Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Silica nanoparticles induce oxidative stress, inflammation, and endothelial dysfunction in vitro via activation of the MAPK/Nrf2 pathway and nuclear factor-κB signaling
Authors Guo C, Xia Y, Niu P, Jiang L, Duan J, Yu Y, Zhou X, Li Y, Sun Z
Received 20 October 2014
Accepted for publication 20 December 2014
Published 20 February 2015 Volume 2015:10(1) Pages 1463—1477
DOI https://doi.org/10.2147/IJN.S76114
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Caixia Guo,1,2 Yinye Xia,1,2 Piye Niu,1,2 Lizhen Jiang,1,2 Junchao Duan,1,2 Yang Yu,1,2 Xianqing Zhou,1,2 Yanbo Li,1,2 Zhiwei Sun1,2
1School of Public Health, 2Beijing Key Laboratory of Environmental Toxicology, Capital Medical University, Beijing, People’s Republic of China
Abstract: Despite the widespread application of silica nanoparticles (SiNPs) in industrial, commercial, and biomedical fields, their response to human cells has not been fully elucidated. Overall, little is known about the toxicological effects of SiNPs on the cardiovascular system. In this study, SiNPs with a 58 nm diameter were used to study their interaction with human umbilical vein endothelial cells (HUVECs). Dose- and time-dependent decrease in cell viability and damage on cell plasma-membrane integrity showed the cytotoxic potential of the SiNPs. SiNPs were found to induce oxidative stress, as evidenced by the significant elevation of reactive oxygen species generation and malondialdehyde production and downregulated activity in glutathione peroxidase. SiNPs also stimulated release of cytoprotective nitric oxide (NO) and upregulated inducible nitric oxide synthase (NOS) messenger ribonucleic acid, while downregulating endothelial NOS and ET-1 messenger ribonucleic acid, suggesting that SiNPs disturbed the NO/NOS system. SiNP-induced oxidative stress and NO/NOS imbalance resulted in endothelial dysfunction. SiNPs induced inflammation characterized by the upregulation of key inflammatory mediators, including IL-1β, IL-6, IL-8, TNFα, ICAM-1, VCAM-1, and MCP-1. In addition, SiNPs triggered the activation of the Nrf2-mediated antioxidant system, as evidenced by the induction of nuclear factor-κB and MAPK pathway activation. Our findings demonstrated that SiNPs could induce oxidative stress, inflammation, and NO/NOS system imbalance, and eventually lead to endothelial dysfunction via activation of the MAPK/Nrf2 pathway and nuclear factor-κB signaling. This study indicated a potential deleterious effect of SiNPs on the vascular endothelium, which warrants more careful assessment of SiNPs before their application.
Keywords: silica nanoparticle, endothelium, oxidative stress, Nrf2, MAPK, NF-κB
Introduction
Nanotoxicology is an emerging field of research as a response to an exponential growth in the development and production of engineered nanoparticles (NPs) worldwide. The interactions of NPs with biological systems, as well as their environmental and human health effects, have not been fully explored.1 There is concern that research on the possible health risks of NPs is not keeping pace with the rapid growth in the number of NP products entering the industry and market. Due to their special physicochemical features, NPs’ biological behavior may differ from that of larger particles. Previous research has shown that nanoscale particles are more toxic than fine particles or bulk materials with the same chemical composition.2,3
Silica NPs (SiNPs) are among the top five commonly used nanomaterials listed in nanotechnology consumer products by the Woodrow Wilson International Center for Scholars.4 They have recently been extensively utilized in biomedical and biotechnological fields for biomolecule detection, diagnosis, imaging, drug delivery, gene therapy, and photodynamic therapy.5,6 As a consequence, human exposure to SiNPs continues to increase in environmental, occupational, and medial settings. Therefore, the environmental and health impact of SiNPs deserve great concern. However, toxicological studies on SiNPs are far behind the speed of their application. Malvindi et al pointed out that SiNPs had good biocompatibility when used in a reasonable concentration range (up to 2.5 nM).7 However, as they are smaller than cellular organelles, it is increasingly recognized that SiNPs could penetrate the cytomembrane, deposit in mitochondria or even the nucleus, and eventually lead to cell death.8–11 It has been confirmed that NPs can systemically translocate to the circulation from the lung.12 Subsequently, translocated particles could mediate cardiovascular effects through direct interaction with vasculature, blood, and the heart. The cardiovascular toxicity of NPs has gradually drawn attention. However, related research is limited, and controversial reports exist.13
Endothelial dysfunction is believed to induce pathological changes in the cardiovascular system, and has been considered as a predictor and an initiating event of atherosclerosis and its complications.14,15 As a biological barrier between circulating blood and the vessel wall, vascular endothelia are extremely important for the maintenance of vascular function and of homeostasis.16 For SiNPs, based on their access to systemic and coronary circulations under inhalation exposure and their wide application for diagnostics or drug delivery through intravenous injection, there would be quite a large probability for them to influence vascular reactivity by directly interacting with the vascular endothelium. A concentration-dependent cytotoxicity of SiNPs on endothelial cell line EA.hy926 has been previously reported.17 There is evidence that SiNPs can induce reactive oxygen species (ROS), impairment of proliferative activity, inflammation, and cell death on vascular endothelial cells.18–20 Our previous study confirmed that SiNPs caused deoxyribonucleic acid (DNA) damage via the Chk1-dependent G2/M-checkpoint signaling pathway on endothelial cells.21 However, understanding of the molecular mechanisms involved in the cytotoxicity of SiNPs on endothelium is still limited.
Based on these issues, a toxicity evaluation of SiNPs on endothelial cell injury and dysfunction was conducted in vitro with human umbilical vein endothelial cells (HUVECs), which have been used as a reference for in vitro studies of endothelial cell function. A series of evaluations, including cell viability, membrane injury, oxidative stress, nitric oxide (NO) release, and inflammatory mediators in HUVECs after exposure to SiNPs were investigated in vitro. Moreover, to understand the observed molecular mechanism of SiNP-induced oxidative stress, in vitro assays were conducted focusing on MAPK cascades, nuclear factor (NF)-κB, and Nrf2.
Materials and methods
Chemicals and reagents
Tetraethyl orthosilicate and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) were obtained from Sigma-Aldrich, USA. Fetal bovine serum and Dulbecco’s modified Eagle’s medium (DMEM) were purchased from Invitrogen, USA. A MiniBest universal ribonucleic acid (RNA) extraction kit, PrimeScript™ RT Master Mix (Perfect Real Time), and SYBR Premix Ex Taq™ II were obtained from Takara, Japan. All other chemicals used were of the highest purity and available from commercial sources.
SiNP preparation and characterization
SiNPs in aqueous suspension were prepared using the Stöber method by the College of Chemistry, Jilin University, People’s Republic of China (PRC). Before use, the particles were isolated by centrifugation (12,000× g, 15 minutes), washed three times with deionized water, and then dispersed in 50 mL of deionized water sterilized by autoclave (0.1 MPa, 120°C, 20 minutes). The size and distribution of SiNPs were assessed by transmission electron microscopy (JEM2100; JEOL, Japan) and ImageJ software. Dynamic light scattering was employed using a Zetasizer (Nano ZS90; Malvern, UK) to examine the hydrodynamic sizes and zeta potential of SiNPs in different dispersion media. The stock suspensions of SiNPs were sonicated for 5 minutes through a sonicator (160 W, 20 kHz, 5 minutes; Bioruptor UDC-200, Belgium) before addition to DMEM or distilled water in order to minimize their aggregation. Meanwhile, inductively coupled plasma atomic emission spectrometry (ICP-AES) (Agilent 720, USA) was used for purity detection, and gel-clot limulus amebocyte lysate (LAL) assay for endotoxin measurement in SiNP suspensions, purchased from Zhan Jiang Bokang Marine Biological (PRC).
Cell culture
HUVECs were obtained from the Cell Resource Center, Shanghai Institutes for Biological Sciences, PRC. Cells were cultured in DMEM supplemented with 10% fetal bovine serum at 37°C in a 5% CO2 humidified incubator.
Cell morphology
After the 24-hour exposure to SiNPs at concentrations of 12.5, 25, 50, and 100 μg/mL, the morphology of HUVECs was observed by using an optical microscope (Olympus IX81, Japan).
Cell-viability assay
The cytotoxicity of SiNPs was determined using the MTT assay as previously described.22 Briefly, cells were seeded in 96-well plates, with approximately 1×104 cells per well. After overnight incubation, the old culture medium was discarded. In order to eliminate the possible reaction between SiNPs and serum in old culture medium, cells were treated by twice rinsing in fresh culture media without serum. Then, the cells were incubated in fresh culture media without serum but containing various concentrations of SiNPs for 24 hours at 37°C: 12.5, 25, 50 and 100 μg/mL. Controls were supplied with an equivalent volume of media without SiNPs or serum. Ultimately, absorbance was measured at 490 nm using a microplate reader (SpectraMax M5; Molecular Devices, USA).
Lactate dehydrogenase-release assay
The release of intracellular lactate dehydrogenase (LDH) into culture medium is an indicator of irreversible cell death due to cell-membrane damage. To determine the effect of SiNPs on the cell membrane, LDH release was assessed using a commercial LDH kit (Jiancheng, PRC) according to the manufacturer’s protocols. Briefly, cells were cultured in 24-well plates, with approximately 8×104 cells per well. After 12 hours of growth, cells were treated by SiNPs at concentrations of 12.5, 25, 50, or 100 μg/mL for 24 hours at 37°C. The supernatants were collected, 100 μL cell medium was used for LDH-activity analysis, and absorbance at 440 nm was measured by an ultraviolet-visible spectrophotometer (Beckman DU-640B, USA). The amount of LDH released is expressed as LDH activity (U/L) in culture media.
