Back to Archived Journals » Research and Reports in Medicinal Chemistry » Volume 5
QSAR and pharmacophore modeling of aminopyridazine derivatives of γ-aminobutyric acid as selective GABA-A receptor antagonists against induced coma
Received 30 June 2015
Accepted for publication 20 August 2015
Published 16 October 2015 Volume 2015:5 Pages 49—60
DOI https://doi.org/10.2147/RRMC.S91485
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Xing-Cong Li
Video abstract presented by Mohd Salman and Sisir Nandi
Views: 3272
Mohd Salman, Sisir Nandi
Division of Pharmaceutical Chemistry, Global Institute of Pharmaceutical Education and Research, Affiliated to Uttarakhand Technical University, Kashipur, Uttarakhand, India
Abstract: Quantitative structure–activity relationship (QSAR) and pharmacophore models have been developed in the present study to predict mode of binding of highly active aminopyridazine derivatives of γ-aminobutyric acid (GABA) acting as selective GABA-A receptor antagonists against induced coma. Descriptor-based QSAR model has been validated internally and by calculating applicability domain. Pharmacophore model predicted the important structural features including hydrophobicity and aromaticity at sixth position and aliphatic hydrophobic substituents must be substituted at fourth position of the aminopyridazine nucleus. Thus, the net aromaticity of the ligand may be enhanced by creating electron-rich environment nearer to the 3-amino cationic center of the aminopyridazine ring that are very much crucial for the inhibition of [3H] GABA-A receptor binding. The aromatic ligand may interact with the aromatic amino acid residues of pentameric structure of target. Finally, structure-based molecular docking study was performed for better interpretation of the mode of binding of the aminopyridazine compounds toward GABA-A target. Theoretical modeling utilizing molecular descriptor-based QSAR, pharmacophore generation, and molecular docking analysis of selective GABA-A receptor antagonists has not yet been reported. Therefore, this study has significant impact for designing of the highly active compounds in this series that are useful for the treatment of coma induced by drugs or chemicals.
Keywords: GABA-A receptor antagonists, QSAR, pharmacophore, molecular docking, induced coma
Introduction
In an attempt to wake comatose patients induced by intoxication of different chemicals, including barbiturates, benzodiazepines, alcohols, and inhalational anesthetics, and caused by different pathophysiological conditions, such as metabolic abnormalities, central nervous system diseases, and acute neurologic injuries, scientists are concentrating in designing and developing the more potent drugs. For the comatose patients, negative potential (Cl−) ions are more inside the neuronal cell membrane. These chloride ions can enter via opening of the ionotropic γ-aminobutyric acid (GABA)-A receptor. GABA is the major inhibitory neurotransmitter in brain. It is responsible for the overall balance between neuronal excitation and inhibition that is vital for normal brain function. Too much inhibition, or too little excitation, can lead to coma, depression, low blood pressure, sedation, or sleep. Too much excitation, or too little inhibition, can result in a range of conditions including convulsions, anxiety, high blood pressure, restlessness, and insomnia.1 GABA can produce its actions by occupying membrane-bound GABA receptors binding sites in the extracellular part of the receptor. There are two classes of GABA receptors including GABA-A and GABA-B. GABA-A receptors, also known as ionotropic receptors, are ligand-gated ion channels, whereas GABA-B receptors are G protein-coupled receptors that are also known as metabotropic receptors.2,3
Comatose patients can be treated by reversing the cause of coma (ie, glucose shock if low sugar), giving medication to stop brain swelling, inducing hypothermia. Inducing hypothermia on comatose patients provides one of the main treatments for patients who suffered from cardiac arrest.4 The comatose patients induced by overdose of the drugs or chemicals that may cause direct activation of Cl− ion channels can be recognized by the fast response of neurons to GABA-A receptor that is blocked by GABA-A receptor antagonists including bicuculline and picrotoxin. Bicuculline acts as a competitive antagonist to the GABA-A receptor. Noncompetitive antagonist picrotoxinin can bind to another site but effectively block the channel gating.2,5 Very few specific GABA-A receptor blockers have been developed yet.6 But there is hardly any quantitative structure–activity relationship (QSAR) and molecular modeling study of selective GABA-A receptor antagonists done.
Therefore, the design and development of GABA-A receptor antagonists are crucial for the treatment of comatose patients. A number of experiments have been carried out for this purpose. But experimental approach needs a lot of time, money, labor, and animal sacrifices. One of the most sophisticated techniques is pharmacoinformatics-based QSAR and drug design for predicting and modeling of the potent GABA-A receptor antagonists via chloride ion channel-blocking action.7
QSAR-based property predictions, pharmacophore modeling, and structure-based molecular docking are used for analyzing mode of binding of the highly active lead compounds. The success of any lead discovery depends on the accuracy of the input data, prediction of appropriate property using statistical tools, and most importantly validation of the developed model. Validation is the process by which the reliability and relevance of a procedure are established for a specific purpose; for pharmacophore models, validation must be mainly for robustness and prediction performances of the models. Prediction of novel property parameters for judging drug likeness of the compound is very important. In the present work, QSAR and pharmacophore modeling of a series of arylaminopyridazine derivative of GABA acting as a selective and competitive GABA-A receptor antagonist have been performed considering the structure of these chemical compounds. Then pharmacophore models have been validated utilizing molecular docking of the highly active compounds that interacted with the active binding sites of human GABA-A receptor target. These observations may help for the designing of potent and selective congeneric GABA-A receptor antagonists against induced coma.
