Back to Archived Journals » Reports in Organic Chemistry » Volume 5
Recent advances in green fluorine chemistry
Received 1 January 2015
Accepted for publication 28 January 2015
Published 14 May 2015 Volume 2015:5 Pages 19—39
DOI https://doi.org/10.2147/ROC.S80171
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sean Kerwin
Yuwen Zeng, Jinbo Hu
Key Laboratory of Organofluorine Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai, People’s Republic of China
Abstract: Fluorinated compounds are intriguing for the development of pharmaceuticals, agrochemicals, and materials, and thus, much effort has been exerted to develop more general and efficient approaches for introducing fluorine atom(s) or fluoroalkyl group(s) into organic molecules. Although many traditional methods usually cannot satisfy the simplicity and cost efficiency for industrial production and laboratorial synthesis, significant progress in green fluorine chemistry has been made in recent years, allowing efficient incorporation of fluorine into complex organic molecules. The main purpose of this review is to describe recent advances in organofluorine chemistry, with an emphasis on generality, selectivity, and environmental friendliness of related methods.
Keywords: green chemistry, organofluorine chemistry, fluorination, perfluoroalkylation, trifluoromethylation
Introduction
Fluorinated organic compounds are of particular interest in the fields of functional materials, pharmaceuticals, and agrochemicals. Notably, nearly 20% of pharmaceutical compounds contain at least one fluorine atom.1 These wide applications can be attributed to the unique metabolic stability, binding affinity, and biological properties of the molecules brought by C–F bonds.2 On the one hand, with an average bond dissociation energy of 105.4 kcal/mol, the C–F bond is one of the strongest bonds in organic chemistry;3 on the other hand, due in some cases to its high activation energy, C–F bond formation is a significant challenge.4
Traditional approaches to achieve fluorination/perfluorination include Balz–Schiemann reaction (Equation 1), halogen exchange (halex) process (Equation 2), Swarts reaction (Equation 3), and electrofluorination (Simons process, Equation 4).

Balz–Schiemann reaction and the halex reaction are used for aromatic fluorination. Balz–Schiemann reaction, which was first reported in the late 1920s,5 represents one of the most general methods for the introduction of a single fluorine into an aromatic ring (Equation 1). Despite most of the commercially available aryl fluorides are synthesized by this approach, the explosive nature of aryl diazonium salts and the requirement of high reaction temperatures retard its application in laboratory. The halex reaction (Equation 2),6 which typically proceeds via an SNAr pathway, is usually carried out at an elevated temperature and limited to electron-deficient aromatic substrates.
Swarts reaction and Simons electrochemical fluorination are applicable for aliphatic fluorination. Swarts reaction was first reported by Belgium chemist Swarts in 1892.7 By treating alkyl chloride with SbF3, chlorinated compounds are converted to the corresponding fluorinated ones (Equation 3). In the early 1930s, HF was found to be an alternative to SbF3 in Swarts reaction.8 This approach has been used for the industrial production of Freons such as HCF2Cl and other fluorinated compounds. However, both SbF3 and HF are hazardous reagents and during this transformation, a large amount of HCl is generated as by-product. Simons electrofluorination, an application of electrosynthesis in organofluorine chemistry, is another foundational method for the preparation of organofluorine compounds (Equation 4). The Simons process, which can be regarded as an improvement in fluorination with dangerous fluorine gas, was developed by Simons in the 1930s but published in 1949.9 This electrochemical fluorination process has been used for the production of perfluorinated amines, ethers, as well as acyl and sulfonyl fluorides. However, compared with fluorination with fluorine gas, this cost-effective process may result in low yields.
One of the main goals of chemical society is to make the current chemical processes “greener” and more atom economic. From this perspective, the generation and use of toxic materials should be avoided. Additionally, conventional fluorination/perfluoroalkylation methods are restricted to those electron-deficient or pre-activated substrates, and they are only suitable for the synthesis of simple fluorinated molecules with a narrow substrate scope. In this regard, fluorination/perfluoroalkylation of unactivated compounds in an eco-friendly way is highly desired by both industry and academia.
In the last 5 years, much effort has been directed toward finding new synthetic approaches to fluorine-containing molecules. These new methods use unactivated substrates and safe, environmentally benign reagents and can readily access desired molecules that are otherwise difficult to synthesize with traditional approaches. Although most of these reactions may not fulfill the ideal green chemistry, they are great improvement over the traditional methods and are somewhat “green”. This review aims to describe recent advances in the efficient fluorination and fluoroalkylation methods, with an emphasis on the green reactions possessing high generality, selectivity, and environmental friendliness.
Advances in fluorination
C–F bond formation for the synthesis of aryl fluorides
In 2006, Hull et al reported the first palladium-catalyzed fluorination of C–H bonds (Figure 1).10 Previous to this work, Grushin demonstrated that C–F bond reductive elimination from PdII center is extremely challenging.11 Ball and Sanford reasoned that this key C–F coupling step can be facilitated when PdII complex is oxidized to PdIV. The presence of PdIV–F complex was confirmed in the subsequent mechanistic studies.12
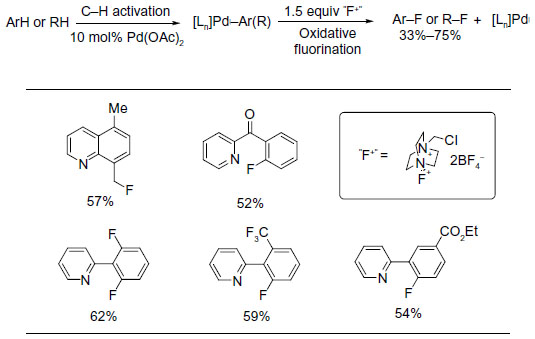 | Figure 1 Palladium-catalyzed fluorination of C–H bonds. |
The first Pd-catalyzed aryl fluorination involving Pd0/PdII was reported in 2009.13 To achieve the reductive elimination process from a PdII complex, a sterically demanding, electron-rich biarylmonophosphine ligand (tBuBrettPhos) is needed (Figure 2). It was believed that this ligand not only promotes reductive elimination of Ar–F because of its large size but also prevents the formation of dimeric complexes [LPdAr(F)]2. It is worth noting that regioisomeric products were formed in a few cases, which indicates that aryne intermediates may be involved in a competing pathway.14,15
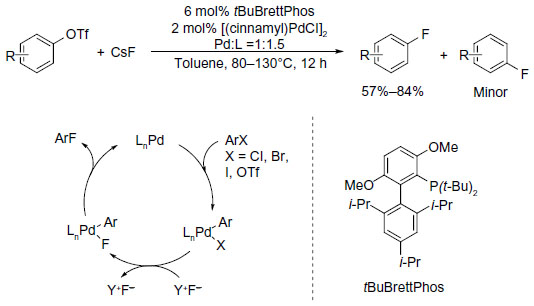 | Figure 2 Palladium-catalyzed formation of aryl fluorides. |
18F-labeled molecules are excellent radiotracers for positron emission tomography (PET).16 However, the short half-life of 18F (110 minutes) requires a rapid construction of the C–F bond, which indicates that the fluorine should be introduced into molecules at a late stage of the synthesis to diminish the decay of 18F species. The 18F-labeled molecules could be synthesized by either electrophilic or nucleophilic fluorination reactions. However, electrophilic fluorination reagents, such as 18F2 and [18F]-N-F reagents, are produced in lower specific activity than [18F]-fluoride.16–18 Additionally, [18F]-fluoride is readily available and easy to handle, which makes it more adaptable in PET imaging.
In 2011, Lee et al reported a new late-stage electrophilic fluorination reagent for PET imaging (Figure 3).19 Treatment of the reagent (a PdIV complex) with potassium fluoride (KF) affords a new PdIV–F complex within 5 minutes, which could further react with Ar-Pd complex to give the corresponding Ar–F in 10 minutes. It should be noted that during this transformation, a net [18F]-fluoride umpolung is realized. This method is used for the synthesis of 18F-labeled aryl fluorides, affording radiochemical yields (RCYs) up to 33%. Additionally, this fluorination reagent is stable at room temperature and can be manipulated briefly in air. Thus, it can be prepared on a large scale, stored, and used when needed.
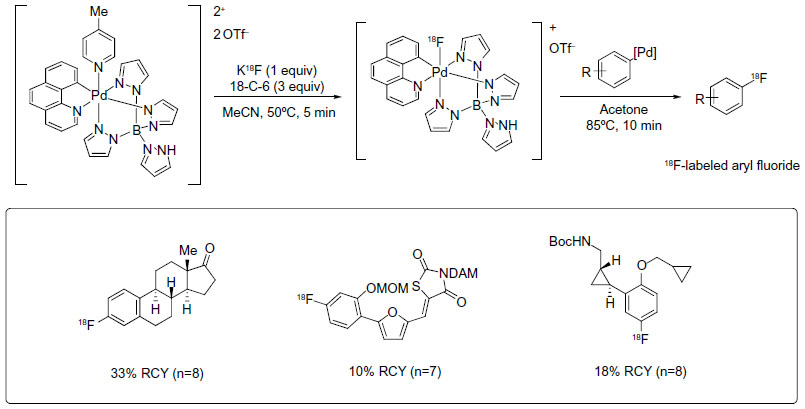 | Figure 3 Palladium-mediated late-stage fluorination for PET imaging. |
Although the method mentioned above is the first oxidative fluorination using fluoride as the fluorine source, this protocol has some drawbacks for 18F-labeled synthesis: the two-step sequence increases reaction time and makes the method less suitable for non-chemists. Moreover, the fluoride must be dry, which makes the reaction procedure not user friendly. To overcome these inherent limitations of the palladium chemistry, Lee et al developed a nickel-mediated oxidative fluorination reaction using the similar [18F]-fluoride umpolung protocol.20 Under the action of an external oxidant, the combination of an aqueous fluoride source and aryl nickel complexes enables practical, one-step access to fluorinated arenes (Figure 4). This reaction proceeds in less than 1 minute and is not only successful for the synthesis of both electron-rich and electron-poor, highly functionalized aryl fluorides but also applicable for the synthesis of alkenyl fluorides. Furthermore, the nickel complexes are moisture- and air-stable solids and can be stored under air.
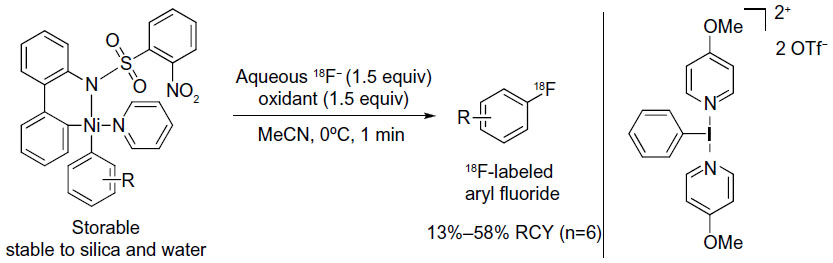 | Figure 4 Nickel-mediated oxidative fluorination for PET imaging. |
Palladium(II)/(III) redox system has also been employed in fluorination. In 2013, Mazzotti et al developed palladiumIII-catalyzed fluorination of aryl boronic acids (Figure 5).21 Unlike the previously reported Pd-mediated fluorination,13,19 the C–F bond formation presented here seems to proceed without the formation of organopalladium complexes. The author proposed a single-electron-transfer (SET) mechanism involving a well-defined PdIII intermediate. First, the PdII complex is oxidized by Selectfluor through an SET pathway to give PdIII intermediate and Selectfluor radical cation, which undergoes fluorine atom transfer with aryl trifluoroborate to form the C–F bond and generate a delocalized aryl radical. SET from this radical to PdIII complex regenerates the PdII catalyst and affords a delocalized aryl cation, which converts to the aryl fluoride after loss of BF3. The Pd-catalyzed fluorination reaction proceeds under mild conditions and is tolerant with moisture. However, this method is ineffective for fluorination of heterocycles. Furthermore, substrates bearing electron-withdrawing groups are likely to give constitutional isomers which are difficult to separate from the desired products.