SiNP uptake
The amount of SiNPs existing in cells was determined using ICP-AES. Briefly, HUVECs were exposed to SiNPs for 24 hours, subsequently washed with phosphate-buffered saline (PBS), and trypsinized. Cell density was determined with a hemocytometer. The cell suspensions were centrifuged at 1,500× g for 5 minutes. In total, 5 mL cell lysate containing HNO3 and H2O2 (3:1) was added in cell pellets to dissolve SiNPs. Lastly, SiNPs that entered cells were measured by detecting the intracellular silicon content using ICP-AES.
Intracellular ROS measurement
The production of intracellular ROS was measured using the 2′,7′-dichlorofluorescein diacetate (DCFH-DA) (Beyotime, PRC) as an oxidation-sensitive probe. Briefly, HUVECs were treated with various concentrations of SiNPs for 24 hours. Then, the cells were incubated with PBS containing 10 mM DCFH-DA for 30 minutes in a CO2 incubator. After incubation, the cells were washed with PBS. DCF fluorescence was observed using a flow cytometer (Becton-Dickinson, USA) and a laser scanning confocal microscope (LSM 710; Zeiss, Germany) by capturing images at 400× magnification.
Assessment of oxidative damage
In addition to the analysis of ROS levels, the malondialdehyde (MDA) content was measured as an end product of lipid peroxidation, which was carried out using commercially available kits (Jiancheng) according to the manufacturer’s instructions. Briefly, after exposure to SiNPs for 24 hours, HUVECs were washed once with ice-cold PBS and lysed in ice-cold radioimmunoprecipitation assay lysis buffer (Ding-Guo, PRC) for 30 minutes. After the lysates were centrifuged at 12,000× g, 4°C for 10 minutes, the supernatants were collected for measurement of the production of MDA. The protein concentrations of these extracts were determined by performing the bicinchoninic acid protein assay (Pierce, USA). The defense systems against free radical attack were assessed by the measurement of both the activities of superoxide dismutase (SOD) and glutathione peroxidase (GSH-Px), using the corresponding commercial kit from Nanjing Jiancheng Bioengineering Institute (Jiancheng).
Glutathione and glutamate cysteine ligase subunit-level estimation
Glutathione (GSH) levels were quantified using a commercial reduced GSH assay kit (Jiancheng), and the amount of GSH was expressed in terms of micromoles of GSH per gram of proteins. Glutamate cysteine ligase (GCL), also known as γ-glutamylcysteine synthetase, is the first rate-limiting enzyme in GSH synthesis, and has both catalytic (GCLC) and modifier (GCLM) subunits. Total GCLM and GCLC levels were quantified by real-time polymerase chain reaction (PCR) according to the methods mentioned in the following sections.
NO, NOS, and ET-1 measurement
NO content in cells was measured using a commercial kit (Jiancheng). Moreover, the messenger RNA (mRNA) expressions of inducible nitric oxide synthase (iNOS), endothelial NOS (eNOS), and ET-1 were quantified by real-time PCR. In brief, total RNA was isolated using the MiniBest universal RNA extraction kit according to the manufacturer’s protocol. Then, complementary DNA was synthesized using the PrimeScript RT Master Mix. To quantify relative mRNA levels of the genes in HUVECs, real-time PCR was performed using the SYBR Premix Ex Taq II following the manufacturer’s instructions on a LightCycler 480 instrument (Roche, Germany). All quantifications were performed with β-actin as an internal standard, and the relative amount of mRNA was calculated using the 2–ΔΔCT method. Three independent experiments were used to determine the average fold changes for each target gene. The primer sets are listed in Table S1.
Quantification of mRNA levels of redox- and inflammation-related factors
The relative amount of redox related factors, including Nrf2 and its downstream genes, such as HMOX1, SOD2, CAT, GPX, GCLC, GCLM, NQO1, TXN1, and TXNRD1, were quantified using real-time PCR as mentioned earlier. Furthermore, inflammatory cytokines, including IL-1β, IL-6, IL-8, and TNFα, and ICAM-1, VCAM-1, and MCP-1, were also measured. The primer sets are listed in Table S1.
Western blot analysis
To analyze whether SiNPs influence the expression of cellular factors involved in the MAPK signaling pathway, we measured the protein levels of Nrf2, heme oxygenase (HO)-1, ERK, p38MAPK, JNK, and NF-κB in HUVECs by Western blot analysis after SiNP treatment for 24 hours. As an internal control, glyceraldehyde 3-phosphate dehydrogenase was also detected. Densitometric analysis of the Western blots was performed using Image Lab™ software (version 3.0; Bio-Rad, USA). The relative values of the samples were measured by normalizing all data to the respective control samples of each experiment.
Statistical analysis
Data are expressed as means ± standard deviations. Significance was determined by using one-way analysis of variance, followed by least significant difference tests to compare differences between groups. Differences were considered significant at P<0.05.
Results
Characterization of SiNPs
Prior to the study of toxicity, the characterization of SiNPs was examined. The morphology and average size of SiNPs were determined by transmission electron microscopy. As shown in Figure 1A, SiNPs had a near-spherical shape and were well dispersed, with an average diameter of 58 nm. Size distribution measured by ImageJ software showed approximately normal distribution (Figure 1B). Additionally, to investigate the possible effect of the dispersion medium on SiNPs, the hydrodynamic sizes and zeta potentials of SiNPs were measured either in DMEM after incubation at 37°C or in distilled water at room temperature at different time points, and these are summarized in Table 1. Owing to the van der Waals force and hydrophobic interaction with surrounding media, SiNPs generally had larger hydrodynamic size in dispersion media than their original size. The polydispersity index, an indicator of dispersion, showed SiNPs with good monodispersity in distilled water or DMEM. Zeta potential is a marker of colloidal stability. In general, when the absolute value of a zeta potential is higher than 30 mV, particles are not prone to aggregation or precipitation. All the results indicated that the SiNPs in dispersion media still possessed uniform shape along with relatively favorable dispersibility and stability. The major trace-metal impurities in SiNPs detected by ICP-AES are summarized in Table 2, suggesting that the purity of SiNPs was better than 99.9%. Additionally, the LAL assay indicated no detectable Gram-negative endotoxin on the SiNPs at concentrations of 0.65, 1.25, 2.5, 5, and 10 mg/mL, indicating SiNPs were endotoxin-free.
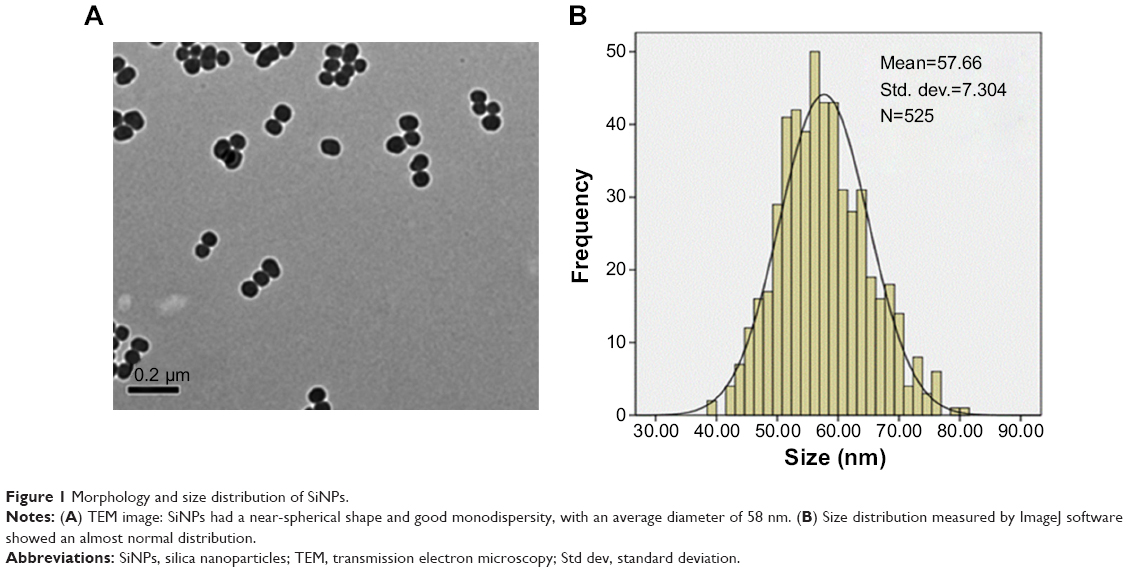 | Figure 1 Morphology and size distribution of SiNPs. |
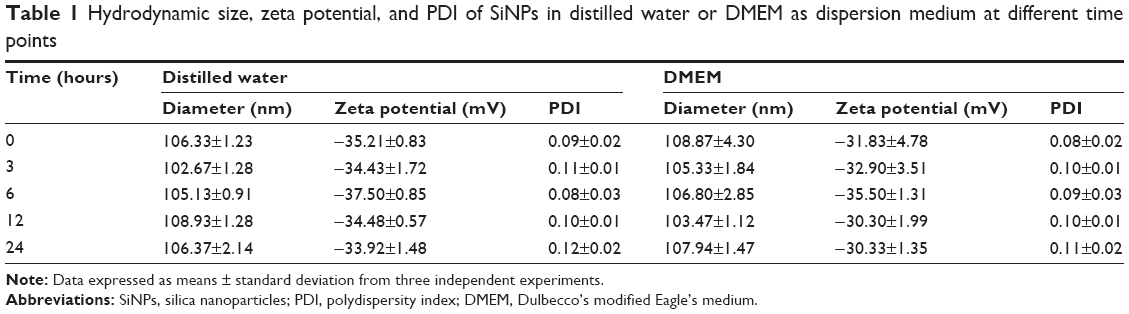 | Table 1 Hydrodynamic size, zeta potential, and PDI of SiNPs in distilled water or DMEM as dispersion medium at different time points |
 | Table 2 Trace-metal impurity levels in SiNPs (μg/10 mg SiO2) |
Cytotoxicity of SiNPs
To evaluate the possible toxicity of SiNPs on endothelial cells, cellular morphology and cell viability were determined after SiNP exposure to HUVECs for 24 hours. The morphological changes of HUVECs became gradually apparent as the dosage of SiNPs increased, and were characterized by cell-density reduction, irregular shape, and cellular shrinkage (Figure 2A). Changes in cellular morphology were directly reflected by cell viability. As indicated in Figure 2B, cell viability decreased remarkably, along with increased SiNP concentration. When cells were exposed to SiNPs at concentrations of 12.5, 25, 50, or 100 μg/mL, cell viability decreased to 92.93%, 81.24%, 42.68%, and 33.44%, respectively, which was significantly lower than that in the control group except for 12.5 μg/mL. Moreover, cell viability declined as time passed, suggesting a dose- and time-dependent effect of SiNP toxicity on HUVECs. Additionally, a significant difference in LDH release was observed when HUVECs were incubated with SiNPs for 24 hours compared with the control (Figure 2C). The LDH-leakage results showed that SiNPs induced injury to the cell membrane, which was strongly in accordance with the result for cell viability. A significant negative correlation was also observed between LDH leakage and MTT cell viability (R2=0.9559, Figure 2D).
