Back to Journals » Research Reports in Clinical Cardiology » Volume 10
Spotlight on sudden arrhythmic death syndrome
Received 25 June 2019
Accepted for publication 27 August 2019
Published 12 September 2019 Volume 2019:10 Pages 57—66
DOI https://doi.org/10.2147/RRCC.S187480
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Richard Kones
David Yuan,1 Hariharan Raju1,2
1Cardiology Department, Concord Repatriation General Hospital, Sydney, Australia; 2Clinical School, Faculty of Medicine and Health, University of Sydney, Sydney, Australia
Correspondence: Hariharan Raju
Cardiology Department, Concord Repatriation General Hospital, Hospital Road, Concord West 2139, Australia
Tel +61 29 812 2900
Fax +61 29 475 1155
Email [email protected]
Abstract: Sudden cardiac death (SCD) is defined as death from a cardiac cause within 1 hr of symptom onset or, if unwitnessed, in a person last seen well within 24 hrs. Sudden arrhythmic death syndrome (SADS) describes cases of SCD with no abnormalities found on expert autopsy attributable as the cause of death. The epidemiology of these conditions has been challenging to study as definitions have changed over time; however, it is apparent that the incidence of SCD increases with age whilst SADS decreases as coronary artery disease becomes more prevalent. Accurate reporting of truly negative autopsies of SCDs has been assisted by guidelines from governing bodies such as The Association for European Cardiovascular Pathology, allowing identification of SADS cases. Primary arrhythmic cardiac conditions like ong QT syndrome, Brugada syndrome and catecholaminergic polymorphic ventricular tachycardia are the predominant etiologies of SADS. When the decedent did not have a known phenotype for these conditions, clinical evaluation using screening tests like echocardiogram, resting and stress electrocardiograms and holter monitoring, followed by specialized testing when appropriate such as cardiac magnetic resonance and pharmacological provocation testing of surviving family members becomes crucial in potentially identifying the cause and guide targeted genetic testing. Although advancement in gene analysis such as next-generation sequencing has also allowed the application of “molecular autopsy” to identify pathogenic variants to establish the cause of death and enable cascade testing and risk stratification of family members, many of the genetic variants identified through this method have been classified as non-pathogenic since the establishment of standards and guidelines by the American College of Medical Genetics. Whilst majority of cases of SADS are still unexplained, there is increasing awareness and understanding of this syndrome allowing appropriate identification of surviving family members at risk and implementation of measures to prevent further premature death.
Keywords: sudden cardiac death, sudden arrhythmic death syndrome, sudden unexplained death, unexplained cardiac death, molecular autopsy
Introduction
Sudden cardiac death (SCD) refers to an unexpected arrest of the cardiovascular system. It is defined as death from a cardiac cause from 1 hr of onset of symptoms or, if unwitnessed, in a person last seen well within 24 hrs.1 This excludes non-cardiac sudden deaths such as aortic dissection, hemorrhagic stroke, pulmonary embolism or other such pathologies.
Sudden arrhythmic death syndrome (SADS) refers to a subset of SCD, where, despite thorough and expert postmortem histological and toxicological examinations, a cause of death cannot be identified.2 This can be likened to sudden infant death syndrome, which is also a diagnosis of exclusion, albeit in infancy.3 There is some overlap in the literature with other terms such as sudden unexplained death (SUD) or unexplained sudden cardiac death (USCD) which are also used to describe similar populations of unexplained death where an autopsy has not been performed.4
Epidemiology
A recent review of the global incidence of SCD in all age groups found significant geographical variation, with lowest rates reported in Japan of 14.9 per 100,000 persons to 110.8 per 100,000 persons reported by The Resuscitation Outcomes Consortium in the United States.1 SADS (otherwise defined as SUD or USCD) accounts for between 25% and 50% of the SCD cases in those between 1 and 35 years of age, based predominantly on retrospective data,5–8 although additional evidence from an Australasian prospective study of 490 SCD cases supports this.9 The annual incidence of SCD in this young population is estimated to be between 1 and 3 per 100,000 persons in western countries.5–9
Age impacts on etiology and incidence of SCD even amongst those 1–35 years of age. Data from the Swedish National Board of Forensic Medicine and the Swedish Cause of Death Registry support an overall incidence of 1.3 SCDs per 100,000 person-years in the 1–35 years of age population which increased to 1.8 per 100,000 person-years in the 15–35 years of age subgroup; SADS was the most common etiology, accounting for 31% of the SCD.8 Similarly, incidence of SCD increased with age to 35 years in the Australasian prospective study by Bagnall et al: overall annual incidence of 1.3 cases per 100,000 persons in a population 1–35 years of age, with the highest annual incidence in the 31–35 years subgroup of 3.2 per 100,000 persons.9 There is a notable decline from the 1–5 year to the 6–10-year-old groups. The authors highlight in the discussion that the high incidence in the youngest group is predominantly in 1- to 2-year-old children with the lowest incidence in the 6- to 10-year-old group and postulate that this is due to the etiologies underlying sudden infant death syndrome which decline throughout early childhood.9 Unexplained SCD was most prevalent in the 16–20 years age group (0.8 cases per 100,000 persons) whilst in the 31–35 years age group, coronary artery disease (CAD) predominated.9 