Back to Journals » Therapeutics and Clinical Risk Management » Volume 12
New developments in the treatment of chemotherapy-induced neutropenia: focus on balugrastim
Authors Ghidini M, Hahne JC, Trevisani F, Panni S, Ratti M, Toppo L, Tomasello G
Received 1 February 2016
Accepted for publication 10 May 2016
Published 24 June 2016 Volume 2016:12 Pages 1009—1015
DOI https://doi.org/10.2147/TCRM.S80732
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Garry Walsh
Video abstract presented Michele Ghidini.
Views: 604
Michele Ghidini,1 Jens Claus Hahne,2 Francesco Trevisani,3 Stefano Panni,1 Margherita Ratti,1 Laura Toppo,1 Gianluca Tomasello1
1Medical Department, Division of Oncology, ASST di Cremona, Ospedale di Cremona, Cremona, Italy; 2Division of Molecular Pathology, The Institute of Cancer Research, London and Sutton, UK; 3Department of Urology, Unit of Urology/Division of Oncology, IRCCS Ospedale San Raffaele, URI, Milan, Italy
Abstract: Neutropenia and febrile neutropenia are two major complications of chemotherapy. Dose reductions, delays in treatment administration, and the use of granulocyte colony-stimulating factors are equally recommended options to preserve absolute neutrophil count in case of chemotherapy regimens bringing a risk of febrile neutropenia of 20% or higher. Recombinant granulocyte colony-stimulating factors, such as filgrastim and lenograstim, have a short elimination half-life (t1/2) and need to be used daily, while others, like pegfilgrastim and lipegfilgrastim, are characterized by a long t1/2 requiring only a single administration per cycle. Balugrastim is a novel long-acting recombinant granulocyte colony-stimulating factor obtained by means of a genetic fusion between recombinant human serum albumin and granulocyte colony-stimulating factor. Albumin binding increases the molecular weight and determines a high plasmatic stability leading to a t1/2 of ~19 days. Balugrastim’s efficacy, safety, and tolerability have been assessed in four different clinical trials involving breast cancer patients treated with doxorubicin and docetaxel. Pegfilgrastim was chosen as a comparator. Balugrastim was noninferior to pegfilgrastim with regard to the reduction of mean duration of severe neutropenia during cycle 1. Moreover, both treatments were comparable in terms of efficacy and safety profile. Balugrastim was well tolerated, with the only related adverse event being mild to moderate bone pain. The aim of this review is to summarize the currently available literature data on balugrastim.
Keywords: G-CSF, granulocyte colony-stimulating factors, albumin, febrile neutropenia, pegfilgrastim
Introduction to balugrastim and management issues in chemotherapy-induced neutropenia
Neutropenia and febrile neutropenia (FN) represent two frequent complications of chemotherapy treatments.1 Neutropenia can be defined as a reduction in the normal count of neutrophils, while FN represents a medical emergency in which the absolute neutrophils count (ANC) falls below the value of 0.5×109/L and the body temperature is equal or superior to 38°C for >1 hour.2 Neutrophils act as early responders against pathogens. Indeed, during mucositis, alterations in the microbial flora and the concomitant disruption of the barrier function of mucosa can lead to sepsis due to the lack of these early responding innate immune cells.3
Having recognized that these conditions can lead to dose reductions, delay in chemotherapy and hospitalization due to the high risk of infections and sepsis, the role of granulocyte colony-stimulating factors (G-CSFs) is fundamental.1 The natural human G-CSF is a glycoprotein produced by monocytes, fibroblasts, and endothelial cells.4–8 G-CSF is not species-specific. It has been shown to primarily affect neutrophil progenitor proliferation within the bone marrow and to stimulate the release of mature neutrophils into the peripheral blood,9–15 and promote differentiation,8,15 and selected end-cell functional activation, for example, enhanced phagocytic activity.16,17