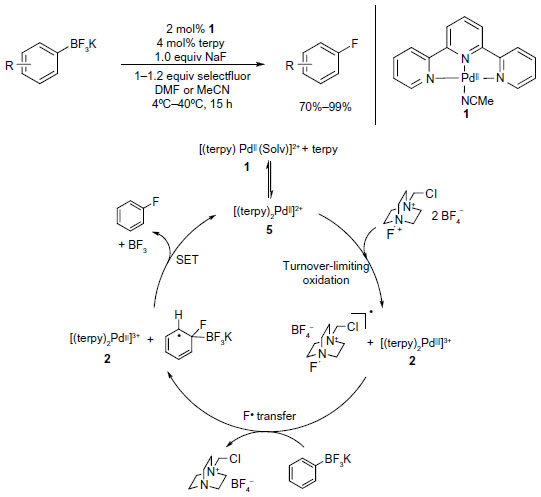 | Figure 5 Palladium(III)-catalyzed fluorination of aryl boronic acids. |
C–F bond reductive elimination from high-valence metal center is not restricted to palladium and nickel. In 2009, Furuya et al described the silver-catalyzed electrophilic fluorination of aryl stannanes (Figure 6, left).22 This catalytic transformation is applicable for late-stage fluorination of natural products and their derivatives. The proposed mechanism is distinct from conventional cross-coupling reactions and features a multinuclear high-valent aryl silver fluoride complex. It was suggested that, compared with mononuclear complexes, the AgII–AgII redox interactions may reduce the energy barrier of C–F reductive elimination.
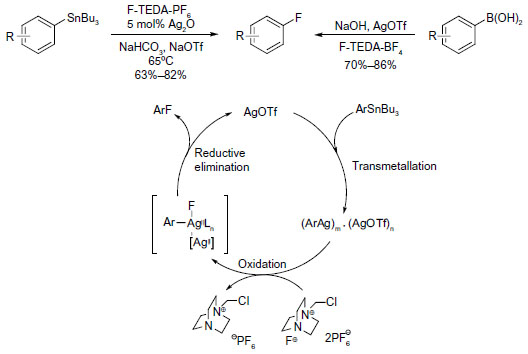 | Figure 6 Silver-mediated fluorination of aryl stannanes and aryl boronic acids. |
Considering that organotin compounds are expensive and toxic, Furuya and Ritter developed a new protocol,23 namely using readily available, nontoxic aryl boronic acids instead of aryl stannanes as aryl sources (Figure 6, right). However, transmetallation from boron to silver is too fast to develop a catalytic cycle; thus, in this case, stoichiometric silver triflate is needed to prepare aryl silver in situ.
Copper has also been introduced in fluorination reactions. In 2002, a CuII-promoted fluorination of arene was reported (Figure 7),24 in which CuF2 served as an electrophilic fluorination reagent and is converted into Cu(0) and HF. After fluorination, a stream of HF/O2 was passed over the catalyst at 350°C–400°C to regenerate the CuF2. Compared with the classical synthetic route (Balz–Schiemann reaction), this method is “greener” and more cost-effective. However, due to its harsh conditions, only structurally simple aryl fluorides, such as fluorobenzenes, fluorotoluenes, and difluorobenzenes, can be synthesized, and the regioselectivity is low.
 | Figure 7 Copper(II) fluoride-mediated fluorination of arenes. |
In 2012, Fier and Hartwig described a copper-mediated fluorination of aryl iodides (Figure 8, left).25 In this approach, silver fluoride is used as both the nucleophilic fluorine source and trapping reagent for iodide ions. However, to achieve a moderate yield, the reaction was carried out at a high temperature (140°C) and with excess of Cu(I) salt and AgF. Furthermore, it is difficult to separate the desired fluorination products from the protonated byproducts. After preliminary mechanistic studies, pathways involving radical intermediates and aryne intermediates were ruled out. The author proposed that this reaction involves oxidative addition to form a Cu(III) intermediate and C–F bond reductive elimination from an ArCu(III)–F intermediate.
 | Figure 8 Copper-mediated fluorination of aryl iodides and aryl boronate esters. |
Only 1 year later, Fier et al reported another copper-mediated fluorination reaction, that is, copper-mediated electrophilic fluorination of aryl boronate esters (Figure 8, right).26 Both electron-rich and electron-deficient aryl boronate esters are suitable for this reaction. Substrates containing ester, ketone, aldehyde, amide, nitrile, halide, and heterocycle functionalities undergo fluorination in moderate-to-good yield. The major side reaction is protodeborylation, which forms the corresponding arenes. Deuterium-labeling experiments suggest that the proton comes from the adventitious water in the reaction system. After extensively studying the reaction mechanism, the authors concluded that fluorination does not occur via the formation of ArCu(I) species. Instead, a cationic Cu(III) fluoride intermediate is in situ generated and reacts with the combination of AgF and aryl boronate esters.
Shortly after, Ye and Sanford published a similar protocol for copper-mediated fluorination of aryl stannanes and aryl trifluoroborates (Figure 9).27 In this method, the electrophilic fluorination reagent severs as both an oxidant for Cu(I) and a fluorine source. Furthermore, the reactions proceed under very mild conditions (at room temperature in many cases).
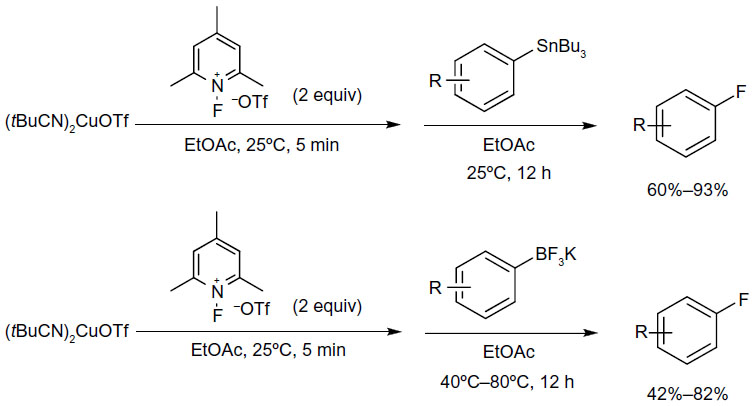 | Figure 9 Copper-mediated fluorination of aryl stannanes and aryl trifluoroborates. |
Although electrophilic fluorination reagents are widely used as both excellent oxidants and fluorine sources in many transition-metal-promoted fluorination reactions, these reagents do have limitations as they are too expensive for large-scale applications and not suitable for PET imaging applications. Ye et al reasoned that the combination of an additional oxidant and a nucleophilic fluorine source could also enable the copper-promoted fluorination of aryl boron derivatives (Figure 10).28 Functional groups including ketones, esters, and aldehydes are compatible with this method. Pyridine derivatives also undergo fluorination, albeit in low yields. However, substrates containing chloride, bromide, and iodide on the aromatic ring are susceptible to undergo competing halodeboronation under the reaction conditions. In this reaction, Cu(OTf)2 is both a transition-metal promoter and an oxidant for C–F bond formation.
 | Figure 10 Copper(II)-mediated fluorination of aryl trifluoroborates with KF. |
Besides late transition metals, main group metals such as magnesium can also be used in fluorination reactions. In 2010, Yamada et al reported an efficient electrophilic fluorination of functionalized aryl and heteroaryl Grignard reagents (Figure 11).29 The functionalized Grignard reagents can be prepared either by halogen–magnesium exchange or by a direct insertion of Mg in the presence of LiCl. The authors found that direct fluorination of Grignard reagents is inefficient. It is believed that the formation of the protonated arene as a side product can be attributed to the formation of a radical intermediate, which abstracts a hydrogen atom from the solvent. Thus, when the original solvent (tetrahydrofuran) was replaced by other solvents with poor hydrogen atom-donating ability, a dramatic improvement in reaction yield was observed. After optimization, the mixture of perfluorodecalin and dichloromethane was found to be a good solvent system, affording the fluorination products in up to 94% yield.
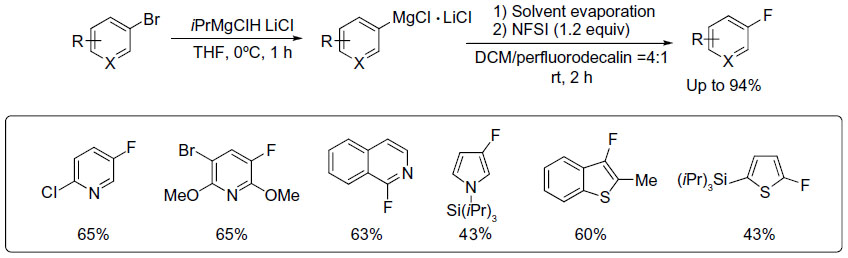 | Figure 11 Electrophilic fluorination of aryl magnesium reagents. |
Phenols are also good substrates for deoxyfluorination. In 2011, Tang et al developed a new deoxyfluorination reagent, Phenfluor, for one-step fluorination of phenols (Figure 12).30 This reagent can be handled in air as a solid but slowly hydrolyzes in a wet atmosphere. Both electron-rich and electron-deficient functional groups are tolerant. However, strong hydrogen-bond donors such as alcohols are not tolerated. Mechanistic studies showed that there is a hydrogen bond between one hydrogen atom of the imidazolium heterocycle and bifluoride counteranion in the crystal structure of reaction intermediate. It is suggested that the hydrogen bonding renders the uronium a better nucleofuge and thus facilitates fluorination reactions.