Overall, USCD was the predominant etiology, reflecting 40% of the cases. Data from the Danish National Patient Registry and Danish Cause of Death Registry pertaining to SCD 1–49 years support a similar shift from predominance of SUD to CAD comparing age groups 1–35 and 36–49 years: annual incidence of SCD increased from 2.3 per 100,000 up to 21.7 per 100,000 persons; proportion of SUD fell from 48% to 25% of SCD; and proportion of SCD attributed to CAD increased from 15% to 44%.5 The proportion of SCD contributed to by SADS/SUD and CAD in the aforementioned studies is summarized in Figure 1.5,8,9 Even in a young athlete population, SADS is still the most prevalent finding among those who have SCD over hypertrophic cardiomyopathy (HCM).10 One meta-analysis including 34 studies with a combined sample size of 4605 subjects showed that in the younger population (age<35 years), SCD with structurally normal hearts on autopsy is more common than from HCM (27% vs 10%).11
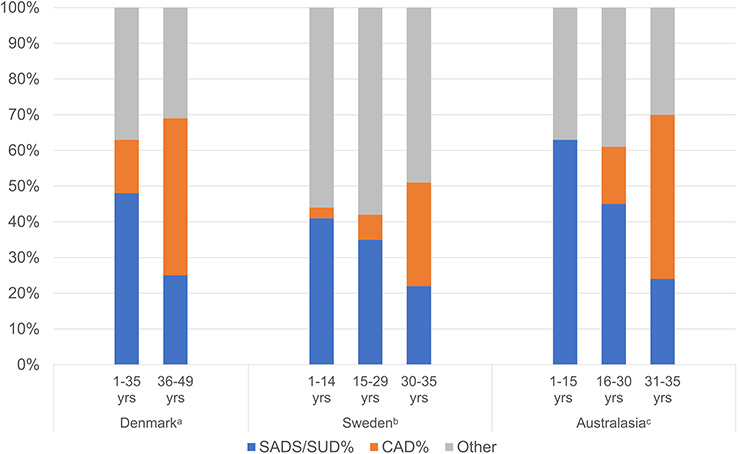 |
Figure 1 Proportion of SCD cases contributed to by SADS/SUD and CAD stratified by age in 3 national studies. (A) Other category comprised hypertrophic heart, arrhythmogenic right ventricular cardiomyopathy and myocarditis. Data from Risgaard et al.5 (B) Other category comprised unspecific cardiomyopathy, congenital heart disease, hypertrophic cardiomyopathy, thoracic aortic aneurysm, dilated cardiomyopathy, arrhythmogenic right ventricular cardiomyopathy and coronary artery anomaly. Data from Wisten et al.8 (C) Other category comprised dilated cardiomyopathy, myocarditis, arrhythmogenic right ventricular cardiomyopathy, hypertrophic cardiomyopathy, aortic dissection and “other”. Data from Bagnall et al.9 |
Several recent reports have indicated that the overall incidence of SCD in all age groups is declining.12–14 A recent study examining the incidence of SCD in those aged 1–35 years also showed a statistically significant reduction from 3.1 per 100,000 person-years in the year 2000 to 2.5 per 100,000 person-years in the year 2009, corresponding to an average annual percent reduction of 3%; there was no trend to change in the contribution to SCD by SADS.15 The authors postulate that this reduction is possibly due to implementation of public health initiatives such as availability of automated defibrillators, cardiopulmonary resuscitation training in schools and when acquiring a driver’s license and reduction in CAD (whilst less than in the elderly, still one of the largest contributors to SCD after SADS).15
Heterogeneity in reported incidence
The reported incidence of SCD is often inconsistent in the literature due to variations in study design and population. As SADS is a subset of SCD, similar challenges apply to determining its true burden. There are significant variations in the incidence of SCD due to location of study populations as shown by a prospective North American study in all age groups where annual incidence was between 40.3 and 86.7 per 100,000 persons across 10 sites in the United States and Canada.16 Further differences arise from inconsistencies of the definition of SCD and how data are obtained. A systematic review of studies on the incidence of SCD in the United States showed a range from 180,000 to >450,000 per annum, with significant differences in how SCD was defined, variations in how data were collected and extrapolated.17 Aside from regional variation of population characteristics, one of the possible explanations for variability in incidence of both SCD and SADS is the rate of autopsy routinely performed in different countries. For example, in Australia and UK, it is a routine practice for all cases of unexpected sudden death to be referred to the coroner for autopsy, whilst in countries such as Denmark and Sweden, this is not the case,5,7,8 with up to 51% of the unexpected sudden deaths not being autopsied.
Furthermore, Wilms et al assessed the completeness of 110 police and autopsy reports in cases of SCD in subjects aged 0–40 years of age against local best practice guidelines in Australia and New Zealand.18 The findings showed that whilst location, activity at time of death and some clinical history were well documented, specific and crucial details such as family history, drug and alcohol use, possible arrhythmic trigger and history of fits/faints or collapses were reported less than 25% of the time.18 From an autopsy point of view, heart weight, valvular, pulmonary and myocardial histology were well reported; however, less than 50% of the reports commented on septal, left and/or right ventricular wall thickness, the site that histology samples were obtained, left and right ventricular or conduction system histology. Coronary artery histology was only reported in 18% of the cases.18 Multidisciplinary teams such as the Trans-Tasman Response Against Sudden Death in the Young (TRAGADY) Network in Australasia have written guidelines with the aim of standardizing the investigation of SCD in the young.19
Autopsy diagnosis
SADS requires the presence of normal macroscopic and microscopic cardiac postmortem examination.4 The Association for European Cardiovascular Pathology updated guidance for cardiac autopsy in 2017 and recommend that postmortem abnormalities are classified in a probabilistic fashion as to the likelihood of being responsible for death.4 Acute myocardial infarction, acute diffuse myocarditis and mitral valve papillary muscle or chordae tendineae rupture with mitral valve incompetence and pulmonary edema are considered certain causes of death;4 chronic ischemic heart disease, obstructive CAD (>75% luminal stenosis), amyloidosis, sarcoidosis and many of the cardiomyopathies (hypertrophic, arrhythmogenic and dilated) are highly probable to be responsible for SCD when identified at autopsy.4 However, other abnormalities are “uncertain” as the cause of death: anomalous right coronary artery, focal myocarditis and moderate aortic sclerosis.4 Furthermore, the guideline also highlights some histological findings that fall between the spectrum of physiological to pathological and are of uncertain significance. These include findings deviating from normal myocardium such as scattered foci of inflammation without necrosis or fatty infiltration without fibrosis but are not diagnostic for focal myocarditis and arrhythmogenic cardiomyopathy, respectively.4