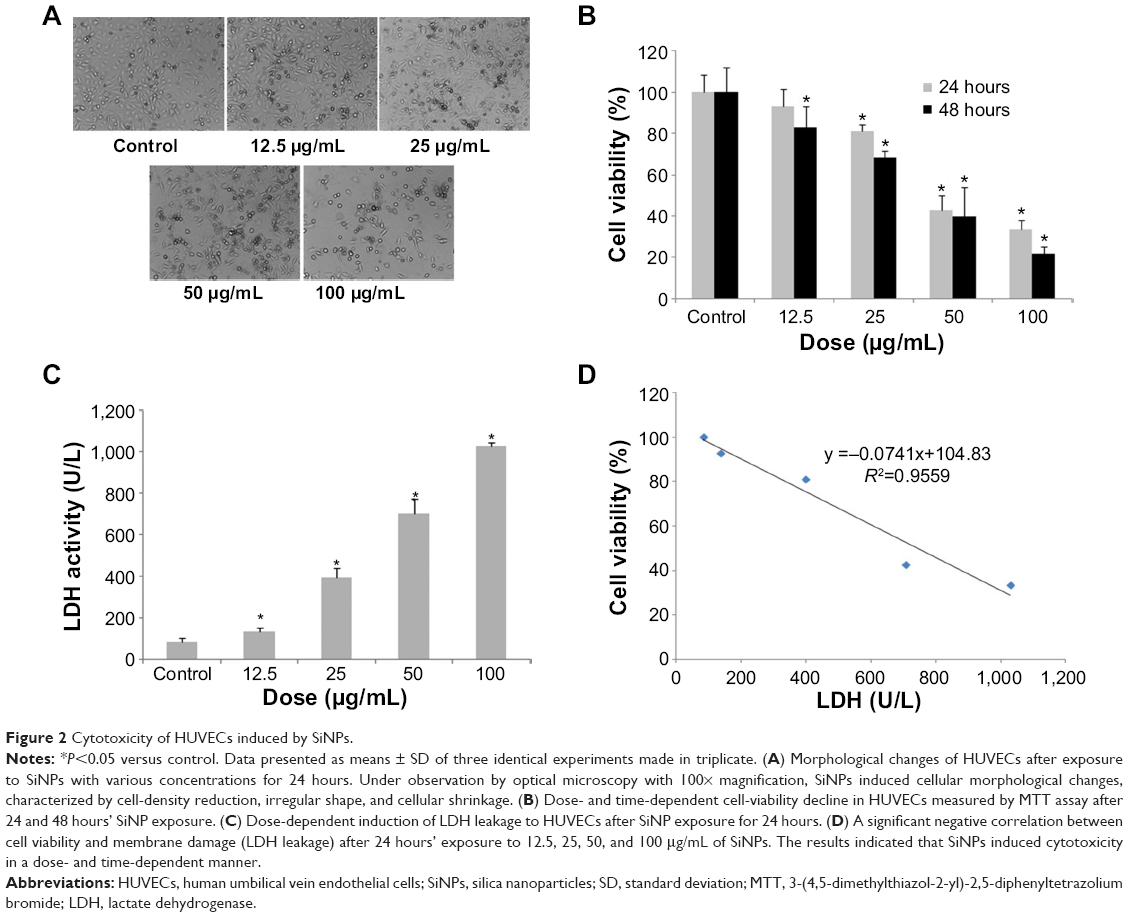 | Figure 2 Cytotoxicity of HUVECs induced by SiNPs. |
Uptake of SiNPs by HUVECs
ICP-AES was applied for the investigation of cellular uptake of SiNPs. The number of internalized SiNPs was calculated via the silicon content in the lysis solution divided by the number of cells, providing a quantitative measurement of SiNP uptake. Results showed that after incubation with SiNPs for 24 hours, the average silicon content (μg) internalized by each 106 cells was increased along with increased SiNP-exposure dosages, which were 0.12±0.04, 2.33±0.47, 5.46±0.85, 13.87±1.24, and 34.24±5.77 at control, 12.5, 25, 50, 100 μg/mL respectively, suggesting a clear dose-dependent increase in the uptake of the SiNPs by HUVECs.
Oxidative stress and damage triggered by SiNPs
To get a closer insight into cytotoxicity induced by SiNPs, the ROS level was determined by using DCFH-DA probe. As shown in Figure 3A, ROS production significantly increased after 24 hours of SiNP exposure. Furthermore, the fluorescence intensity was enhanced with the increase in the SiNP concentration, suggesting a significant increase in ROS production (Figure 3B). In the 100 μg/mL SiNP-treated group, the fluorescence intensity was significantly elevated (about 3.12-fold higher than that of control). Results demonstrated that SiNPs induced intracellular ROS generation increased in a dose-dependent manner. The overproduction of ROS would break down the balance of the oxidative/antioxidative system, resulting in lipid peroxidation, which is closely related to reduction of antioxidative capacity. Figure 3C shows that the intracellular level of MDA, an indicator of lipid peroxidation, was significantly increased in HUVECs exposed to SiNPs for 24 hours when compared to that in the control group. Furthermore, SiNPs inhibited the activity of GSH-Px (Figure 3E), and upregulated SOD activity (Figure 3D) and intracellular GSH content (Figure 3G) in HUVECs. In addition, the mRNA expressions of SOD, GSH-Px, and the GCL subunits GCLC and GCLM were measured. Significant change was confirmed in the mRNA expressions of SOD2 and GSH-Px, which was perfectly consistent with that in their activities (Figure 3F). Figure 3H showed that GCLC mRNA expression was upregulated by SiNPs, while the levels of GCLM mRNA were not altered. All these results revealed that SiNPs induced oxidative stress via ROS overproduction followed by disorder in the oxidant/antioxidant system and production of MDA.
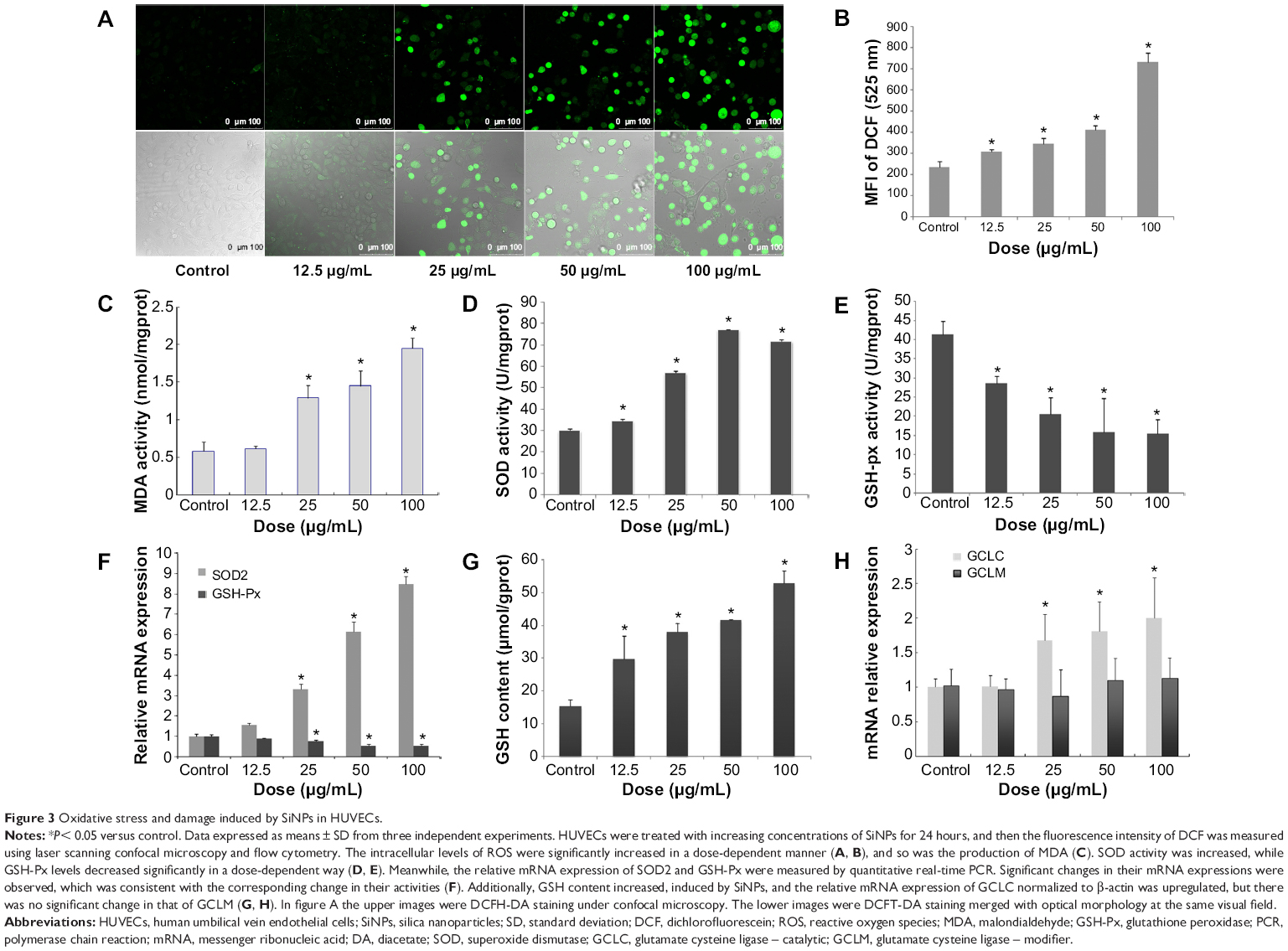 | Figure 3 Oxidative stress and damage induced by SiNPs in HUVECs. |
Imbalanced NO/NOS in HUVECs caused by SiNPs
Results showed that SiNP treatment in HUVECs stimulated a rapid increase in NO content and iNOS mRNA levels, especially the high concentration of 50 or 100 μg/mL (Figure 4A and B). Interestingly, SiNPs produced a decline in eNOS and ET-1 mRNA levels (Figure 4B and C).