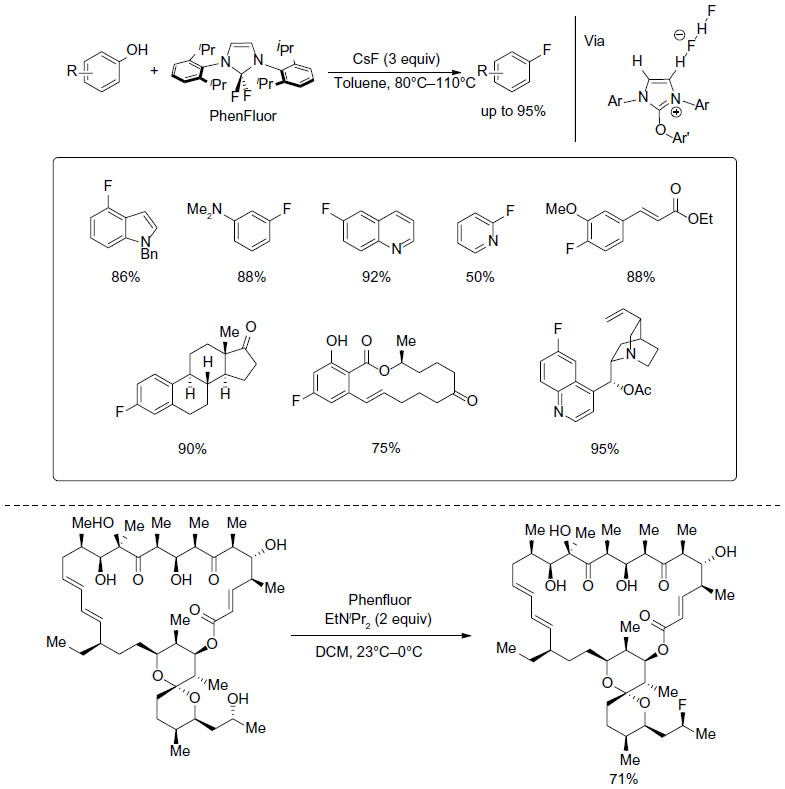 | Figure 12 Deoxyfluorination of phenols and alcohols. |
Although Phenfluor was developed for deoxyfluorination of phenols, after appropriate modification of the reaction conditions, this reagent can also be used in late-stage deoxyfluorination of alcohols.31 In this reaction, chiral secondary alcohols can typically be fluorinated with inversion of the stereogenic carbon center, which is consistent with an SN2 mechanism. The fluorination process is highly chemoselective, and thus, tertiary alcohols and hydroxyl groups engaging in hydrogen bonding are not reactive.
In contrast to Ritter’s fluoride umpolung strategy, Gao et al established a new strategy for fluorination of phenols with [18F]-fluoride (Figure 13).32 This metal-free oxidative fluorination features an aryl umpolung protocol. Although, in 2002, Bienvenu et al had established the 19F version of this oxidative fluorination,33 the potential use of this fluorination in 18F radiochemistry had not been examined before. However, for transferring the method to 18F labeling, use of [18F]-HF•Py is disfavored, because in radiochemistry, carrier-added fluorine sources lead to labeled products with low specific activity.34 After optimization, [18F]-4-fluorophenol can be obtained in 21% decay-corrected RCY using [18F]-tetrabutylammonium fluoride (TBAF) as a fluoride source. In this method, the tert-butyl group is essential because a good electrofuge is needed in acid-catalyzed aromatization.
 | Figure 13 Metal-free oxidative fluorination of phenols with [18F]-fluoride. |
Fluorinated heterocycles such as 2-fluoropyridines are prevalent in pharmaceuticals, agrochemicals, and materials. Traditional approaches to these compounds require pre-functionalized substrates, are limited in scope, and can be hazardous. In 2013, Hartwig et al reported a silver(II) fluoride-mediated selective C–H fluorination reaction for vicinal position of pyridines and diazines (Figure 14).35 This reaction was an analog to the Chichibabin reaction and can be adopted for a broad range of substituted nitrogen heterocycles. During the course of the reaction, AgF2 is consumed, while yellow solid AgF is formed and could be observed by 19F NMR. Further mechanistic studies revealed that AgF2 serves not only as a fluorination reagent but also as an oxidant.
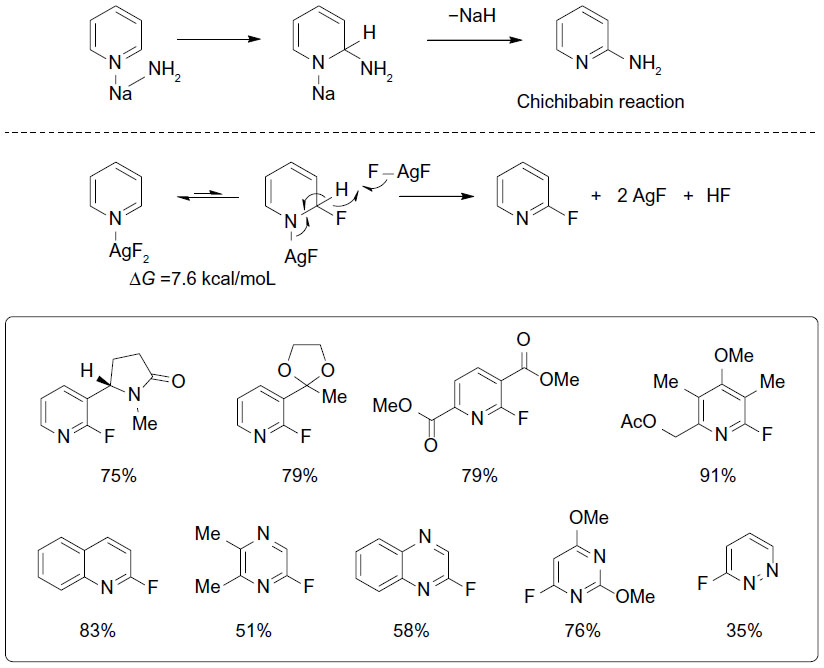 | Figure 14 Silver(II) fluoride-mediated selective C–H fluorination of pyridines. |
Synthesis of alkyl fluorides
Early methods for fluorination mainly focused on activated substrates containing sp3C–H bond, such as carbonyl derivatives, sulfones, and phosphate esters (Figure 15).36–38 However, catalytic fluorination of unactivated sp3C–H bonds remains a challenge.4,39
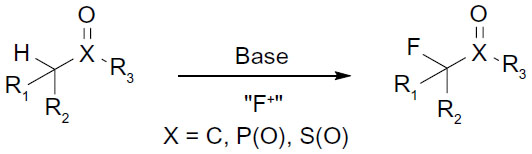 | Figure 15 Fluorination of activated Csp3–H bonds. |
One attractive strategy for alkyl fluoride synthesis is vicinal fluorocyclization of alkenes.40 During this transformation, feedstock olefins can be easily converted into cyclic fluorinated molecules. Both electrophilic and nucleophilic fluorine sources can be employed in this strategy. In general, these fluorocyclization reactions are divided into two categories: fluorination followed by cyclization and cyclization followed by fluorination (Figure 16).
 | Figure 16 Fluorocyclization of alkenes. |
One representative example for the fluorination–cyclization comes from Kong et al (Figure 17).41 In this reaction, chiral ArIIIIF2 was employed both as oxidant and fluorine source. Terminal alkenes undergo intramolecular and regio- and enantioselective aminofluorination to afford six-membered ring product. A reaction mechanism involving oxidation of sulfonamide rather than alkene was proposed. In an elegant study, Suzuki et al demonstrated that this method can be performed catalytically, by generating the chiral ArIIIIF2 reagent in situ in the presence of (R)-binaphthyl diiodide, mCPBA, and aqueous HF.42
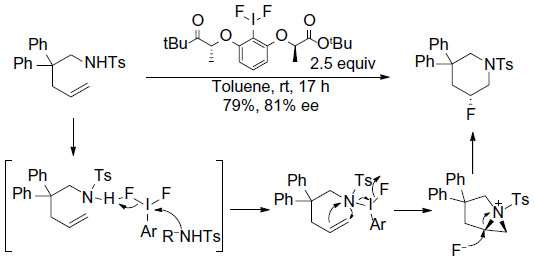 | Figure 17 Regio- and enantioselective aminofluorination of alkenes. |
More examples on fluorination–cyclization are available. In 2011, Rauniyar et al reported asymmetric electrophilic fluorination using an anionic chiral phase-transfer catalyst (Figure 18).43 The authors reasoned that a unique “charge-inverted” phase-transfer catalytic process may be achieved by exchanging the BF4− ion of Selectfluor with a bulky lipophilic chiral anion. The newly formed soluble chiral fluorination reagent undergoes an asymmetrical electrophilic fluorination–cyclization process to afford a fluorocyclization product. A study on nonlinear relationship suggested that both of the two BF4− anions are exchanged for chiral phosphates before reacting with the substrate. Even unactivated olefins with only alkyl substituents can deliver fluorinated products in good yields, albeit with reduced enantioselectivity.
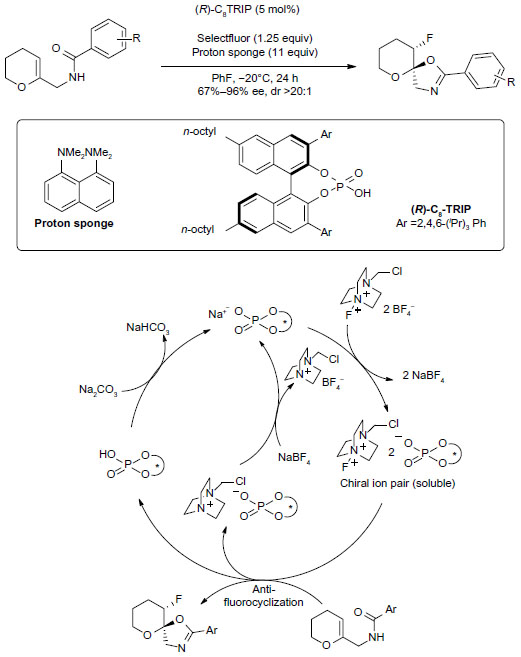 | Figure 18 Fluorocyclization under chiral ion-pairing catalysis. |
While traditional approaches to alkyl fluorides rely on the use of either electrophilic or nucleophilic fluorination reagents, radical fluorination has been restricted to hazardous, explosive, and uncontrollable reagents F2, hypofluorites, and XeF2.44–46 Recent computational studies found that the C–F bond homolytic dissociation energies of Selectfluor and N-fluorobenzenesulfonimide (NFSI) are 60–62 kcal/mol and 63 kcal/mol, respectively, suggesting that these reagents are suitable for fluorine atom transfer (Figure 19).47
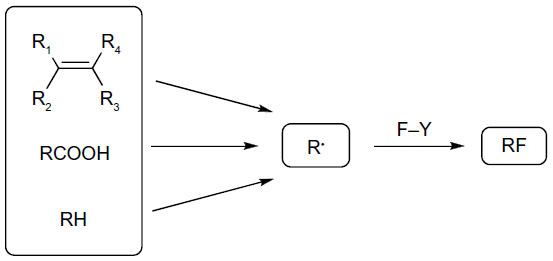 | Figure 19 Fluorine transfer to alkyl radicals. |
In 2012, Rueda et al reported a new aliphatic fluorination approach via fluorination of alkyl radicals with NFSI as a fluorine radical source.47 Peroxyesters undergo thermal decarboxylation to give alkyl radical intermediates, and the intermediates are subsequently fluorinated by NFSI (Figure 20, left). In the following studies, a complementary photochemical protocol was developed (Figure 20, right).48 However, this protocol is limited to carboxylic acids bearing aryl or aryloxy groups at the α position.