Significance of uncertain histological findings
These findings of uncertain significance are potential causes of overdiagnosis, thereby impacting on evaluation of surviving family members. These findings may be misclassified as pathological, leading to erroneous diagnosis of cardiomyopathies such as arrhythmogenic right ventricular cardiomyopathy (ARVC) and HCM.10
Papadakis et al summarized the postmortem findings of uncertain significance for several conditions responsible for SCD.20 The study defined “findings of uncertain significance” for conditions including HCM (left ventricular hypertrophy and/or myocardial fibrosis in the absence of myocardial disarray), ARVC (fatty infiltration of the right ventricular wall in the absence of fibrosis), dilated cardiomyopathy (DCM) (mild ventricular dilatation in the absence of significant fibrosis or myocardial inflammation), coronary atherosclerosis (atherosclerosis with estimated ≤50% luminal narrowing of the coronary arteries or 2 mm probe patent in the absence of acute or chronic infarction) and myocarditis (scattered lymphocytic inflammatory foci with no fibrosis or myocyte necrosis).20 The UK study reports the diagnostic yield of assessing both the relatives of subjects classified as SADS and those with postmortem histopathological findings of uncertain significance was similar (47% and 51%) when looking for inherited cardiac disorders.20 Furthermore, despite the main study cohort being one of the patients with some degree of abnormality on their postmortem histology, primary arrhythmic disorders were still by far the most common diagnosis, with only 2 out of the 41 families subsequently being diagnosed with an inherited cardiomyopathy disorder.20 Raju et al conducted a similar study in an Australian population which again, showed no significant difference in the rate of diagnosis of cardiogenetic disorders between family members of SADS cases and SUD cases with isolated non-diagnostic cardiac histology on postmortem.21 In addition to this, the study demonstrated a significantly higher rate of uncertain autopsy findings in SCD cases compared with matched non-cardiac premature death controls, suggesting that although the uncertain findings were unlikely to be primarily responsible for the SCD, they could not be considered innocent bystanders.21
Emerging role of imaging
Computer tomography (CT) and cardiac magnetic resonance (CMR) have become increasingly important in diagnostic pathways and have also been explored in the postmortem setting.22,23 Of interest, Puranik et al reported that CMR was able to accurately diagnose ARVC, HCM, myocardial infarction/CAD, ruptured aortic aneurysm, pulmonary embolus and myocarditis as the cause of death in 12 out of 12 subjects who went on to have confirmatory autopsy.23 There were particular limitations noted such as the potential to overdiagnose conditions such as HCM due to myocardial edema or death during systole which artificially increase the ventricular septal thickness or ARVC whereby right ventricular dilatation is used as a diagnostic criteria, but the systolic dysfunction and regional dyskinesis diagnostic components are not met. Of relevance, a recent study utilizing CMR in the diagnostic work up of out of hospital cardiac arrest patients showed that CMR performed within 7 days of presentation identified a diagnosis in all 44 patients, but more importantly was able to reclassify 11 out of the 26 cases without obstructive coronary disease after initial assessment with coronary angiography and echocardiography.24 The study reported that during a mean of 36 months of follow-up, all those with myocardial edema on CMR had an uncomplicated course, whilst 35% of those without myocardial edema experienced either death or appropriate defibrillation.24 CMR may also be useful as a second tier screening tool in surviving relatives of the proband if there is any clinical suspicion for cardiomyopathies such as ARVC or HCM.25 In addition to cardiomyopathies, mitral valve prolapse (MVP) and mitral annular disjunction (MAD), which can be diagnosed on CMR, have been shown to be associated with increased rates of ventricular arrhythmia and SCD.26,27
Etiology of sudden arrhythmic death syndrome
When postmortem autopsy is negative, the term SADS is used to describe the cause of death. This is under the assumption that in the absence of structural cardiac disease the primary disorder leading to death is arrhythmic. Whilst malignant arrhythmias and sudden death occur in conditions such as Wolff Parkinson White28,29 and vasospastic angina30 and also yield negative autopsies, they have no impact on surviving family members in contrast to the inheritable primary arrhythmic conditions described in this review.
The conditions that contribute to the majority of these diagnoses are long QT syndrome (LQTS), Brugada syndrome (BrS) and catecholaminergic polymorphic ventricular tachycardia (CPVT), with other diagnoses such as early repolarization syndrome (ERS) being significantly less common.31,32 Studies seeking to uncover the cause of SADS have consistently shown that around one-third of cases of SADS can be attributed to these primary ion channelopathy arrhythmic syndromes which are subsequently diagnosed in close relatives.9,31,33,34 Further clinical evaluation of surviving family members may also uncover cardiomyopathic diagnoses which may have been undetected in the proband.
