Back to Journals » International Journal of Nephrology and Renovascular Disease » Volume 11
Is the light at the end of the tunnel nigh? A review of ADPKD focusing on the burden of disease and tolvaptan as a new treatment
Authors Barnawi RA ![]() , Attar RZ, Alfaer SS, Safdar OY
, Attar RZ, Alfaer SS, Safdar OY ![]()
Received 7 March 2017
Accepted for publication 4 November 2017
Published 1 February 2018 Volume 2018:11 Pages 53—67
DOI https://doi.org/10.2147/IJNRD.S136359
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Pravin Singhal
Rashid A Barnawi,1 Rahaf Z Attar,1 Sultan S Alfaer,1 Osama Y Safdar2
1Faculty of Medicine, King Abdulaziz University, Jeddah, Saudi Arabia; 2Pediatric Nephrology Center of Excellence, Faculty of Medicine, King Abdulaziz University, Jeddah, Saudi Arabia
Abstract: Autosomal dominant polycystic kidney disease (ADPKD) causes pathological cystic changes to the kidney and is characterized by numerous renal and systemic manifestations. ADPKD is the fourth most common renal disease requiring renal replacement therapy. In this report, we present a detailed review of ADPKD, with a particular focus on its major economic, psychological, and social burden in affected patients. Treatment of this disease has been based on prophylactic and supportive measures. However, in recent years, new drugs have emerged as promising agents that may retard the progression of ADPKD, such as tolvaptan. In this report, we provide an in-depth discussion of tolvaptan, which has shown an effect in decreasing annual total kidney volume growth and renal function decline, thus slowing disease progression. The mechanism of action, side effects, and available data on cost-effectiveness are discussed together with the results of the first clinical trials and the most recent trials with regard to its efficacy and safety. Tolvaptan has recently received approval and been granted marketing authorization in Japan, Canada, Korea, Switzerland, and Europe. A demand for widely accepted guidelines for its use has emerged since its approval. The currently available series of recommendations and guidelines as to when to start treatment with tolvaptan, as well as which patients should be treated, are also reviewed in this report. We lastly offer some considerations for future trials, and raise unanswered questions.
Keywords: autosomal dominant polycystic kidney disease, quality of life, economic burden, tolvaptan, tolvaptan cost-effectiveness, tolvaptan use recommendations
Introduction
Polycystic kidney disease (PKD) causes pathological cystic changes to the kidney1 and is part of a heterogeneous group of disorders characterized by the development of numerous renal cysts as well as renal and systemic manifestations2 (Figures 1 and 2), as opposed to single or multiple simple benign renal cysts that are commonly encountered incidentally during imaging of the kidneys and adrenals. Such simple cysts tend to be asymptomatic because they do not enlarge the kidneys, distort normal kidney structure, or cause reduced renal function, unlike in PKD.1,3,4
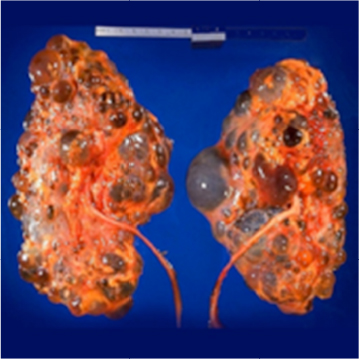 | Figure 1 Gross pathology of a polycystic kidney from an autopsy specimen of bilateral ADPKD. Image courtesy of CDC/Dr. Edwin P. Ewing, Jr. Retrieved from the case, courtesy of Prof. Frank Gaillard, Radiopaedia.org, rID: 9719. Abbreviations: ADPKD, autosomal dominant polycystic kidney disease; CDC, Centers for Disease Control and Prevention. |
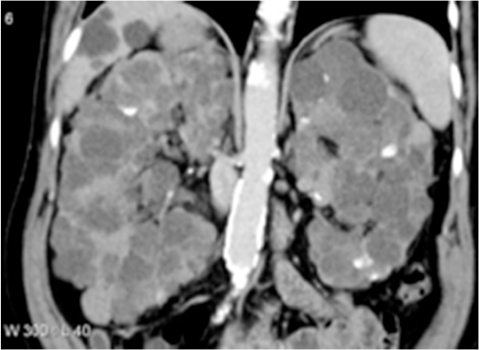 | Figure 2. CT of the abdomen (coronal reformats) showing enlarged kidneys with innumerable cysts ranging in size. There are also cysts in the liver. Retrieved from the case courtesy of Prof. Frank Gaillard, Radiopaedia.org, rID: 5203. Abbreviation: CT, computed tomography. |