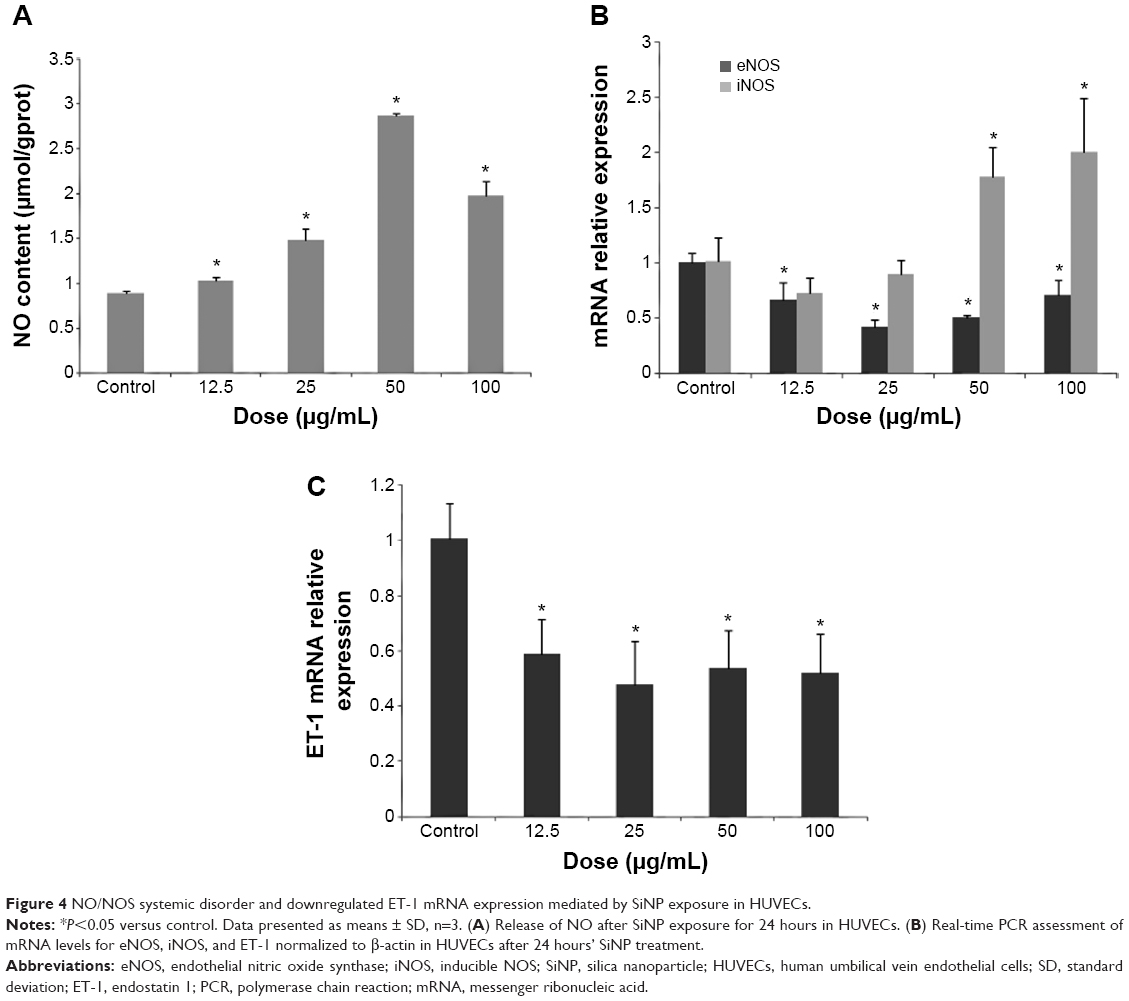 | Figure 4 NO/NOS systemic disorder and downregulated ET-1 mRNA expression mediated by SiNP exposure in HUVECs. |
Inflammatory response induced by SiNPs
To further explore the effect of SiNPs on HUVECs at the molecular level, expressions of key inflammatory cytokines, adhesion molecules, and chemokines were detected. Results demonstrated that the expressions of mRNA for IL-1β, IL-6, IL-8, TNFα, ICAM-1, VCAM-1, and MCP-1 were all upregulated with SiNP exposure, indicating SiNP treatment produced a robust inflammation response in HUVECs (Figure 5). Of interest, ICAM-1 mRNA expression was only significantly upregulated by SiNPs with lower doses (12.5 or 25 μg/mL), but there was no obvious alteration at higher doses (50 or 100 μg/mL). These phenomena might be associated with the severe inhibition in cell viability mediated by SiNPs.
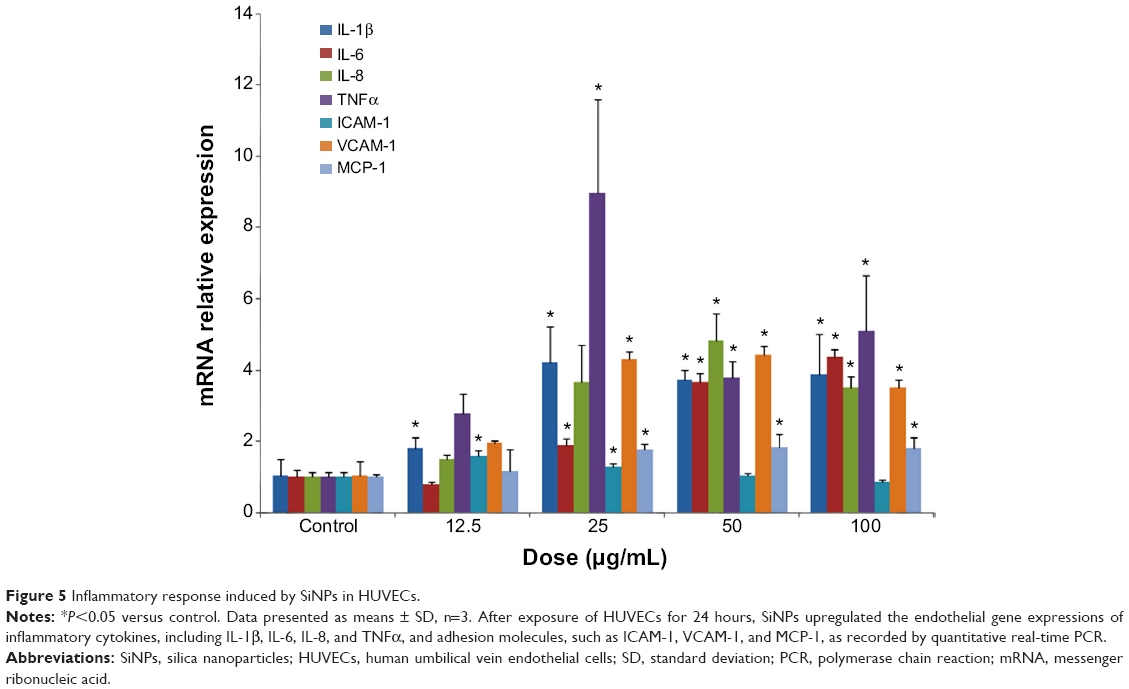 | Figure 5 Inflammatory response induced by SiNPs in HUVECs. |
Activation of Nrf2, NF-κB, and MAPK signaling pathways
To understand the possible mechanism involved in SiNP-induced oxidative stress and injury in HUVECs, the Nrf2-mediated oxidative signaling pathway was investigated by performing quantitative real-time PCR and Western blot analysis. The mRNA expressions of Nrf2 and its key downstream genes tested were upregulated with SiNP exposure in HUVECs (Figure 6A). Consistent with this observation, SiNPs increased the protein expression of Nrf2 and HO-1 (Figure 6B). To further clarify the possible upstream oxidative signaling pathway involved in Nrf2 activation, leading to HO-1 induction, the activation of three key MAPKs – ERK, JNK, and p38 MAPK – was examined. Results demonstrated that expressions of the unphosphorylated forms of MAPKs were constant, regardless of SiNP exposure, whereas expressions of the phosphorylated forms of JNK and p38 MAPK were strongly induced by SiNPs, and that of ERK was suppressed (Figure 6B). Similarly, NF-κB in HUVECs was increased after exposure to SiNPs (Figure 6B).