A diagnosis is usually made through a thorough investigation of the proband’s surviving family members, starting with a clinical evaluation. Behr et al conducted one of the first studies to employ this strategy, utilizing specific history taking, cardiovascular physical examination, 12 lead electrocardiogram (ECG), echocardiogram and holter monitoring.2 More frequent use of exercise stress tests, signal-averaged ECG and provocation testing has improved the diagnostic yield of clinical evaluation.32,35,36 Some early studies report up to 50% diagnostic yield through the clinical evaluation of surviving family members alone.2,37,38 Contemporary studies report a lower yield from clinical evaluation alone9,36,39 and may be attributed to an increase in reporting and referral patterns of cases of SCD for further investigation, creating a more heterogeneous population compared to earlier studies. In addition to this, molecular genetic testing of targeted genes linked to clinical phenotype has been used to increase the overall diagnostic yield.3,20,25,33–35,39,40 An important concept termed signal-to-noise ratio refers to the positive predictive value of a genetic variant derived from dividing the prevalence of the variant in confirmed cases by its prevalence in a control population.41 The positive predictive value is considered high when the signal-to-noise ratio is >10:1.41 The approach of clinical evaluation of surviving family members followed by targeted genetic testing is currently supported by both European Society of Cardiology and American Heart Association/American College of Cardiology/Heart Rhythm Society (HRS) guidelines in the evaluation of first degree relatives of probands of sudden arrhythmic death.42,43
Long QT syndrome
Congenital LQTS has a reported prevalence of around 1 in 2000 persons.44 When screening tests such as resting or continuous ECGs are negative, further testing can be performed in the form of exercise stress ECGs or adrenaline (epinephrine) challenge as this may unmask subclinical QT prolongation.42–44 Subtypes 1–3 are the most prevalent and defined by the mutated risk-gene: genes which encode potassium channels (KCNQ1 in LQT1; KCNH2 in LQT2) and cardiac sodium channel (SCN5A in LQT3).44 However, there are hundreds of mutations that have been identified in 14 different genes which encode components of cardiac ion channels.33 Approximately 85% of those with genotypically confirmed LQTS have an inherited genetic variant, with the remaining minority being de novo mutations.44 The major LQTS genes have a high yield of 75% with a favorable “signal-to-noise” ratio of 19:1.33,41 This has resulted in the HRS guideline recommending that those with a confirmed pathogenic mutation are diagnosed with LQTS regardless of phenotypical QT duration.
Brugada syndrome
A recent meta-analysis reported a worldwide prevalence of BrS of around 0.5 per 1000 persons.45 The highest prevalence was seen in Southeast Asia with a Type 1 Brugada pattern on ECG in up to 1.8 in 1000 persons, and being 9 times more common in Asians than Caucasians.45 The diagnostic criteria for BrS has been revised several times since its original description and was most recently revisited in the 2015 J-wave syndromes expert consensus conference report. The consensus agrees that the diagnosis of BrS is made if a Type 1 pattern is found in more than one of the right precordial leads either spontaneously or if induced by pharmacological provocation, must also be in the presence of one of the following: documented VF or polymorphic VT, syncope of probable arrhythmic cause, a family history of SCD at <45 years old with negative autopsy, coved-type ECGs in family members or nocturnal agonal respiration.46 The sensitivity of the ECG diagnosis of Type 1 Brugada pattern can be increased by further steps such as shifting the right precordial leads to higher intercostal spaces (typically 2nd or 3rd) and by procainamide/flecainide/ajmaline challenge.36,42,43,47 Other factors such as drugs or medications, fever and diurnal variation also increase the prevalence of the Type 1 pattern.45,47 SCN5A is the most commonly affected gene in BrS but is only affected in around 20% of clinically diagnosed BrS cases.47 Unlike in LQTS, genetic testing for the diagnosis of BrS has added little due to variable expressivity, reduced genetic penetrance, oligogenic inheritance, a yield of 20% and a less favorable signal-to-noise ratio of 10:1.33,41
Catecholaminergic polymorphic ventricular tachycardia
The prevalence of CPVT is estimated at 1:10,000 persons48 and with pathogenesis attributed to mishandling of intracellular calcium due to mutations in the RYR2 (autosomal dominant inheritance) and CASQ2 (autosomal recessive inheritance) genes causing unregulated release during diastole.33 The diagnosis is made in the presence of bidirectional or polymorphic ventricular tachycardia on ECG, the diagnostic yield of which can be increased with the use of exercise testing or adrenaline (epinephrine) drug challenge.33,48 However, like LQTS, guidelines support the diagnosis of CPVT if a pathogenic mutation is identified in the RYR2 or CASQ2 genes regardless of phenotype due to its high yield of 60% and favorable genetic signal-to-noise ratio of 20:1.41,42
Strategies to improve the sensitivity of clinical evaluation
In the evaluation of surviving family members with normal investigations on initial screening, provocation testing with drugs such as sodium blockers for the diagnosis of BrS and adrenaline (epinephrine) for LQTS and CPVT increases the diagnostic yield.42,43 The use of ajmaline has been shown to be more sensitive than flecainide and procainamide in provoking a Type 1 pattern on drug challenge.49,50 Papadakis et al demonstrated the clinical utility of high right parasternal leads (HRPL) during the assessment of 911 relatives of probands of SADS and ajmaline challenge in 670 relatives who were without a diagnosis after initial investigations.36 During initial evaluation, only 4 (0.4%) of relatives had a spontaneous Type 1 pattern (2 of which were only detected using HRPL).36 Ajmaline testing was required to unmask 97% of the blood relatives eventually diagnosed with BrS.36 The use of HRPLs during ajmaline challenge increased the diagnostic yield by a further 16% (an additional 49 families recognized).36 The use of drug provocation testing is controversial given the high rate (27%) of positive tests in patients with the benign arrhythmia atrioventricular nodal reentrant tachycardia (AVNRT), raising concerns regarding overdiagnosis.51 This data contrasts with limited data on diagnostic yield of ajmaline provocation pertaining to a control population, where the “false positive” rate for ajmaline provocation is 5%.36,51 Adrenaline infusions have been used as a surrogate for exercise stress testing in the diagnosis of CPVT with disappointing results, adding little to the diagnostic yield of exercise stress testing.52 Similarly, early studies using adrenaline testing to diagnose and genotype LQTS were promising;53,54 however, exercise stress testing is preferred and is included in the clinical diagnostic Schwartz score.55,56
Genetic testing strategies
Clinical evaluation and targeted genetic testing
Clinical evaluation of the proband’s surviving family members can give clues to the genotypes of interest based on phenotypic diagnoses. Known pathogenic genetic variants can then be tested for in other family members for risk stratification and can also be tested in the proband should genetic material be available. Many studies have taken this approach with varying rate of diagnosis due to differences in population, availability of autopsy and expertise, and the rigorousness of the clinical evaluation protocol.33 The mean diagnostic yield from these studies is around 30% from clinical evaluation alone.2,34,37,38,57–59 Of the studies which utilized genetic testing, 23–47% of those with a clinical phenotype went on to also have a molecular diagnosis.34,37,38,58 This diagnostic yield does not seem to have changed in recent years, as shown by Raju et al in 2019 whilst comparing SADS and SUD with uncertain histopathological findings, reporting a clinical diagnostic yield 25% and genetic yield in those with a clinical phenotype of 40%.21 The pooled results from several studies utilizing clinical familial evaluation followed by targeted phenotype driven genetic evaluation of probands and their families are shown in Figure 22,9,34,37–39,57,60,61 and the overall diagnostic yield appears to be 20–30%.