Recombinant granulocyte colony-stimulating factors (rG-CSFs) act as endogenous G-CSF and are clinically indicated in order to preserve ANC in the case of chemotherapy regimens associated with a risk of FN ≥20%.18 Some of these regimens are used in breast cancer (eg, cisplatin, doxorubicin, and cyclophosphamide [PAC], docetaxel, doxorubicin, cyclophosphamide [TAC], and dose dense 5-fluorouracil, epirubicin, and cyclophosphamide [FEC]).19 Rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone, used in non-Hodgkin’s lymphomas, is associated with a 20% risk of FN.20 Docetaxel–carboplatin doublet, used in non-small-cell lung cancer, doxorubicin and ifosfamide for sarcomas and docetaxel, cisplatin, and 5-fluororuracil increase the risk of FN by >20% as well.19 rG-CSFs can be administered both for primary and secondary prophylaxis. In the first case, G-CSF is used to prevent FN at the beginning of a “high-risk” treatment, while the use in secondary prophylaxis happens after a first neutropenic event.19 rG-CSFs, such as filgrastim and lenograstim, have a short elimination half-life (t1/2), thus requiring administration on a daily basis.2 On the other hand, long-acting rG-CSFs, such as pegfilgrastim and lipegfilgrastim, are pegylated molecules of filgrastim.2 Because of this stabilizing modification, they have a reduced renal clearance and are mainly metabolized by neutrophils and require a single administration per cycle.2
A new generation of long-acting rG-CSFs has been recently developed by combining G-CSF with a protein or peptide (fusion proteins) in order to increase molecular weight and lengthen t1/2.21 Balugrastim is a long-acting rG-CSF produced in the yeast Saccharomyces cerevisiae obtained by means of a genetic fusion between recombinant human serum albumin and G-CSF.21,22 By combining the N-terminus of G-CSF to the C-terminus of albumin, balugrastim has a high plasmatic stability and can be administered in a once-per-cycle fixed dosage.21 So far, few studies have investigated balugrastim’s safety and activity. This review aims at summarizing the available literature data on this novel molecule, focusing on pharmacology, pharmacokinetics, toxicity, and efficacy.
Pharmacology, mechanism of action, and pharmacokinetics of balugrastim
Balugrastim is a 759 amino acid single polypeptide chain with a molecular mass of ~85 kDa.23 The biological active compound of balugrastim is G-CSF, a naturally occurring cytokine that has several effects on hematopoietic progenitor cells.24 The natural human G-CSF is a glycoprotein composed of a single polypeptide chain of 175 amino acids and is glycosylated at threonine 133.25 By fusion of G-CSF to human serum albumin, the turnover rate of G-CSF is reduced and it has a longer duration of action.26 The t1/2 of balugrastim is ~19 days in humans, and the G-CSF fusion protein is highly soluble.26 Balugrastim can be used as a long-lasting adjunct therapy to patients receiving myelosuppressive chemotherapy for reducing incidence and duration of chemotherapy-induced neutropenia and FN.26
Balugrastim exhibits nonlinear pharmacokinetics.27 Clearance of balugrastim is dependent on its concentration and neutrophil count. Normally, G-CSF is cleared by the kidney,28 but the human serum albumin component of balugrastim not only acts as a carrier of G-CSF, it also extends its plasma half-life by minimizing renal clearance.29 Therefore, the main elimination of balugrastim is via receptor-mediated neutrophil endocytosis.29
Subcutaneous administration of 3.45 and 11.5 μg/kg of balugrastim resulted in maximum serum concentrations of 4 and 49 ng/mL, respectively, within 2–8 hours.11,30 After intravenous administration, the volume of distribution averaged 150 mL/kg and the elimination half-life was ~3.5 hours.31
Intravenous (1–70 μg/kg twice daily) as well as subcutaneous administration (1–3 μg/kg once daily, or by continuous subcutaneous infusion 3–11 μg/kg/day) of G-CSF resulted in a dose-dependent increase in neutrophil counts over the dose range of 1–70 μg/kg/day.32–34 One study described the subcutaneous use of 450 μg/kg balugrastim without severe side effects in breast cancer patients.35