Materials and methods
Biological activity data of aminopyridazine compounds
Biological activity is measured in terms of bioassays to calculate the inhibitory concentration against specific microorganism or binding affinity toward the receptor target proteins. Drug discovery often involves the use of QSAR to identify chemical structures that could have good inhibitory effects on specific targets and have low toxicity (nonspecific activity). In the present study, a series of 33 aminopyridazine derivatives of GABA acting as selective GABA-A antagonists have been considered from the published literature.8 These compounds were synthesized by substituting different aromatic groups at sixth position of pyridazine nucleus. Six-membered carbon skeleton aromatic rings (such as phenyl) with substituents introduced at 2, 3, or 4 position of the aromatic ring are more active than five-membered heteroaromatic ring system (such as thienyl). The substituents introduced at 2, 3, or 4 position of the aromatic ring are Cl, F, NO2, OH, OCH3, and CH3. Cl, F, NO2, and CH3 are electron withdrawing in nature, whereas OH and OCH3 are electron donating in nature.
These compounds have specific binding affinity toward GABA-A receptor antagonism. The biological activity of the above 33 compounds are expressed in terms of the concentration of the inhibitors where the response binding affinity (Ki) for the inhibition of [3H] GABA-A receptor binding (μM) is reduced by half. These Ki values were converted into pKi which is defined as negative log of Ki. All the structures along with compound’s biological activity values are given at Table 1.
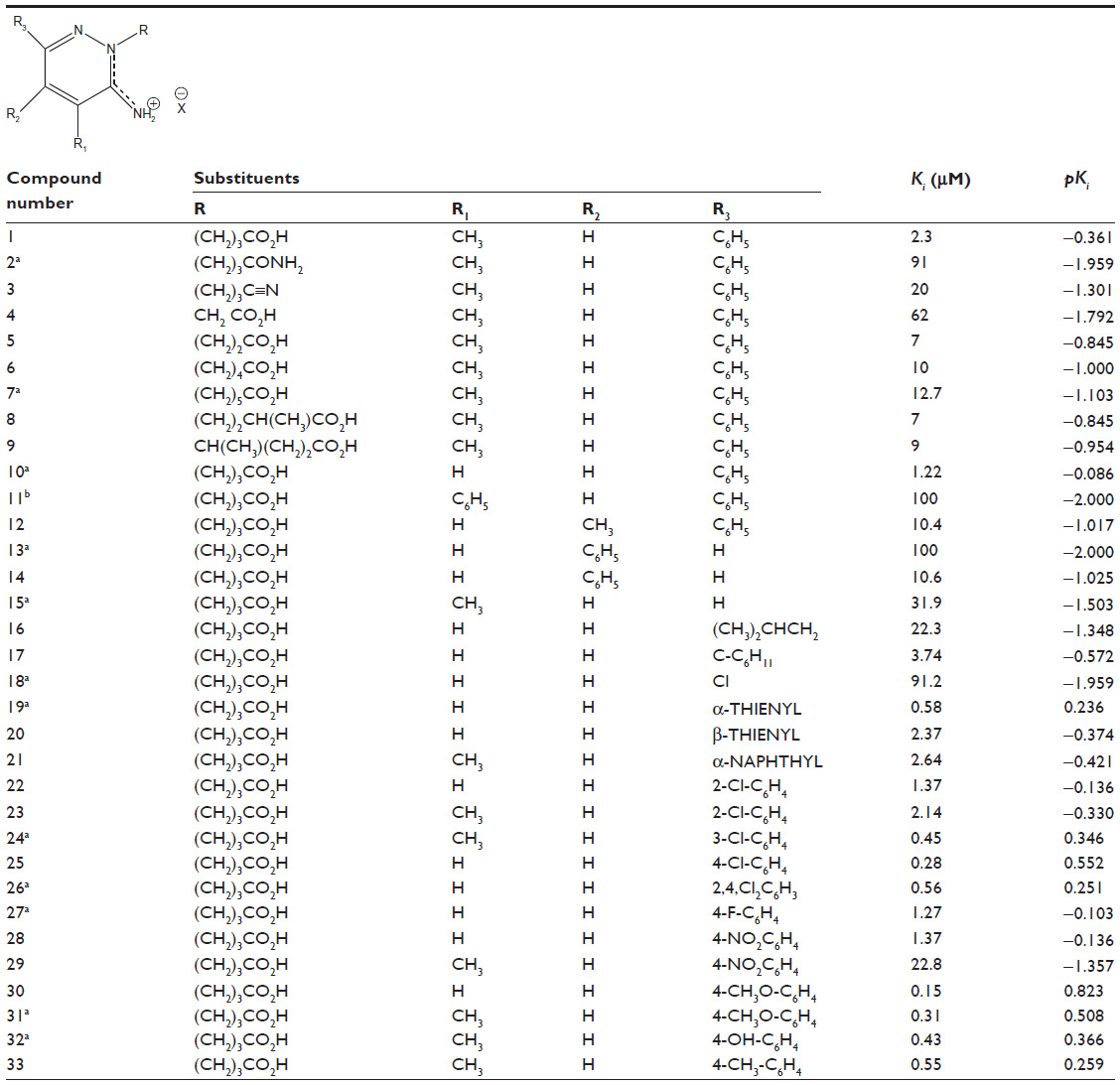 | Table 1 Biological activity data |
Computational section
Optimization of chemical structures
Structure optimization is performed to obtain the most energetically stable geometry of chemical structures. The structures of 33 aminopyridazine derivatives of GABA were drawn using 2D Chemdraw. The drawn structures were then converted into three-dimensional (3D) modules, and the 3D geometries of all compounds were fully optimized using MM2 force field using a value of 0.01 as dielectric constant considering Chem3D Ultra.9
These energetically minimized stable conformations were then taken into consideration for the computation of theoretical structural descriptors for further QSAR modeling.
