suggesting the nonporous or macroporous solid characteristics. The mean surface area, pore volume, and pore size of SBA-15 were calculated as 595.01 m2/g, 1.02 cm3/g, and 9.19 nm, respectively. In contrast, the Brunauer-Emmett-Teller surface area of Aerosil 200 was only 155.93 m2/g, and no peak was recorded in the range of 2–50 nm (Figure 1B) for corresponding pore size distribution. These results suggested that the porous structure of SBA-15 played an important role in the solidification process. The mesoporous structure of SBA-15 was clearly observed by transmission electron microscopy; the straight lattice fringes at the parallel orientation (Figure 2A) and the well-ordered hexagonal arrays at the vertical orientation (the insert in Figure 2A) confirmed the existence of hexagonal arrays of mesochannels.
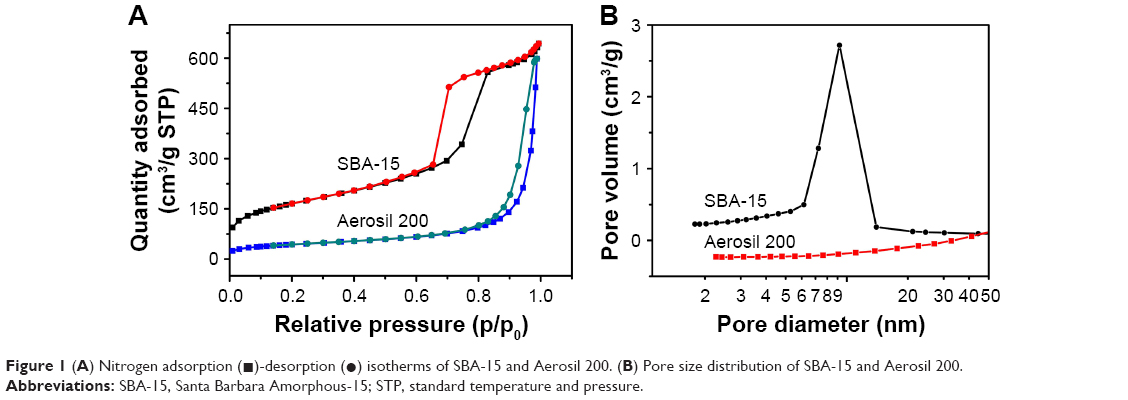 | Figure 1 (A) Nitrogen adsorption (◼)-desorption (•) isotherms of SBA-15 and Aerosil 200. (B) Pore size distribution of SBA-15 and Aerosil 200. |
 | Figure 2 Images of different samples. |
The solid self-emulsifying matrix was prepared by adopting a fixed ratio (1:3, w/w) of SBA-15 to liquid SEDDS. As shown in Figure 2B, the obtained product appeared as white powder. The powder was further investigated for surface morphology using scanning electron microscopy. The crystals of FNB were found to be irregular with a smooth surface (Figure 2C), while the SSEDDS appeared as rough-surfaced particles (Figure 2D). No crystals of FNB were observed in the SSEDDS matrix, indicating that the liquid SEDDS was completely absorbed by mesoporous silica SBA-15. Additionally, the X-ray diffraction results showed that the diffraction peaks representing FNB crystal completely disappeared in the SSEDDS matrix, confirming that FNB in the matrix was in an amorphous or molecular state (Figure 3).