Clearance rates of balugrastim were ~0.5–0.7 mL/min/kg.27 Single parenteral doses or daily intravenous doses over a 14-day period resulted in comparable half-lives.27 The half-lives were similar for intravenous administration (231 minutes, following doses of 34.5 μg/kg) and for subcutaneous administration (210 minutes, following G-CSF dosages of 3.45 μg/kg).27 Continuous 24-hour intravenous infusions of 20 μg/kg over an 11–20-day period produced steady-state serum concentrations of balugrastim with no evidence of drug accumulation over the time period investigated.27
Firstly, balugrastim safety was evaluated in a Phase I/IIa study.35 The results of this study must be considered very carefully because of the very limited statistical power.35 Pharmacokinetic analysis demonstrated that balugrastim serum concentrations were detectable across different dose groups (50, 150, 300, and 450 μg/kg) in most patients (45 out of 50 patients) for at least 144 hours post dose.35 Drug exposure was higher in cycle 1 compared to cycle 0, most likely because chemotherapy reduces the number of neutrophils, which play an important role in the clearance of balugrastim.35 In cycle 1, balugrastim was detected longer than 144 hours in most patients (45/50 sampled) only in the 150, 300, and 450 μg/kg dose groups, supporting once-per-cycle dosing.35 The median t1/2, of balugrastim in cycle 1 was ~36 hours for the 300 μg/kg dose group and 30 hours for the 450 μg/kg dose group.35
In one multicenter Phase II study, safety and efficacy of balugrastim versus pegfilgrastim were further evaluated in breast cancer patients scheduled to receive myelosuppressive chemotherapy.1 In this study, three doses (30, 40, and 50 mg) were considered.1 Results from the compartmental modeling analysis indicated that balugrastim was slowly absorbed from the subcutaneous injection site, with the majority of the medication absorbed within 24–36 hours.1 The mean absorption t1/2 for balugrastim ranged in this study between 8.1 and 15.4 hours and tended to increase with escalations in dose.1 For most patients, 80% of dose absorption occurred within the first 24–36 hours.1 Mean coefficient of variation (Cmax) values for patients treated with balugrastim 30, 40, or 50 mg were 408, 1,580, and 1,200 ng/L, respectively.1 Median time to Cmax (Tmax) value was 12.0 hours for the balugrastim 30 mg group and 24.0 hours for both the higher dose group.1
Recently, the results of two Phase III studies comparing the effects of balugrastim treatment versus pegfilgrastim in breast cancer patients receiving chemotherapy have been published.18,26 Pegfilgrastim is another recombinant G-CSF produced in Escherichia coli.36 The t1/2 of pegfilgrastim is extended by attachment of polyethylene glycol moiety. Such modification facilitates once-per-chemotherapy-cycle dosing.36 In a double-blind randomized Phase III study, 40 mg of balugrastim and 6 mg of pegfilgrastim per chemotherapy cycle were used and compared in patients with breast cancer receiving chemotherapy.23 These two drugs showed comparable plasma t1/2 in cycle 1 of 38.7 and 41.7 hours, respectively.26 The other trial was a multicenter, randomized, open-label, Phase III noninferiority study.18 In this study, the efficacy and safety of once-per-cycle 40 mg or 50 mg of balugrastim and 6 mg of pegfilgrastim were compared again only in patients with breast cancer receiving chemotherapy with doxorubicin and docetaxel.18 In this patient cohort, the median terminal elimination t1/2 was found to be 37.4 hours for 40 mg and 35.5 hours for 50 mg of balugrastim.18 The mean Cmax (coefficient of variation [CV]%) for 40 and 50 mg of balugrastim was 875 (76.3%) and 975 (74.1%) ng/mL, respectively.1,18 The median Tmax was ~24 hours for both treatment groups.18 Mean estimates for apparent total clearance were 1.34 and 1.18 L/hour for the 40 mg and 50 mg balugrastim groups, respectively.18 Mean estimates for the apparent volume of distribution were 80.1 (for 40 mg balugrastim group) and 69.2 (for 50 mg balugrastim group).18
Differences in pharmacology and pharmacokinetics between balugrastim and pegfilgrastim are listed in Table 1.