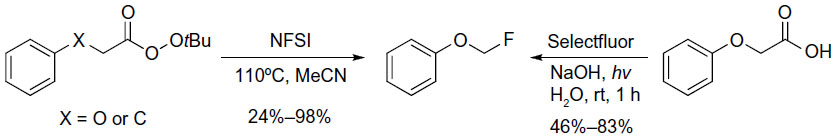 | Figure 20 Decarboxylative radical fluorination. |
Barker and Boger investigated Fe(III)/NaBH4-mediated free radical hydrofluorination of unactivated alkenes (Figure 21).49 This reaction shows exclusive Markovnikov regioselectivity and excellent functional group compatibility. Furthermore, the availability of NaBD4 and NaBT4 provides the opportunity for the synthesis of isotopic and radiolabeled compounds.
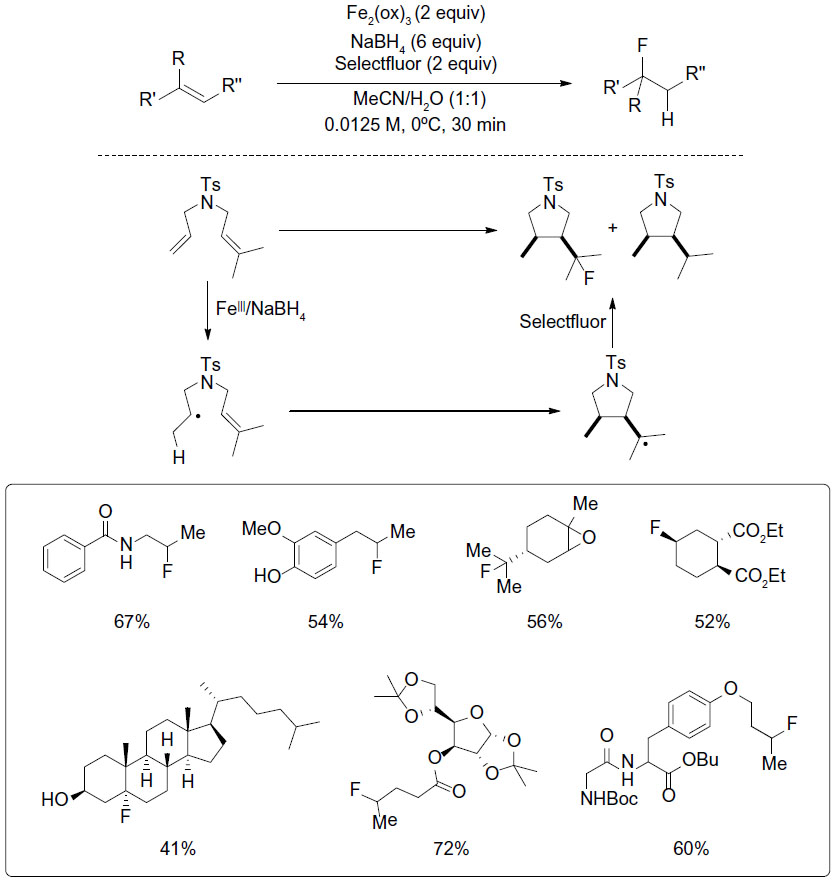 | Figure 21 Fe(II)/NaBH4-mediated radical hydrofluorination of unactivated alkenes. |
Yin et al have also employed Selectfluor to realize a Hunsdiecker-type oxidative fluorodecarboxylation of carboxylic acids (Figure 22).50 A tentative mechanism involving AgII–F and AgIII–F intermediates has been proposed. SET between carboxylic acids and AgIII–F generates AgII–F complexes and alkyl radicals, and the alkyl radicals in turn abstract fluorine atoms from AgII–F complexes to give the fluorinated compounds.
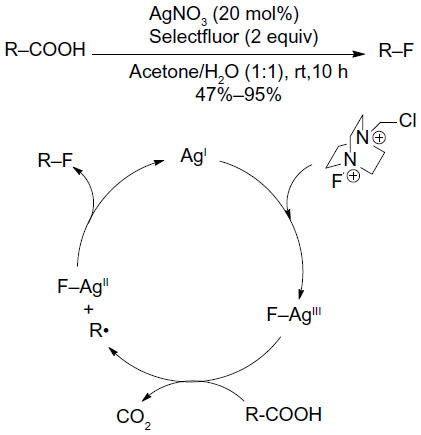 | Figure 22 Silver-catalyzed decarboxylative fluorination of aliphatic carboxylic acids. |
The selective fluorination of unactivated Csp3–H bonds has been elegantly demonstrated by two groups using different organometallic systems. Liu et al have developed a manganese-porphyrin-catalyzed oxidative aliphatic C–H fluorination reaction (Figure 23).51 In this reaction, the bulky pre-catalyst MnIII (tetra-mes-prophyrin [TMP])Cl reacts with AgF to form the real catalyst MnIII(TMP)F, which is oxidized by PhIO to give the highly reactive MnV-oxo species. This intermediate then abstracts a hydrogen atom from R–H to form the C-centered radical, which in turn abstracts a fluorine atom from MnIV(TMP)F2 to give the fluorinated product. This key step has been demonstrated by the reaction of cyclooctane with independently prepared MnIV(TMP)F2, which produces cyclooctyl fluoride in 43% yield.
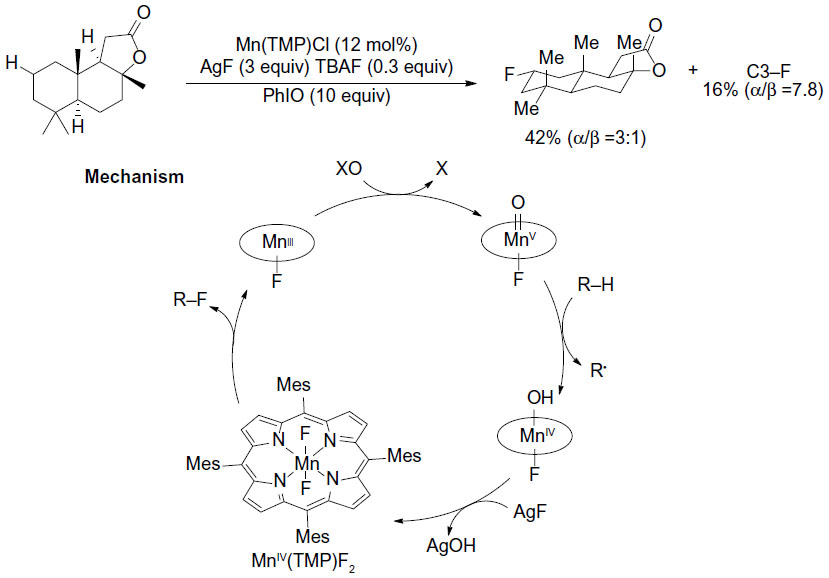 | Figure 23 Manganese porphyrin-catalyzed oxidative aliphatic C–H fluorination. |
Blooms et al reported a copper-catalyzed radical fluorination of various aliphatic substrates in the presence of Selectfluor (Figure 24).52 The phase-transfer catalyst KB(C6F5)4 was added to improve the solubility of Selectfluor, and N-hydroxyphthalimide was found to be a good co-catalyst. When the reaction is performed in the presence of 2,2,6,6-tetramethyl-1-oxylpiperidine (TEMPO), only trace amounts of products were observed. This control experiment, together with other observations, suggests a radical pathway during fluorination.
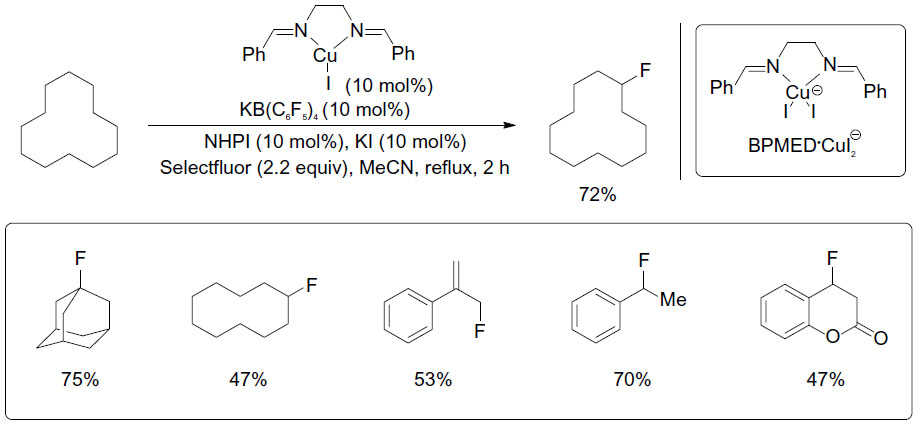 | Figure 24 CuI-catalyzed aliphatic, allylic, and benzylic fluorination. |
Other advances in recent green fluorination reactions include transition-metal-mediated fluorination of C–H bonds. In 2013, Braun and Doyle reported a palladium-catalyzed fluorination of allylic C–H bond (Figure 25).53 By using the electrophilic Pd(II)-sulfoxide catalyst system developed by White and co-workers,54 allylic C–H fluorination was achieved under mild conditions. Many functional groups such as esters, amides, and heterocycles were tolerated in this transformation, and the regioselectivity could be achieved up to 7.8:1 (branched:linear). Furthermore, this C–H fluorination approach is also useful for late-stage fluorination of natural products or drug candidates.
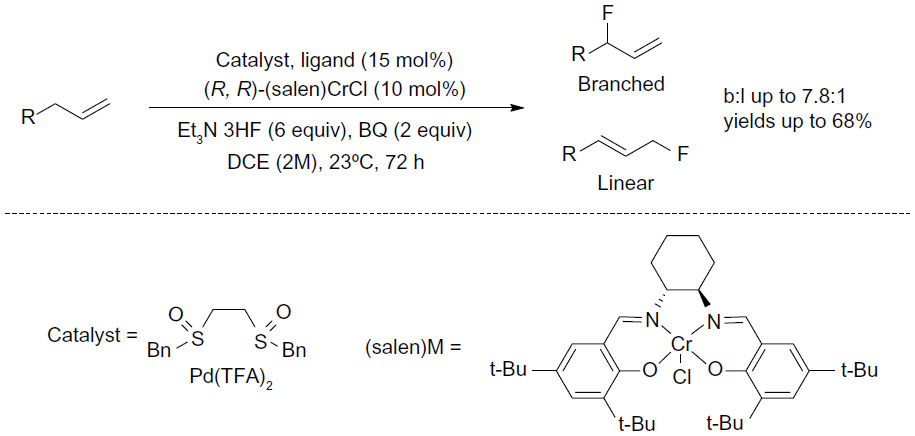 | Figure 25 Palladium-catalyzed allylic C–H fluorination. |
Advances in perfluoroalkylation
Synthesis of perfluoroalkylated arenes
Perfluoroalkylated arenes have been recognized as privileged structure in the fields of biochemistry and materials science. For example, introducing a CF3 group can dramatically enhance the metabolic stability, lipophilicity, and bioavailability of the parent molecule.55 Despite the widespread utility of perfluoroalkyl (Rf)-containing arenes, their application is usually limited by the synthetic availability. To address this problem, recent research has been directed to transition-metal-mediated cross-coupling with aromatic compounds and Rf sources. Among transition metals used in cross-coupling reactions, the late transition metal copper has been demonstrated to be the most promising metal. As early as the late 1960s, McLoughlin and Thrower found that perfluoroalkylation of iodoarenes can be achieved by heating Rf–I, aryl iodides, and copper powder in N,N-dimethyl formamide (DMF) at elevated temperature.56 This transformation requires a stoichiometric amount of copper.57 The failure in developing a catalytic reaction may due to the mismatching in reaction rate between the formation of Cu–Rf from Rf–I and Cu and the following perfluoroalkylation.