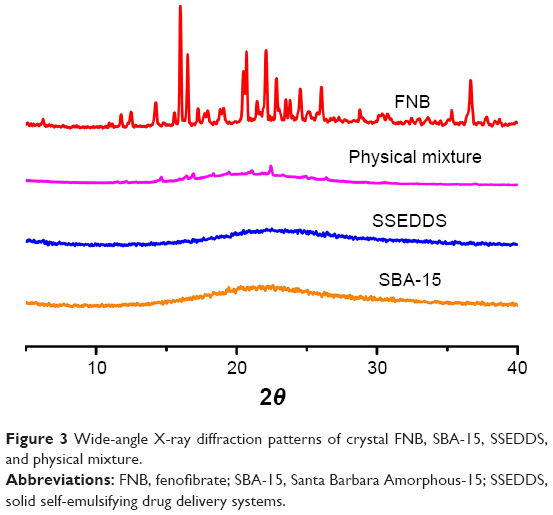 | Figure 3 Wide-angle X-ray diffraction patterns of crystal FNB, SBA-15, SSEDDS, and physical mixture. |
Effect of various precipitation inhibitors on supersaturation assay
In developing an effective supersaturated formulation, it is essential to use a precipitation inhibitor to maintain the drug in supersaturated state in aqueous medium. In this study, several pharmaceutical polymers such as HPMC E3, HPMC E5, HPMC E15, PVP K30, PVP K90, PVP VA64, and Soluplus were screened as potential candidates for precipitation inhibitors. The selection of polymers was motivated by their performance as precipitation inhibitors in several previous reports.16,29 In order to evaluate the apparent FNB concentration–time profile and duration of the supersaturated state, in vitro dissolution test was conducted under a non-sink condition. The total volume of the medium chosen here was 200 mL, based on physiologic consideration of the total volume of the residual stomach fluid. Also, a solvent-shift method was used to generate FNB supersaturation. As shown in Figure 4A, Soluplus significantly inhibited the precipitation of FNB, showing a consistently higher apparent FNB concentration–time profile throughout the whole period of 2 h. In the case of other polymers including HPMC E3, HPMC E5, HPMC E15, PVP K30, PVP K90, and PVP VA64, the concentration of FNB decreased rapidly due to precipitation. To evaluate the supersaturation effect more specifically, the AUCss was calculated according to the linear trapezoidal rule. As shown in Figure 4B, the supersaturation capability of the precipitation inhibitors was in the sequence: Soluplus >> HPMC E5> HPMC E3–HPMC E15> PVP VA64> PVP K30–PVP K90. The AUCss of the Soluplus group was 10.01 times higher than that of HPMC E5. These results clearly demonstrated that a small amount of Soluplus could retard FNB precipitation effectively and sustain a higher apparent concentration over ~120 min.
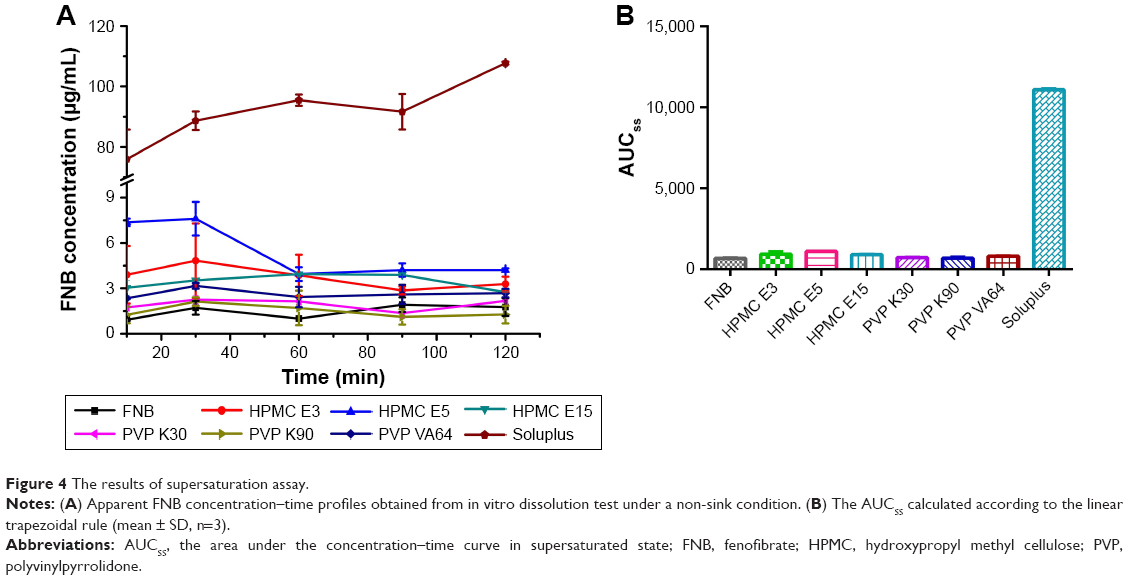 | Figure 4 The results of supersaturation assay. |
Effect of Soluplus amount on drug dissolution from SSEDDS
In order to investigate the influence of Soluplus amount upon the supersaturated state as a function of time, a series of solid self-emulsifying formulations containing the precipitation inhibitor at various polymer/drug ratios were evaluated. The apparent FNB concentration in aqueous medium increased with increase in Soluplus proportion (Figure 5). When the Soluplus:FNB ratio reached 1:1, the precipitation inhibition effect was amplified. However, a further increase in Soluplus:FNB ratio from 1:1 to 10:1 had a little effect, yielding similar apparent FNB concentration–time profiles. Based on these results, the optimized formula for FNB and Soluplus at a ratio of 1:1 was finally established as the super-SSEDDS of FNB.