 | Table 1 Comparison of pharmacology and pharmacokinetics between balugrastim and pegfilgrastim |
Efficacy studies
Balugrastim’s efficacy has been mainly evaluated in Phase II and in two Phase III trials (Table 2).1,18,26 In all cases, only patients affected by breast cancer and receiving chemotherapy with doxorubicin 60 mg/m2 and docetaxel 75 mg/m2 every 21 days were enrolled. The choice of this systemic therapy was determined by the high risk of FN given by the combination therapy (33%).1,18,26 Gladkov et al1,18 enrolled 78 patients for the Phase II and 256 patients for Phase III trials. In the Phase II study, patients were treated with escalating doses of balugrastim (30, 40, or 50 mg) or the standard dose of pegfilgrastim (6 mg).1 Similarly, in the open-label randomized Phase III study, patients were randomly assigned to 40 or 50 mg of balugrastim or 6 mg of pegfilgrastim (1:1:1 ratio).18 Conversely, Volovat et al26 designed a two-phase study: first, a double-blind session (n=304) with a randomization to balugrastim 40 mg or pegfilgrastim 6 mg (1:1 ratio) and second, an open-blind single-arm phase in which patients (n=77) further received balugrastim. In both studies, patients received a subcutaneous injection of either balugrastim or pegfilgrastim 24 hours after chemotherapy for up to four cycles.1,18,26
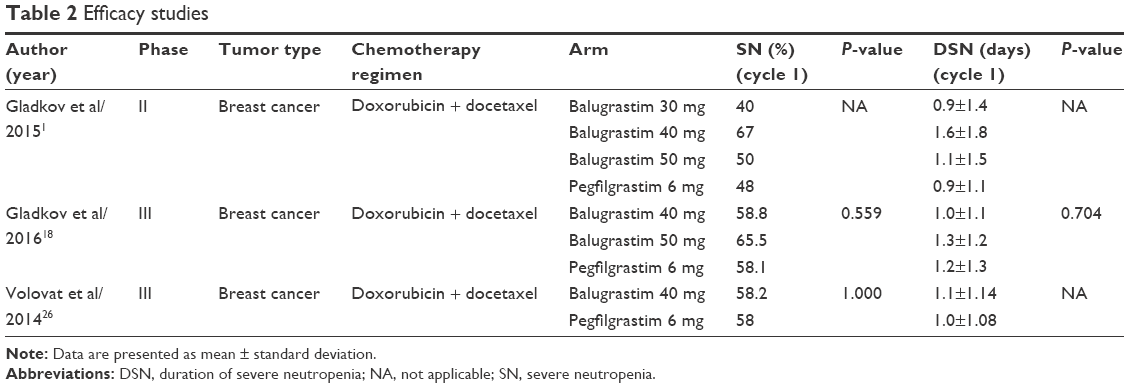 | Table 2 Efficacy studies |
The Phase II study was aimed at investigating different doses of balugrastim with a dose-escalation system in order to select two dosages for the Phase III trial.1 Time to ANC recovery (days between ANC nadir and ANC >0.5×109/L) for cycle 1 was similar for balugrastim 40 mg (2.6 days) and 50 mg (2.0 days), pegfilgrastim (2.4 days), and longer for balugrastim 30 mg (3.1 days).1 Moreover, in the same cycle, patients treated with balugrastim 30 mg suffered from a 20% incidence of FN, which was a higher result than the rates for balugrastim 40 mg (9.5%) and 50 mg (10%), and pegfilgrastim (8%).1 Based on these results, dosages of 40 and 50 mg were chosen for comparison with pegfilgrastim in the subsequent randomized Phase III.18
The two Phase III studies shared the same primary endpoint, which was the duration of severe neutropenia (DSN) in cycle 1, defined as the amount of days with ANC inferior to 0.5×109/L.18,26 Gladkov et al18 reported a nonsignificant difference in DSN between the different treatment groups during cycle 1. Indeed, mean DSNs for balugrastim 40 and 50 mg, and pegfilgrastim were 1.0, 1.3, and 1.2 days (P=0.704), respectively.18 Similarly, in Volovat’s study, mean DSN in the balugrastim 40 mg group was 1.1 days compared to 1.0 day obtained by using pegfilgrastim (95% two-sided confidence interval: −0.13 to 0.37 days).26 In the end, balugrastim was assumed to be noninferior to pegfilgrastim.18,26 Moreover, while the mean DSN was shorter in cycles 2–4 than in cycle 1, no statistically significant difference in the mean DSN between balugrastim and pegfilgrastim was reported in these cycles as well.18,26
As far as secondary efficacy endpoints are concerned, incidence of severe neutropenia (ANC <0.5×109/L), incidence of grade 3/4 neutropenia and its duration did not differ between balugrastim and pegfilgrastim, but decreased in rate and length from cycle 1 to cycles 2–4.18,26 Similarly, incidence of FN in cycle 1 was similar between pegfilgrastim and balugrastim in both studies: rates were 3.5% for balugrastim 40 mg, 6.0% for balugrastim 50 mg, and 2.3% in the pegfilgrastim group (P=0.398),18 versus 2% for balugrastim 40 mg and 4% for pegfilgrastim, P=0.446.26