Calculation of structural descriptors
The fully optimized structures of all aminopyridazine compounds were incorporated into molecular descriptor calculation module of PreADMET software,10 which is a web-free leading provider of tools in the area of structure prediction, structural proteomics, bioinformatics, chemoinformatics, and rational drug design. Theoretical molecular descriptors are the mathematical quantification of molecules that play a fundamental role in chemistry, pharmaceutical drug design, environmental protection, and computational and structural biology.11–13 A total of 201 molecular descriptors, including constitutional, geometrical, and physicochemical, are calculated solely from the structure of the molecules. Prior to the model development, these were reduced to 71. The reduction in the descriptors was due to keeping a constant value for, or nearly, all of the compounds and for those that perfectly correlated (r=1.0) with other descriptors. These descriptors are well-known properties for understanding the spatial orientation of molecules, pharmacokinetic profiles, and their interactions with the biological substrates and drug-like molecules at the atomic level.14 Table 2 contains calculated descriptors data that are used for the QSAR modeling utilizing regression analysis.
 | Table 2 Molecular descriptors used in this study |
Statistical analysis
To make a correlation between response and predictor variables, regression analysis is carried out. In the present study, compound’s binding affinity toward [3H] GABA-A receptor is taken as the response variable, whereas structural descriptors solely calculated from the chemical structure of 33 aminopyridazine derivatives of GABA were considered as predictor invariants. As the number of calculated descriptors is not so larger than the number of compounds, multiple linear regression is suitable in this study for QSAR modeling.15 MINITAB software16 has been used to develop QSAR equation taking forward selection as variable selection tools considering F-value =4.0. This QSAR model can explain the influences of descriptors toward ligand’s GABA-A receptor binding affinity.
Pharmacophore modeling
To focus on the mode of binding of these congeneric ligands toward antagonism of GABA-A receptor by blocking chloride ion channels, pharmacophore for the highly active molecules has been generated. The pharmacophore model can depict the assessment of 3D features encoding bioactivity of a series of active molecules having common binding mode to the biological target.17–19 Therefore, 3D pharmacophore is based upon the assumption that all the structurally diverse molecules can bind in a common binding mode to the biological target. These 3D properties include six pharmacophoric features such as hydrogen bond donor, hydrogen bond acceptor, hydrophobicity of the moiety, aromatic rings, positive ionization properties (cation), and negative ionization properties (anion).20–23
Pharmacophore modeling attempt for the aminopyridazine compounds was carried out using the software Portable InteLigand Scout (Version 2.02). Ligand Scout is a software tool that allows the rapid and transparently deriving 3D chemical feature-based pharmacophores from structural data of macromolecule ligand complexes in a fully automated and convenient way.24
Molecular docking
Very recently, X-ray crystal structure of human GABA-A receptor has been solved (Protein Data Bank [PDB] code 4COF).25 Therefore, these compounds were further subjected to molecular docking study to explore the interactions between aminopyridazine ligand and human GABA-A target. Molecular docking helps to study the ligand–receptor interaction for identifying the active binding sites of target protein. It helps to obtain the most energetically stable geometry of ligand–receptor complex, so that the energy of interactions between ligand and receptor would be minimal. The minimum energy of interaction can be represented by different scoring functions such as dock score, piecewise linear potential score, potential of mean force score, and steric and electrostatic score. This score is used to predict the binding affinity of a ligand toward receptor. This utility allows screening a set of compounds for lead optimization.26–29 In the present article, molecular docking calculations were carried out by using ArgusLab 4.0.1 dock engine,30,31 which is a freeware. It uses Ascore as scoring function.32 Ascore represents a probability-based score that considers different parameters such as hydrogen bond donors, hydrogen bond acceptors, π-stacking, and nonpolar hydrophobic interactions. ArgusLab docking tool is designed to enable flexible docking of ligands to perform a full conformational and positional search within a rigid binding site. This structure-based methods support QSAR and pharmacophore modeling for predictions of promising structural requirements necessary for the design of inhibitors.
The crystal structure of human GABA-A receptor used in the present exercise (PDB code: 4COF) was freely accessed and downloaded from the Brookhaven Protein Databank (http://www.rcsb.org). Water molecules and all HET bound molecules except BEN A 500 (benzamidine) were removed from the PDB file of the target protein. This cocrystallized HET benzamidine molecule was considered as a reference to make the binding site for the ligand X-ray group. The generated binding cavity consists of all active residues having at least one atom within 3.5 Å from any atom in the cocrystallized ligand X-ray group. The optimized highly active molecule number 30 was docked into active binding cavity of GABA-A target considering grid resolution (angle) of 0.4 degrees as default value. ArgusLab allows free rotation of the ligand inside the cavity so as to generate a number of 150 poses. The best complex pose with minimal interaction energy of −9.71 kcal/mol has been taken into consideration for better explanation of mode of interaction between the ligand and active amino acid residues of the receptor protein.
Results and discussion
QSAR modeling
QSAR model for the whole data set of 33 aminopyridazine derivatives of GABA has been developed using a combination of constitutional, geometrical, and physicochemical descriptors calculated solely from the structure of these chemical compounds.
The QSAR model in terms of multiple linear regression is given as

At N=33, R2=0.344, PRESS =25.6141, and S=0.778, where N is the number of observations, R is the square root of multiple R2 for regression, PRESS is predictive sum of squared deviation, and S is the standard error of estimation.
The descriptor-based QSAR model can explain only 34.4% of the variances of GABA-A receptor antagonism for the studied compounds. The result is not so satisfactory. Therefore, the data set has been studied for further statistical validation by calculating normal probability distribution of the residuals (Figure 1) and residual versus fitted plot of the response data (Figure 2), respectively.