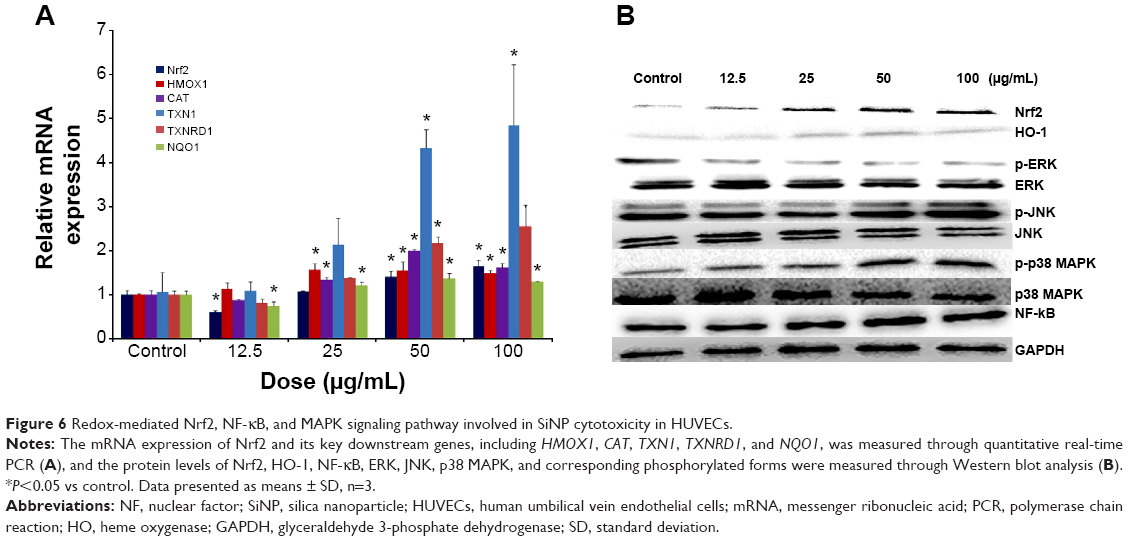 | Figure 6 Redox-mediated Nrf2, NF-κB, and MAPK signaling pathway involved in SiNP cytotoxicity in HUVECs. |
Discussion
Nanomedicine, the use of nanotechnology for biomedical applications, has the potential to change the landscape of the diagnosis and therapy of many diseases. Along with the huge advancement in nanotechnology and material science, taking advantage of unique properties amenable for in vivo applications, SiNPs have emerged as a promising drug-delivery platform to realize the potential of nanomedicine.23 Therefore, it is very likely for SiNPs to enter the bloodstream, and for endothelial cells to be exposed. Liu and Sun demonstrated that SiNPs can induce dysfunction of endothelial cells.24 However, a complete understanding of how SiNPs interact with endothelial cells, particularly at the molecular level, remains unclear and poorly understood. Our findings demonstrated that direct exposure of HUVECs to SiNP-induced intracellular ROS generation led to oxidative stress, imbalance in redox status, and inflammation through the activation of Nrf2, MAPK, and NF-κB pathways, and subsequently to the dysfunction and injury of endothelial cells.
Currently, the cell uptake of NPs is an important issue in designing suitable cell-tracking and drug-carrier nanomaterial systems. Phagocytosis and nonphagocytic endocytosis were involved in the cellular uptake of NPs.10,25 It is increasingly recognized that SiNPs can enter cells easily by endocytosis.19,26 We also confirmed that SiNPs were internalized via the endocytic pathway,27 and mainly accumulated in the cytoplasm.27,28 Besides, the endothelial caveolae, clathrin-coated pits, and macropinocytosis were also involved.29 Cellular or subcellular distribution of NPs has a serious influence on protein or gene expression, and cytotoxicity.30 As indicated in Figure 2, SiNPs induced change in cell morphology, decline in cell viability, and loss of membrane integrity, indicated by LDH release, suggesting SiNPs did result in cytotoxicity in HUVECs. Meanwhile, the endothelial cytotoxicity mediated by SiNPs was increased in a dose- and time-dependent manner. This may be correlated with the amount of silicon taken up by HUVECs, since as the concentrations of SiNPs grew, more SiNPs localizing inside would initiate more deleterious cytotoxicity.
It has been suggested that NP–cell interactions may lead to oxidative stress and oxidant injury.31 NPs not only induce spontaneous ROS generation at the surface, owing to their chemical and surface characteristics, but also lead to free radical generation after their interaction with cellular constituents and components, eg, mitochondrial damage.32 Indeed, previous studies have reported oxidative stress as the toxic mechanism of SiNPs in various cell types.33–35 Oxidative stress arises from a significant increase in concentrations of ROS to levels that are toxic to biomolecules like DNA, proteins, and lipids. Subsequently, the abnormal accumulation of intracellular ROS could induce oxidative stress and cause oxidative damage, followed by the production of lipid peroxidation and inhibition of antioxidant activities, resulting in cells failing to maintain normal physiological redox-regulated functions.
Our previous studies suggested oxidative stress induced by SiNPs is possibly mediated by ROS-dependent pathways in hepatocellular carcinoma cells in vitro.27 In the present study, direct measurement of ROS, as well as the induction of MDA, activities of antioxidant enzymes (eg, SOD, GSH-Px), and GSH, provided strong evidence for the involvement of oxidative stress in SiNP-induced toxicity (Figure 3). MDA, one of the final products of lipid oxidation by free radicals and ROS, is commonly known as a marker of oxidative stress. Lipid peroxidation further gave rise to more free radicals, and then damage biomolecules and cause injury to the cell membrane and cytotoxicity in conjunction with ROS, as indicated by an increased level of LDH enzyme release and inhibition of cell viability (Figure 2). Notably, our results demonstrated an increase in either SOD activity or GSH content, inconsistent with some reports finding inhibition of SOD activity and depletion of reduced GSH once oxidative stress occurred. However, the elevated activities of antioxidant enzymes, like SOD and CAT, and a decrease in GSH level were involved in nickel NP-induced oxidative stress in human skin epidermal cells.36 Meanwhile, the increased SOD activity was consistent with its upregulated expression in mRNA levels.
Research has demonstrated that GCL activity can be rapidly increased following exposure to oxidative stress due to an increase in GCL holoenzyme formation.37 Increased expressions of GCLC and GCLM mRNA can be a direct marker of oxidative stress.38 In our study, GCLC was transcriptionally upregulated by SiNPs, which may further increase GSH synthesis. Weldy et al observed increased levels in endothelial GCLC and GCLM as well as total GSH after diesel exhaust-particulate exposure.39 This phenomenon might be correlated with exposure dosage, time, particle size, and other physicochemical properties of NPs, as well as cell type, etc.
NO plays a key role in endothelial function, and endothelial dysfunction is associated with and may contribute to an increased risk of cardiovascular events.40 Furthermore, it is very critical in proinflammatory responses, and is overproduced in abnormal physiological conditions. ROS induced by SiNPs may trigger the generation of NO,34 consistent with our data, as reflected in Figure 4. Similarly, Corbalan et al demonstrated that SiNPs penetrate the plasma membrane of endothelial cells and stimulate NO release in vitro.19 An increase in NO generation can also lead to higher microvascular permeability.41 Conversely, it has been postulated that the nanomechanics and cytoskeletal rearrangements in endothelial cells result in altered NO production,42 consistent with our observed morphological change induced by SiNPs, as shown in Figure 2. Consequently, the process of increased and prolonged NO production leads to depletion of enzymatic substrates and cofactors and uncoupling of eNOS eventually.43 Uncoupled eNOS can generate superoxide anion radical, O2–•,44 which rapidly reacts with NO to produce peroxynitrite (ONOO−), and change the balance of [NO]/[ONOO−] unfavorably. Certainly, the generated ROS like O2–• mediated by SiNPs could also be involved in the production of ONOO− in the presence of NO. ONOO– can further react with CO2, lipids, etc, to yield the hydroxyl radical •OH and eventually oxidative stress. Another study by Corbalan et al also showed that SiNPs induced a low [NO]/[ONOO−] ratio in human platelets, consequently leading to platelet aggregation.45 Our previous in vivo study suggested SiNPs could lead to endothelial NO/NOS system disorder followed by endothelial dysfunction.46
Upon analysis of endothelial gene expressions, we observed that SiNPs caused a decrease in eNOS mRNA levels while simultaneously upregulating iNOS mRNA expression, as reflected in Figure 4. It was demonstrated that a large amount of NO accumulation produced by iNOS overexpression in damaged tissue forms ONOO−.47 Moreover, the inhibition of eNOS has been shown to impair microvascular barrier function, characterized by the increase in microvascular fluid and protein flux.48,49 Simultaneously, the transcriptional regulation of iNOS is largely mediated by the transcription factor NF-κB, whereas the downregulation of eNOS possibly might be due to negative feedback caused by an increase in available NO, likely iNOS-derived.50 It is becoming increasingly clear that an imbalance between NO and ET-1 is a characteristic of endothelial dysfunction and important in the progression of vascular disease. Numerous reports have demonstrated an increase in plasma ET-1 as well as its mRNA in the lung and heart after particle exposure.51–53 However, a previous report showed a decline in ET-1 expression after diesel exhaust-particulate exposure.39 Our observed downregulating effect of SiNPs on ET-1 expression in HUVECs might have been due to NO-mediated inhibition on EDN1 gene expression.54
There is a growing body of evidence that SiNPs cause toxic and inflammatory effects due to their unique physicochemical profile.55–57 At present, the regulatory mechanism regarding inflammation response mediated by SiNPs exposure is not totally understood. It has been confirmed that ROS generated by SiNPs trigger proinflammatory responses.34,58 Moreover, inflammatory factors are capable of inducing activation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidases and ROS production, subsequently resulting in oxidative stress and impairment in vascular reactivity.59–61 TNFα plays an important role in the promotion of inflammatory response, IL-1β and IL-8 are important for regulation of inflammatory response, and IL-6 is crucial for induction of acute-phase reactions. Excessive production of any of these factors contributes to harmful inflammatory reactions. Strikingly, we observed that endothelial cells had dramatically increased response to SiNPs for the expressions of IL-1β, -6, and -8 and TNFα, suggesting a robust proinflammatory response occurred after SiNP treatment.