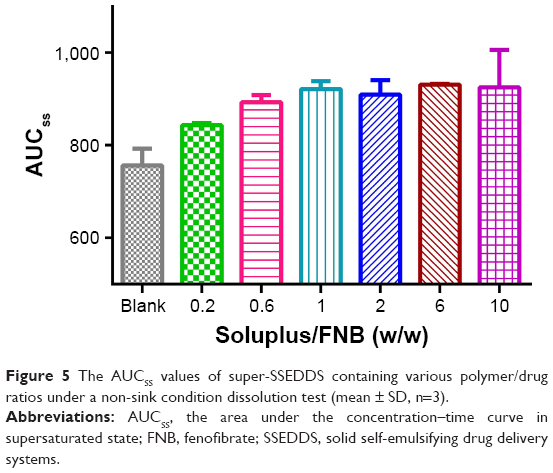 | Figure 5 The AUCSS values of super-SSEDDS containing various polymer/drug ratios under a non-sink condition dissolution test (mean ± SD, n=3). |
To evaluate the reconstitution property of the solid self-emulsifying formulations, the appearance, droplet size, and PDI of the reconstituted emulsions were evaluated. As shown in Figure 6, clear and slightly bluish emulsions with obvious Tyndall effect were observed. The mean droplet size and PDI of SSEDDS were recorded as 125.23±1.50 nm and 0.19±0.01, respectively, which suggest the well-retained self-emulsifying ability in the solid matrix. The mean droplet size of the Soluplus-incorporated formulation was slightly larger than that of conventional SSEDDS due to the absorption of Soluplus on the particle surfaces and the enhanced steric stabilization generated by the strongly hydrated polymer chain.
 | Figure 6 Characterization of the reconstituted emulsion. |
Mechanism of precipitation inhibition
Drug precipitation is normally separated into two stages: nucleation and crystal growth. At the first stage, dissolved drug molecules in solution start to aggregate into small clusters. The clusters continue to grow, which eventually leads to the formation of nuclei that can act as centers of crystallization and move to the second stage, crystal growth.30 Crystal growth is the stage where growth units gather into the surface of the existing nuclei and ultimately arrange into the crystal structure.31,32
Drug molecules tend to precipitate after supersaturation induction.22 The concept of generation and maintenance of a metastable supersaturated state is commonly described by the “Spring and Parachute” theory.33 A higher energy form of drug molecule in supersaturated solution is usually thermodynamically unstable (a spring). The degree of supersaturation (S = C/Cs, also known as supersaturation ratio, where C is the total drug concentration and Cs is the saturation concentration of the drug in the solvent) is the driving force for the precipitation. The higher the initial degree of supersaturation, the more rapid precipitation will happen.34 Recently, a series of pharmaceutical excipients have been successfully explored as precipitation inhibitors (“parachutes”) for temporary inhibition of drug precipitation.35 Inhibition of drug precipitation can be achieved thermodynamically by reducing the degree of supersaturation, or kinetically by inhibiting or retarding the drug precipitation in supersaturated systems.29
In supersaturated solution, when the concentration of surfactants exceeds their critical micelle concentration, an improvement in drug solubility will inhibit the precipitation by reducing the degree of supersaturation. Soluplus has the potential to act as a surfactant due to hydrophilic polyethylene glycol and hydrophobic polycaprolactam groups. The saturation solubility of FNB in a series of Soluplus solutions was measured. As shown in Table 1, the equilibrium solubility of FNB in deionized water was recorded as only 0.22±0.01 μg/mL. As the concentration of Soluplus increased from 0.1 to 5.0 mg/mL, the saturation solubility of FNB gradually increased, indicating that FNB was incorporated in the hydrophobic regions of the micelles. In this study, the final concentration of Soluplus for super-SSEDDS in the dissolution medium was 0.5 mg/mL, which was above its critical micelle concentration (7.6 μg/mL).36 Therefore, the increased saturation solubility could reduce the degree of supersaturation, which ultimately results in retarded precipitation.
