Back to Journals » Eye and Brain » Volume 11
Neuro-Ophthalmological Manifestations Of Septo-Optic Dysplasia: Current Perspectives
Authors Ganau M, Huet S ![]() , Syrmos N
, Syrmos N ![]() , Meloni M, Jayamohan J
, Meloni M, Jayamohan J
Received 21 June 2019
Accepted for publication 24 September 2019
Published 18 October 2019 Volume 2019:11 Pages 37—47
DOI https://doi.org/10.2147/EB.S186307
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Margaret Wong-Riley
Mario Ganau,1 Sibel Huet,1 Nikolaos Syrmos,2 Marco Meloni,3 Jayaratnam Jayamohan1
1Department of Neurosurgery, Oxford University Hospitals NHS Foundation Trust, Oxford, UK; 2Department of Neurosurgery, School of Medicine, Aristotle University of Thessaloniki, Macedonia, Greece; 3Department of Medicine and Surgery, Bicocca University, Milan, Italy
Correspondence: Mario Ganau
Department of Neurosurgery, Oxford University Hospitals NHS Foundation Trust, Oxford, UK
Email [email protected]
Abstract: Septo-optic dysplasia (SOD), also known as de Morsier syndrome, is a rare congenital disorder belonging to the group of mid-line brain malformations. Despite the highly variable phenotypic penetration, its classical triad include a) optic nerve hypoplasia (ONH), b) agenesis of septum pellucidum and corpus callosum, and c) hypoplasia of the hypothalamo-pituitary axis. SOD has stringent diagnostic criteria requiring 2 or more features of the classic triad, therefore it represents a separate entity from other conditions such as ONH and achiasmia syndromes which share only some of these aspects, or SOD plus syndrome which is characterized by additional cortical abnormalities. Starting from its etiology and epidemiology, this narrative review focuses on the management of SOD patients, including their diagnosis, treatment and follow-up. To date, SOD is not curable; nonetheless, many of its symptoms can be improved through a tailored approach, consisting of hormonal replacement, corrective ophthalmological surgery and neuropsychological support.
Keywords: septo-optic dysplasia, optic nerve hypoplasia, hypopituitarism, achiasmia, hydrocephalus, congenital visual loss
Plain Language Summary
Septo-optic dysplasia (SOD) syndrome is a rare congenital disorder belonging to the mid-line brain malformations group; even though the phenotypic penetration can be highly variable resulting in a wide heterogeneity, its classical triad includes optic nerve hypoplasia (ONH), agenesis of septum pellucidum and corpus callosum, hypoplasia of the hypothalamo-pituitary axis. Initial patients’ referrals for suspected cases of SOD are due to loss of vision in one of both eyes, developmental delay, seizures, sleep disturbances and precocious puberty. Following an in-depth description of the frequency and causes of this syndrome, this narrative review will focus on its clinical presentation. Although SOD is not curable, many aspects of SOD syndrome can be improved through a tailored approach which may include hormonal replacement, corrective ophthalmological surgery and active neuropsychological support.
Introduction
Septo-optic dysplasia (SOD) syndrome, a congenital disorder belonging to the mid-line brain malformations group, was firstly described in 1956 by the French-Swiss neurologist Georges de Morsier.1 Even though the phenotypic penetration can be highly variable resulting in a wide heterogeneity, its classical triad includes the following:
- Optic nerve hypoplasia (OHN)
- Agenesis of midline structures (namely septum pellucidum and corpus callosum)
- Hypoplasia of the hypothalamo-pituitary axis
SOD has stringent diagnostic criteria requiring 2 or more features of the classic triad, so that only up to 30–47% of patients diagnosed with this condition will present all the features. Additionally, ONH may occur in the context of ONH syndrome, which does not fulfill the SOD definition provided above despite being oftentimes wrongly used synonymously, and this certainly results in a misnomer and represents the source of confusion. In fact ONH tends to be more frequent and seems to have different etiology.2 On the other hand, patients with SOD can also present other specific brain abnormalities (such as hydrocephalus, polymicrogyria, grey matter heterotopias, etc.) on top of those described above; for this reason, some authors have proposed different definitions and classifications. Due to this complexity, the scientific community is still debating about the most appropriate nomenclature to standardize the diagnosis of this disorder, and the term “SOD plus syndrome” (for cases with associated cortical brain development such as schizencephaly) or “SOD spectrum” (for cases showing a much wider range of congenital abnormalities including for instance cleft palate) has been proposed.4
The clinical relevance of this condition is epitomized by the fact that SOD is among the top causes of congenital visual loss in developed countries.3–5 Although this review focuses primarily on the neuro-ophthalmological features of SOD, it will also attempt to comprehensively cover the critical aspects of its global management at both diagnostic and therapeutic stages.
Methodology
In the absence of guidelines for narrative reviews, similar to those used when reporting systematic reviews (ie PRISMA, http://www.prisma-statement.org/), we followed the “best-evidence synthesis” approach described by Green et al6 to analyze various sources of knowledge and provide a summary of the most valuable contributions produced so far by international research groups and clinicians with expertise on SOD. We started from a Medline search (https://www.ncbi.nlm.nih.gov/pubmed/) which was conducted using the key words: “septo-optic dysplasia” + “epidemiology” or “management”. This initial step allowed to identify 479 articles, which were screened according to the following criteria: large epidemiological studies, clinical trials, and systematic reviews were given priority in terms of their strength of scientific evidence as per Oxford Centre for Evidence Based Medicine (OCEBM, https://www.cebm.net/2016/05/ocebm-levels-of-evidence/), whereas case series were taken into account to verify working hypothesis, or incidence/prevalence of both common and rare clinical features of this condition. The second step included a review of the Online Mendelian Inheritance in Man (OMIM, https://www.omim.org/) and Genetic Home Reference (GHR, https://ghr.nlm.nih.gov/) databases to verify genetic correlations with the disorders discussed in the manuscript (not only SOD but also ONH syndrome), their penetration and possible phenotypes. Furthermore, the clinicaltrial.gov database was searched to identify completed and ongoing clinical trials on SOD patients. During the third step, we accessed institutional websites from world-renowned Universities, Hospitals, Foundations and Associations with a specific page dedicated to SOD patients. Additionally, it is worth mentioning that the summary of the current evidence drawn on this condition also results from a discussions with experts in the field of SOD and our professional experience in diagnosing and treating pediatric patients presenting with clinical features of SOD, SOD plus or SOD spectrum disorders.