There are two types of PKD: autosomal dominant PKD (ADPKD) and autosomal recessive PKD (ARPKD). ARPKD is most commonly encountered in infants and children and is far less common than ADPKD, which is the most common inherited cause of kidney disease and is our primary focus here. ADPKD affects all races and has no gender or age predilection. In the United States, it is estimated that ADPKD affects 1 in every 400 to 1,000 live births, with 5,000–6,000 new cases diagnosed each year and approximately 600,000 Americans affected by the disease. More than 2,000 patients start renal replacement therapy (RRT) due to cystic kidney disease annually. ADPKD is, however, less prevalent in other countries, such as France (1 per 1,111), Wales (1 per 2,459), and Japan (1 per 4,033).2,5–7
ADPKD is a systemic disease with multiple renal and extra-renal manifestations. Patients with ADPKD may develop end-stage renal disease (ESRD) at some point in their lives, and ADPKD is considered the fourth most common renal disease requiring RRT.8 The mean age of onset of ESRD for the PKD1 variant is 54.3 years, whereas that for the PKD2 variant is 74 years.2 Renal manifestations are produced by the progressive and continuous enlargement and proliferation of fluid-filled cysts, leading to enlargement of the kidney up to five times the normal volume in the years prior to the development of kidney failure. Thus, total kidney volume is the most important predictor for the development of renal insufficiency and progression of ADPKD.9 It is important to note that, although the glomerular filtration rate (GFR) declines as the total kidney volume increases, the creatinine level only rises in advanced stages of the disease after the non-cystic parenchyma of the kidney has been irreversibly damaged. Therefore, the measurement of GFR does not aid in the detection of kidney damage early on in the disease course.10 Other renal manifestations include hypertension, urinary tract infection, an inability to concentrate urine, hematuria, renal stones, and acute or chronic flank and abdominal pain, either due to kidney enlargement or kidney stones.2,9,11–13 Moreover, kidney enlargement can reach a stage in which the kidney obstructs the iliac vein or inferior vena cava, with possible thrombus formation and pulmonary embolism.13
The most common extra-renal manifestation is polycystic liver disease (PLD).10,14 It is more common in women than in men. In addition, it appears earlier in women and they tend to have larger and more numerous cysts. Although liver cysts are usually asymptomatic, they may – if they grow substantially – produce pressure effects and manifest as abdominal pain or discomfort, dyspnea, early satiety, gastroesophageal reflux, mechanical lower back pain, hepatic venous outflow obstruction, or bile duct compression presenting as obstructive jaundice. Other serious complications include cyst hemorrhage, infection, and torsion or rupture. Moreover, liver cysts are estrogen-dependent; therefore, they can be more problematic in women. Cysts can, furthermore, grow in the pancreas, seminal vesicle, arachnoid membrane, and spinal meninges. Other extra-renal manifestations include cerebral and coronary artery aneurysms, cardiac valve disease, colonic diverticula, abdominal wall and inguinal hernias, and bronchiectasis.2,10,11,14–16
The burden of this disease is considerable, which necessitates conducting more research and studies to find methods to slow its progression. In the next sections, a detailed review of ADPKD is presented, focusing on its economic, psychological, and social burden on the patient. We will, in addition, discuss in depth the newly developed medication tolvaptan for the treatment of ADPKD.