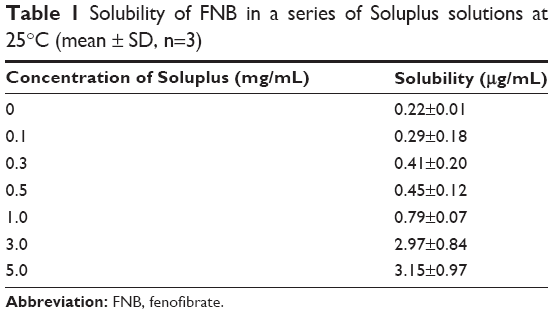 | Table 1 Solubility of FNB in a series of Soluplus solutions at 25°C (mean ± SD, n=3) |
It was reported that the nucleation activation energy was increased by hydrogen bonding between polymers and drug molecules, leading to delayed nucleation and crystal growth.37,38 In this study, FNB has two carbonyl groups (hydrogen-bond acceptor) that can form hydrogen bonds. Since Soluplus has two hydroxyl groups (hydrogen-bond donor) and HPMC has only one hydroxyl group per monomer unit, Soluplus–FNB association is expected to be stronger than HPMC–FNB, leading to effective precipitate inhibition. In contrast, both PVP and PVP VA64 are rich in carbonyl groups (hydrogen-bond acceptor) and have lower tendency to form hydrogen bonds with FNB, thus resulting in poor inhibition ability. Similar observations have also been reported previously, suggesting that polymers rich in hydrogen-bond donors are more suitable for drugs with hydrogen-bond acceptors in order to construct an effective supersaturated drug delivery system.16,20
Based on the results obtained, it may be concluded that inhibition of FNB precipitation could be achieved both thermodynamically and kinetically by Soluplus. Thermodynamically, Soluplus increased the apparent saturation solubility and decreased the extent of supersaturation. Kinetically, it might also interact with FNB by hydrogen bonding. This combinational mechanism resulted in the enhanced Spring and Parachute effect, consequently leading to a better inhibition effect (Figure 7).
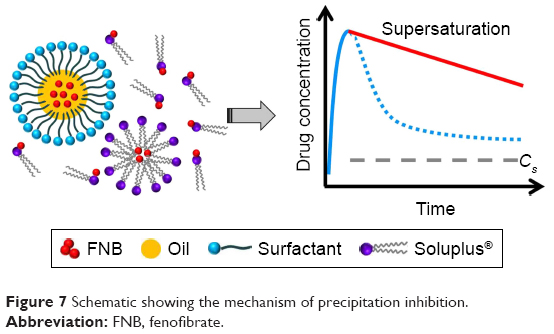 | Figure 7 Schematic showing the mechanism of precipitation inhibition. |
In vitro cytotoxicity of SEDDS, SBA-15, and Soluplus
The biosafety of the excipients must be considered before their application. Caco-2 cells have been widely used as a cellular model to simulate the human intestinal epithelium.39 Herein, liquid SEDDS formulation, mesoporous silica SBA-15, and Soluplus were incubated with Caco-2 for 48 h at a broad concentration range of 10–400 μg/mL and their cytotoxicity was evaluated by MTT assay. As shown in Figure 8, all the samples showed negligible cytotoxicity as the cell viabilities of all samples remained above 90%, demonstrating that the excipients used in this study are biocompatible for oral drug delivery.