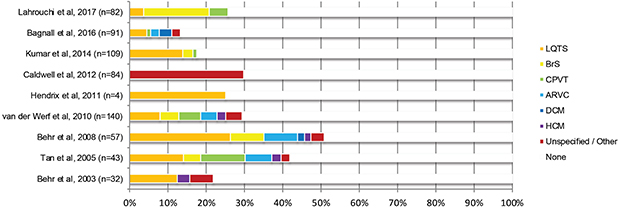 |
Figure 2 Diagnostic yield of genetic cardiovascular conditions from clinical evaluation with targeted genetic analysis series following sudden unexplained death and/or SADS of a proband. Data from references 2, 9, 34, 37–39, 57, 60 and 61. |
The molecular autopsy
Molecular autopsy panels are a combination of predetermined genes, with associations to cardiac conditions, that are considered pathogenic or highly probable to be pathogenic. The term molecular autopsy is used when this panel of genes are analyzed in the retained DNA of a proband with no known pre-existing phenotype and a normal postmortem examination (ie, SADS). Identified variants are adjudicated to be pathogenic, likely pathogenic or variants of uncertain significance (VUS) based on their minor allele frequency (MAF).9,33,39 Early studies utilizing this technique focused on specific LQTS (KCNQ1, KCNH2, SCN5A) and CPVT (RYR2) genes with diagnostic yields of up to 30% in specific referred cases. The yield for LQTS and CPVT has been around 15% and 10%, respectively,3,33,62 in these studies. Subsequent studies applying more stringent diagnostic criteria for gene variants found even lower yield (13% for LQTS and 11% for CPVT).33 A large prospective study by Bagnall et al included 490 SCD cases and applied the 4 commonly utilized aforementioned genes applied as a molecular autopsy panel. A clinically relevant gene mutation in the molecular autopsy panel was found in only 9% of the 113 cases where DNA was available and family consent was obtained.9 The authors postulated that previous retrospective studies were subject to ascertainment and referral bias, whilst their study took a population approach resulting in a comparatively lower diagnostic yield.9 Beyond the techniques utilized in previous studies, a panel of 55 genes linked to arrhythmia and cardiomyopathies was analyzed using “next-generation sequencing” in an attempt to identify further potential etiologies of the unexplained sudden death cases.9 The overall yield was 27% when including these pathogenic and probably pathogenic variants.9 One criticism of this approach is that the commonly used MAF threshold of 1:1000 may be too lenient and lead to misclassification of non-pathogenic variants as being clinically significant and consequently inaccurate labeling of cause of death followed by inappropriate clinical and genetic evaluation of family members. The fluid definition of pathogenicity due to expanding understanding of variants has been a common problem such as for investigators in New Zealand who reviewed molecular autopsies performed on 365 sudden unexplained death in the young (SUDY) cases between 2006 and 2013 for variants in LQTS genes.63 Whilst 27 cases of gene variants were identified over an 8-year period, with the change in understanding of pathogenicity over time, only 13 of these remained as likely pathogenic with the rest being downgraded to VUS or polymorphisms by the Cardiac Inherited Disease Group.63 The authors highlight the challenge of studying this population in the absence of whole exome control populations and the difficulty of defining pathogenicity.63
In 2015, the American College of Medical Genetics (ACMG) published guidelines to tackle the challenge of a rapidly growing number of variants being detected through application of high-throughput next-generation sequencing.64 They sought to standardize the terminology used to describe a variant’s pathogenicity and provide guidance on how to assign variants into “pathogenic”, “likely pathogenic”, “uncertain significance”, “likely benign” and “benign” categories. Since then, many previously describe variants thought to be attributable to disease have had their pathogenicity downgraded to “uncertain significance” or less. Following the publication of these guidelines, Lahrouchi et al applied a panel of 77 cardiac arrhythmia and cardiomyopathy genes to 302 expertly evaluated SADS cases and despite the large number of genes analyzed, only found a pathogenic or likely pathogenic gene variant in only 13% of the cases.39 The authors used a strict MAF of 1:10,000 and classified the variant pathogenicity using the ACMG guidelines to achieve a more robust variant classification.39 These were the proposed reasons for why, despite using an even larger panel of genes than Bagnall et al,9 their overall diagnostic yield was significantly lower.39 This study also highlights the challenge of finding a large number of VUS associated with cardiomyopathies with a prevalence of 97% and a “VUS to pathogenic/probably pathogenic” ratio of 28:1.39
Current practice
The American Heart Association published a statement in 2019 supporting the establishment of specialized cardiovascular genetics clinics for the investigation, diagnosis and management of inheritable diseases including channelopathies.65 Such specialized clinics provide a setting for targeted history taking, physical examination, interpretation of available imaging results and performance of further specialized imaging, arrhythmia monitoring and functional testing. The combined expertise of clinical cardiologists, pathologists and geneticists with in-depth understanding of these inherited diseases is crucial for having informed discussions with patients and families regarding the implications of existing and potential further investigations such as genetic testing.65