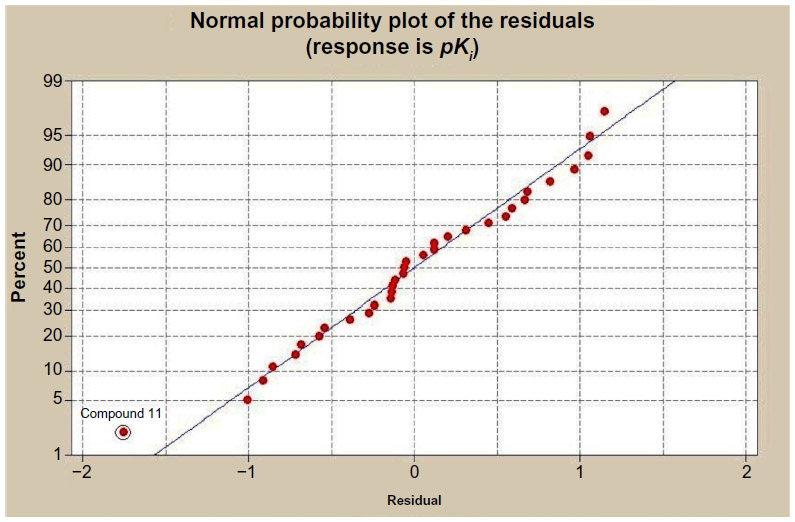 | Figure 1 Normal probability distribution of the residuals. |
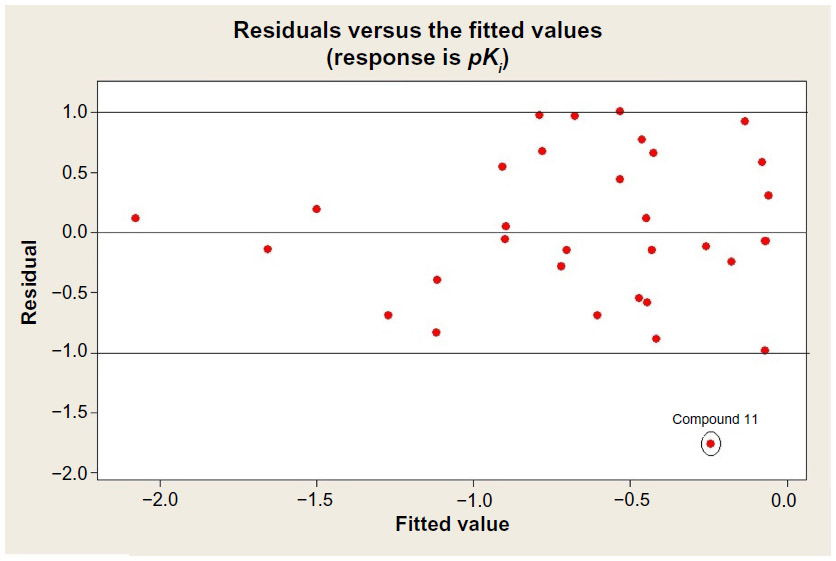 | Figure 2 Residual versus fitted plot of the response data. |
Normal probability distribution of the residuals graph has shown that compound 11 is bit a far from the regression line. The statistical analyses predicted that the compound 11 is out of fit in the applicability domain (−1.0 to 1.0) of the response data. The applicability domain of a QSAR is the physicochemical, structural, or biological space, knowledge, or information on which the model has been developed and for which it is applicable to make predictions for new compounds. The applicability domain of a QSAR should be described in terms of the most relevant parameters, that is, usually those that are descriptors of the model. This result gives some input for further in-depth structural study about compound number 11.
In a next attempt, compound number 11 has been deleted from the data set and again QSAR was modeled which follows as

At N=32, R2=0.50, PRESS =21.955, and S=0.683. Values of structural invariants modeled in the QSARs are given in Table 3.
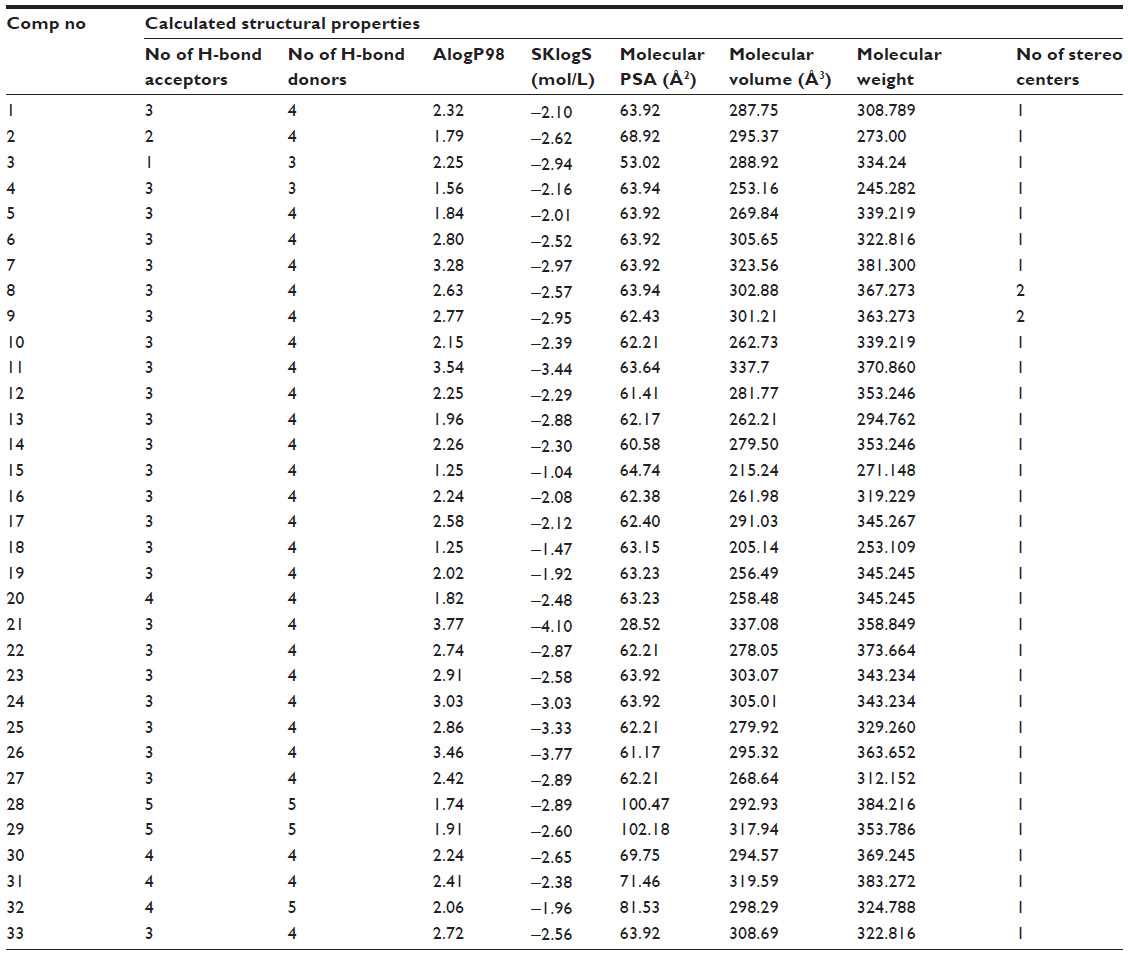 | Table 3 Calculated structural invariants of a series of aminopyridazine derivatives of γ-aminobutyric acid |
From the QSAR Equation 2, it is clear that R2 value is dramatically increased to 0.50 if compound number 11 is not taken into consideration. Descriptors with positive coefficients, such as number of H-bond acceptors, molecular AlogP98, SKlogS, topological polar surface area (PSA), and molecular weight, give positive impacts upon GABA-A receptor antagonism, whereas number of H-bond donors, molecular volume, and number of stereo centers having negative coefficients can produce reverse effects on the biological activity. Increasing the values of H-bond acceptors, molecular AlogP98, SKlogS, molecular topological PSA, and molecular weight may increase binding affinity of the ligand toward receptor cavity. H-bond acceptors and topological PSA are responsible for enhancing the polarity character of the molecule. Molecular AlogP98, SKlogS, and molecular weight contribute lipid solubility of the ligand. Another important statistical metric is the T-value associated with the model, defined as the descriptor coefficient divided by its standard error.33,34 Descriptors with large |T| values are important in the predictive model and, as such, can be examined in order to gain some understanding of the nature of property or activity of interest (Table 4).
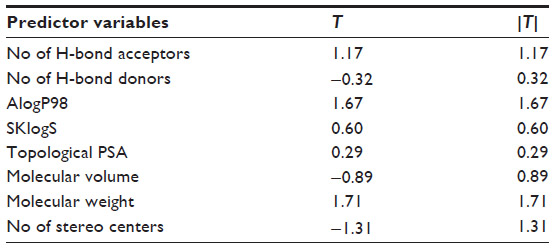 | Table 4 T-values of the modeled parameters |