Values of mean ANC nadir and time to ANC nadir did not differ between pegfilgrastim and balugrastim treatments in both studies.18,26 Mean ANC nadir in cycle 1 was considerably lower than in subsequent cycles, with values of 0.7, 0.6, and 0.7×109/L, respectively, for balugrastim 40 mg and 50 mg, and pegfilgrastim 6 mg, (P=0.423).18 Between cycles 2 and 4, mean ANC nadir reached the lowest point of 1.2 in cycle 3 with balugrastim 50 mg and a highest value of 1.6×109/L in cycle 4 with balugrastim 40 mg.18 Similar values were reported by Volovat et al26 regarding mean ANC nadir for cycle 1 (0.8 for both pegfilgrastim and balugrastim, P=0.763). Mean times to ANC nadir in cycle 1 were 6.4, 6.7, and 6.5 days, respectively, for balugrastim 40 mg and 50 mg, and pegfilgrastim (P=0.610), and slightly increased in further cycles, with a minimum of 6.8 days in cycle 1 with balugrastim 40 mg and a maximum of 7.8 days with pegfilgrastim, both in cycles 3 and 4.18 The randomized double-arm study registered a mean time to ANC nadir of 6.7 days for pegfilgrastim and of 6.8 days for balugrastim 40 mg (P=0.963).26 Mean time to ANC recovery was significantly longer in the cohort treated with pegfilgrastim (2.6 days) compared to balugrastim 40 mg (2.0 days) and 50 mg (2.1 days), (P=0.005).18 The effect of balugrastim was lost in the following cycles, with no significant difference in mean time to ANC recovery between different treatments and cycles.18 Similarly, no significant difference in mean time to ANC recovery for cycle 1 was found in the other study considered, with 2.1 days for pegfilgrastim and 2.0 days for balugrastim 40 mg (P=0.259).26 Overall survival was not affected by the different treatments administered: 91.8% patients treated with balugrastim 40 mg were alive after a 1-year follow-up period compared to 84.5% for balugrastim 50 mg and 90.7% for pegfilgrastim.18
Taken together, the results of the efficacy analysis accounted for the noninferiority of balugrastim treatment over pegfilgrastim. Additionally, no differences in primary and secondary endpoints were found between the two different dosages of balugrastim that were investigated. Balugrastim showed a unique advantage over pegfilgrastim in shortening the mean time to ANC recovery.1,18,26
Safety and tolerability
Balugrastim’s safety and tolerability were mainly tested in a Phase I/IIa and Phase II study.1,35 In the first one, Avisar et al35 administered escalating doses of balugrastim (50, 150, 300, and 450 g/kg) in cycle 0 (2 weeks before the beginning of chemotherapy) and in cycles 1 and 2. Thirteen patients were treated with doxorubicin 50 mg/m2 and docetaxel 75 mg/m2 every 21 days.35 Since the balugrastim dose was escalated safely to the dosage of 450 g/kg without any limiting toxicity, the following Phase IIa trial included further 51 patients who were randomized to receive balugrastim 300 or 450 g/kg, or pegfilgrastim 6 mg (2:2:1 ratio).1 The assessment of safety performed before the beginning of chemotherapy in the Phase I trial reported only six total adverse events (AEs) experienced by four patients.35 For the balugrastim 300 g/kg cohort, two patients experienced grade 1 urinary tract infection and pyrexia, while two patients treated with balugrastim 450 g/kg suffered from grade 2 bone pain, grade 2 headache (twice), and grade 3 hypertension.35 Most of the other AEs reported during cycles 1 and 2 of the Phase I study and Phase II were reasonably ascribable to chemotherapy and not to balugrastim.35 Considering AEs were most likely related to balugrastim, bone pain occurred five times in four patients treated with balugrastim during Phase II therapy: one event was related to balugrastim 300 g/kg (grade 1), while the remaining four cases occurred during balugrastim 450 g/kg (grade 2).35 Two cases of headache, one episode of hypertension, one case of pyrexia, and one injection site reaction, all grade 1, were registered with balugrastim 450 g/kg.35 Moreover, a grade 2 rash occurred with the same dosage.35
In the Phase II dose-finding study, bone pain occurred in two patients (grade 1 and grade 2) treated with balugrastim 50 mg (10%), while it was not observed for the other dosages.1 The same AE was found in two patients (grade 1) in the balugrastim cohort (7.7%).1 Phase III studies confirmed these findings.18,26 Gladkov et al18 reported a higher amount of grade 4 AEs in the 50 mg balugrastim cohort (46.4%) than in the 40 mg group (34.1%) and pegfilgrastim (36%). Similarly, Volovat et al26 showed a slightly higher amount of AEs after balugrastim treatment (19.6% for balugrastim, 18.7% in pegfilgrastim treatment in the double-blind phase). Bone pain and related symptoms amounted to 11.8% for balugrastim versus 10.7% for balugrastim in the double-blind phase and 18.2% in the balugrastim open-label phase.26