SOD And Its Etiology
While the precise causes of SOD remain unknown, a combination of genetic predisposition and prenatal exposure to environmental factors are believed to play major roles in its occurrence.7 Acers was the first author to provide a seminal contribution by suggesting how environmental risk factors such as drugs consumption, viral infections and maternal diabetes could be possible causes for malformations typical of SOD syndrome.8 Other studies have strengthened the link between SOD and these factors, while also defining young maternal age (usually age at conception/delivery < 22 years), and primiparity as the most important causative factors.2,5,7–11 More recently, the link between drugs, alcohol, viral infections, maternal diabetes, smoking and SOD came under scrutiny, and the general consensus is that environmental agents during pregnancy should not be underestimated nor merely considered concomitant predisposing factors.
On the other hand, it is nowadays widely accepted that specific genes are involved in the development of SOD, the most important ones being HESX1, SOX2/SOX3 and OTX2.7 Evidence for the role of those genes can be found in their involvement in embryonic development of the eyes, optic nerves and pituitary gland. For instance, HESX1, which is located in the short (p) arm of chromosome 3 at position 14.3, belongs to a family of homeobox genes and is an important marker for the early differentiation of the forebrain and adenohypophysis. The HESX1 mutations can lead to different endocrine deficiencies including those recognized under the umbrella of the Combined Pituitary Hormone Deficiency syndrome and those seen in SOD patients.7 At least five mutations in the HESX1 gene have been directly correlated to SOD, some of them involve only single base pairs, while others involve deletion or duplication of genetic material altering the HESX1 protein and silencing its activity. SOX2, SOX3 and OTX2 are other genes known to be implicated in the development of SOD: they all encode proteins which act as transcription factors regulating the activity of other genes. Hence, by specific binding to other DNA regions, those transcription factors play a pivotal role in the fate of the fetus and in particular in the early formation of many different tissues and organs. OTX2 and SOX2 are critical for the embryonic development of the optic nerves and may be associated with more severe phenotypes that include severe bilateral eye defects and malformations of the corpus callosum and infundibulum.7,12,13
Of note, the OMIM and GHR databases clearly demonstrate how the genes involved in SOD differ from those directly linked with the pathogenesis of other syndromes (ie PAX6 and PROKR2 for ONH syndrome, TUBA8 for ONH with polymicrogyria, etc.). Finally, although SOD is usually seen as a sporadic disorder, familial cases, mostly with autosomal recessive inheritance, have been described, and in rare instances, an autosomal dominant pattern has also been identified. These aspects are certainly in keeping with the wide spectrum of genetic predispositions and related phenotypes with variability of each component of the triad mentioned above.
Theoretical Frameworks
Given the above three theoretical frameworks can be considered regarding the pathogenesis of septo-optic dysplasia. Acknowledging that a genetic predisposition is not always inherited but could also result from de novo mutations is essential; the other main theories proposed pinpoint the importance of an early triggering event: either a prenatal vascular insult or a perinatal secondary degeneration. A vascular disruptive sequence similar to porencephaly or hydranencephaly, possibly involving the territorial distribution of the proximal trunk of the anterior cerebral artery (ACA), has been proposed by Lubinsky.9 Although early intrauterine vascular injuries to the central nervous system may lead to a complete tissue resorption with no residual signs of infarction, this hypothesis does not take into account the wide variability of the intrauterine events leading to SOD.3 Another limitation of this vascular theory is related to the fact that not all anatomical structures affected are within the territorial distribution of the ACA. As all affected components arise from different tissues and alter embryological processes at different stages of fetus development, a perinatal event caused by traumatic, infectious or toxic insults would justify the different penetration and heterogeneity of dysplasia identifiable in SOD patients.10,11,13
Epidemiology Of SOD
The EUROCAT study was the first attempt to delve deeper into the epidemiology of SOD in Europe: this collaborative effort gathered and analyzed data pertaining to an overall population of 6.4 million births over an observational period of 10 years (2005–2014) concluding that the prevalence of this syndrome was between 1.9 and 2.5/100,000.14 These figures are remarkably lower compared to a retrospective regional study conducted in Canada, in the State of Manitoba, where the annual incidence reached 53/100,000 (represented in 55% of cases by SOD plus syndrome) for the same time frame (2011–2016). Although the authors identified a clustering effect in two northern provinces (Churchill, with 66.9 cases per 100,000 youths <19 years, and Nunavut, 37.1 per 100,000), that study drew significant attention mostly because it pinpointed a striking 1.11-fold annual increase (95% confidence interval 1.07 to 1.16), which translates for ~800% increase over two decades.11
It can be therefore argued that the stricter the inclusion criteria (SOD versus SOD plus syndrome), the lower the calculated incidence and prevalence; additionally, it should be noted that these epidemiological studies seem to reveal specific regional clustering. For instance, authors of the EUROCAT study disclosed that SOD prevalence was significantly higher in UK registries compared with other 28 European registries (P = 0.021 in the multilevel model) and that also the additional risk for younger mothers was significantly greater in the UK compared to the rest of Europe (P = 0.027). In fact, a study of the occurrence of SOD/ONH in the Greater Manchester and Lancashire region of Northwest England revealed an incidence of 10.9/100,000 per year.16 A second study conducted between 1998 and 2009 always in England, but in the adjacent region of the West Midlands, found similar figures and highlighted that the median maternal/paternal ages in SOD were 21 and 23.5 years, compared to UK means of 29.3 and 32.4 years (p < 0.001).17 Also, they reported that first trimester bleeding was markedly increased at 25% compared to 0.07% in the UK (p < 0.001), and ethnicity showed a non-significant higher prevalence in Afro-Caribbean and mixed race groups and significantly lower prevalence (p = 0.004) in South Asian groups.17 In both regions, the significantly higher standardized incidence ratios were found to be associated to the high rates of unemployment and high prevalence of teenage conception and pregnancies.