T-values of the modeled parameters denote that number of H-bond acceptors, AlogP98, molecular weight, and number of stereo centers are important predictors responsible for producing binding affinity toward receptor.
Further robustness of the QSAR modeling has been done by dividing the data set (N=32) into training and test set approach and also by formulating several QSAR models using different types of descriptors, and results were compared in terms of prediction accuracy and statistical parameters including R2. Out of the 32 analogs, 62.5% of total compounds (20) have been considered as training set and remaining 37.5% of the total compounds (12) have been placed in the test set for the validation of derived models. Test molecules are randomly selected and marked by “a” as given in Table 1. It was observed that the training model based on the combination of constitutional, geometrical, and physicochemical indices contributes maximum impact on the GABA-A receptor antagonism activity obtained in terms of R2=0.511, whereas QSAR models under the framework of constitutional, geometrical, and physicochemical indices alone produces lower to moderate impacts in terms of R2 values of 0.471, 0.460, and 0.445, respectively. The validated training QSAR model framed by the combination of constitutional, geometrical, and physicochemical indices is given as follows.

At N=20, R2=0.511, PRESS =41.210, and S=0.630.
The model 3 can explain maximum variance of 51.1% of the GABA-A receptor antagonism of the studied compounds. The model 3 was then used to predict the binding affinity of the test compounds. Observed and predicted binding affinities for the test compounds were plotted and represented in Figure 3. It is clear that all the points except two are seemed to be nearer to the regression line. Therefore, good prediction was done by the training model.
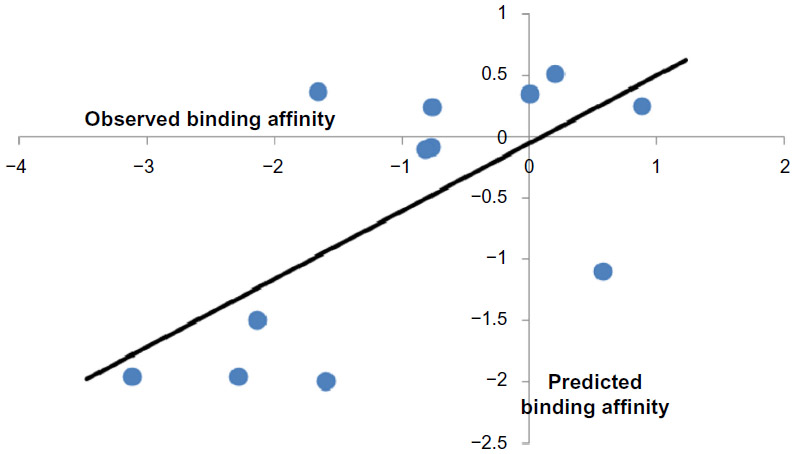 | Figure 3 Plot of observed versus predicted binding affinity of the test compounds. |
Prediction of mode of binding using pharmacophore modeling
In the present study, highly active compounds including 25, 30, 31, and 32 have been selected for pharmacophore model development considering highest active compound (number 30) as reference. A unique lowest energy 3D structure was generated for each mentioned ligand with the help of Chem3D Ultra, where appropriate hydrogen was added to all the structures and was subsequently subjected to energy minimization using MM2 force field with a constant dielectric of 0.01. Ligand-based pharmacophore model generation was performed with Ligand Scout using default settings. The pharmacophore of the groups and subgroups has been superimposed in order to get the common pharmacophore of highly active aminopyridazine derivatives of GABA acting as selective GABA-A receptor antagonists (Figure 4).