Pathogenesis
Mutation spectrum
Genetic mutation plays a major role in the pathogenesis of most cases of ADPKD. A mutation involving the PKD1 gene (~85% of cases) and the PKD2 gene (~15% of cases) leads to cyst formation and its progressive growth.5 The PKD1 gene is a complex gene that is composed of 46 exons and is located on chromosome 16p13.17 The PKD2 gene is composed of 15 exons and is located on chromosome 4q21. PKD1 as well as PKD2 display marked allelic heterogeneity, with approximately 200 different PKD1 and more than 50 different PKD2 mutations described. Most of these mutations are private and unique to a single-family pedigree.17–19 The vast majority of these mutations truncate the protein, due to frame-shifting deletion or insertion, nonsense mutations, or splicing defects, but in-frame and missense (non-truncating) mutations have also been described.17,18 PKD1 mutations are mostly found in the 3’ region of the gene; however, mutations can occur throughout both genes.18 Recent genotype/phenotype correlations for PKD1 propose that not all mutations lead to a similar phenotypic outcome. A study that was done in Mayo Clinic included 434 patients with ADPKD, of whom 221 patients (50.9%) had truncating PKD1, 141 patients (32.5%) had non-truncating PKD1, and 72 patients (16.6%) had a PKD2 mutation. Patients with truncating PKD1 developed ESRD at a younger age (median age 56.5) when compared to patients with non-truncating PKD1 and PKD2 (median age 68.4 and 79.4 years, respectively; all p < 0.001).20 Further, studies have shown that 7%–10% of ADPKD-affected families are genetically unresolved (GUR). Families are defined as GUR when no mutations of PKD1 and PKD2 can be detected from Sanger sequencing or multiplex ligation-dependent probe amplification. Approximately, 3% of GUR ADPKD-affected familial disease is caused by mutation in the GANAB gene that encodes the Glucosidase II Alpha Subunit (GIIa). This mutation is most likely caused by a defect in polycystin-1 maturation.21
There has been some recent evidence suggesting that a threshold/dosage model best explains how cysts develop in ADPKD.22,23 Cytogenesis arises when the function of polycystin-1 or -2 falls below a certain critical level (cystogenic threshold; explained in the next section). This may occur following a somatic mutation affecting the other allele (two-hit hypothesis)24,25 or other factors such as environmental effects and renal damage.22
Mechanism of cyst formation
Mutations in PKD1 and PKD2 genes lead to loss of the normal function of the two transmembrane proteins polycystin-1 (encoded by the PKD1 gene) and polycystin-2 (encoded by the PKD2 gene). Calcium-containing urinary flow in renal tubules stimulates polycystin-1, which in turn interacts with polycystin-2 and forms a complex that regulates the intracellular levels of calcium and cyclic adenosine monophosphate (cAMP). A defect in the function of aforementioned proteins leads to disruption of intracellular calcium homeostasis, and subsequently increases the level of cAMP, which promotes cellular proliferation and fluid secretion in ADPKD. Additionally, polycystin-1 binds and activates G-protein–coupled membrane receptors that, when activated, affect the activity of adenylyl cyclase, which leads to an increase in intracellular cAMP levels. Another route by which cell proliferation is induced is through the mammalian target of rapamycin (mTOR) pathway. Polycystin-1 indirectly upregulates the kinase activity of the mTOR pathway, which promotes cell hypertrophy, cell division, and cell survival. Understanding this pathogenesis is essential to comprehending how pathogenesis-based therapeutics actually modulate ADPKD progression.7,11–13,15,23,25,26 Figure 3 illustrates key mechanisms by which cysts can develop in ADPKD as well as targets of potential treatments.
 | Figure 3 Schematic illustration of the key mechanisms of ADPKD pathogenesis and targets of potential treatments. Polycystin-1 and -2 expressed in different subcellular locations and regulate (1) proliferation, (2) fluid secretion, (3) ciliary function, (4) cell–cell adhesion, and (5) cell–matrix interaction of renal epithelial cells. Dysfunction of polycystin-1 or -2 results in aberrant signaling pathways including: (A) activation of cAMP, (B) decreased intracellular calcium concentrations, and (C) activation of mTOR. These changes lead to transformation of normal cells to a “cystic phenotype” and promote cyst formation. The targets of candidate drugs are depicted as blue boxes. Reproduced with permission from Chang MY, Ong AC. New treatments for autosomal dominant polycystic kidney disease. Br J Clin Pharmacol. 2013;76(4):524–535. © 2013 The Authors. British Journal of Clinical Pharmacology © 2013 The British Pharmacological Society.26 Abbreviations: ADPKD, autosomal dominant polycystic kidney disease; cAMP, cyclic adenosine monophosphate; CFTR, cystic fibrosis transmembrane regulator; ER, endoplasmic reticulum; ERK, extracellular-signal regulated kinase; GlcCer, glucosylceramide; HDAC, histone deacetylase; IL-6R, interleukin-6 receptor; MEK, mitogen-activated protein kinase; mTOR, the mammalian target of rapamycin; PC, polycystin; PDE, phosphodiesterase; PKA, protein kinase A; SR, somatostatin receptor; TSC, tuberous sclerosis; V2R, vasopressin V2 receptor. |
Burden of ADPKD
Economic burden
It is well documented that the majority of patients with ADPKD will eventually develop ESRD and require RRT.27,28 The healthcare costs and the economic burden of ESRD are enormous for affected individuals and for governments or private enterprises, even in developed countries.29–32 Moreover, the serious health consequences of ESRD have a huge impact on patients emotionally, physically, and psychosocially as well as on their quality of life (QOL).33–35 Herein, we intend to elucidate the burden of ADPKD specifically on the finances and QOL of patients.