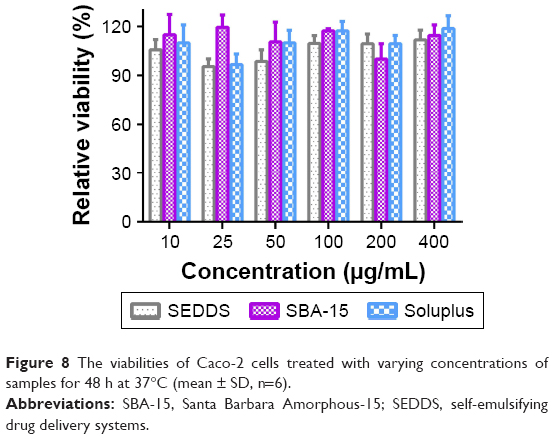 | Figure 8 The viabilities of Caco-2 cells treated with varying concentrations of samples for 48 h at 37°C (mean ± SD, n=6). |
In vivo pharmacokinetic study
The formulations of super-SSEDDS and SSEDDS were orally administered to six healthy beagle dogs, with the dose of FNB being 100 mg. FNB is hydrolyzed into the main active metabolite fenofibric acid by plasma and tissue esterases, and the profiles of mean plasma concentrations of fenofibric acid vs time are presented in Figure 9. The drug plasma concentrations of super-SSEDDS were higher than those of solid self-emulsifying matrix without precipitation inhibitor at all the indicated time points. The pharmacokinetic parameters were calculated and are summarized in Table 2. Both SSEDDS formulations showed rapid absorption with a short Tmax around 2 h, and the super-SSEDDS showed a higher AUC value of 45.05 μg h/mL as compared to that of the SSEDDS formulation (32.74 μg h/mL).
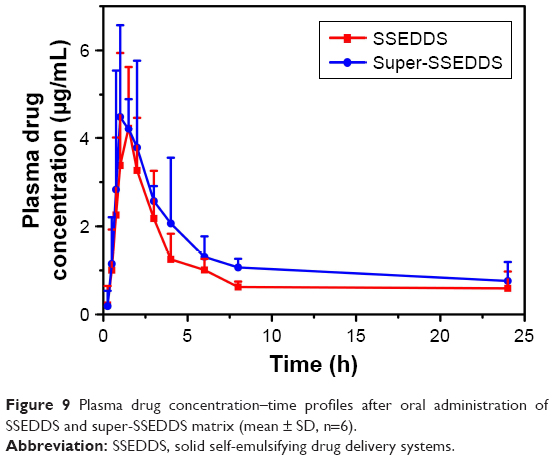 | Figure 9 Plasma drug concentration–time profiles after oral administration of SSEDDS and super-SSEDDS matrix (mean ± SD, n=6). |
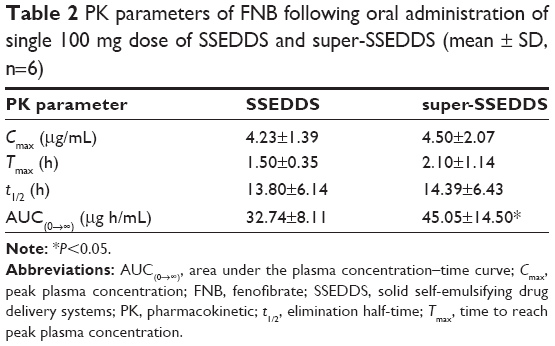 | Table 2 PK parameters of FNB following oral administration of single 100 mg dose of SSEDDS and super-SSEDDS (mean ± SD, n=6) |
It is reported that SEDDS can offer remarkable improvement in the solubility and bioavailability of poorly water-soluble drugs.40 The spontaneous formation of oil-in-water emulsion upon drug release process presents the drug in a dissolved form with nano-scale droplets. These small droplets provide a huge interfacial area favorable for drug absorption.41 The self-emulsifying ability of the obtained SSEDDS was well retained, and a uniform O/W microemulsion was formed when introduced to the aqueous media under mild agitation. Additionally, the super-SSEDDS could further improve the oral absorption of FNB, showing even 40% greater relative bioavailability than that of SSEDDS. These results were in good agreement with the in vitro results described above, implying that the supersaturable formulation was able to induce a higher free drug concentration in the GI tract over a longer period of time. Moreover, Soluplus has been reported to be an effective absorption enhancer for inhibiting the biologic activity of P-glycoprotein.42,43 Therefore, the P-glycoprotein inhibition afforded by Soluplus cannot be excluded and may account for the increased oral absorption.