In 2009, Oishi et al descried a copper-catalyzed trifluoromethylation of aryl iodides (Figure 26).58 To solve the rate-mismatching problem, TESCF3 was used to slow down the formation rate of CuCF3. At the same time, a chelating diamine ligand was introduced to accelerate the trifluoromethylation step, which ensures the regeneration of sufficient amount of reusable copper complexes. This method is well suited for electron-deficient aryl iodides; however, electron-neutral and electron-rich substrates give lower yields.
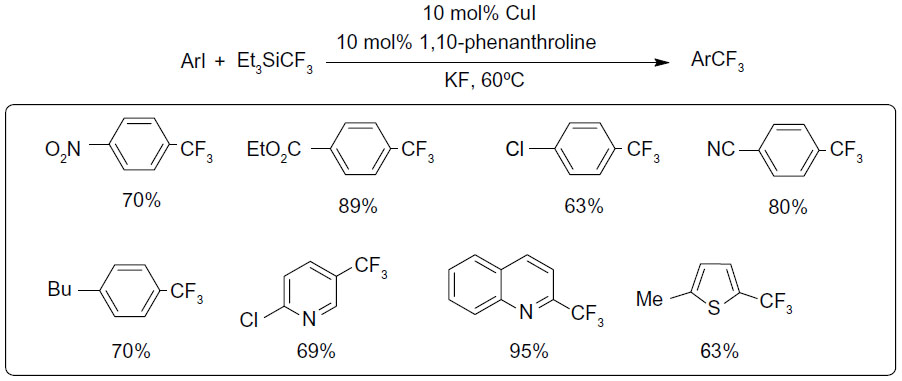 | Figure 26 Copper-catalyzed aromatic trifluoromethylation. |
In 2010, Chu and Qing descried copper-mediated oxidative trifluoromethylation of both terminal alkynes and boronic acids (Figure 27).59,60 For trifluoromethylation of terminal alkynes, air was used as oxidant. To minimize the formation of alkyne homo-coupling product, the alkyne substrate was added to the reaction mixture over 4 hours via a syringe pump. In contrast, for trifluoromethylation of aryl boronic acids, stoichiometric amount of Ag2CO3 is required.
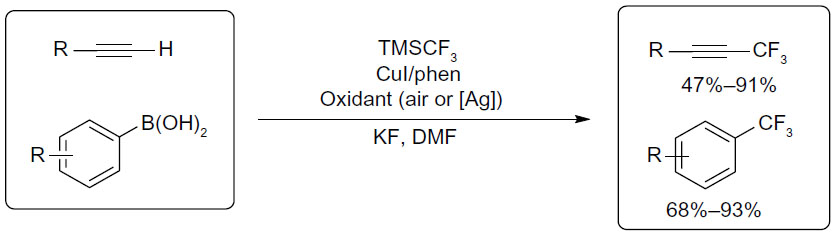 | Figure 27 Copper-mediated oxidative trifluoromethylation of terminal alkynes and aryl boronic acids. |
Trifluoromethylation of aryl boronic acids can also be achieved using electrophilic CF3 reagent and catalytic amount of copper. In 2011, Xu et al61 and Liu et al62 reported two similar methods for trifluoromethylation of aryl boronic acids (Figure 28). Umemoto and Togni reagents were used as electrophilic CF3 sources. In addition to aryl boronic acids, alkenyl boronic acids also undergo these transformations in reasonable yields, with partial isomerization of the alkenes.
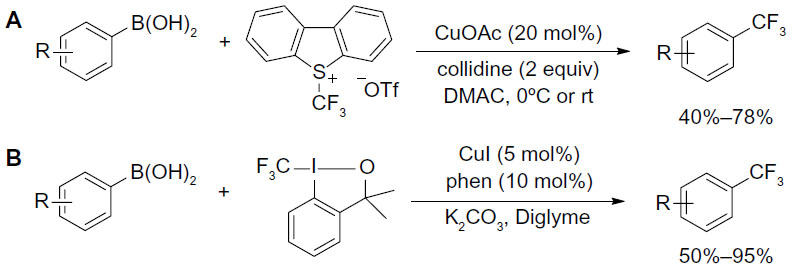 | Figure 28 Copper-catalyzed trifluoromethylation of aryl boronic acids with CF3+ reagents. |
Fluoroform (CF3H) is a nontoxic gas and is not an ozone depleter. It is cheap and available in large quantities. From the view of atom economy, CF3H is an ideal source to prepare stable CF3 metal derivatives.63 However, direct deprotonation of CF3H by strong bases usually leads to decomposition of CF3 anion to give difluorocarbene at room temperature.64 In 2011, Zanardi et al found that a mixture of CuCl and tBuOK in 1:2 ratio can effectively cuprate CF3H (Figure 29).65 The resulting CuCF3 readily reacts with ArI or ArB(OH)2 to give the corresponding ArCF3. This protocol has been extended to C2F5H; however, this direct cupration reaction failed in the cases of other higher perfluoroalkanes.66 It was proposed that HF elimination is preferred for higher perfluoroalkanes with more than two carbons because the olefinic products RfCF=CF2 are more stable than CF2=CF2.
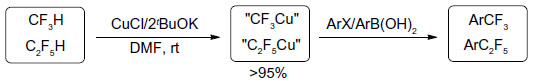 | Figure 29 Direct cupration of CF3H and C2F5H. |
Due to the high activation energy, the reductive elimination of Ar–Rf from PdII complexes, PdII(Ar)(Rf) complexes, is more challenging than their non-fluorinated analogs. Although this elementary reaction has been demonstrated by Grushin through conversion of Xantphos-chelated PdII(Ph)(CF3) to PhCF3 upon heating at 80°C,67 the catalytic reaction could not occur with Xantphos as the ligand. In 2010, Cho et al developed the first Pd-catalyzed coupling of aryl chlorides with TESCF3 (Figure 30).68 Both electron-poor and electron-rich aryl chlorides are suitable substrates; however, functional groups such as aldehyde, ketone, and unprotected OH or NH are not tolerated in this reaction. Furthermore, minor amounts of hydrogenated products were usually observed and could not be separated from desired products.
 | Figure 30 Palladium-catalyzed trifluoromethylation of aryl chlorides. |
However, similar to Pd-catalyzed C–F coupling, C–CF3 bond reductive elimination from high-valent PdIV complexes is much easier than that from PdII complexes. The catalytic trifluoromethylation of arenes via PdII-mediated C–H activation, proceeding via oxidation with an electrophilic CF3+ source followed by reductive elimination from PdIV(Ph)(CF3) complexes, has been reported by Wang et al (Figure 31).69 However, this method is limited to substrates with directing groups.
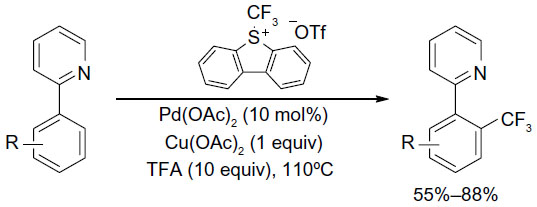 | Figure 31 Pd(II)-catalyzed C–H trifluoromethylation of arenes. |
In 2013, Dai et al,70 Danoun et al,71 and Wang et al72 reported the Sandmeyer-type trifluoromethylation of aryl diazonium salts (Figure 32). Although these reactions are carried out similarly, different mechanisms have been proposed in their reports. In Fu’s reaction, Cu powder reacts with aryl diazonium salt and Umemoto reagent to give Ar• and CuCF3, respectively. Then, trifluoromethylation takes place between these two key intermediates. In Goossen’s work, trifluoromethylation reaction proceeds analogously to Sandmeyer halogenations, which occurs between ArN2+ and CuCF3. On the contrary, preliminary mechanistic investigations suggest that aryl radical is not likely involved in Wang’s one-pot, two-step reaction. Thus, the author proposed an oxidative addition/reductive elimination pathway at Ag center. Both of the methods proceed at low temperature and tolerate various functional groups. Moreover, aryl amines are readily available substrates, and the amino group can be introduced at the late stage of complex molecule synthesis.
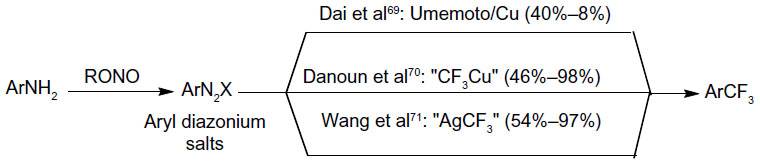 | Figure 32 Sandmeyer trifluoromethylation of aryl diazonium salts. |
Although nucleophilic/electrophilic trifluoromethylation of arenes requires pre-functionalized substrates, transformations via radical pathway demonstrate synthetic advantages in direct trifluoromethylation of non-functionalized arenes.73 From a synthetic viewpoint, the poor site selectivity in many cases may be a major limitation of radical reactions. However, they are particularly suitable in the field of pharmaceuticals, where the synthesis of drug candidates with a CF3 group at different positions around the aromatic ring is needed.
In 2011, Loy and Sanford described a palladium-catalyzed C–H perfluoroalkylation of arenes using relatively inexpensive RfI as Rf source (Figure 33).74 Significantly low yields were obtained for aromatic substrates bearing electron-withdrawing groups, and isomers were observed when asymmetrical arenes were used as substrates. The authors suggested that caged or Pd-associated radical intermediates are involved in this perfluoroalkylation reaction. It is worth mentioning that CF3I is not as efficient as other RfIs in this transformation, affording only 26% yield of PhCF3 as determined by gas chromatography.
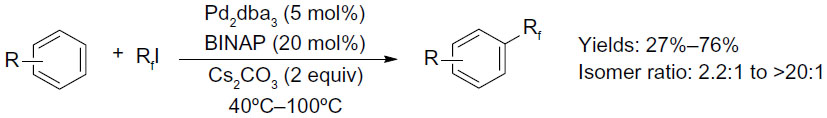 | Figure 33 Palladium-catalyzed C–H perfluoroalkylation of arenes. |
Ji et al reported a protocol for innate C–H trifluoromethylation of heterocycles (Figure 34).75 Using Langlois et al reagent76 as a benchtop stable CF3 radical source, a variety of electron-deficient and electron-rich heteroarenes undergo facile trifluoromethylation. This result is different from pioneering studies by Langlois, in which electrophilic CF3 radical undergoes radical aromatic substitution reactions only with electron-rich substrates. Note that this reaction is operationally simple, which readily takes place in aqueous medium at room temperature (without solvent purification or an inert atmosphere).