The current guideline-driven strategy is to perform focused genetic analysis based on the clinical phenotype identified in the surviving family members.42,43 Specific genes associated with the phenotype, as discussed previously, are analyzed in the affected family members and/or the proband. If a pathogenic variant is identified from the genetic material of the proband, which needs to be harvested at the time of postmortem examination, this can be useful in efficiently risk-stratifying family members who may have subclinical phenotype through use of cascade testing.42,43 Current guidelines give moderate level recommendations to applying molecular autopsy techniques with the main benefit, should a clinically attributable variant be identified, being cascade testing of family members to identify those at risk.42,43 It is currently not recommended to perform large panel genetic testing on family members without abnormalities on clinical evaluation.42
The implications of genetic testing and their results must be adequately discussed with families both before and after genetic tests are performed. The specialized cardiovascular genetic clinic environment allows genetic counselors to do this with the support of other medical, nursing and psychology staff.65
Conclusion
Through growing awareness of SADS as an important diagnostic entity, there have been improvements in the data collection and referral pattern for postmortem investigations of SCD probands. Diagnostic protocols defined in international guidelines help clinicians to methodically investigate for the underlying diagnoses using histological, clinical and genetic analysis of SCD cases and relatives. Future developments and refinement of imaging and molecular analytical diagnostic criteria will hopefully further improve the overall diagnostic yield of inheritable cardiac disorders that underlie these deaths. This will aid in risk modification and prevention of SCD in surviving family members of the probands, who may harbor subclinical forms of the same cardiogenetic condition.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Wong CX, Brown A, Lau DH, et al. Epidemiology of sudden cardiac death: global and regional perspectives. Heart Lung Circ. 2019;28(1):6–14. doi:10.1016/j.hlc.2018.08.026
2. Behr E, Wood DA, Wright M, et al. Cardiological assessment of first-degree relatives in sudden arrhythmic death syndrome. Lancet. 2003;362(9394):1457–1459. doi:10.1016/s0140-6736(03)14692-2
3. Raju H, Behr ER. Unexplained sudden death, focussing on genetics and family phenotyping. Curr Opin Cardiol. 2013;28(1):19–25. doi:10.1097/HCO.0b013e32835b0a9e
4. Basso C, Aguilera B, Banner J, et al. Guidelines for autopsy investigation of sudden cardiac death: 2017 update from the Association for European Cardiovascular Pathology. Virchows Arch. 2017;471(6):691–705. doi:10.1007/s00428-017-2221-0
5. Risgaard B, Winkel BG, Jabbari R, et al. Burden of sudden cardiac death in persons aged 1 to 49 years: nationwide study in Denmark. Circ Arrhythm Electrophysiol. 2014;7(2):205–211. doi:10.1161/CIRCEP.113.001421
6. Margey R, Roy A, Tobin S, et al. Sudden cardiac death in 14- to 35-year olds in Ireland from 2005 to 2007: a retrospective registry. Europace. 2011;13(10):1411–1418. doi:10.1093/europace/eur161
7. Winkel BG, Holst AG, Theilade J, et al. Nationwide study of sudden cardiac death in persons aged 1–35 years. Eur Heart J. 2011;32(8):983–990. doi:10.1093/eurheartj/ehq428
8. Wisten A, Krantz P, Stattin EL. Sudden cardiac death among the young in Sweden from 2000 to 2010: an autopsy-based study. Europace. 2017;19(8):1327–1334. doi:10.1093/europace/euw249
9. Bagnall RD, Weintraub RG, Ingles J, et al. A prospective study of sudden cardiac death among children and young adults. N Engl J Med. 2016;374(25):2441–2452. doi:10.1056/NEJMoa1510687
10. D’Silva A, Papadakis M. Sudden cardiac death in athletes. Eur Cardiol. 2015;10(1):48–53. doi:10.15420/ecr.2015.10.01.48
11. Ullal AJ, Abdelfattah RS, Ashley EA, Froelicher VF. Hypertrophic cardiomyopathy as a cause of sudden cardiac death in the young: a meta-analysis. Am J Med. 2016;129(5):486–496 e482. doi:10.1016/j.amjmed.2015.12.027
12. Feng JL, Nedkoff L, Knuiman M, et al. Temporal trends in sudden cardiac death from 1997 to 2010: a data linkage study. Heart Lung Circ. 2017;26(8):808–816. doi:10.1016/j.hlc.2016.11.021
13. Niemeijer MN, van Den Berg ME, Leening MJ, et al. Declining incidence of sudden cardiac death from 1990–2010 in a general middle-aged and elderly population: the rotterdam study. Heart Rhythm. 2015;12(1):123–129. doi:10.1016/j.hrthm.2014.09.054
14. Messner T, Lundberg V. Trends in sudden cardiac death in the northern Sweden MONICA area 1985–99. J Intern Med. 2003;253(3):320–328.
15. Lynge TH, Nielsen JL, Blanche P, et al. Decline in incidence of sudden cardiac death in the young: a 10-year nationwide study of 8756 deaths in Denmark. Europace. 2019;21(6):909–917. doi:10.1093/europace/euz022
16. Nichol G, Thomas E, Callaway CW, et al. Regional variation in out-of-hospital cardiac arrest incidence and outcome. JAMA. 2008;300(12):1423–1431. doi:10.1001/jama.300.12.1423
17. Kong MH, Fonarow GC, Peterson ED, et al. Systematic review of the incidence of sudden cardiac death in the United States. J Am Coll Cardiol. 2011;57(7):794–801. doi:10.1016/j.jacc.2010.09.064
18. Wilms HR, Midgley DJ, Morrow P, Stables S, Crawford J, Skinner JR. Evaluation of autopsy and police reports in the investigation of sudden unexplained death in the young. Forensic Sci Med Pathol. 2012;8(4):380–389. doi:10.1007/s12024-012-9340-3
19. TRAGADY. Post-mortem in Sudden Unexpected Death in the Young: Guidelines on Autopsy Practice. The Royal College of Pathologists of Australasia; 2008.