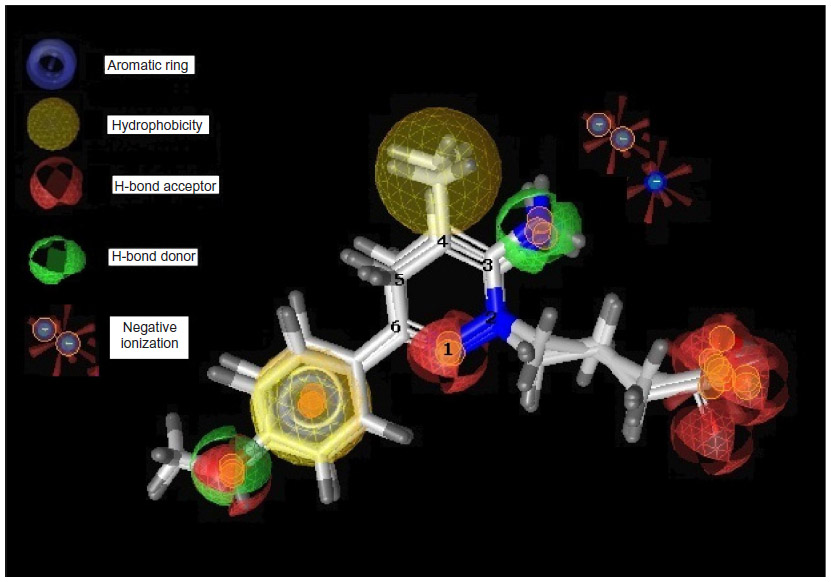 | Figure 4 Pharmacophore model of highly active compounds. |
The earlier model can predict the mode of binding of highly active congeners acting as GABA-A receptor antagonist via Cl− ion channel-blocking action. GABA-A receptor–Cl− ion channel complex is composed of five subunits such as α1, 2β2, and 2γ2 pentamers. The subunits are thought to be arranged around the channel like a rosette, and α-subunit usually bears the GABA-binding sites. Pharmacophore model clears that the highly active predicted ligand would be aromatic in nature. The aromatic ligand may interact with the aromatic amino acid residues of the pentameric subunits. The sixth position of the aminopyridazine moiety should be substituted by aromatic and hydrophobic groups; the fourth position of this aromatic group associated to the sixth position of the aminopyridazine nucleus should be connected with hydrogen bond donor or hydrogen bond acceptor group. The fourth position of the aminopyridazine should be substituted by hydrophobic moiety; the third position of the aminopyridazine should be substituted by hydrogen bond donating groups (such as amino group). This amino group contributed positively charged center, which is in proximity with the negatively charged ionization of anionic group as well as electron-rich environment contributed by π electron delocalization of aromatic group and 2-butyryl side chain of the aminopyridazine moiety. Linking a butyric side chain to the N(2) nitrogen of a aminopyridazine nucleus may contribute hydrogen bond acceptor property necessary for the receptor antagonism. Hydrogen bond acceptor is one of the important properties for producing necessary biological activity for these congeners. It has already been predicted by the QSAR model Equation 2. QSAR modeling and statistical analysis of these congeners assume intensive structural study of compound number 11. Therefore, pharmacophore model of this compound has been done for predicting the structural features as shown in Figure 5.
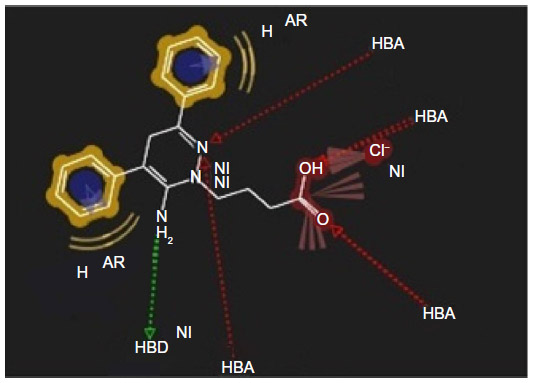 | Figure 5 Pharmacophore model of low active compound number 11. |
Substitution of aromatic group at the fourth position of the aminopyridazine nucleus produces more electron density or negative ionization nearer to the cationic center of the aminopyridazine nucleus. This may contribute detrimental effects to the GABA-A receptor antagonism. Therefore, fourth position of the aminopyridazine must be substituted by aliphatic hydrophobic moiety instead of aromatic group.
The pharmacophore models were developed based on two important assumptions: 1) the free and receptor-bound conformations of all 33 aminopyridazine derivatives of GABA are identical and 2) the ligand conformations obtained by ab initio modeling followed by energy minimization are unique, but these concepts are not always true, sometimes the same ligand adopting multiple, equally probable conformations. Therefore, both these assumptions should be better justified by molecular docking study of aminopyridazine and human GABA-A target.