Both the in vitro dissolution test under supersaturation condition and the in vivo pharmacokinetic performance clearly indicate that the super-SSEDDS containing Soluplus is remarkably effective in inhibiting FNB precipitation and improving the rate and extent of drug absorption.
Conclusion
In summary, a novel super-SSEDDS formulation was successfully designed by incorporation of amphiphilic polymer Soluplus as a precipitation inhibitor. An optimized SSEDDS containing FNB was developed by using mesoporous silica as the insert reservoir. The in vitro dissolution test under supersaturation condition clearly demonstrated that Soluplus effectively suppressed the drug precipitation and maintained a metastable supersaturated state. The in vivo pharmacokinetic study in beagle dogs indicated that the super-SSEDDS significantly improved the oral absorption of FNB, providing about 40% greater relative bioavailability when compared to SSEDDS. In a nutshell, the super-SSEDDS formulation containing Soluplus provides an effective approach to improve the oral absorption of poorly water-soluble drugs.
Acknowledgments
This work was supported by the National Natural Science Funds of China (grant No 81473155), Public Research Platform for Production Technology of Novel Pharmaceutical Formulations, Science and Technology Foundation Guangzhou (grant No 201509030006), and Innovative Scientific Research Team Introducing Project of Zhongshan City (grant No 2015-224).
Disclosure
The authors report no conflicts of interest in this work.
References
Tang B, Cheng G, Gu JC, Xu CH. Development of solid self-emulsifying drug delivery systems: preparation techniques and dosage forms. Drug Discov Today. 2008;13(13–14):606–612. | ||
Feeney OM, Crum MF, McEvoy CL, et al. 50 years of oral lipid-based formulations: Provenance, progress and future perspectives. Adv Drug Del Rev. 2016;101:167–194. | ||
Taupitz T, Dressman JB, Buchanan CM, Klein S. Cyclodextrin-water soluble polymer ternary complexes enhance the solubility and dissolution behaviour of poorly soluble drugs. Case example: itraconazole. Eur J Pharm Biopharm. 2013;83(3):378–387. | ||
Zu Y, Wu W, Zhao X, et al. Enhancement of solubility, antioxidant ability and bioavailability of taxifolin nanoparticles by liquid antisolvent precipitation technique. Int J Pharm. 2014;471(1–2):366–376. | ||
Rashid R, Kim DW, Yousaf AM, et al. Comparative study on solid self-nanoemulsifying drug delivery and solid dispersion system for enhanced solubility and bioavailability of ezetimibe. Int J Nanomedicine. 2015;10:6147–6159. | ||
Sposito PA, Mazzeti AL, de Oliveira Faria C, et al. Ravuconazole self-emulsifying delivery system: in vitro activity against Trypanosoma cruzi amastigotes and in vivo toxicity. Int J Nanomedicine. 2017;12:3785–3799. | ||
Lee DR, Ho MJ, Jung HJ, et al. Enhanced dissolution and oral absorption of tacrolimus by supersaturable self-emulsifying drug delivery system. Int J Nanomedicine. 2016;11:1109–1117. | ||
Oh DH, Kang JH, Kim DW, et al. Comparison of solid self-microemulsifying drug delivery system (solid SMEDDS) prepared with hydrophilic and hydrophobic solid carrier. Int J Pharm. 2011;420(2):412–418. | ||
Iosio T, Voinovich D, Grassi M, et al. Bi-layered self-emulsifying pellets prepared by co-extrusion and spheronization: influence of formulation variables and preliminary study on the in vivo absorption. Eur J Pharm Biopharm. 2008;69(2):686–697. | ||