20. Papadakis M, Raju H, Behr ER, et al. Sudden cardiac death with autopsy findings of uncertain significance: potential for erroneous interpretation. Circ Arrhythm Electrophysiol. 2013;6(3):588–596. doi:10.1161/CIRCEP.113.000111
21. Raju H, Parsons S, Thompson TN, et al. Insights into sudden cardiac death: exploring the potential relevance of non-diagnostic autopsy findings. Eur Heart J. 2019;40(10):831–838. doi:10.1093/eurheartj/ehy654
22. Roberts IS, Benamore RE, Benbow EW, et al. Post-mortem imaging as an alternative to autopsy in the diagnosis of adult deaths: a validation study. Lancet. 2012;379(9811):136–142. doi:10.1016/S0140-6736(11)61483-9
23. Puranik R, Gray B, Lackey H, et al. Comparison of conventional autopsy and magnetic resonance imaging in determining the cause of sudden death in the young. J Cardiovasc Magn Reson. 2014;16:44. doi:10.1186/1532-429X-16-44
24. Zorzi A, Susana A, De Lazzari M, et al. Diagnostic value and prognostic implications of early cardiac magnetic resonance in survivors of out-of-hospital cardiac arrest. Heart Rhythm. 2018;15(7):1031–1041. doi:10.1016/j.hrthm.2018.02.033
25. Semsarian C, Ingles J, Wilde AA. Sudden cardiac death in the young: the molecular autopsy and a practical approach to surviving relatives. Eur Heart J. 2015;36(21):1290–1296. doi:10.1093/eurheartj/ehv063
26. Basso C, Perazzolo Marra M, Rizzo S, et al. Arrhythmic mitral valve prolapse and sudden cardiac death. Circulation. 2015;132(7):556–566. doi:10.1161/CIRCULATIONAHA.115.016291
27. Dejgaard LA, Skjolsvik ET, Lie OH, et al. The mitral annulus disjunction arrhythmic syndrome. J Am Coll Cardiol. 2018;72(14):1600–1609. doi:10.1016/j.jacc.2018.07.070
28. Klein GJ, Bashore TM, Sellers TD, Pritchett EL, Smith WM, Gallagher JJ. Ventricular fibrillation in the wolff-parkinson-white syndrome. N Engl J Med. 1979;301(20):1080–1085. doi:10.1056/NEJM197911153012003
29. Munger TM, Packer DL, Hammill SC, et al. A population study of the natural history of wolff-parkinson-white syndrome in Olmsted County, Minnesota, 1953–1989. Circulation. 1993;87(3):866–873. doi:10.1161/01.cir.87.3.866
30. Kundu A, Vaze A, Sardar P, Nagy A, Aronow WS, Botkin NF. Variant angina and aborted sudden cardiac death. Curr Cardiol Rep. 2018;20(4):26. doi:10.1007/s11886-018-1000-0
31. Hayashi M, Shimizu W, Albert CM. The spectrum of epidemiology underlying sudden cardiac death. Circ Res. 2015;116(12):1887–1906. doi:10.1161/CIRCRESAHA.116.304521
32. Mellor G, Nelson CP, Robb C, et al. The prevalence and significance of the early repolarization pattern in sudden arrhythmic death syndrome families. Circ Arrhythm Electrophysiol. 2016;9(6). doi:10.1161/CIRCEP.116.003960.
33. Miles CJ, Behr ER. The role of genetic testing in unexplained sudden death. Transl Res. 2016;168:59–73. doi:10.1016/j.trsl.2015.06.007
34. Kumar S, Peters S, Thompson T, et al. Familial cardiological and targeted genetic evaluation: low yield in sudden unexplained death and high yield in unexplained cardiac arrest syndromes. Heart Rhythm. 2013;10(11):1653–1660. doi:10.1016/j.hrthm.2013.08.022
35. Krahn AD, Healey JS, Chauhan V, et al. Systematic assessment of patients with unexplained cardiac arrest: Cardiac Arrest Survivors With Preserved Ejection Fraction Registry (CASPER). Circulation. 2009;120(4):278–285. doi:10.1161/CIRCULATIONAHA.109.853143
36. Papadakis M, Papatheodorou E, Mellor G, et al. The diagnostic yield of brugada syndrome after sudden death with normal autopsy. J Am Coll Cardiol. 2018;71(11):1204–1214. doi:10.1016/j.jacc.2018.01.031
37. Tan HL, Hofman N, van Langen IM, van der Wal AC, Wilde AA. Sudden unexplained death: heritability and diagnostic yield of cardiological and genetic examination in surviving relatives. Circulation. 2005;112(2):207–213. doi:10.1161/CIRCULATIONAHA.104.522581
38. Behr ER, Dalageorgou C, Christiansen M, et al. Sudden arrhythmic death syndrome: familial evaluation identifies inheritable heart disease in the majority of families. Eur Heart J. 2008;29(13):1670–1680. doi:10.1093/eurheartj/ehn219
39. Lahrouchi N, Raju H, Lodder EM, et al. Utility of post-mortem genetic testing in cases of sudden arrhythmic death syndrome. J Am Coll Cardiol. 2017;69(17):2134–2145. doi:10.1016/j.jacc.2017.02.046
40. Anastasakis A, Papatheodorou E, Ritsatos K, et al. Sudden unexplained death in the young: epidemiology, aetiology and value of the clinically guided genetic screening. Europace. 2018;20(3):472–480. doi:10.1093/europace/euw362
41. Ackerman MJ, Priori SG, Willems S, et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies: this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Europace. 2011;13(8):1077–1109. doi:10.1093/europace/eur245
42. Priori SG, Blomstrom-Lundqvist C, Mazzanti A, et al. ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: the task force for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur Heart J. 2015;36(41):2793–2867. doi:10.1093/eurheartj/ehv316