Antibodies to balugrastim were reported in two out of 169 patients, while antibodies against the albumin domain of balugrastim were detected in six cases. However, the antibody response to balugrastim was low titer, transient, and had no neutralizing effect.18
Neither increases in spleen dimensions nor splenic ruptures have been reported during balugrastim administration,1,18,26,35 while a transient increase in spleen volume during G-CSF administration and rare cases of splenic rupture have been noticed both in healthy peripheral blood stem cell donors and cancer patients treated with G-CSF, granulocyte macrophage colony-stimulating factor, or pegylated G-CSF.3,37
Similarly, we did not find any reported case of acute myeloid leukemia (AML) or myelodysplasia secondary to balugrastim use.1,18,26,35 Although rare events, the incidence of both AML and myelodysplasia was found to have increased significantly with G-CSF use in patients treated with chemotherapy.3,37 On the other hand, a nonsignificant correlation between G-CSF use and onset of secondary AML or myelodysplasia was reported in healthy peripheral blood stem cell donors.3,37
In conclusion, balugrastim’s administration was shown to be extremely safe. Bone pain was described as the most frequent AE directly connected to the subcutaneous injection. Its onset was more frequent with higher doses of balugrastim (eg, 50 mg and 450 g/kg).1,18,35
Quality of life, patient satisfaction, and acceptability
From the first-in-human Phase I study to the recent Phase III randomized trials, balugrastim-related AEs did not require an interruption or discontinuation of treatment. No toxic death occurred in any of the considered studies.1,18,26,35 Bone pain was easily managed with common analgesics only.26 Consent withdrawals were registered for five patients in the Phase II dose-finding study (two in the balugrastim 40 mg, three in the balugrastim 50 mg cohort),1 seven patients in the Phase III two-arms trial (five patients treated with balugrastim and two with pegfilgrastim)26 and seven patients in the Phase III three-arms study (four patients treated with balugrastim 40 mg, two patients with balugrastim 50 mg, and one patient with pegfilgrastim).18 As a whole, treatment with balugrastim did not cause any severe AEs, and the quality of life was not compromised at all by its use.
Conclusion and place in therapy
Balugrastim is a new long-lasting rG-CSF created by linking G-CSF to recombinant human albumin in S. cerevisiae. Its t1/2 of ~19 days makes it possible to be conveniently administered in a once-per-cycle subcutaneous injection. Phase I and II trials reported a good safety and tolerability profile in breast cancer patients treated with doxorubicin and docetaxel for breast cancer. Two Phase III studies compared balugrastim with pegfilgrastim demonstrating a comparable efficacy between the two treatments and the noninferiority of balugrastim with regard to the reduction in mean DSN during the first cycle of chemotherapy. The dose of 40 mg was associated with better efficacy parameters compared to the 50 mg dose, such as reduced mean DSN and minor incidence of FN during cycle 1. Moreover, balugrastim 40 mg registered fewer grade 4 AEs than 50 mg.
Secondary AMLs and myelodysplasias were not described after balugrastim use. This is probably due to the recent development of the drug and the short period of follow-up available. Indeed, leukemogenic events caused by G-CSF were reported after a median follow-up of at least 4 years. Therefore, long-term studies are necessary to adequately evaluate balugrastim safety.
Balugrastim represents a valid alternative to pegfilgrastim when a long-acting rG-CSF is required in clinical practice. Since all studies were performed in homogeneous populations of breast cancer patients treated with docetaxel and doxorubicin, further research involving different tumor types and chemotherapy combinations is definitely needed.
Acknowledgment
The authors would like to thank Michael Davies (Unique Languages) for linguistic revision.