On a separate note, the rates reported in the literature appear obviously higher when the analysis was focused only on the presence of ONH regardless of its occurrence within a formal diagnosis of SOD, and it is also worth mentioning that many authors have noticed a progressive increase in the incidence of OHN over the last decades.5,15 An example can be provided by a study conducted on the Swedish database of visually impaired children which reported an increased prevalence from 1.8 to 6.3/100,000 over 4 decades between 1970 and early 2000 in the population aged 0–19 years.15 Nonetheless, authors also confirmed that in only 10% of the patients included ONH was part of a SOD syndrome. This review of the epidemiological studies identified through our research strategy highlights the role that international registries play in providing a snapshot of the incidence and prevalence of rare diseases. As such, the role of hospitals, foundations and associations in supporting patients and fostering the compliance with registries is of the upmost importance, hence the decision to provide a list of useful links at the bottom of this manuscript.
Clinical Presentation
For a long time, SOD syndrome has been sporadically described or its clinical relevance taken into account only when ONH was severe enough to cause blindness.7 It is now well accepted that patients with SOD spectrum can present with relatively preserved vision and a wide range of endocrine or midline defects. As such, mild SOD cases can well counterbalance, in a broad range of clinical presentations, and the other extreme represented by cases of achiasmia, facial abnormalities, and panhypopituitarism.18
For this reason, besides separately describing each anatomical abnormality, we found appropriate to analyze in detail any possible symptom associated with the SOD triad and stress the notion that the clinical presentation may well vary with age and severity of the syndrome (see Table 1).
 |
Table 1 SOD And Its Clinical Features (two Or More Features Of The Triad Are Needed To Establish A Diagnosis Of SOD) |
Since ophthalmological aspects are the earliest that can be identified in SOD patients, and the main object of the present review, we will start introducing them and only afterwards we will describe its endocrinological and neurological symptoms.2,20
Visual Disturbances
Although ONH is the most common presenting symptom, a review of the literature reveals that visual impairment accounts only for 23% of overall reported symptoms in SOD patients and may include also very mild cases of astigmatism.5,7,18–21 ONH can be bilateral or unilateral, with the latter being less severe and less common.2,19,23 Of note, the frequency reported by Ryabets-Lienhard et al for bilateral ONH were 55–80%, of which 2/3 demonstrated an asymmetrical presentation, similarly to those figures the frequency for unilateral ONH initially described by Borchert was 20%.5,19 Regardless of the many different neuro-ophthalmological presentations, the current consensus seems to be that newborn could be difficult to diagnose with SOD, because they may not manifest any visual disturbances until 1–3 months of age.
The chief clinical finding in newborns with SOD is nystagmus, which usually can be appreciated by the first three months, especially in cases of bilateral ONH. In terms of temporal presentation, nystagmus is often followed by occurrence of strabismus in the first year. This is typical of both children with bilateral and asymmetric ONH or unilateral ONH. Either during infancy or adulthood, many SOD patients will experience progressive loss in visual acuity, with 80% of the cases with bilateral SOD becoming legally blind at some point of their life.19,24 Clinicians should be aware that unilateral cases of ONH tend to be diagnosed significantly later than bilateral ones: Riedl et al found that patients with bilateral ONH usually presented for the first time during the first 6 months of life with delayed visual maturation and nystagmus, whereas children with unilateral ONH usually present many years later, during pre-school age, often with an initial complaint of isolated strabismus.20 The course of the disease will therefore be longer and more severe in patients with bilateral ONH.5,19,21 This said, an interesting theory proposed by Borchert in his reappraisal of ONH suggests that the optic nerve myelination occurring in the first years of life can tamponade and delay onset of visual disturbances by improving visual acuity in children with SOD regardless of the severity of their anatomical malformations.19
Hypopituitarism
Regarding symptoms related to developmental abnormalities of the pituitary gland, many studies show that out of the three features of SOD, hypopituitarism is the most commonly reported one, ranging between 62% and 80%. Specific hormonal deficits, mostly affecting GH and TSH production, seem to be more frequent than gonadotropic dysregulation, nonetheless also ACTH deficiency, hypothyroidism, and adrenal insufficiency are commonly described. Interestingly the involvement of neurohypophysis with diabetes insipidus is rarely reported and this seems in keeping with the different embryologic development of anterior and posterior portions of the pituitary gland. Since congenital defects of the hypothalamo-pituitary axis can lead to early obesity, excessive or asymmetrical growth of facial bones and limbs, altered tone of the voice and development of genital organs, some authors have concluded that the phenotypic presentation of SOD is heavily influenced by the severity of its endocrine symptoms.22,23 Others have highlighted that even rare manifestations can play a significant role in the clinical diagnosis: the occurrence of either hypoglycemia, presence of micropenis and/or cryptorchidism with hypoplastic testes, prolonged jaundice or manifestation of diabetes insipidus usually lead to earlier referrals and further investigations.
Neurological Aspects
Beside rare severe cases characterized by low Apgar scores, prolonged jaundice, cryptorchidism, lethargy, poor feeding, cerebral palsy and irritability, neurological signs and symptoms are usually late manifestations of SOD syndrome.24
Findings can be highly variable, nonetheless they are seen in up to 70% of patients with this diagnosis; they may initially present with either recurring seizures, developmental delay and/or tetraparesis. The etiology of seizures may be complex: hypoglycemia can often represent a confounding factor, delaying a definitive diagnosis of infantile spasm. Patients usually require antiepileptic drugs coverage, but the number of reports regarding drug resistant epilepsy in patients with SOD is increasing over time, confirming its therapeutic challenges as well as the previously mentioned increase in the prevalence of this condition. Patients who present with seizures are highly likely to have neuroanatomical abnormalities such as schizencephaly, polymicrogyria and cortical dysplasia often in a more complex picture of SOD-plus syndrome, but this might not always be the case. On the other hand, those abnormalities are always identified in patients with spastic paresis.21
Another confounding factor for developmental and psychomotor delay is certainly the frequent presence of congenital hydrocephalus, whereas the origin of neurobehavioral and neuropsychological disturbances, which can range from severe mental retardation to autism with otherwise normal intellectual status, seem to be more unspecific.5,24,25 Finally, sleeping disorders are some of the least described clinical features of SOD syndrome, nonetheless their cause can be easily identified in the impaired development of midline structures and their role in circadian rhythm.25
Diagnostic Stage
While in the previous section we presented each clinical feature according to the degree of severity and age at presentation, we will now focus on the first stage of their management which obviously starts with the appropriate diagnostic tests.