43. Al-Khatib SM, Stevenson WG, Ackerman MJ, et al. 2017 AHA/ACC/HRS guideline for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Rhythm Society. Heart Rhythm. 2018;15(10):e73–e189. doi:10.1016/j.hrthm.2017.10.036
44. Mizusawa Y, Horie M, Wilde AA. Genetic and clinical advances in congenital long QT syndrome. Circ J. 2014;78(12):2827–2833. doi:10.1253/circj.cj-14-0905
45. Vutthikraivit W, Rattanawong P, Putthapiban P, et al. Worldwide prevalence of brugada syndrome: a systematic review and meta-analysis. Acta Cardiol Sin. 2018;34(3):267–277. doi:10.6515/ACS.201805_34(3).20180302B
46. Antzelevitch C, Yan GX, Ackerman MJ, et al. J-wave syndromes expert consensus conference report: emerging concepts and gaps in knowledge. J Arrhythm. 2016;32(5):315–339. doi:10.1016/j.joa.2016.07.002
47. Antzelevitch C, Brugada P, Borggrefe M, et al. Brugada syndrome: report of the second consensus conference: endorsed by the Heart Rhythm Society and the European Heart Rhythm Association. Circulation. 2005;111(5):659–670. doi:10.1161/01.CIR.0000152479.54298.51
48. Napolitano C, Bloise R, Memmi M, Priori SG. Clinical utility gene card for: Catecholaminergic polymorphic ventricular tachycardia (CPVT). Eur J Hum Genet. 2014;22:1. doi:10.1038/ejhg.2013.55
49. Wolpert C, Echternach C, Veltmann C, et al. Intravenous drug challenge using flecainide and ajmaline in patients with Brugada syndrome. Heart Rhythm. 2005;2(3):254–260. doi:10.1016/j.hrthm.2004.11.025
50. Cheung CC, Mellor G, Deyell MW, et al. Comparison of ajmaline and procainamide provocation tests in the diagnosis of brugada syndrome. JACC Clin Electrophysiol. 2019;5(4):504–512. doi:10.1016/j.jacep.2019.01.026
51. Hasdemir C, Payzin S, Kocabas U, et al. High prevalence of concealed brugada syndrome in patients with atrioventricular nodal reentrant tachycardia. Heart Rhythm. 2015;12(7):1584–1594. doi:10.1016/j.hrthm.2015.03.015
52. Marjamaa A, Hiippala A, Arrhenius B, et al. Intravenous epinephrine infusion test in diagnosis of catecholaminergic polymorphic ventricular tachycardia. J Cardiovasc Electrophysiol. 2012;23(2):194–199. doi:10.1111/j.1540-8167.2011.02188.x
53. Shimizu W, Noda T, Takaki H, et al. Diagnostic value of epinephrine test for genotyping LQT1, LQT2, and LQT3 forms of congenital long QT syndrome. Heart Rhythm. 2004;1(3):276–283. doi:10.1016/j.hrthm.2004.04.021
54. Vyas H, Hejlik J, Ackerman MJ. Epinephrine QT stress testing in the evaluation of congenital long-QT syndrome: diagnostic accuracy of the paradoxical QT response. Circulation. 2006;113(11):1385–1392. doi:10.1161/CIRCULATIONAHA.105.600445
55. Schwartz PJ, Crotti L. QTc behavior during exercise and genetic testing for the long-QT syndrome. Circulation. 2011;124(20):2181–2184. doi:10.1161/CIRCULATIONAHA.111.062182
56. Wong JA, Gula LJ, Klein GJ, Yee R, Skanes AC, Krahn AD. Utility of treadmill testing in identification and genotype prediction in long-QT syndrome. Circ Arrhythm Electrophysiol. 2010;3(2):120–125. doi:10.1161/CIRCEP.109.907865
57. van der Werf C, Hofman N, Tan HL, et al. Diagnostic yield in sudden unexplained death and aborted cardiac arrest in the young: the experience of a tertiary referral center in The Netherlands. Heart Rhythm. 2010;7(10):1383–1389. doi:10.1016/j.hrthm.2010.05.036
58. McGorrian C, Constant O, Harper N, et al. Family-based cardiac screening in relatives of victims of sudden arrhythmic death syndrome. Europace. 2013;15(7):1050–1058. doi:10.1093/europace/eus408
59. Mellor G, Raju H, de Noronha SV, et al. Clinical characteristics and circumstances of death in the sudden arrhythmic death syndrome. Circ Arrhythm Electrophysiol. 2014;7(6):1078–1083. doi:10.1161/CIRCEP.114.001854
60. Caldwell J, Moreton N, Khan N, et al. The clinical management of relatives of young sudden unexplained death victims; implantable defibrillators are rarely indicated. Heart. 2012;98(8):631–636. doi:10.1136/heartjnl-2011-300924
61. Hendrix A, Borleffs CJ, Vink A, et al. Cardiogenetic screening of first-degree relatives after sudden cardiac death in the young: a population-based approach. Europace. 2011;13(5):716–722. doi:10.1093/europace/euq460
62. Skinner JR, Crawford J, Smith W, et al. Prospective, population-based long QT molecular autopsy study of postmortem negative sudden death in 1 to 40 year olds. Heart Rhythm. 2011;8(3):412–419. doi:10.1016/j.hrthm.2010.11.016
63. Marcondes L, Crawford J, Earle N, et al. Long QT molecular autopsy in sudden unexplained death in the young (1–40 years old): Lessons learnt from an eight year experience in New Zealand. PLoS One. 2018;13(4):e0196078. doi:10.1371/journal.pone.0196078
64. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. doi:10.1038/gim.2015.30
65. Ahmad F, McNally EM, Ackerman MJ, et al. Establishment of specialized clinical cardiovascular genetics programs: recognizing the need and meeting standards: a scientific statement from the American Heart Association. Circulation. 2019;12(6):e000 054. doi:10.1161/HCG.0000000000000054
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.