Aminopyridazine and human GABA-A target docking
From the docking study (Figure 6), it is clear that the sixth position of the aminopyridazine moiety is surrounded by hydrophobic and aromatic residues, such as LEU 99, GLU 155, and TYR 97, respectively. The sixth aromatic substituent has formed a π-stacking interaction with TYR 1493. The third position substituent is connected with TYR 205 (blue stick model) by a hydrogen bond with a distance of 2.49 Å, and this moiety is interacted with hydrophobic amino acid residues including GLY 158 and THR 202. Carboxylic group of N(2)-butyryl side chain has interacted with the GLN 1495 (pink stick model) and TYR 157 (white stick model) by three hydrogen bondings having distances of 1.5 Å, 1.87 Å, and 2.24 Å, respectively. Hydrogen bond, hydrophobic, aromatic, and π-stacking interactions play a major role for producing GABA-A receptor antagonism activity. These findings are perfectly correlated with the pharmacophore developed in the present models for the active ligands of aminopyridazine derivatives of GABA.
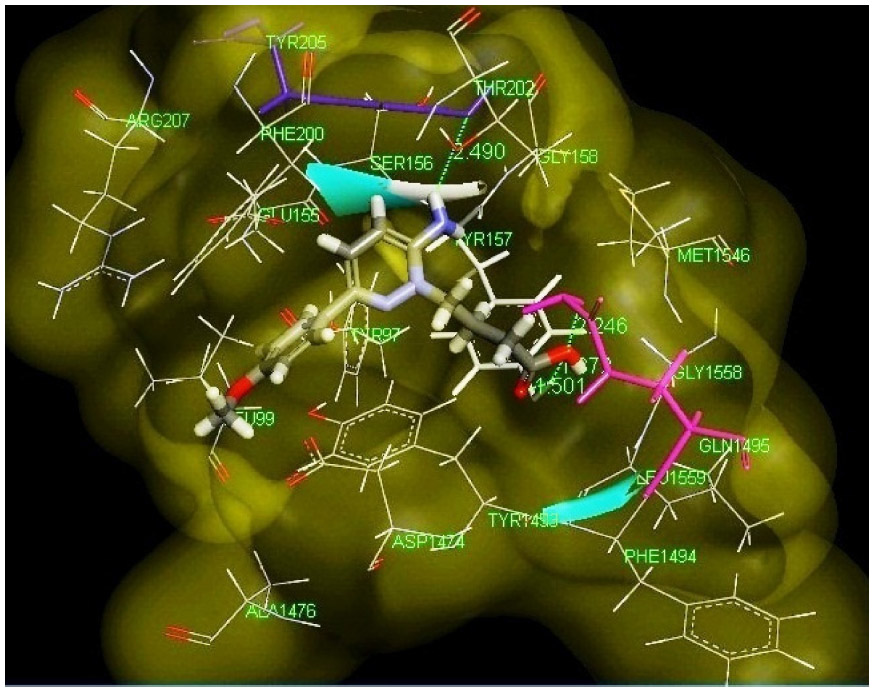 | Figure 6 Best pose of highly active ligand 30 (stick model) docked with the human GABA-A target cavity represented by molecular surface. |
Conclusion
In the present work, an attempt has been made for QSAR, pharmacophore modeling, and structure-based molecular docking to predict the mode of binding of active aminopyridazine congeners of GABA acting as selective GABA-A receptor antagonist. Pharmacophore and docking models are correlated that the sixth position of the 3-aminopyridazine system should be replaced by aromatic and hydrophobic groups that can contribute to aromaticity and hydrophobic interactions with the residues such as LEU 99, GLU 155, and TYR 97, respectively. The sixth aromatic substituent also produced a π-stacking interaction with TYR 1493. Therefore, π electron delocalization occurs in the pyridazine system of positively charged center present at 3-amino position. This may create electron-rich environment in proximity of the cationic 3-amino center of the pyridazine system that may fast enhance the binding affinity of the ligand toward GABA-A receptor-mediated chloride ion channel-blocking activity. N(2)-butyryl side chain formed three hydrogen bonds with GLN 1495 and TYR 157 that are very crucial for producing biological activity. Statistical analyses detected outlier property of compound number 11. It has been further searched the reason behind this. It was predicted from the pharmacophore model that the fourth position of the aminopyridazine nucleus must be replaced by the aliphatic hydrophobic group instead of aromatic moiety that may impart negative effect toward binding affinity of the ligand toward receptor site. The study in this direction may help the synthetic chemist in designing the promising aminopyridazine derivatives of GABA that are beneficial for the treatment of induced coma utilizing the following necessary model (Figure 7) formulated using pharmacophoric features of highest active compound number 30.
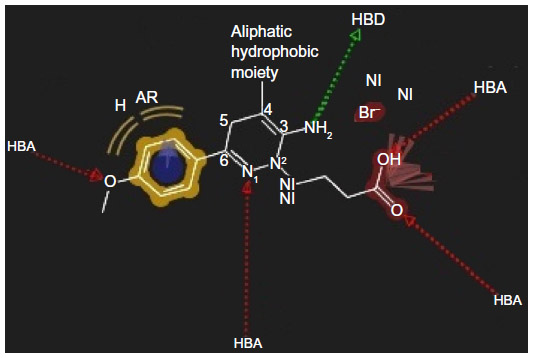 | Figure 7 Structural requirements for designing highly active congeneric compounds. |
Acknowledgments
SN sincerely thanks the National Institute of Chemistry, Slovenia, for availing Ligand Scout software used in the present work. Salman shows deep sense of gratitude to his supervisor Dr Sisir Nandi for the guidance and motivation. Salman acknowledges Mr Rajeshwar KK Arya, Department of Pharmaceutical Sciences, Kumaun University, Bhimtal Campus, India, for his constructive suggestions in this project.
Disclosure
The authors report no conflicts of interest in this work.