The incidence of ESRD caused by ADPKD differs among countries, varying from 4.8 (in Japan) to 7.9 (in the United States) and from 3.9 to 15.3 (in Europe) cases per million individuals per year.36 These figures may not seem striking, but several studies have shown that ADPKD has a high cost for patients and constitutes an economic burden to the healthcare system relative to the general population.36–39 In a retrospective study in a large dialysis organization in the United States that included 1,274 patients with ADPKD and aimed to quantify healthcare expenditures and costs for ADPKD patients with ESRD, the total healthcare costs for those patients were considerable – at US$4,254 per patient per month or US$51,048 per patient-year.37 However, the same study showed that the total costs for ESRD patients other than those with ADPKD were higher. Another study in the United States that reviewed the administrative records of a private health insurer and included 1,913 patients with ADPKD demonstrated that higher charges are associated with advanced kidney disease, as the annual medical charges (unadjusted) increased from US$24,497 in patients with an estimated glomerular filtration rate (eGFR) greater than 90 mL/min to US$134,784 in those with an eGFR of greater than 15 mL/min (p < 0.0001). This suggests that strategies that prevent decline of renal function in patients with ADPKD may cause reduction in medical charges.39 In Europe, the European Renal Association and European Dialysis and Transplant Association (ERA-EDTA) estimated that, in 2010, approximately 50,000 patients with ADPKD were on RRT in the 27 EU countries, representing a high cost of €1.5 billion per year. This estimation did not include costs of medical care for outpatients or hospital admissions for complications and comorbidities.36
Impact on patient quality of life
The impact of the disease does not end at the financial burden to patients; in addition, it greatly affects QOL of patients, even for those who do not reach ESRD.40–44 In Sao Paulo, Brazil, in a study that attempted to evaluate the frequency of anxiety and depression, and QOL in patients with ADPKD, 38 patients with ADPKD completed the questionnaires of the State Trait Anxiety Inventory (STAI), Beck Depression Inventory (BDI), and QOL-Short-Form SF-36, and were also assigned for interviews. The results demonstrated that 60% of those patients had depression, a mild level of state anxiety and a moderate level of trait anxiety, and they also had low scores for functional capacity, pain, and physical and mental health. Although the impact of ADPKD and its comorbidities seems severe in those patients, it is important to note that some patients had low levels of schooling and low economic status, which was associated with higher scores of trait anxiety. Therefore, these factors may have had an impact on their QOL. Furthermore, the sample size was relatively small, which may limit the broad adoption of their results.40 However, another study of 1,043 patients specified that patients with a low eGFR of 20–44 mL/min/1.73 m2 were significantly more likely to report pain that impacts their daily lives, and they had lower SF-36 scores than patients with an eGFR of more than 45 mL/min/1.73 m2.35 This finding is also supported by a thematic synthesis of 21 studies from six countries (Australia, Canada, Germany, Sweden, United Kingdom, and United States) which reported that the unpredictable onset of pain impacts daily lives of patients with ADPKD and prevents them from establishing long-term life goals. Moreover, patients carried the burden of guilt and shame of having the disease and transferring it to their children, isolated themselves, and were in a state of constant worry as they were more conscious of their mortality. Several patients decided to seize opportunities because the future was uncertain for them. For other patients, ADPKD meant facing probable discrimination with employers or in obtaining health insurance even before they developed symptoms.41
It is notable that previous studies used the non–ADPKD-specific SF-36 QOL questionnaire exclusively or in addition to other questionnaires that are not specific for ADPKD; thus, results may not have reflected an accurate picture of the burden of the disease. Therefore, certain studies have designed specific questionnaires to capture a wider spectrum of QOL issues.42,44 In one study that included 158 patients registered at the Sheffield Kidney Institute in the United Kingdom who had chronic kidney disease (CKD) but were not on RRT, the authors used both validated and novel instruments: a kidney disease quality-of-life short form (KDQOL SF1.3), a nine-item patient health questionnaire (PHQ9), a multidimensional scale of perceived social support (MSPSS), and a novel genetic psychosocial risk instrument (GPRI-ADPKD) designed to address the specific psychosocial effects of living with ADPKD. The results revealed that patients with a lower eGFR (< 30 mL/min) or larger kidneys (mean length on ultrasound ≥ 17 cm) displayed reduced QOL and increased psychosocial risks. In addition, 62% of patients felt guilty about transferring ADPKD to their children, and clinically significant depression was reported in 22% of patients. Patients with lower eGFR and large kidneys were significantly more likely to score lower in physical functioning, general health, sexual function, and vitality (energy/fatigue).42 Another prospective study in Japan used both the SF-36 and a novel 12-item questionnaire. This study involved a total of 219 patients, of whom 108 were on dialysis, and reported a similar trend. The physical component summary score (PCS), mental component summary score (MCS), and role/social component summary score (RCS) of the SF-36 questionnaire of all patients were markedly lower than mean scores for the Japanese population. Furthermore, hemoglobin level, serum albumin level, ascites, and cerebrovascular disease all had a notable impact on the PCS; mental disease had a significant influence on the MCS; and serum albumin, which reflects liver function and renal function and, therefore, patients’ lifestyle, significantly influenced the RCS. Overall, ADPKD symptoms and complications (pain, sleep disturbances, heartburn, fever, gross hematuria, and anorexia) were all found to affect QOL.44 This finding should prompt us to address the disease with more than just attempts to develop new medications. Furthermore, awareness campaigns, support groups, and educational programs should play a role in the management of patients with ADPKD.