Disclosure
The authors report no conflicts of interest in this work.
References
Gladkov O, Moiseyenko V, Bondarenko IN, et al. Phase II dose-finding study of balugrastim in breast cancer patients receiving myelosuppressive chemotherapy. Med Oncol. 2015;32(6):623. | ||
Pfeil AM, Allcott K, Pettengell R, von Minckwitz G, Schwenkglenks M, Szabo Z. Efficacy, effectiveness and safety of long-acting granulocyte colony-stimulating factors for prophylaxis of chemotherapy-induced neutropenia in patients with cancer: a systematic review. Support Care Cancer. 2015;23(2):525–545. | ||
Bennett CL, Djulbegovic B, Norris LB, Armitage JO. Colony-stimulating factors for febrile neutropenia during cancer therapy. N Engl J Med. 2013;368(12):1131–1139. | ||
Zsebo KM, Yuschenkoff VN, Schiffer S, et al. Vascular endothelial cells and granulopoiesis: interleukin-1 stimulates release of G-CSF and GM-CSF. Blood. 1988;71(1):99–103. | ||
Koeffler HP, Gasson J, Ranyard J, Souza L, Shepard M, Munker R. Recombinant human TNF alpha stimulates production of granulocyte colony-stimulating factor. Blood. 1987;70(1):55–59. | ||
Metcalf D. The Haematopoietic Colony Stimulating Factors. Amsterdam: Elsevier; 1984:55–92. | ||
Seelentag WK, Mermod JJ, Montesano R, Vassalli P. Additive effects of interleukin 1 and tumour necrosis factor-alpha on the accumulation of the three granulocyte and macrophage colony-stimulating factor mRNAs in human endothelial cells. EMBO J. 1987;6(8):2261–2265. | ||
Souza LM, Boone TC, Gabrilove J, et al. Recombinant human granulocyte colony-stimulating factor: effects on normal and leukemic myeloid cells. Science. 1986;232(4746):61–65. | ||
Duhrsen U. In vitro growth patterns and autocrine production of hemopoietic colony stimulating factors: analysis of leukemic populations arising in irradiated mice from cells of an injected factor-dependent continuous cell line. Leukemia. 1988;2(6):334–342. | ||
Duhrsen U, Metcalf D. A model system for leukemic transformation of immortalized hemopoietic cells in irradiated recipient mice. Leukemia. 1988;2(6):329–333. | ||
Duhrsen U, Villeval JL, Boyd J, Kannourakis G, Morstyn G, Metcalf D. Effects of recombinant human granulocyte colony-stimulating factor on hematopoietic progenitor cells in cancer patients. Blood. 1988;72(6):2074–2081. | ||
Eyles JL, Hickey MJ, Norman MU, et al. A key role for G-CSF-induced neutrophil production and trafficking during inflammatory arthritis. Blood. 2008;112(13):5193–5201. | ||
Molineux G, Kinstler O, Briddell B, et al. A new form of filgrastim with sustained duration in vivo and enhanced ability to mobilize PBPC in both mice and humans. Exp Hematol. 1999;27(12):1724–1734. | ||
Roberts AW, Metcalf D. Granulocyte colony-stimulating factor induces selective elevations of progenitor cells in the peripheral blood of mice. Exp Hematol. 1994;22(12):1156–1163. | ||
Welte K, Bonilla MA, Gillio AP, et al. Recombinant human granulocyte colony-stimulating factor. Effects on hematopoiesis in normal and cyclophosphamide-treated primates. J Exp Med. 1987;165(4):941–948. | ||
Cerci C, Ergin C, Eroglu E, et al. Effects of granulocyte-colony stimulating factor on peritoneal defense mechanisms and bacterial translocation after administration of systemic chemotherapy in rats. World J Gastroenterol. 2007;13(18):2596–2599. | ||
Weisbart RH, Kacena A, Schuh A, Golde DW. GM-CSF induces human neutrophil IgA-mediated phagocytosis by an IgA Fc receptor activation mechanism. Nature. 1988;332(6165):647–648. | ||
Gladkov O, Moiseyenko V, Bondarenko IN, et al. A phase iii study of balugrastim versus pegfilgrastim in breast cancer patients receiving chemotherapy with doxorubicin and docetaxel. Oncologist. 2016;21(1):7–15. | ||