Shanmugam S, Baskaran R, Balakrishnan P, Thapa P, Yong CS, Yoo BK. Solid self-nanoemulsifying drug delivery system (S-SNEDDS) containing phosphatidylcholine for enhanced bioavailability of highly lipophilic bioactive carotenoid lutein. Eur J Pharm Biopharm. 2011;79(2):250–257. | ||
Setthacheewakul S, Kedjinda W, Maneenuan D, Wiwattanapatapee R. Controlled release of oral tetrahydrocurcumin from a novel self-emulsifying floating drug delivery system (SEFDDS). AAPS Pharm Sci Tech. 2011;12(1):152–164. | ||
Qi X, Qin J, Ma N, Chou X, Wu Z. Solid self-microemulsifying dispersible tablets of celastrol: formulation development, charaterization and bioavailability evaluation. Int J Pharm. 2014;472(1–2):40–47. | ||
Quan G, Wu Q, Zhang X, et al. Enhancing in vitro dissolution and in vivo bioavailability of fenofibrate by solid self-emulsifying matrix combined with SBA-15 mesoporous silica. Colloid Surface B Biointerfaces. 2016;141:476–482. | ||
Kataoka M, Sugano K, da Costa Mathews C, et al. Application of dissolution/permeation system for evaluation of formulation effect on oral absorption of poorly water-soluble drugs in drug development. Pharm Res. 2012;29(6):1485–1494. | ||
Williams HD, Sassene P, Kleberg K, et al; LFCS Consortium. Toward the establishment of standardized in vitro tests for lipid-based formulations, part 3: understanding supersaturation versus precipitation potential during the in vitro digestion of type I, II, IIIA, IIIB and IV lipid-based formulations. Pharm Res. 2013;30(12):3059–3076. | ||
Bevernage J, Hens B, Brouwers J, Tack J, Annaert P, Augustijns P. Supersaturation in human gastric fluids. Eur J Pharm Biopharm. 2012;81(1):184–189. | ||
Lee DH, Yeom DW, Song YS, et al. Improved oral absorption of dutasteride via Soluplus (R)-based supersaturable self-emulsifying drug delivery system (S-SEDDS). Int J Pharm. 2015;478(1):341–347. | ||
Wei Y, Ye X, Shang X, et al. Enhanced oral bioavailability of silybin by a supersaturatable self-emulsifying drug delivery system (S-SEDDS). Colloid Surface A. 2012;396:22–28. | ||
Song WH, Park JH, Yeom DW, et al. Enhanced dissolution of celecoxib by supersaturating self-emulsifying drug delivery system (S-SEDDS) formulation. Arch Pharm Res. 2013;36(1):69–78. | ||
Gao P, Akrami A, Alvarez F, et al. Characterization and optimization of AMG 517 supersaturatable self-emulsifying drug delivery system (S-SEDDS) for improved oral absorption. J Pharm Sci. 2009;98(2):516–528. | ||
Thomas N, Holm R, Mullertz A, Rades T. In vitro and in vivo performance of novel supersaturated self-nanoemulsifying drug delivery systems (super-SNEDDS). J Control Release. 2012;160(1):25–32. | ||
Yu H, Xia D, Zhu Q, Zhu C, Chen D, Gan Y. Supersaturated polymeric micelles for oral cyclosporine A delivery. Eur J Pharm Biopharm. 2013;85(3):1325–1336. | ||
Zhang K, Yu H, Luo Q, et al. Increased dissolution and oral absorption of itraconazole/Soluplus extrudate compared with itraconazole nanosuspension. Eur J Pharm Biopharm. 2013;85(3 Pt B):1285–1292. | ||
Bevernage J, Brouwers J, Brewster ME, Augustijns P. Evaluation of gastrointestinal drug supersaturation and precipitation: Strategies and issues. Int J Pharm. 2013;453(1):25–35. | ||