References
Chebib M, Johnston GAR. GABA-activated ligand gated ion channels: medicinal chemistry and molecular biology. J Med Chem. 2000;43: 1427–1447. | |
Bormann J. The ‘ABC’ of GABA receptors. Trends Pharmacol Sci. 2000;21:16–19. | |
Wagner DA, Czajkowski C. Structure and dynamics of the GABA binding pocket: a narrowing cleft that constricts during activation. J Neurosci. 2001;21:67–74. | |
Baldursdottir S, Sigvaldason K, Karason S, Valsson F, Sigurdsson GH. Induced hypothermia in comatose survivors of asphyxia: a case series of 14 consecutive cases. Acta Anaesthesiol Scand. 2010;54: 821–826. | |
Sigel GJ. In: Delorey TM, Olsen RW, editors. Basic Neurochemistry. New York: Raven Press; 1994:398–399. | |
Schwartz DR, Suzdak PD, Paul SM. γ-Aminobutyric acid (GABA)- and barbiturate mediated Cl- uptake in rate brain synaptoneurosomes: evidence for rapid desensitization of the GABA receptor-coupled chloride ion channel. Mol Pharmacol. 1986;30:419–426. | |
Nantasenamat C, Isarankura-Na-Ayudhya C, Prachayasittikul V. Advances in computational methods to predict the biological activity of compounds. Expert Opin Drug Discov. 2010;5:633–654. | |
Wermuth CG, Bourguignon JJ, Schlewer G, et al. Synthesis and structure-activity relationships of a series of aminopyridazine derivatives of γ-aminobutyric acid acting as selective GABA-A antagonists. J Med Chem. 1987;30:239–249. | |
Mills N. ChemDraw ultra 10.0. J Am Chem Soc. 2006;128: 13649–13650. | |
Pre Admet [homepage on the Internet]. Available from: http://preadmet.bmdrc.org/. Accessed October 2, 2015. | |
Todeschini R, Consonni V. Molecular Descriptors for Chemoinformatics. Vol 2. Wiley-VCH, Weinheim; 2009. | |
Karelson M. Molecular Descriptors in QSAR/QSPR. John Wiley & Sons, New York; 2000. | |
Devillers J, Balaban AT, editors. Topological Indices and Related Descriptors in QSAR and QSPR. Taylor and Francis, Cambridge; 2000. | |
Warren J. Drug discovery: lessons from evolution. Br J Clin Pharmacol. 2011;71:497–503. | |
Katritzky AR, Petrukhin R, Tatham D, et al. Interpretation of quantitative structure-property and -activity relationships. J Chem Inf Comput Sci. 2001;41:679–685. | |
Minitab® 17 [homepage on the Internet]. Available from: http://www.minitab.com. Accessed September 24, 2015. | |
Leach AR, Gillet VJ, Lewis RA, Taylor R. Three dimensional pharmacophore methods in drug discovery. J Med Chem. 2009;53: 539–558. | |
Shoichet BK. Virtual screening of chemical libraries. Nature. 2004; 432:862. | |
Mason JS, Good AC, Martin EJ. 3-D pharmacophores in drug discovery. Curr Pharm Des. 2001;7:567–597. | |
Langer T, Krovat EM. Chemical feature-based pharmacophores and virtual library screening for discovery of new leads. Curr Opin Drug Discov Dev. 2003;6:370–376. | |
Koes DR, Camacho CJ. Pharmer: efficient and exact pharmacophore search. J Chem Inf Model. 2011;51:1307–1314. | |
Nandi S, Bagchi MC. Activity prediction of some non tested anticancer compounds using GA based PLS regression models. Chem Biol Drug Des. 2011;78:587–595. | |
Batra A, Nandi S, Bagchi MC. QSAR and pharmacophore modeling of indole-based C-3 pyridone compounds as HCV NS5B polymerase inhibitors utilizing computed molecular descriptors. Med Chem Res. 2015;24:2432–2440. | |
Wolber G, Langer T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J Chem Inf Model. 2005;45:160–169. | |
Miller PS, Aricescu AR. Crystal structure of a human gamma-aminobutyric acid receptor, the GABA(A)R-beta3 homopentamer. Nature. 2014;512:270–275. | |
Kitchen DB, Decornez H, Furr JR, Bajorath J. Docking and scoring in virtual screening for drug discovery: methods and applications. Nat Rev Drug Discov. 2004;3:935–948. | |
Teague SJ. Implications of protein flexibility for drug discovery. Nat Rev Drug Discov. 2003;2:527–541. | |
Stahl M, Rarey M. Detailed analysis of scoring functions for virtual screening. J Med Chem. 2001;44:1035–1042. | |
Nandi S, Bagchi MC. 3D-QSAR and molecular docking studies of 4-anilinoquinazoline derivatives: a rational approach to anticancer drug design. Mol Divers. 2010;14:27–38. | |
Thompson MA. ArgusLab 4.0.1. Seattle, WA: Planaria Software LLC. Available from: http://www.arguslab.com. | |
Thompson MA, Zerner MC. A theoretical examination of the electronic structure and spectroscopy of the photosynthetic reaction center from Rhodopseudomonas viridis. J Am Chem Soc. 1990;113: 8210–8215. | |
Beausoleil SA, Villen J, Gerber SA, Rush J, Gygi SP. A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat Biotechnol. 2006;24:1285–1292. | |
Bagchi MC, Mills D, Basak SC. Quantitative structure-activity relationship (QSAR) studies of quinolone antibacterials against M. fortuitum and M. smegmatis using theoretical molecular descriptors. J Mol Model. 2007;13:111–120. | |
Nandi S, Bagchi MC. QSAR of aminopyrido[2,3-d]pyrimidin-7-yl derivatives: anticancer drug design by computed descriptors. J Enzyme Inhib Med Chem. 2009;24:937–948. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.