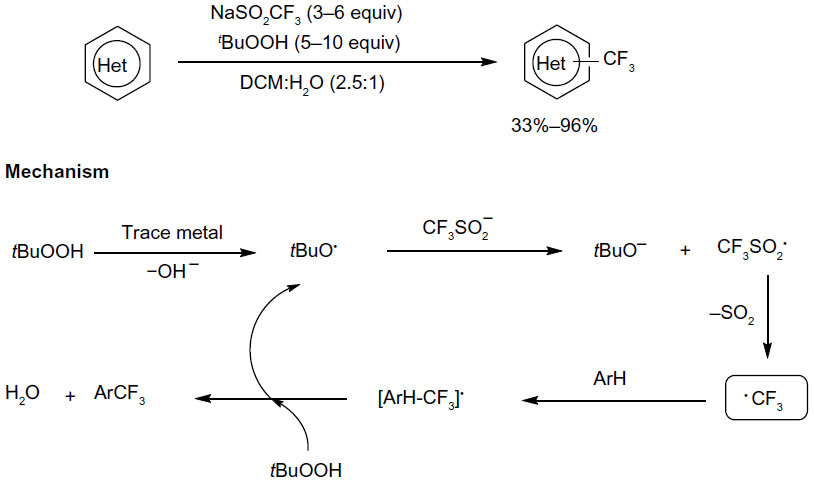 | Figure 34 Innate C–H trifluoromethylation of heterocycles. |
Almost at the same time, Nagib and MacMillan reported a photoredox-catalyzed trifluoromethylation of arenes and heteroarenes (Figure 35).77 TfCl (triflyl chloride), whose more electron-deficient nature can enable a more facile reduction, was used as a better CF3 radical source than CF3I. This transformation proceeds at room temperature and occurs at the electron-rich position(s) of aromatic ring. The synthetic value of this transformation has also been demonstrated by trifluoromethylation of biologically active molecules.
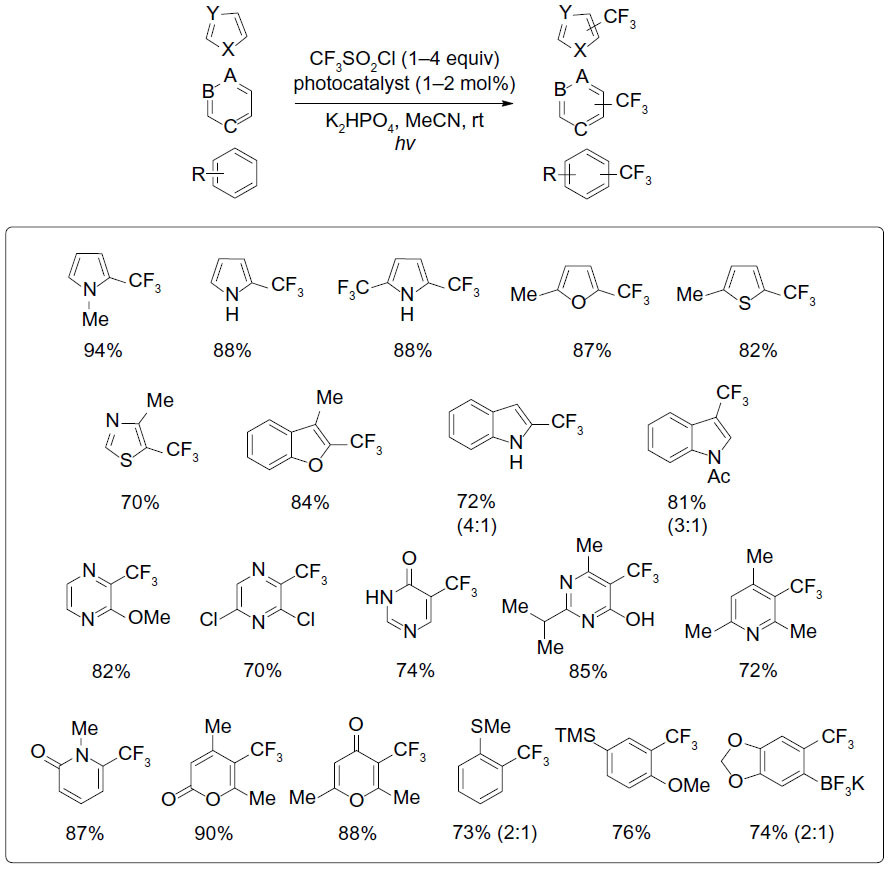 | Figure 35 Photoredox-catalyzed trifluoromethylation of arenes and heteroarenes. |
Most of the trifluoromethylated arenes were synthesized using preformed arenes; however, in 2013, Zhang et al described a cyclization method to obtain 6-trifluoromethyl-phenanthridines through radical trifluoromethylation of isonitriles (Figure 36).78 In this new protocol, the arene core is constructed during the trifluoromethylation process. In this reaction, the iodide initiator reacts with the Togni reagent to form iodine and trifluoromethyl radicals, and the latter is trapped by aryl isonitrile to generate an imidoyl radical. Subsequently, a sequence of radical cyclization/deprotonation/single-electron reduction of Togni reagent affords the final product and a CF3 radical, which reenters the next reaction cycle. Additionally, the reaction can also be applied to the synthesis of other perfluoroalkylated compounds in good yields.
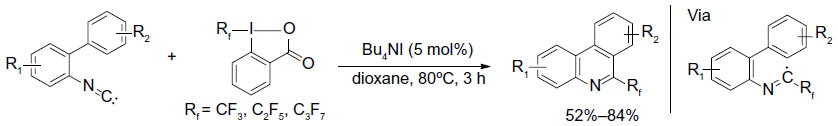 | Figure 36 Synthesis of trifluoromethylated phenanthridines through radical trifluoromethylation of isonitriles. |
A similar protocol has been developed by Zeng et al, in which arene cores were formed during the trifluoromethylation of aryne intermediates (Figure 37).79 Using AgCF3 as a soft CF3 transfer reagent, arynes were trifluoromethylated to generate vicinal CF3-aryl silver intermediates, which in situ react with 1-iodophenylacetylene to give the product and silver phenylacetylide. 2,2,6,6-Tetramethylpiperidine was found to be an efficient ligand for promoting this transformation. It should be noted that compared to other monofunctionalization (trifluoromethylation) reactions, this method features a vicinal difunctionalization of arenes. Furthermore, the o-trifluoromethylated iodobenzenes can sever as excellent o-trifluoromethylated arene-building blocks which are otherwise difficult to synthesize.
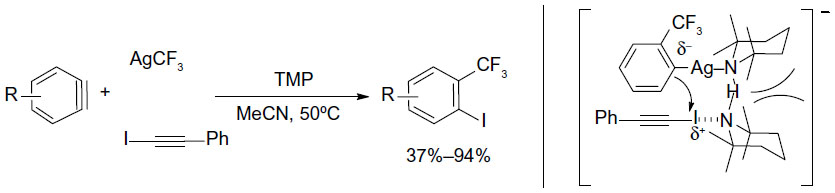 | Figure 37 Silver-mediated trifluoromethylation–iodination of arynes. |
Despite much effort has been directed to introduce 18F into organic molecules, there are few general methods for [18F]-trifluoromethylation of arenes.80–82 In 2013, Huiban et al reported the direct [18F]-trifluoromethylation of aryl and heteroaryl iodides (Figure 38).83 In the presence of [18F]-fluoride and copper(I) salt, the in situ-generated difluorocarbene is transformed to [18F]-CuCF3, which further undergoes cross-coupling with ArI to give a [18F]-trifluoromethylated arene product. Additionally, this difluorocarbene/[18F]-fluoride/CuI strategy also finds application in [18F]-trifluoromethylation of pharmaceutical candidates to facilitate drug development.
 | Figure 38 [18F]-trifluoromethylation of aryl and heteroaryl iodides. |
Synthesis of alkenyl and alkyl trifluorides
In 2011, Parsons et al,84 Xu et al,85 and Wang et al86 reported the trifluoromethylation of unactivated olefins with a copper(I) salt and either Umemoto or Togni reagent (Figure 39). Although these reactions provide accesses to compounds bearing CF3 at allylic positions, these transformations are restricted to monosubstituted terminal olefins. Different mechanisms were proposed for these methods, including radical addition of CF3 to alkenes (Parsons et al)84 and migration insertion of CuIII–CF3 to double bond (Xu et al).85
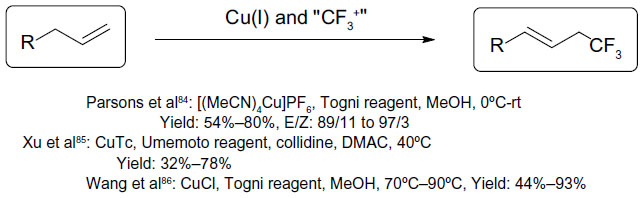 | Figure 39 Copper-catalyzed trifluoromethylation of unactivated olefins. |
Allylsilanes are also good substrates for copper-catalyzed trifluoromethylation to give trifluoromethylated products. Shimizu et al found that different types of allylsilanes undergo facile trifluoromethylation to afford either gem-disubstituted terminal olefins or vinylsilanes bearing a CF3 on the allylic position (Figure 40).87 Due to the higher nucleophilicity of allylsilanes, this reaction can proceed under very mild conditions (room temperature).
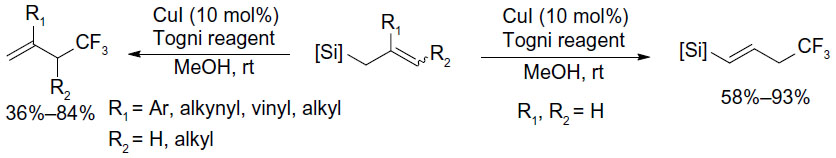 | Figure 40 Copper-catalyzed trifluoromethylation of allylsilanes. |
Parsons et al rationalized that if a boron-based functional group is placed geminal to the CF3 group in reaction intermediate, the boron group may be eliminated prior to β-hydrogen, thus generating a different product, that is, vinyl-CF3-containing product (Figure 41).88 After a quick screening, the authors found that the ratio of E/Z isomers was dependent on the rate of elimination of the boron-based leaving group; thus, the poor leaving ability of BF3 leads to the highest E/Z product ratio. Although the reaction conditions are mild, this method has some limitations, as electron-deficient and trisubstituted vinyl-BF3K salts furnish only trace amounts of products. Preliminary mechanistic studies suggest the involvement of a carbocationic intermediate generated by Lewis acid catalysis; however, a radical pathway cannot be ruled out at present.
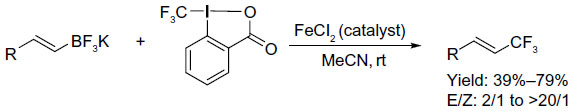 | Figure 41 Iron-catalyzed trifluoromethylation of potassium vinyltrifluoroborates. |
Carboxylic acids are ubiquitous compounds and easy to obtain in large scales. In 2012, He et al described copper-catalyzed di- and trifluoromethylation of α,β-unsaturated carboxylic acids (Figure 42).89 This protocol provides an opportunity to synthesize vinyl-Rf-containing molecules. Both electron-rich and electron-deficient unsaturated carboxylic acids are viable substrates. Notably, reactions are highly stereoselective, with E/Z ratios ranging from 92:8 to more than 99:1. Mechanistic studies revealed that the carboxylic acid group plays a crucial role for the success of the efficient C–Rf bond formation.