When an initial index of suspicion for SOD is raised, a thorough clinical examination is warranted to identify other physical abnormalities typical of this syndrome and reach a definitive diagnosis.24 Besides pediatricians, the first specialists involved in the management of those patients are neuro-ophthalmologists, and we will therefore start with the crucial aspects of ophthalmological assessment before considering the laboratory investigations and the most appropriate neuroradiology protocols.
Ophthalmological Assessment
The initial approach to impaired vision will entail a clinical testing of the presence and degree of ONH and associated motility disturbances must be evaluated.24
- Direct pupillary responses: The initial clinical assessment of SOD patients may demonstrate pupils equals and reactive to light, nonetheless pupillary responses may well be absent or severely sluggish.9,28 Normal pupillary response is not predictive of good visual acuity: Borchert et al found that 46% of patients with eyes with normally reactive pupils had visual acuity less than 20/200.26 The opposite is however not true: poor pupillary responses are always associated with poor visual acuity.26
- Visual Axes: Visual axes in patients with SOD can be either normal or there may be presence of esotropia, exotropia or even shift pattern (esotropia to exotropia).28 Most of those aspects can correlate with a loss of ganglion cells and subsequent reduction in visual function. Abnormal alignment of visual axes can be seen concomitantly with strabismus.2 Nystagmus and strabismus are common findings in patients with hypoplastic optic nerve.18,28 Although nystagmus is not typically a congenital symptom, it represents a clearly detectable abnormality for referral after 3–6 months age and may be one of the first presenting feature of SOD from a ophthalmological perspective. The nystagmus can be univectorial, bivectorial, rotatory or multivectorial. Jerk and pendular nystagmus can also be seen.28 The presence of nystagmus is usually associated with poor visual acuity.26 There may also be abnormal fixation, pursuit and absence of saccadic movements; a preference for particular head and gaze positions may also be present.28
- Presence and degree of ONH: Per definition ONH is characterized by small-appearing optic disc/s and, as a general rule, SOD patients have always a smaller optic nerve area than the general population (see Figure 1). In many cases, although not invariably, this is associated with a pigmented ring around the disc, tilting of the disc, abnormal nerve fiber layer, and tortuosity of the retinal vessels.26 Clinical confirmation requires direct video-ophthalmoscopy assessment demonstrating the initial findings and ruling out the possible bias of inter-observer variability. Labeling ONH by simply referring to the optic disc size has been historically hindered because of the difficulty in obtaining absolute measurements of optic discs in living individuals, particularly children, since measurements depend on magnification properties such as axial length of the eye and refractive error.26 Over 20 years ago, Borchert et al proposed the relative disc size (ratio of the horizontal disc diameter to the disc-macula distance) as a possible objective criteria for ONH and demonstrated that patients with a ratio ≤ 0.30 have very poor visual acuity (on average 20/200), while a ratio ≤ 0.15 indicated visual acuity less than or equal to light perception.26 Zeki et al confirmed that the lower limit of normal relative disk size should be 0.41; however, the general consensus seems to be that normal range of the ratio of the horizontal disc diameter to the disc-macula distance is from 0.47 to 0.63, and ratio inferior than 0.40 should imperatively be investigated for ONH. Other measurements have also been proposed and support such clinical approach: for instance, Awan suggested to measure the distance from the center of the disc to the center of the macula and compared it to the horizontal disc diameter, finding that the normal ratio was 2.1 to 3.2, which converts algebraically to a relative disc size of 0.037 to 0.63. Of note, fundus photographs should always be correlated with neuroradiological findings on MRI brain to confirm the presence of congenital abnormalities affecting the optic nerves and chiasm.5 This is particularly important because one limitation of clinical examinations alone is the difficulty to diagnose patients with mild/moderate ONH, whereas some authors have suggested that MRI may be a superior diagnostic tool as it can detect non-clinically symptomatic hypoplastic optic nerves.27 In fact, small pale optic discs may be misdiagnosed as normal-sized discs with optic atrophy because the distinction between the optic disc and the hypopigmented scleral canal can be challenging.7 Furthermore, other morphological findings should also be taken into account while assessing those patients, including pale optic disc, macular abnormalities, reduced retinal vessel diameter, tortuous retinal arterioles/venules/both, uncommonly straight vessels with decreased branching.5,24,28 Although not pathognomonic (since it can be found even in patients with myopia), another common morphological abnormality in SOD patients is the so-called “double-ring sign”: a hypopigmented or hyperpigmented peripapillary halo surrounding the disc.7 Overall, it should be noted that the presence of those additional morphological findings has been reported to be a negative prognostic factor.28
- Optical coherence tomography (OCT) has nowadays replaced fundus photography in clinical practice and is easier to perform. One of the negative aspects of OCT is that patients often need to be sedated; nonetheless, OCT results are superior and cheaper than MRI for examining ganglion cells and their axons. Additionally, OCT appears helpful in detecting optic atrophy versus reduced number of axons.