Ratti M, Tomasello G. Lipegfilgrastim for the prophylaxis and treatment of chemotherapy-induced neutropenia. Expert Rev Clin Pharmacol. 2015;8(1):15–24. | ||
Pettengell R, Johnsen HE, Lugtenburg PJ, et al. Impact of febrile neutropenia on R-CHOP chemotherapy delivery and hospitalizations among patients with diffuse large B-cell lymphoma. Support Care Cancer. 2012;20(3):647–652. | ||
Rogers B, Dong D, Li Z, Li Z. Recombinant human serum albumin fusion proteins and novel applications in drug delivery and therapy. Curr Pharm Des. 2015;21(14):1899–1907. | ||
Halpern W, Riccobene TA, Agostini H, et al. Albugranin, a recombinant human granulocyte colony stimulating factor (G-CSF) genetically fused to recombinant human albumin induces prolonged myelopoietic effects in mice and monkeys. Pharm Res. 2002;19(11):1720–1729. | ||
Avisar N, Pukac L, Adar L, et al. Recombinant albumin-partnering technology: development of balugrastim, a novel long-acting granulocyte colony-stimulating factor. Blood. 2013;122(21):4854. | ||
Molineux G. Granulocyte colony-stimulating factors. Cancer Treat Res. 2011;157:33–53. | ||
Hill CP, Osslund TD, Eisenberg D. The structure of granulocyte-colony-stimulating factor and its relationship to other growth factors. Proc Natl Acad Sci U S A. 1993;90(11):5167–5171. | ||
Volovat C, Gladkov OA, Bondarenko IM, et al. Efficacy and safety of balugrastim compared with pegfilgrastim in patients with breast cancer receiving chemotherapy. Clin Breast Cancer. 2014;14(2):101–108. | ||
Molineux G, Foote M, Arvedson T, editors. Twenty Years of G-CSF: Clinical and Nonclinical Discoveries. Basel: Springer Science; 2012. | ||
Wiczling P, Lowe P, Pigeolet E, Ludicke F, Balser S, Krzyzanski W. Population pharmacokinetic modelling of filgrastim in healthy adults following intravenous and subcutaneous administrations. Clin Pharmacokinet. 2009;48(12):817–826. | ||
Buchner A, Lammerich A, Avisar N, inventors; Teva Pharmaceutical Industries Ltd, assignee; Recombinant Human Albumin-Human Granulocyte Colony Stimulating Factor for the Prevention of Neutropenia in Pediatric Patients. United States Patent US 20140271538A1; 2014. | ||
Morstyn G, Campbell L, Duhrsen U, et al. Clinical studies with granulocyte colony stimulating factor (G-CSF) in patients receiving cytotoxic chemotherapy. Behring Inst Mitt. 1988;(83):234–239. | ||
Helms RA, Quan DJ, editors. Textbook of Therapeutics: Drug and Disease Management. Philadelphia, PA: Wolters Kluwer; 2006. | ||
Gabrilove JL, Jakubowski A, Fain K, et al. Phase I study of granulocyte colony-stimulating factor in patients with transitional cell carcinoma of the urothelium. J Clin Invest. 1988;82(4):1454–1461. | ||
Morstyn G, Campbell L, Souza LM, et al. Effect of granulocyte colony stimulating factor on neutropenia induced by cytotoxic chemotherapy. Lancet. 1988;1(8587):667–672. | ||
Bronchud MH, Scarffe JH, Thatcher N, et al. Phase I/II study of recombinant human granulocyte colony-stimulating factor in patients receiving intensive chemotherapy for small cell lung cancer. Br J Cancer. 1987;56(6):809–813. | ||
Avisar N, Adar L, Bock J, et al. First-in-human, phase I/IIa dose-escalation and safety study of balugrastim in breast cancer patients receiving myelosuppressive chemotherapy. Cancer Chemother Pharmacol. 2015;75(5):929–939. | ||
Yang BB, Kido A. Pharmacokinetics and pharmacodynamics of pegfilgrastim. Clin Pharmacokinet. 2011;50(5):295–306. | ||
Tigue CC, McKoy JM, Evens AM, Trifilio SM, Tallman MS, Bennett CL. Granulocyte-colony stimulating factor administration to healthy individuals and persons with chronic neutropenia or cancer: an overview of safety considerations from the research on adverse drug events and reports project. Bone Marrow Transplant. 2007;40(3):185–192. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.