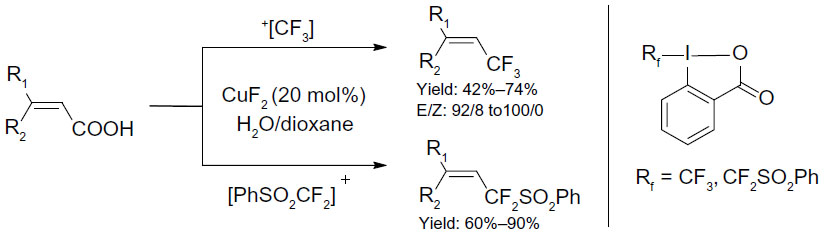 | Figure 42 Decarboxylic trifluoromethylation of unsaturated carboxylic acids. |
In 2013, Wu et al90 and Mizuta et al91 described two methods for hydrotrifluoromethylation of unactivated alkenes (Figure 43). In Qing’s work, the nucleophilic CF3 anion generated in situ from TMSCF3 and NaOAc was oxidized by PhI(OAc)2 to give CF3 radical. The addition of CF3 radical to unactivated alkene forms a new alkyl radical, which is quenched by 1,4-CHD (1,4-cyclohexadiene), an excellent H donor. It was found that a catalytic amount of Ag(I) salt could facilitate this reaction. However, the role of silver has not been revealed. In Gouverneur’s work, CF3 radical is generated during the reduction of Umemoto reagent under photoredox conditions. Then, the newly formed alkyl radical reacts with MeOH and the oxidative RuIII species to afford the trifluoromethylated product. Isotope-labeling experiments demonstrated that the α-H of MeOH serves as the hydrogen atom source. It is noteworthy that these two methods are also adapted for hydrotrifluoromethylation of alkynes. Under their reaction conditions, trifluoromethylated alkenes are obtained as mixtures of E/Z isomers.
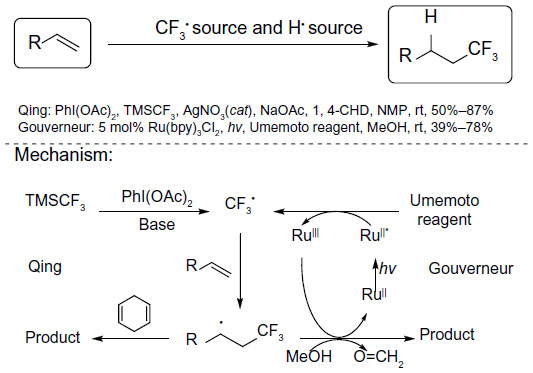 | Figure 43 Hydrotrifluoromethylation of unactivated alkenes. |
Summary and outlook
In recent years, the chemical society has witnessed a rapid development in fluorination and perfluoroalkylation reactions. Most of these improvements are based on either transition-metal chemistry or radical chemistry. Additionally, electrophilic fluorination/perfluoroalkylation reagents were frequently used in these methods as they are both reactive oxidants and excellent F/Rf sources. While traditional methods are only suitable for simple fluorinated molecule synthesis and have a narrow substrate scope, these newly developed methods show great promise for preparing F/Rf-containing molecules with diversified substitution.
Despite these advances, achieving fluorination/perfluoroalkylation processes that have both high efficiency and selectivity remains a great challenge. In the view of green chemistry, the use of F+ and Rf+ is not atom economic, and these reagents are not adaptable for 18F-labeled compounds synthesis. Furthermore, reactions through radical pathways frequently give inseparable regioisomers.
For both academia and industry, developing efficient, flexible, and mild fluorination/perfluoroalkylation processes that employ low-cost nucleophilic F/Rf-transferring reagents, such as alkali metal fluorides and fluoroform, is highly desirable. Recent research has directed to transition-metal-catalyzed direct C–H fluorination/perfluoroalkylation of unactivated substrates, which may be the most conceptually attractive pathway in fluorine-containing molecule synthesis. Although these investigations are still in their infancy, we believe that significant advances in this field can be achieved in the near future.
Acknowledgments
This work was supported by the National Basic Research Program of China (2015CB931900, 2012CB215500), the National Natural Science Foundation of China (21372246, 21421002), Shanghai QMX program (13QH1402400), and the Chinese Academy of Sciences. We thank Dr Chuanfa Ni for his proofreading of this manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
Thayer AM. Fabulous fluorine. Chem Eng News. 2006;84(23):15–24. | |
Stahl T, Klare HFT, Oestreich M. Main-group Lewis acids for C–F bond activation. ACS Catal. 2013;3(7):1578–1587. | |
O’Hagan D. Understanding organofluorine chemistry. An introduction to the C–F bond. Chem Soc Rev. 2008;37(2):308–319. | |
Furuya T, Kamlet AS, Ritter T. Catalysis for fluorination and trifluoromethylation. Nature. 2011;473(7348):470–477. | |
Balz G, Schiemann G. On aromatic fluoric compounds, I A new method for its representation. Ber Dtsch Chem Ges. 1927;60:1186–1190. | |
Finger GC, Kruse CW. Aromatic fluorine compounds.7. Replacement of aromatic –Cl and –NO2 groups by –F. J Am Chem Soc. 1956;78(23):6034–6037. | |
Swarts F. Étude sur le fluo chloroforme [A study on fluorinated chloroform]. Acad. Roy. Belg. 1892;24:474. | |
Daudt HW, Youker MA, inventors; Kinetic Chemicals Inc, assignee. Preparation of carbon compounds containing fluorine. United States Patent US 2005705. September 20, 1930. | |
Simons JH, Harland WJ. The electrochemical process for the production of fluorocarbons. J Electrochem Soc. 1949;95:47–66. | |
Hull KL, Anani WQ, Sanford MS. Palladium-catalyzed fluorination of carbon-hydrogen bonds. J Am Chem Soc. 2006;128(22):7134–7135. | |
Grushin VV. The organometallic fluorine chemistry of palladium and rhodium: studies toward aromatic fluorination. Acc Chem Res. 2010;43(1):160–171. | |
Ball ND, Sanford MS. Synthesis and reactivity of a mono-sigma-aryl palladium(iv) fluoride complex. J Am Chem Soc. 2009;131(11):3796–3797. | |
Watson DA, Su M, Teverovskiy G, et al. Formation of ArF from LPdAr(F): catalytic conversion of aryl triflates to aryl fluorides. Science. 2009;325(5948):1661–1664. | |
Lee HG, Milner PJ, Buchwald SL. Pd-catalyzed nucleophilic fluorination of aryl bromides. J Am Chem Soc. 2014;136(10):3792–3795. | |
Milner PJ, Kinzel T, Zhang Y, Buchwald SL. Studying regioisomer formation in the pd-catalyzed fluorination of aryl triflates by deuterium labeling. J Am Chem Soc. 2014;136(44):15757–15766. | |
Ametamey SM, Honer M, Schubiger PA. Molecular imaging with PET. Chem Rev. 2008;108(5):1501–1516. | |
Miller PW, Long NJ, Vilar R, Gee AD. Synthesis of C-11, F-18, O-15, and N-13 radiolabels for positron emission tomography. Angew Chem Int Ed Engl. 2008;47(47):8998–9033. | |
Littich R, Scott PJH. Novel strategies for fluorine-18 radiochemistry. Angew Chem Int Ed Engl. 2012;51(5):1106–1109. | |
Lee E, Kamlet AS, Powers DC, et al. A fluoride-derived electrophilic late-stage fluorination reagent for PET imaging. Science. 2011; 334(6056):639–642. | |
Lee E, Hooker JM, Ritter T. Nickel-mediated oxidative fluorination for PET with aqueous [F-18] fluoride. J Am Chem Soc. 2012;134(42):17456–17458. | |
Mazzotti AR, Campbell MG, Tang PP, Murphy JM, Ritter T. Palladium(III)-catalyzed fluorination of arylboronic acid derivatives. J Am Chem Soc. 2013;135(38):14012–14015. | |
Furuya T, Strom AE, Ritter T. Silver-mediated fluorination of functionalized aryl stannanes. J Am Chem Soc. 2009;131(5):1662–1663. | |
Furuya T, Ritter T. Fluorination of boronic acids mediated by silver(I) triflate. Org Lett. 2009;11(13):2860–2863. | |
Subramanian MA, Manzer LE. A “greener” synthetic route for fluoroaromatics via copper (II) fluoride. Science. 2002;297(5587):1665–1665. | |
Fier PS, Hartwig JF. Copper-mediated fluorination of aryl iodides. J Am Chem Soc. 2012;134(26):10795–10798. | |
Fier PS, Luo JW, Hartwig JF. Copper-mediated fluorination of arylboronate esters. Identification of a Copper(III) Fluoride Complex. J Am Chem Soc. 2013;135(7):2552–2559. | |
Ye YD, Sanford MS. Mild copper-mediated fluorination of aryl stannanes and aryl trifluoroborates. J Am Chem Soc. 2013;135(12):4648–4651. | |
Ye YD, Schimler SD, Hanley PS, Sanford MS. Cu(OTf)(2)-mediated fluorination of aryl trifluoroborates with potassium fluoride. J Am Chem Soc. 2013;135(44):16292–16295. | |
Yamada S, Gavryushin A, Knochel P. Convenient electrophilic fluorination of functionalized aryl and heteroaryl magnesium reagents. Angew Chem Int Ed Engl. 2010;49(12):2215–2218. | |
Tang PP, Wang WK, Ritter T. Deoxyfluorination of phenols. J Am Chem Soc. 2011;133(30):11482–11484. | |
Sladojevich F, Arlow SI, Tang PP, Ritter T. Late-stage deoxyfluorination of alcohols with phenofluor. J Am Chem Soc. 2013;135(7):2470–2473. | |
Gao ZH, Lim YH, Tredwell M, et al. Metal-free oxidative fluorination of phenols with [F-18]fluoride. Angew Chem Int Ed Engl. 2012;51(27):6733–6737. | |
Bienvenu A, Barthelemy A, Boichut S, Marquet B, Billard T, Langlois BR. Synthesis of 4-fluorophenols from 4-tert-butylphenols and fluoride sources under oxidative conditions. Collect Czech Chem C. 2002; 67(10):1467–1478. | |
Josse O, Labar D, Georges B, Gregoire V, Marchand-Brynaert J. Synthesis of [F-18]-labeled EF3 [2-(2-nitroimidazol-1-yl)-N-(3,3,3-trifluoropropyl)-acetamide], a marker for PET detection of hypoxia. Bioorgan Med Chem. 2001;9(3):665–675. | |
Fier PS, Hartwig JF. Selective C–H fluorination of pyridines and diazines inspired by a classic amination reaction. Science. 2013;342(6161):956–960. | |
Ma JA, Cahard D. Update 1 of: asymmetric fluorination, trifluoromethylation, and perfluoroalkylation reactions. Chem Rev. 2008;108(9):r1–r43. | |
Cahard D, Xu XH, Couve-Bonnaire S, Pannecoucke X. Fluorine and chirality: how to create a nonracemic stereogenic carbon-fluorine centre? Chem Soc Rev. 2010;39(2):558–568. | |