- Visual function: Patients with SOD can present with astigmatism, myopia, hyperopia, and associated refractive errors; contrast sensitivity is likely to be reduced but hopefully this is not always the case.28 As anticipated above, SOD patients may present with unilateral or bilateral involvement, and a visual acuity ranging from absent or minimal light perception to relatively normal findings.9,24,30 The presence of unilateral or bilateral ONH has remarkable prognostic implications because patients can preserve a normal visual acuity bilaterally if they present with unilateral ONH.28 Additionally, Signorini et al found that children with ONH but relatively good visual acuity are less likely to have associated endocrine and neurological pathologies.28 Visual acuity is known to proportionally increase with the optic disc size; however, normal visual acuity does not warrant a normal visual function, since peripheral visual field defects may be present.23
- Visual field studies: Assessment will start on manual examination with a confrontational visual field test; the examiner may find several possible deficits in order of frequency: bitemporal hemianopia, generalized constriction, central, binasal or altitudinal defects. While in SOD patients with unilateral ONH the visual fields may be normally preserved in the uninvolved eye, some authors have suggested that objective retinal function should be evaluated qualitatively and quantitatively by full field electroretinography (FF-ERG) and focal macular ERG (FM-ERG). By studying ONH patients with spectral-domain optical coherence tomography analysis of the circumpapillary region, Katagiri et al found a significant thinning in the retinal nerve fiber layer and from inner nuclear layer to the retinal pigment epithelium.29 FF-ERG analysis showed significantly reduced amplitudes and preserved implicit time in the photopic negative response, whereas the amplitudes and implicit times of the other FF-ERG components did not differ significantly. FM-ERG analysis showed significantly reduced amplitudes but preserved implicit times in all components. The authors concluded that the change of retinal structure responsible for visual field defects in patients with ONH are mostly secondary to decreased retinal ganglion cells and their axons, but also foveal abnormalities, with a relatively preserved peripheral retina.
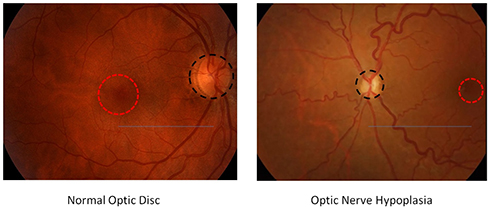 |
Figure 1 For a proper comparison between normal optic disc and optic nerve hypoplasia (ONH), the examiner should take into account the size of optic disc (disc diameter shown as a dashed black circle) compared to the macula (macula diameter shown as a dashed red circle) as well as the relative disc size (ratio accounting for the distance between optic disc and macula shown as a continuous blue line). As a general rule, ONH is diagnosed when the ratio is < 0.4. |
Furthermore, SOD patients can also present with vertical tilt-conus-ectasia syndrome or horizontal tilt-conus syndrome associated with amblyopia or bilateral decreased visual acuity. In those cases, as reported by Alt et al visual field defects will correspond to the direction of the tilt.9
- Electrophysiological testing: Visual evoked potentials (VEP) and electro-oculographic studies may be pathological or within normal range in SOD patients.26,30 Abnormal VEPs can document either bilateral or unilateral delayed latencies.26 Given this wide range of possible findings, Coupland and Sarnat concluded that VEP should not be used as a diagnostic criterion of severe dysplasias, but rather considered as a supplementary tool for detecting and correlating the associated abnormalities of brain development with affected visual pathways.31
Neuroradiological Protocols
As previously mentioned, ophthalmological findings should be correlated with the presence of developmental abnormalities of midline structures. Any patient with evidence of ONH should therefore undergo MRI brain to demonstrate or rule out the presence/extent of the agenesis of the midline structures and other intracranial abnormalities (usually administration of contrast media such Gadolinium is not needed).7,32–34 Hellström et al24 suggested a systematic stepwise approach when analyzing the MRI of a possible SOD patient, and neuroradiologists should address the following questions:
(1) Is the anterior pituitary of normal size?
(2) Is the posterior pituitary present, and what is its location?
(3) Is the pituitary stalk present, and what is its size?
(4) What are the sizes of optic nerves and optic chiasm?
(5) Is the septum pellucidum present?
(6) Are there other midline abnormalities (eg corpus callosum dysgenesis)?
MRI scan (see Figure 2) can and should be used to rule out abnormalities of septum pellucidum/corpus callosum, which are normal in patients with SOD-like syndrome; and detect associated cortical malformations, which are particularly relevant in the context of SOD plus syndrome. Alt et al demonstrated that only 6% of individual with septum pellucidum/corpus callosum dysgenesis actually fulfill the other radiologic criteria for SOD, 18% of patients in fact fall into the so-called SOD-like category, whereas ¾ of patients can be classified under the umbrella of SOD-plus syndrome, which appears to be extremely more common.7,10
 |
Figure 2 Eight-year-old ♀, presenting with macrocephaly and congenital visual loss. She also has a history of hypopituitarism, on replacement medications. Examination showed stable signs of likely “burnt out” or stable hydrocephalus. Images confirm absent chiasm, and monoventricle resulting from absence of septum pellucidum as seen in many patients with SOD. |
Cortical abnormalities such as polymicrogyria, schizencephaly, and cortical dysplasia are of remarkable predictive value (see Table 2), hence the clinical team would in fact be expected to provide a clinical correlation for all those radiological findings and potentially predict the possible onset of seizures and developmental delays.19
Endocrinological Tests
In the absence of specific blood markers for SOD syndrome, laboratory investigations with full endocrine screening are of paramount importance to assess the extent of the hypopituitarism.35–39 Most studies are in agreement that patients with suspected SOD should undergo the investigations reported in Table 3.