It has been reported that decreased viability of endothelial cells may result in the loss of endothelial cell integrity and an increase in vascular permeability, thus leading to an increase in the migration of monocytes and macrophages into vessels in vivo.14,62 Monocyte migration and adhesion to endothelial cells are considered to be one of the early events in the initiation of atherosclerosis, which is caused by the upregulation of adhesion molecules in endothelial cells and the increased expression of chemokines.63 SiNPs can significantly augment proinflammatory and procoagulant responses through CD40-CD40L-mediated monocyte–endothelial cell interactions.64 VCAM-1 and ICAM-1 contributed to the inflammatory process by facilitating the adhesion of monocytes to endothelial cells. Exposure to SiNPs induces protein expression of ICAM-1 and VCAM-1.65 Our data also indicated significantly upregulated mRNA expression in VCAM-1, while ICAM-1 only increased after 12.5 or 25 μg/mL SiNP exposure on HUVECs. Enhanced expression of ICAM-1 was provoked by several kinds of NPs, but varied. For instance, ZnO NPs significantly upregulate ICAM-1 expression at a dose greater than 10 μg/mL,66 while such upregulation requires greater than 20 μg/mL for iron oxide NPs in endothelial cells.14 MCP-1 is known to exacerbate inflammation and has influence on the progression of atherosclerosis.67 It can regulate the transmigration of adhered monocytes across the endothelial barrier into the subendothelial layer.68 Despite belonging to macrophage-derived factor, MCP-1 mRNA expression in endothelial cells was significantly increased under SiNP exposure once the dosage was greater than 25 μg/mL. Our results suggest that SiNPs may have the potential to initiate monocyte recruitment, adhesion, and migration into the subendothelial layer of the intima, representing a considerable risk factor for promoting early events in the development of atherosclerosis.
The role of oxidative stress in inflammatory and endothelial dysfunction is still unclear, but it is a main research area in nanotoxicology. Oxidative stress could trigger the induction of the MAPK signaling pathway, and of transcription factors, such as Nrf2 and NF-κB. These transcription factors could induce mRNA expression of proinflammatory mediators, and finally cause inflammation and related diseases. Therefore, to further elucidate the molecular mechanism of the observed oxidative stress and related physiological alteration by SiNP exposure, the evaluation of toxicity needs to be focused on the oxidative stress-responding signal-transduction pathway (ie, the MAPK pathway) and transcription factors (ie, NF-κB and Nrf2).
Nrf2 has been recognized as a transcription factor that controls mechanisms of cellular defense response against oxidative stress. Under oxidative stress, Nrf2 is stabilized and translocated to the nucleus, where it can transactivate a large battery of antioxidant response element-regulated genes, including endogenous antioxidants (ie, SOD, CAT, GSH-Px, and Trx reductase [TrxR]), phase II detoxifying enzymes (ie, GCL, NAD[P]H:quinone oxidoreductase [NQO]-1, and HO-1), and transporters. The induction of Nrf2 and its downstream gene expression is an adaptive intracellular response to protect against NP-induced oxidative stress and related oxidative damage. For instance, SiNP exposure, which exerts toxic effects, alters the protein regulation of such antioxidant proteins as TrxR in human keratinocyte cells30 and HO-1 in human endothelial cells17 or human bronchial cells.58 Furthermore, human lung fibroblast cells could counteract the oxidative stress induced by SiNPs via the induction of enzymatic activities, such as CAT and GSH S-transferase, an enzyme that catalyzes reactions in order to detoxify peroxidized lipids.69 As indicated in Figure 6A, SiNP-induced activation of Nrf2 was observed, which further triggered expressions of antioxidant genes, such as HO1, CAT, TXN1, TXNRD1, and NQO1, to restore oxidative homeostasis. In addition, the GCLC and GCLM genes also contain antioxidant-response elements within their 5′-promoter region, and can be transcriptionally regulated by the activation of Nrf2.70–72 Therefore, the increase in GCLC mRNA expression might have been Nrf2-mediated, as well as the upregulated SOD mRNA expression referred to in Figure 3F.
To further clarify the possible upstream oxidative signaling pathway involved in Nrf2 activation, we examined MAPK, which is involved in cell survival against oxidative stress through the Nrf2 signaling pathway. The MAPK signaling pathway is classified into three components: ERK1/2, JNK, and p38 MAPK. As seen in Figure 6, SiNPs activated the MAPK signaling pathway via phosphorylation, especially p38 MAPK and JNK. The activation of MAPK signaling molecules could ultimately activate the transcription factor NF-κB,73 suggesting that SiNP-induced upregulated NF-κB expression may occur via the MAPK-Nrf2 signaling pathway. It has been confirmed that the activation of NF-κB and induction of HO-1 via the p38-Nrf2 signaling pathway are involved in oxidative stress induced by ceria NPs,74 the ERK-Nrf2 pathway in that by SiNPs in human bronchial epithelial cells,58 the p38-Nrf2 pathway in the oxidative damage induced by TiO2 NPs,75,76 and the p53 pathway in oxidative stress by ZnO NPs.77 Additionally, NF-κB is a known modulator of inflammation, and its activation is responsive to various oxidant stimuli. For the inflammatory process, NF-κB binds to cognate DNA sequences in the regulatory region of genes involved in inflammatory response, leading to release of immunorelated cytotoxic factors (eg, iNOS) and proinflammatory cytokines, as mentioned earlier through κB sites located in their promoter region,78 implying the strike inflammation response induced by SiNPs in this study might have been related to NF-κB activation to some extent.
Conclusion
In summary, the present study demonstrated that SiNPs induced ROS generation and caused redox imbalance, oxidative stress, and NO/NOS system disorder, leading to oxidative damage and inflammation response, which consequently resulted in endothelial cytotoxicity and endothelial dysfunction via the MAPK-Nrf2 and NF-κB signaling pathways. Further studies on the mechanisms by which SiNPs induce the MAPK-Nrf2 signaling pathway are warranted to better understand NP-induced cytotoxicity by oxidative stress, as are studies with inhibitors and in vivo animal studies. All in all, our findings suggest that SiNP exposure could be a hazardous factor for vascular homeostasis, and more studies need to focus on SiNP exposure, adverse effects, and biological mechanisms in considering the safety of the application of NPs.
Acknowledgments
The authors would like to thank to Professor Wensheng Yang from Jilin University for the preparation of SiNPs. This work was supported by the National Natural Science Foundation of China (81102095, 81202242, 81230065), Project for Youth Talents in Beijing Colleges and Universities (YETP1670), the Opening Project of Beijing Key Laboratory of Clinical Epidemiology (2013LCLB04), and the Beijing Natural Science Foundation Program and Scientific Research Key Program of the Beijing Municipal Commission of Education (KZ201410025022).
Disclosure
The authors report no conflicts of interest in this work.