Jannin V, Musakhanian J, Marchaud D. Approaches for the development of solid and semi-solid lipid-based formulations. Adv Drug Deliv Rev. 2008;60(6):734–746. | ||
Wei-hong T, Min-chang G, Zhen X, Jie S. Pharmacological and pharmacokinetic studies with vitamin D-loaded nanoemulsions in asthma model. Inflammation. 2014;37(3):723–728. | ||
Trana PH, Tran TT, Piao ZZ, et al. Physical properties and in vivo bioavailability in human volunteers of isradipine using controlled release matrix tablet containing self-emulsifying solid dispersion. Int J Pharm. 2013;450(1–2):79–86. | ||
Balakrishnan P, Lee BJ, Oh DH, et al. Enhanced oral bioavailability of dexibuprofen by a novel solid self-emulsifying drug delivery system (SEDDS). Eur J Pharm Biopharm. 2009;72(3):539–545. | ||
Xu S, Dai WG. Drug precipitation inhibitors in supersaturable formulations. Int J Pharm. 2013;453(1):36–43. | ||
Erdemir D, Lee AY, Myerson AS. Nucleation of crystals from solution: classical and two-step models. Acc Chem Res. 2009;42(5):621–629. | ||
Burda C, Chen X, Narayanan R, El-Sayed MA. Chemistry and properties of nanocrystals of different shapes. Chem Rev. 2005;105(4):1025–1102. | ||
Lindfors L, Forssen S, Westergren J, Olsson U. Nucleation and crystal growth in supersaturated solutions of a model drug. J Colloid Interface Sci. 2008;325(2):404–413. | ||
Guzman HR, Tawa M, Zhang Z, et al. Combined use of crystalline salt forms and precipitation inhibitors to improve oral absorption of celecoxib from solid oral formulations. J Pharm Sci. 2007;96(10):2686–2702. | ||
Patel DD, Joguparthi V, Wang Z, Anderson BD. Maintenance of supersaturation I: indomethacin crystal growth kinetic modeling using an online second-derivative ultraviolet spectroscopic method. J Pharm Sci. 2011;100(7):2623–2641. | ||
Brouwers J, Brewster ME, Augustijns P. Supersaturating drug delivery systems: the answer to solubility-limited oral bioavailability? J Pharm Sci. 2009;98(8):2549–2572. | ||
Thomas R. Solubility enhancement with BASF pharma polymers: solubilizer compendium. Lampertheim: BASF (Ed); 2011. | ||
Miller DA, DiNunzio JC, Yang W, McGinity JW, Williams RO 3rd. Enhanced in vivo absorption of itraconazole via stabilization of supersaturation following acidic-to-neutral pH transition. Drug Dev Ind Pharm. 2008;34(8):890–902. | ||
Balani PN, Wong SY, Ng WK, Widjaja E, Tan RB, Chan SY. Influence of polymer content on stabilizing milled amorphous salbutamol sulphate. Int J Pharm. 2010;391(1–2):125–136. | ||
Artursson P, Palm K, Luthman K. Caco-2 monolayers in experimental and theoretical predictions of drug transport. Adv Drug Del Rev. 2001;46(1–3):27–43. | ||
Agrawal AG, Kumar A, Gide PS. Self emulsifying drug delivery system for enhanced solubility and dissolution of glipizide. Colloid Surface B Biointerfaces. 2015;126:553–560. | ||
Mu H, Holm R, Mullertz A. Lipid-based formulations for oral administration of poorly water-soluble drugs. Int J Pharm. 2013;453(1):215–224. | ||
Linn M, Collnot EM, Djuric D, et al. Soluplus (R) as an effective absorption enhancer of poorly soluble drugs in vitro and in vivo. Eur J Pharm Sci. 2012;45(3):336–343. | ||
Jin X, Zhou B, Xue L, San W. Soluplus (R) micelles as a potential drug delivery system for reversal of resistant tumor. Biomed Pharmacother. 2015;69:388–395. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.