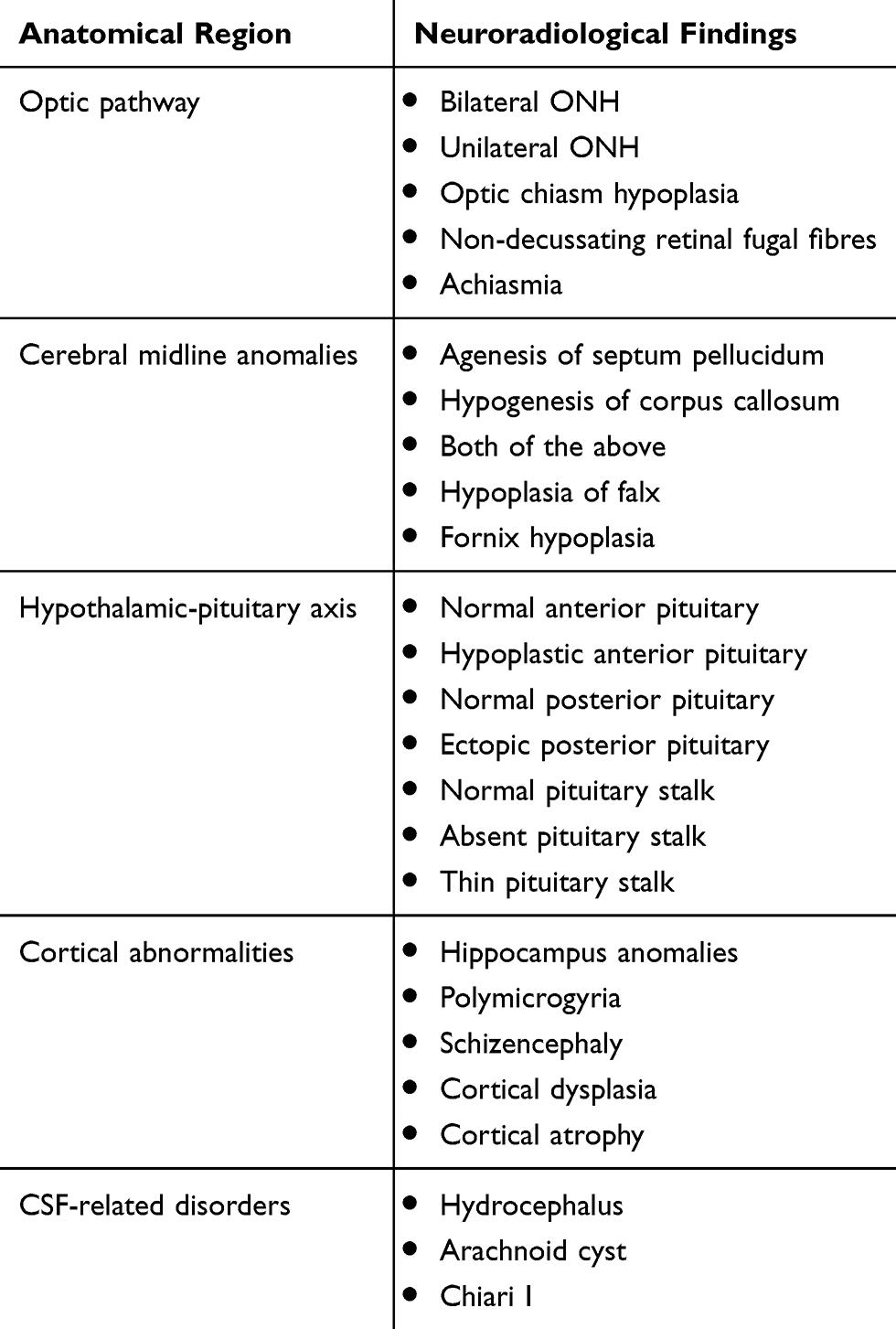 |
Table 2 SOD And Its Neuroradiological Features |
 |
Table 3 Testing Endocrine Dysfunctions In SOD Patients |
Testing of pituitary function should be conducted fairly regularly, at least every six months, similarly to what happens in the management of endocrinological tumors.36,38,39
Qian et al have reported sensitivity and specificity of brain MRI as signs of endocrinopathy are 67.9% and 83.3%, respectively. This study has determined that abnormal MRI findings do not have the sensitivity to predict endocrinopathy, nor does a normal MRI rule out possible endocrine abnormalities. When patients with ONH present with normal neurological examinations, normal endocrine workup and normal developmental milestones, a MRI of the brain may be deferred until new indications arise.40
Additional Investigations
When evaluating a patient with SOD, the presence of clinically evident or subclinical seizures, focal deficits, neurodevelopmental disabilities and other associated morbidities such as cerebral palsy, autism, and sleep-cycle disorders should be assessed.25 EEG are often normal, and Hellström et al reported that this is the case in at least 50% of SOD patients.24 Autism spectrum disorders can be present to up to 30–33% in these patients.41,42 They frequently present with behavioral and emotional problems, such as attention deficit disorder, and should therefore be examined by a psychiatrist/neuropsychologist. The importance of a complete neuro-ophthalmological assessment could not be emphasized enough because visual impairment is known to foster the psychosocial and behavioral delay in those children.43 Finally, the degree of hypothyroidism in children with SOD may not be overtly diagnosed during the first years of their life, and it has been demonstrated that this can worsen their neurodevelopmental delay.23
Clinical And Surgical Management
As highlighted in this review, SOD patients require a multidisciplinary approach at any stage of their diagnosis, treatment and long-term management. It is important that their pathognomonic clinical and radiological features are identified early because appropriate hormonal replacement has to be started as soon as possible. The type of replacement needed by those patients obviously vary according to the type of hypopituitarism they present with, of note all the trials on SOD patients (NCT00140413, NCT00825591 and NCT02558829) revolve around optimization of endocrinological therapies. Whereas some studies have focused on the monitoring throughout early childhood, it should be noted that many specialists believe that a lifelong monitoring and surveillance should be provided.7
Although ONH is not a treatable condition in itself, associated ophthalmological findings (ie amblyopia, refractive errors) should be identified, treated and/or followed up on a regular basis, ideally every year.7 Eye patching has been proposed by some groups, but the overall consensus is that it is not helpful in most cases, including those of severely asymmetrical ONH.7,19 Similarly, strabismus surgery has been advocated to promote a better visual outcome in selected cases; nonetheless, many studies support the theory that corrective surgery should be postponed until it becomes psychosocially challenging for the patient.7,19 Psychomotor delay, autism and attention deficit disorders would require a tailored therapeutic approach focused on mobilization, speech therapy, and psychological support.7
Prognosis And Outcome
Unfortunately, SOD is not a curable disease, but its outcome can be optimized, as seen above, with adequate hormonal replacement, neurological follow-up and multidisciplinary support. Children with SOD tend to be shorter than the average population, especially if growth hormone deficit is not tackled in time.20,36,37 In fact, adequate treatment with growth hormone significantly increases the growth rate of these children and only less than 1% report adverse events related to the treatment, such as scoliosis, carbohydrate metabolism abnormalities, injection site complications and vomiting.36
Children with SOD show an increased tendency towards obesity which prevalence appears to be up to 44% despite hormonal therapy. A balanced diet and weight control is therefore recommended. Children with SOD may also have varying cardiopulmonary problems, such as asthma.7,36
The degree of psychomotor delay if often unpredictable and together with visual deficits represents the main outcome influencing features of SOD. Finally, although it should be acknowledged that it is extremely unusual for the affected gene/s to be identified in an individual without family history of SOD, therefore explaining why genetic counseling is not usually helpful, it should be noted that those with consanguinity should be tested to identify genetic background and counseling should be provided.32 At the same time, it is important to be reminded that, in light of the current scientific knowledge regarding the effects of environmental causes or vascular events in triggering a prenatal/perinatal pathological cascade, healthcare professionals should in principle reassure affected families because the series of events leading to SOD are considered highly unlikely to recur in future pregnancies.