Yang X, Wu T, Phipps RJ, Toste FD. Advances in catalytic enantioselective fluorination, mono-, di-, and trifluoromethylation, and trifluoromethylthiolation reactions. Chem Rev. 2015;115(2):826–870. | |
Furuya T, Kuttruff CA, Ritter T. Carbon-fluorine bond formation. Curr Opin Drug Disc. 2008;11(6):803–819. | |
Wolstenhulme JR, Gouverneur V. Asymmetric fluorocyclizations of alkenes. Acc Chem Res. 2014;47(12):3560–3570. | |
Kong WQ, Feige P, de Haro T, Nevado C. Regio- and enantioselective aminofluorination of alkenes. Angew Chem Int Ed Engl. 2013;52(9):2469–2473. | |
Suzuki S, Kamo T, Fukushi K, et al. Iodoarene-catalyzed fluorination and aminofluorination by an Ar-I/HF.pyridine/mCPBA system. Chem Sci. 2014;5(7):2754–2760. | |
Rauniyar V, Lackner AD, Hamilton GL, Toste FD. Asymmetric electrophilic fluorination using an anionic chiral phase-transfer catalyst. Science. 2011;334(6063):1681–1684. | |
Grakausk V. Aqueous fluorination of carboxylic acid salts. J Org Chem. 1969;34(8):2446–2450. | |
Rozen S. Elemental fluorine as a legitimate reagent for selective fluorination of organic-compounds. Acc Chem Res. 1988;21(8):307–312. | |
Patrick TB, Khazaeli S, Nadji S, Heringsmith K, Reif D. Mechanistic studies of fluorodecarboxylation with xenon difluoride. J Org Chem. 1993;58(3):705–708. | |
Rueda-Becerril M, Sazepin CC, Leung JC, et al. Fluorine transfer to alkyl radicals. J Am Chem Soc. 2012;134(9):4026–4029. | |
Leung JCT, Chatalova-Sazepin C, West JG, Rueda-Becerril M, Paquin JF, Sammis GM. Photo-fluorodecarboxylation of 2-aryloxy and 2-aryl carboxylic acids. Angew Chem Int Ed Engl. 2012;51(43):10804–10807. | |
Barker TJ, Boger DL. Fe(III)/NaBH4-mediated free radical hydrofluorination of unactivated alkenes. J Am Chem Soc. 2012;134(33):13588–13591. | |
Yin F, Wang ZT, Li ZD, Li CZ. Silver-catalyzed decarboxylative fluorination of aliphatic carboxylic acids in aqueous solution. J Am Chem Soc. 2012;134(25):10401–10404. | |
Liu W, Huang XY, Cheng MJ, Nielsen RJ, Goddard WA, Groves JT. Oxidative aliphatic C–H fluorination with fluoride ion catalyzed by a manganese porphyrin. Science. 2012;337(6100):1322–1325. | |
Bloom S, Pitts CR, Miller DC, et al. A polycomponent metal-catalyzed aliphatic, allylic, and benzylic fluorination. Angew Chem Int Ed Engl. 2012;51(42):10580–10583. | |
Braun MG, Doyle AG. Palladium-catalyzed allylic C-h fluorination. J Am Chem Soc. 2013;135(35):12990–12993. | |
Chen, MS, White, MC. A sulfoxide-promoted, catalytic method for the regioselective synthesis of allylic acetates from monosubstituted olefins via C–H oxidation. J Am Chem Soc. 2004;126(5):1346−1347. | |
Purser S, Moore PR, Swallow S, Gouverneur V. Fluorine in medicinal chemistry. Chem Soc Rev. 2008;37(2):320–330. | |
McLoughlin VC, Thrower J. A route to fluoroalkyl-substituted aromatic compounds involving fluoroalkylcopper intermediates. Tetrahedron. 1969;25(24):5921–5940. | |
Chen QY, Wu SW. Methyl fluorosulfonyldifluoroacetate – a new trifluoromethylating agent. J Chem Soc Chem Comm. 1989;11:705–706. | |
Oishi M, Kondo H, Amii H. Aromatic trifluoromethylation catalytic in copper. Chem Commun. 2009;14:1909–1911. | |
Chu LL, Qing FL. Copper-mediated oxidative trifluoromethylation of boronic acids. Org Lett. 2010;12(21):5060–5063. | |
Chu LL, Qing FL. Copper-mediated aerobic oxidative trifluoromethylation of terminal alkynes with Me3SiCF3. J Am Chem Soc. 2010;132(21):7262–7263. | |
Xu J, Luo DF, Xiao B, et al. Copper-catalyzed trifluoromethylation of aryl boronic acids using a CF3+ reagent. Chem Commun. 2011;47(14):4300–4302. | |
Liu TF, Shen QL. Copper-catalyzed trifluoromethylation of aryl and vinyl boronic acids with an electrophilic trifluoromethylating reagent. Org Lett. 2011;13(9):2342–2345. | |
Tomashenko OA, Grushin VV. Aromatic trifluoromethylation with metal complexes. Chem Rev. 2011;111(8):4475–4521. | |
Symons EA, Clermont MJ. Hydrogen isotope exchange between fluoroform (CF3H) and water.1. Catalysis by hydroxide ion. J Am Chem Soc. 1981;103(11):3127–3130. | |
Zanardi A, Novikov MA, Martin E, Benet-Buchholz J, Grushin VV. Direct cupration of fluoroform. J Am Chem Soc. 2011;133(51):20901–20913. | |
Lishchynskyi A, Grushin VV. Cupration of C2F5H: isolation, structure, and synthetic applications of [K(DMF)2][(t-BuO)Cu(C2F5)]. Highly efficient pentafluoroethylation of unactivated aryl bromides. J Am Chem Soc. 2013;135(34):12584–12587. | |
Grushin VV, Marshall WJ. Facile Ar-CF3 bond formation at Pd. Strikingly different outcomes of reductive elimination from [(Ph3P)(2)Pd(CF3)Ph] and [(Xantphos)Pd(CF3)Ph]. J Am Chem Soc. 2006;128(39):12644–12645. | |
Cho EJ, Senecal TD, Kinzel T, Zhang Y, Watson DA, Buchwald SL. The palladium-catalyzed trifluoromethylation of aryl chlorides. Science. 2010;328(5986):1679–1681. | |
Wang XS, Truesdale L, Yu JQ. Pd(II)-catalyzed ortho- trifluoromethylation of arenes using TFA as a promoter. J Am Chem Soc. 2010;132(11):3648–3649. | |
Dai JJ, Fang C, Xiao B, et al. Copper-promoted Sandmeyer trifluoromethylation reaction. J Am Chem Soc. 2013;135(23):8436–8439. | |
Danoun G, Bayarmagnai B, Grunberg MF, Goossen LJ. Sandmeyer trifluoromethylation of arenediazonium tetrafluoroborates. Angew Chem Int Ed Engl. 2013;52(31):7972–7975. | |
Wang X, Xu Y, Mo F, et al. Silver-mediated trifluoromethylation of aryldiazonium salts: conversion of amino group into trifluoromethyl group. J Am Chem Soc. 2013;135(28):10330–10333. | |
Studer AA. “Renaissance” in radical trifluoromethylation. Angew Chem Int Ed Engl. 2012;51(36):8950–8958. | |
Loy RN, Sanford MS. Palladium-catalyzed C–H perfluoroalkylation of arenes. Org Lett. 2011;13(10):2548–2551. | |
Ji Y, Brueckl T, Baxter RD, et al. Innate C–H trifluoromethylation of heterocycles. Proc Natl Acad Sci U S A. 2011;108(35):14411–14415. | |
Langlois BR, Laurent E, Roidot N. Trifluoromethylation of aromatic-compounds with sodium trifluoromethanesulfinate under oxidative conditions. Tetrahedron Lett. 1991;32(51):7525–7528. | |
Nagib DA, MacMillan DWC. Trifluoromethylation of arenes and heteroarenes by means of photoredox catalysis. Nature. 2011;480(7376):224–228. | |
Zhang B, Muck-Lichtenfeld C, Daniliuc CG, Studer A. 6-trifluoromethyl- phenanthridines through radical trifluoromethylation of isonitriles. Angew Chem Int Ed Engl. 2013;52(41):10792–10795. | |
Zeng Y, Zhang L, Zhao Y, Ni C, Zhao J, Hu J. Silver-mediated trifluoromethylation-iodination of arynes. J Am Chem Soc. 2013;135(8):2955–2958. | |
Angelini G, Speranza M, Shiue CY, Wolf AP. H18f + Sb2o3 – a new selective radiofluorinating agent. J Chem Soc Chem Comm. 1986;12:924–925. | |
Prabhakaran J, Underwood MD, Parsey RV, et al. Synthesis and in vivo evaluation of [F-18]-4-[5-(4-methylphenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl]benzenesulfonamide as a PET imaging probe for COX-2 expression. Bioorgan Med Chem. 2007;15(4):1802–1807. | |
Mizuta S, Stenhagen IS, O’Duill M, et al. Catalytic decarboxylative fluorination for the synthesis of tri- and difluoromethyl arenes. Org Lett. 2013;15(11):2648–2651. | |
Huiban M, Tredwell M, Mizuta S, et al. A broadly applicable [F-18]trifluoromethylation of aryl and heteroaryl iodides for PET imaging. Nat Chem. 2013;5(11):941–944. | |
Parsons AT, Buchwald SL. Copper-catalyzed trifluoromethylation of unactivated olefins. Angew Chem Int Ed Engl. 2011;50(39):9120–9123. | |
Xu J, Fu Y, Luo DF, et al. Copper-catalyzed trifluoromethylation of terminal alkenes through allylic C–H bond activation. J Am Chem Soc. 2011;133(39):15300–15303. | |
Wang X, Ye Y, Zhang S, et al. Copper-catalyzed C(sp(3))-C(sp(3)) bond formation using a hypervalent iodine reagent: an efficient allylic trifluoromethylation. J Am Chem Soc. 2011;133(41):16410–16413. | |
Shimizu R, Egami H, Hamashima Y, Sodeoka M. Copper-catalyzed trifluoromethylation of allylsilanes. Angew Chem Int Ed Engl. 2012;51(19):4577–4580. | |
Parsons AT, Senecal TD, Buchwald SL. Iron(II)-catalyzed trifluoromethylation of potassium vinyltrifluoroborates. Angew Chem Int Ed Engl. 2012;51(12):2947–2950. | |
He ZB, Luo T, Hu MY, Cao YJ, Hu JB. Copper-catalyzed di- and trifluoromethylation of alpha, beta-unsaturated carboxylic acids: a protocol for vinylic fluoroalkylations. Angew Chem Int Ed Engl. 2012;51(16):3944–3947. | |
Wu XY, Chu LL, Qing FL. Silver-catalyzed hydrotrifluoromethylation of unactivated alkenes with CF3SiMe3. Angew Chem Int Ed Engl. 2013;52(8):2198–2202. | |
Mizuta S, Verhoog S, Engle KM, et al. Catalytic hydrotrifluoromethylation of unactivated alkenes. J Am Chem Soc. 2013;135(7):2505–2508. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.