References
Gwinn MR, Vallyathan V. Nanoparticles: health effects – pros and cons. Environ Health Perspect. 2006;114(12):1818–1825. | ||
Bakand S, Hayes A, Dechsakulthorn F. Nanoparticles: a review of particle toxicology following inhalation exposure. Inhal Toxicol. 2012;24(2):125–135. | ||
Gatoo MA, Naseem S, Arfat MY, Dar AM, Qasim K, Zubair S. Physicochemical properties of nanomaterials: implication in associated toxic manifestations. Biomed Res Int. 2014;2014:498420. | ||
Hansen SF, Michelson ES, Kamper A, Borling P, Stuer-Lauridsen F, Baun A. Categorization framework to aid exposure assessment of nanomaterials in consumer products. Ecotoxicology. 2008;17(5):438–447. | ||
Yang Y, Li J. Lipid, protein and poly(NIPAM) coated mesoporous silica nanoparticles for biomedical applications. Adv Colloid Interface Sci. 2014;207:155–163. | ||
Mai WX, Meng H. Mesoporous silica nanoparticles: a multifunctional nano therapeutic system. Integr Biol (Camb). 2013;5(1):19–28. | ||
Malvindi MA, Brunetti V, Vecchio G, Galeone A, Cingolani R, Pompa PP. SiO2 nanoparticles biocompatibility and their potential for gene delivery and silencing. Nanoscale. 2012;4(2):486–495. | ||
Liang H, Jin C, Tang Y, Wang F, Ma C, Yang Y. Cytotoxicity of silica nanoparticles on HaCaT cells. J Appl Toxicol. 2014;34(4):367–372. | ||
Lu X, Qian J, Zhou H, et al. In vitro cytotoxicity and induction of apoptosis by silica nanoparticles in human HepG2 hepatoma cells. Int J Nanomedicine. 2011;6:1889–1901. | ||
Zhu J, Liao L, Zhu L, et al. Size-dependent cellular uptake efficiency, mechanism, and cytotoxicity of silica nanoparticles toward HeLa cells. Talanta. 2013;107:408–415. | ||
Fruijtier-Polloth C. The toxicological mode of action and the safety of synthetic amorphous silica – a nanostructured material. Toxicology. 2012;294(2–3):61–79. | ||
Shannahan JH, Kodavanti UP, Brown JM. Manufactured and airborne nanoparticle cardiopulmonary interactions: a review of mechanisms and the possible contribution of mast cells. Inhal Toxicol. 2012;24(5):320–339. | ||
Nemmar A, Holme JA, Rosas I, Schwarze PE, Alfaro-Moreno E. Recent advances in particulate matter and nanoparticle toxicology: a review of the in vivo and in vitro studies. Biomed Res Int. 2013;2013:279371. | ||
Zhu MT, Wang B, Wang Y, et al. Endothelial dysfunction and inflammation induced by iron oxide nanoparticle exposure: risk factors for early atherosclerosis. Toxicol Lett. 2011;203(2):162–171. | ||
Bai N, Khazaei M, van Eeden SF, Laher I. The pharmacology of particulate matter air pollution-induced cardiovascular dysfunction. Pharmacol Ther. 2007;113(1):16–29. | ||
Albini A, Mussi V, Parodi A, et al. Interactions of single-wall carbon nanotubes with endothelial cells. Nanomedicine. 2010;6(2):277–288. | ||
Napierska D, Thomassen LCJ, Rabolli V, et al. Size-dependent cytotoxicity of monodisperse silica nanoparticles in human endothelial cells. Small. 2009;5(7):846–853. | ||
Bauer AT, Strozyk EA, Gorzelanny C, et al. Cytotoxicity of silica nanoparticles through exocytosis of von Willebrand factor and necrotic cell death in primary human endothelial cells. Biomaterials. 2011; 32(33):8385–8393. | ||
Corbalan JJ, Medina C, Jacoby A, Malinski T, Radomski MW. Amorphous silica nanoparticles trigger nitric oxide/peroxynitrite imbalance in human endothelial cells: inflammatory and cytotoxic effects. Int J Nanomedicine. 2011;6:2821–2835. | ||
Peters K, Unger RE, Kirkpatrick CJ, Gatti AM, Monari E. Effects of nano-scaled particles on endothelial cell function in vitro: studies on viability, proliferation and inflammation. J Mater Sci Mater Med. 2004; 15(4):321–325. | ||
Duan JC, Yu YB, Li Y, et al. Toxic effect of silica nanoparticles on endothelial cells through DNA damage response via Chk1-dependent G2/M checkpoint. Plos One. 2013;8(4):e62087. | ||
Li YB, Zhang HX, Guo CX, et al. Cytotoxicity and DNA damage effect of TGA-capped CdTe quantum dots. Chem Res Chin Univ. 2012; 28(2):276–281. | ||
Tang L, Cheng J. Nonporous silica nanoparticles for nanomedicine application. Nano Today. 2013;8(3):290–312. | ||
Liu X, Sun JA. Endothelial cells dysfunction induced by silica nanoparticles through oxidative stress via JNK/P53 and NF-κB pathways. Biomaterials. 2010;31(32):8198–8209. | ||
Rampazzo E, Voltan R, Petrizza L, et al. Proper design of silica nanoparticles combines high brightness, lack of cytotoxicity and efficient cell endocytosis. Nanoscale. 2013;5(17):7897–7905. | ||
He Q, Zhang Z, Gao Y, Shi J, Li Y. Intracellular localization and cytotoxicity of spherical mesoporous silica nano- and microparticles. Small. 2009;5(23):2722–2729. | ||
Sun L, Li Y, Liu XM, et al. Cytotoxicity and mitochondrial damage caused by silica nanoparticles. Toxicol In Vitro. 2011;25(8):1619–1629. | ||
Yu Y, Duan J, Li Y, et al. Silica nanoparticles induce autophagy and autophagic cell death in HepG2 cells triggered by reactive oxygen species. J Hazard Mater. 2014;270:176–186. | ||
Nishikawa T, Iwakiri N, Kaneko Y, et al. Nitric oxide release in human aortic endothelial cells mediated by delivery of amphiphilic polysiloxane nanoparticles to caveolae. Biomacromolecules. 2009;10(8):2074–2085. | ||
Yang X, Liu J, He H, et al. SiO2 nanoparticles induce cytotoxicity and protein expression alteration in HaCaT cells. Part Fibre Toxicol. 2010;7:1. | ||
Li N, Xia T, Nel AE. The role of oxidative stress in ambient particulate matter-induced lung diseases and its implications in the toxicity of engineered nanoparticles. Free Radic Bio Med. 2008;44(9):1689–1699. | ||
Singh S, Shi TM, Duffin R, et al. Endocytosis, oxidative stress and IL-8 expression in human lung epithelial cells upon treatment with fine and ultrafine TiO2: role of the specific surface area and of surface methylation of the particles. Toxicol Appl Pharm. 2007;222(2):141–151. | ||
Lin W, Huang YW, Zhou XD, Ma Y. In vitro toxicity of silica nanoparticles in human lung cancer cells. Toxicol Appl Pharmacol. 2006;217(3): 252–259. | ||
Park EJ, Park K. Oxidative stress and pro-inflammatory responses induced by silica nanoparticles in vivo and in vitro. Toxicol Lett. 2009; 184(1):18–25. | ||
Nemmar A, Beegam S, Yuvaraju P, Yasin J, Shahin A, Ali BH. Interaction of amorphous silica nanoparticles with erythrocytes in vitro: role of oxidative stress. Cell Physiol Biochem. 2014;34(2):255–265. | ||
Alarifi S, Ali D, Alakhtani S, Al Suhaibani ES, Al-Qahtani AA. Reactive oxygen species-mediated DNA damage and apoptosis in human skin epidermal cells after exposure to nickel nanoparticles. Biol Trace Elem Res. 2014;157(1):84–93. | ||
Krejsa CM, Franklin CC, White CC, Ledbetter JA, Schieven GL, Kavanagh TJ. Rapid activation of glutamate cysteine ligase following oxidative stress. J Biol Chem. 2010;285(21):16116–16124. | ||
Thompson SA, White CC, Krejsa CM, et al. Induction of glutamate-cysteine ligase (γ-glutamylcysteine synthetase) in the brains of adult female mice subchronically exposed to methylmercury. Toxicol Lett. 1999;110(1–2):1–9. | ||
Weldy CS, Wilkerson HW, Larson TV, Stewart JA, Kavanagh TJ. DIESEL particulate exposed macrophages alter endothelial cell expression of eNOS, iNOS, MCP1, and glutathione synthesis genes. Toxicol In Vitro. 2011;25(8):2064–2073. | ||