Conclusion
SOD is a rare and heterogeneous congenital disorder. With this review, we attempted to increase the awareness regarding the red flags for SOD and deliver the message that whenever the suspicion is raised patients should be referred appropriately to centers specializing in congenital disorders. SOD cannot be considered as a uniform disease entity and requires differential diagnosis with other overlapping conditions, hence the indications for a comprehensive clinical, radiological and laboratory assessment to establish a tailored approach. Early diagnosis, adequate hormone replacement treatment and a thorough neuro-ophthalmological follow-up can improve the overall prognosis radically. Last but not least, it should be kept in mind that the burden of this syndrome is remarkable not only for the patients but also for their family, therefore social and neuropsychological support are to be considered integral pillars in their management.
Disclosure
The authors report no conflicts of interest in this work.
References
1. De Morsier G. Studies on malformation of cranioencephalic sutures. III. Agenesis of the septum lucidum with malformation of the optic tract. Schweiz Arch Neurol Psychiatr. 1956;77(1–2):267–292.
2. Izenberg N, Rosenbtum M, Parks J. The endocrine spectrum of septo-optic dysplasia. Clin Pediatr (Phila). 1984;23(11):632–636. doi:10.1177/000992288402301105
3. Brodsky MC. Hypothesis: septo-optic dysplasia is a vascular disruption sequence. Surv Ophthalmol. 1998;42(5):489–490. doi:10.1016/s0039-6257(97)00131-8
4. Miller SP, Shevell MI, Patenaude Y, Poulin C, O’Gorman AM. Septo-optic dysplasia plus: a spectrum of malformations of cortical development. Neurology. 2000;54(8):1701–1703. doi:10.1212/wnl.54.8.1701
5. Ryabets-Lienhard A, Stewart C, Borchert M, Geffner M. The optic nerve hypoplasia spectrum. Adv Pediatr. 2016;63(1):127–146. doi:10.1016/j.yapd.2016.04.009
6. Green BN, Johnson CD, Adams A. Writing narrative literature reviews for peer-reviewed journals: secrets of the trade. J Chiropr Med. 2006;5(3):101–117. doi:10.1016/S0899-3467(07)60142-6
7. McCabe M, Alatzoglou K, Dattani M. Septo-optic dysplasia and other midline defects: the role of transcription factors: HESX1 and beyond. Best Pract Res Clin Endocrinol Metab. 2011;25(1):115–124. doi:10.1016/j.beem.2010.06.008
8. Acers TE. Optic nerve hypoplasia: septo-optic-pituitary dysplasia syndrome. Trans Am Ophthalmol Soc. 1981;79:425–457.
9. Lubinsky MS. Hypothesis: septo-optic dysplasia is a vascular disruption sequence. Am J Med Genet. 1997;69(3):235–236. doi:10.1016/j.survophthal.2013.02.004
10. Alt C, Shevell M, Poulin C, Rosenblatt B, Saint-Martin C, Srour M. Clinical and radiologic spectrum of septo-optic dysplasia: review of 17 cas es. J Child Neurol. 2017;32(9):797–803. doi:10.1177/0883073817707300
11. Khaper T, Bunge M, Clark I, et al. Increasing incidence of optic nerve hypoplasia/septo-optic dysplasia spectrum: geographic clustering in Northern Canada. Paediatr Child Health. 2017;22(8):445–453. doi:10.1093/pch/pxx118
12. Kelberman D, Dattani M. Genetics of septo-optic dysplasia. Pituitary. 2007;10(4):393–407. doi:10.1007/s11102-007-0055-5
13. Kelberman D, Dattani M. Septo-optic dysplasia – novel insights into the aetiology. Horm Res. 2008;69(5):257–265. doi:10.1159/000114856
14. Garne E, Rissmann A, Addor M, et al. Epidemiology of septo-optic dysplasia with focus on prevalence and maternal age – A EUROCAT study. Eur J Med Genet. 2018;61(9):483–488. doi:10.1016/j.ejmg.2018.05.010
15. Tornqvist K, Ericsson A, Kallen B. Optic nerve hypoplasia: risk factors and epidemiology. Acta Ophthalmol Scand. 2002;80(3):300–304. doi:10.1034/j.1600-0420.2002.800313.x
16. Patel L, McNally R, Harrison E, Lloyd I, Clayton P. Geographical distribution of optic nerve hypoplasia and septo-optic dysplasia in Northwest England. J Pediatr. 2006;148(1):85–88. doi:10.1016/j.jpeds.2005.07.031
17. Atapattu N, Ainsworth J, Willshaw H, et al. Septo-optic dysplasia: antenatal risk factors and clinical features in a regional study. Horm Res Paediatr. 2012;78(2):81–87. doi:10.1159/000341148
18. Siatkowski R, Sanchez J, Andrade R, Alvarez A. The clinical, neuroradiographic, and endocrinologic profile of patients with bilateral optic nerve hypoplasia. Ophthalmology. 1997;104(3):493–496. doi:10.1016/s0161-6420(97)30286-3
19. Borchert M. Reappraisal of the optic nerve hypoplasia syndrome. J Neuro-Ophthalmol. 2012;32(1):58–67. doi:10.1097/wno.0b013e31824442b8
20. Riedl S, Müllner-Eidenböck A, Prayer D, Bernert G, Frisch H. Auxological, ophthalmological, neurological and MRI findings in 25 Austrian patients with Septo-Optic Dysplasia (SOD). Horm Res Paediatr. 2002;58(3):16–19. doi:10.1159/000066484
21. Riedl S, Vosahlo J, Battelino T, et al. Refining clinical phenotypes in septo-optic dysplasia based on MRI findings. Eur J Pediatr. 2008;167(11):1269–1276. doi:10.1007/s00431-007-0666-x
22. Polizzi A, Pavone P, Iannetti P, Manfré L, Ruggieri M. Septo-optic dysplasia complex: a heterogeneous malformation syndrome. Pediatr Neurol. 2006;34(1):66–71. doi:10.1016/j.pediatrneurol.2005.07.004
23. Ferran K, Paiva I, Gilban D, et al. Septo-optic dysplasia. Arq Neuropsiquiatr. 2010;68(3):400–405. doi:10.1590/s0004-282x2010000300014
24. Hellström A, Aronsson M, Axelson C, et al. Children with septo-optic dysplasia – how to improve and sharpen the diagnosis. Horm Res Paediatr. 2000;53(1):19–25. doi:10.1159/000053200
25. Yates J, Troester M, Ingram D. Sleep in children with congenital malformations of the central nervous system. Curr Neurol Neurosci Rep. 2018;18(7). doi:10.1007/s11910-018-0850
26. Borchert M, Mccullogh D, Rother C, Stout A. Clinical assessment, optic disk measurements, and visual-evoked potential in optic nerve hypoplasia. Am J Ophthalmol. 1995;120(5):605–612. doi:10.1016/s0002-9394(14)72207-x
27. Lenhart P, Desai N, Bruce B, Hutchinson A, Lambert S. The role of magnetic resonance imaging in diagnosing optic nerve hypoplasia. J Am Assoc Pediatr Ophthalmol Strabismus. 2013;17(1):e21. doi:10.1016/j.jaapos.2012.12.074
28. Signorini S, Decio A, Fedeli C, et al. Septo-optic dysplasia in childhood: the neurological, cognitive and neuro-ophthalmological perspective. Dev Med Child Neurol. 2012;54(11):1018–1024. doi:10.1111/j.1469-8749.2012.04404.x
29. Katagiri S, Nishina S, Yokoi T, et al. Retinal structure and function in eyes with optic nerve hypoplasia. Sci Rep. 2017;7(1). doi:10.1038/srep42480
30. McCulloch D, Garcia-Filion P, Fink C, Chaplin C, Borchert M. Clinical electrophysiology and visual outcome in optic nerve hypoplasia. Br J Ophthalmol. 2009;94(8):1017–1023. doi:10.1136/bjo.2009.161117
31. Coupland SG, Sarnat HB. Visual and auditory evoked potential correlates of cerebral malformations. Brain Dev. 1990;12(5):466–472. doi:10.1016/s0387-7604(12)80209-9
32. Ganau M, Talenti G, D’Arco F. Teaching NeuroImages: radiologic features of septo-optic dysplasia plus syndrome. Neurology. 2018;91(23):e2200–e2201. doi:10.1212/wnl.0000000000006631
33. Webb E, Dattani M. Septo-optic dysplasia. Eur J Hum Genet. 2009;18(4):393–397. doi:10.1038/ejhg.2009.125
34. Ganau M, Syrmos N, D’arco F, et al. Enhancing contrast agents and radiotracers performance through hyaluronic acid-coating in neuroradiology and nuclear medicine. Hell J Nucl Med. 2017;20(2):166–168. doi:10.1967/s002449910558
35. Secco A, Allegri AE, Di Iorgi N, et al. Posterior pituitary (PP) evaluation in patients with anterior pituitary defect associated with ectopic PP and septo-optic dysplasia. Eur J Endocrinol. 2011;165(3):411–420. doi:10.1530/EJE-11-0437
36. Parker K, Hunold J, Blethen S. Septo-optic dysplasia/optic nerve hypoplasia: data from the National Cooperative Growth Study (NCGS). J Pediatr Endocrinol Metab. 2002;15(Supplement). doi:10.1515/jpem.2002.15.s2.697
37. Ganau L, Prisco L, Ligarotti G, Ambu R, Ganau M. Understanding the pathological basis of neurological diseases through diagnostic platforms based on innovations in biomedical engineering: new concepts and theranostics perspectives. Medicines. 2018;5(1):22. doi:10.3390/medicines5010022
38. Poliga G, Olla L, Bellisano G, et al. Solitary extramedullary plasmacytoma of the thyroid gland: a case report and review of literature. Internet J Head Neck Surg. 2007;2(1). doi:10.5580/15ad
39. Vedin A, Karlsson H, Fink C, Borchert M, Geffner M. Presenting features and long-term effects of growth hormone treatment of children with optic nerve hypoplasia/septo-optic dysplasia. Int J Pediatr Endocrinol. 2011;2011(1). doi:10.1186/1687-9856-2011-17
40. Qian X, Fouzdar Jain S, Morgan LA, Kruse T, Cabrera M, Suh DW. Neuroimaging and endocrine disorders in paediatric optic nerve hypoplasia. Br J Ophthalmol. 2018;102(7):906–910. doi:10.1136/bjophthalmol-2017-310763
41. Jutley-Neilson J, Harris G, Kirk J. The identification and measurement of autistic features in children with septo-optic dysplasia, optic nerve hypoplasia and isolated hypopituitarism. Res Dev Disabil. 2013;34(12):4310–4318. doi:10.1016/j.ridd.2013.09.004
42. Parr J, Dale N, Shaffer L, Salt A. Social communication difficulties and autism spectrum disorder in young children with optic nerve hypoplasia and/or septo-optic dysplasia. Dev Med Child Neurol. 2010;52(10):917–921. doi:10.1111/j.1469-8749.2010.03664
43. Sonksen P, Dale N. Visual impairment in infancy: impact on neurodevelopmental and neurobiological processes. Dev Med Child Neurol. 2007;44(11):782–791. doi:10.1111/j.1469-8749.2002.tb00287.x
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.