Bonetti PO, Lerman LO, Lerman A. Endothelial dysfunction: a marker of atherosclerotic risk. Arterioscler Thromb Vasc Biol. 2003;23(2): 168–175. | ||
Durán WN, Breslin JW, Sánchez FA. The NO cascade, eNOS location, and microvascular permeability. Cardiovasc Res. 2010;87(2):254–261. | ||
Fels J, Callies C, Kusche-Vihrog K, Oberleithner H. Nitric oxide release follows endothelial nanomechanics and not vice versa. Pflugers Arch. 2010;460(5):915–923. | ||
Huk I, Nanobashvili J, Neumayer C, et al. L-arginine treatment alters the kinetics of nitric oxide and superoxide release and reduces ischemia/reperfusion injury in skeletal muscle. Circulation. 1997;96(2):667–675. | ||
Kalinowski L, Dobrucki IT, Malinski T. Race-specific differences in endothelial function – predisposition of African Americans to vascular diseases. Circulation. 2004;109(21):2511–2517. | ||
Corbalan JJ, Medina C, Jacoby A, Malinski T, Radomski MW. Amorphous silica nanoparticles aggregate human platelets: potential implications for vascular homeostasis. Int J Nanomedicine. 2012;7:631–639. | ||
Du Z, Zhao D, Jing L, et al. Cardiovascular toxicity of different sizes amorphous silica nanoparticles in rats after intratracheal instillation. Cardiovasc Toxicol. 2013;13(3):194–207. | ||
Sugawara T, Chan PH. Reactive oxygen radicals and pathogenesis of neuronal death after cerebral ischemia. Antioxid Redox Sign. 2003;5(5):597–607. | ||
Kubes P. Nitric oxide affects microvascular permeability in the intact and inflamed vasculature. Microcirculation. 1995;2(3):235–244. | ||
Whittle BJ. Nitric oxide – a mediator of inflammation or mucosal defence. Eur J Gastroenterol Hepatol. 1997;9(11):1026–1032. | ||
Zhen J, Lu H, Wang XQ, Vaziri ND, Zhou XJ. Upregulation of endothelial and inducible nitric oxide synthase expression by reactive oxygen species. Am J Hypertens. 2008;21(1):28–34. | ||
Zhao J, Xie Y, Qian X, Jiang R, Song W. Acute effects of fine particles on cardiovascular system: differences between the spontaneously hypertensive rats and Wistar Kyoto rats. Toxicol Lett. 2010;193(1):50–60. | ||
Peretz A, Sullivan JH, Leotta DF, et al. Diesel exhaust inhalation elicits acute vasoconstriction in vivo. Environ Health Perspect. 2008;116(7):937–942. | ||
Thomson EM, Kumarathasan P, Calderón-Garcidueñas L, Vincent R. Air pollution alters brain and pituitary endothelin-1 and inducible nitric oxide synthase gene expression. Environ Res. 2007;105(2):224–233. | ||
Weng YH, Kuo CY, Chiu YW, Kuo ML, Liao SL. Alteration of nitric oxide gas on gene expression of endothelin-1 and endothelial nitric oxide synthase by a time- and dose-dependent manner in human endothelial cells. Chin J Physiol. 2009;52(2):59–64. | ||
Kasper J, Hermanns MI, Bantz C, et al. Inflammatory and cytotoxic responses of an alveolar-capillary coculture model to silica nanoparticles: xomparison with conventional monocultures. Part Fibre Toxicol. 2011;8(1):6. | ||
Yang L, Yan QQ, Zhao J, et al. The role of potassium channel in silica nanoparticle-induced inflammatory effect in human vascular endothelial cells in vitro. Toxicol Lett. 2013;223(1):16–24. | ||
Mendoza A, Torres-Hernandez JA, Ault JG, Pedersen-Lane JH, Gao D, Lawrence DA. Silica nanoparticles induce oxidative stress and inflammation of human peripheral blood mononuclear cells. Cell Stress Chaperones. 2014;19(6):777–790. | ||
Eom HJ, Choi J. Oxidative stress of silica nanoparticles in human bronchial epithelial cell, Beas-2B. Toxicol In Vitro. 2009;23(7):1326–1332. | ||
Kampfrath T, Maiseyeu A, Ying ZK, et al. Chronic fine particulate matter exposure induces systemic vascular dysfunction via NADPH oxidase and TLR4 pathways. Circ Res. 2011;108(6):716–726. | ||
Sun QH, Yue PB, Ying ZK, et al. Air pollution exposure potentiates hypertension through reactive oxygen species-mediated activation of Rho/ROCK. Arterioscler Thromb Vasc Biol. 2008;28(10):1760–1766. | ||
Gao X, Belmadani S, Picchi A, et al. Tumor necrosis factor-alpha induces endothelial dysfunction in Lepr(db) mice. Circulation. 2007;115(2):245–254. | ||
Bobryshev YV. Monocyte recruitment and foam cell formation in atherosclerosis. Micron. 2006;37(3):208–222. | ||
Libby P. Inflammation in atherosclerosis. Nature. 2002;420(6917):868–874. | ||
Liu X, Xue Y, Ding TT, Sun J. Enhancement of proinflammatory and procoagulant responses to silica particles by monocyte-endothelial cell interactions. Part Fibre Toxicol. 2012;9:36. | ||
Napierska D, Quarck R, Thomassen LC, et al. Amorphous silica nanoparticles promote monocyte adhesion to human endothelial cells: size-dependent effect. Small. 2013;9(3):430–438. | ||
Gojova A, Guo B, Kota RS, Rutledge JC, Kennedy IM, Barakat AI. Induction of inflammation in vascular endothelial cells by metal oxide nanoparticles: effect of particle composition. Environ Health Persp. 2007;115(3):403–409. | ||
Deshmane SL, Kremlev S, Amini S, Sawaya BE. Monocyte chemoattractant protein-1 (MCP-1): an overview. J Interferon Cytokine Res. 2009;29(6):313–326. | ||
Choudhury RP, Lee JM, Greaves DR. Mechanisms of disease: macrophage-derived foam cells emerging as therapeutic targets in atherosclerosis. Nat Clin Pract Cardiovasc Med. 2005;2(6):309–315. | ||
Munteanu MC, Radu M, Hermenean A, et al. Antioxidative response induced by SiO2 nanoparticles in MRC5 cell line. Rom Biotechnol Lett. 2010;15(1):5000–5007. | ||
Bea F, Hudson FN, Chait A, Kavanagh TJ, Rosenfeld ME. Induction of glutathione synthesis in macrophages by oxidized low-density lipoproteins is mediated by consensus antioxidant response elements. Circ Res. 2003;92(4):386–393. | ||
Bea F, Hudson FN, Neff-LaFord H, et al. Homocysteine stimulates antioxidant response element-mediated expression of glutamate-cysteine ligase in mouse macrophages. Atherosclerosis. 2009;203(1):105–111. | ||
Ungvari Z, Bailey-Downs L, Gautam T, et al. Adaptive induction of NF-E2-related factor-2-driven antioxidant genes in endothelial cells in response to hyperglycemia. Am J Physiol Heart Circ Physiol. 2011;300(4):H1133–H1140. | ||
Liu HE, Chang AS, Teng CM, Chen CC, Tsai AC, Yang CR. Potent anti-inflammatory effects of denbinobin mediated by dual inhibition of expression of inducible NO synthase and cyclooxygenase 2. Shock. 2011;35(2):191–197. | ||
Eom HJ, Choi J. Oxidative stress of CeO2 nanoparticles via p38-Nrf-2 signaling pathway in human bronchial epithelial cell, Beas-2B. Toxicol Lett. 2009;187(2):77–83. | ||
Ze YG, Zheng L, Zhao XY, et al. Molecular mechanism of titanium dioxide nanoparticles-induced oxidative injury in the brain of mice. Chemosphere. 2013;92(9):1183–1189. | ||
Wang J, Li N, Zheng L, et al. P38-Nrf-2 signaling pathway of oxidative stress in mice caused by nanoparticulate TiO2. Biol Trace Elem Res. 2011;140(2):186–197. | ||
Setyawati MI, Tay CY, Leong DT. Effect of zinc oxide nanomaterials-induced oxidative stress on the p53 pathway. Biomaterials. 2013;34(38):10133–10142. | ||
Hoesel B, Schmid JA. The complexity of NF-κB signaling in inflammation and cancer. Mol Cancer. 2013;12:86. |
Supplementary material
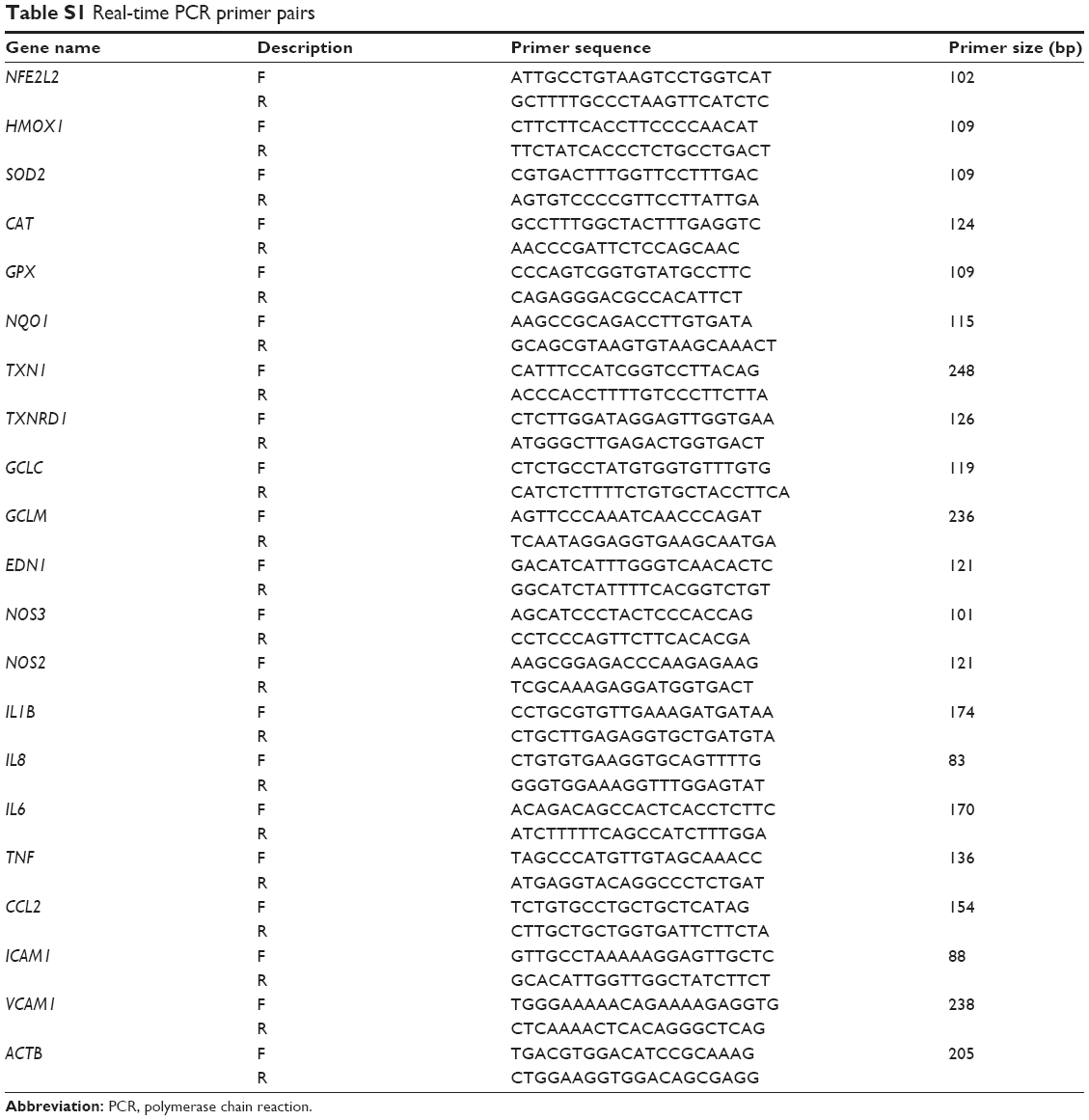 | Table S1 Real-time PCR primer pairs |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.