Tolvaptan as a new treatment for ADPKD
Conventionally, treatment measures for ADPKD have been nonspecific, such as controlling pain and hypertension, bed rest, and supportive therapy. In other words, treatment is based on prophylactic and supportive measures.2,27,45 As discussed earlier, the burdens of ADPKD and RRT are serious and should not be disregarded. For this reason, there have been attempts to develop medications to slow the progression of the disease and to reduce the requirement for RRT. Recently, several trials have described several medications that could possibly halt or slow the progression of the disease. These medications include mTOR inhibitors and somatostatin analogs, which have conflicting evidence and limited safety and efficacy data.2,13,26,27,45 Tolvaptan, which is our main focus, has been introduced by several studies as a promising agent. This drug is discussed in detail in the following sections. In addition, there are a number of proposed agents that are still in the preclinical study phase.2,26 It should be borne in mind that even with the introduction of new treatments, ADPKD remains incurable and patients should be treated holistically considering their other issues, such as psychosocial complications.
Mechanism of action of tolvaptan
Tolvaptan acts as a selective arginine vasopressin V2 receptor antagonist. Arginine vasopressin (antidiuretic hormone) is a neuropeptide hormone produced by the hypothalamus and secreted into systemic circulation by the posterior pituitary gland. There are three primary types of vasopressin receptors – V1A, V1B, and V2 – which are responsible for vasoconstriction, platelet aggregation, glycogenolysis, adrenocorticotropic hormone release by the anterior pituitary gland, osmoregulation, and body fluid regulation. Here, we are concerned with V2 receptors, which are located in the renal collecting ducts and, when bound and stimulated by vasopressin – which is continuously present in the systemic circulation, likely at a higher level in patients with ADPKD46 – increase cAMP and subsequently lead to the upregulation of aquaporin-2 channels and increased water reabsorption. The affinity of V2 receptors for tolvaptan is 1.8 times the affinity for native vasopressin. Thus, when tolvaptan is administered, it antagonizes the action of vasopressin in the collecting ducts and leads to aquaresis, fluid loss, increased osmolality of the urine, and increased serum sodium levels. Moreover, tolvaptan leads to a decrease in cAMP, resulting in a reduced rate of kidney cyst cell proliferation and increased/enhanced fluid secretion, as discussed previously.7
Tolvaptan is protein bound (99%), orally active (with 40% oral bioavailability), and is minimally affected by food. It is metabolized by cytochrome P450 isoenzyme 3A4 (CYP3A4) and excreted from the body along with fecal matter, with an elimination half-life of 12 hours. As tolvaptan is a sensitive substrate for CYP3A4, and both a substrate and a competitive inhibitor of P glycoprotein (P-gp), there are significant drug–drug interactions related to its use. Therefore, it is contraindicated in patients using strong CYP3A4 inhibitors, recommended not to be used with moderate CYP3A4 inhibitors, and should be used cautiously with P-gp inhibitors. Further, tolvaptan is not recommended for use if the creatinine clearance is less than 10 mL/minute or if the patient is not responsive to the medication and is anuric.7
Clinical trials on the safety and efficacy of tolvaptan
There have been many phase II clinical trials but only one phase III trial investigating the efficacy of tolvaptan in ADPKD.7 Table 1 summarizes the findings of the clinical studies of tolvaptan for ADPKD.
 | Table 1 Summary of clinical trials of tolvaptan for autosomal dominant polycystic kidney disease. Note: *(CrCl > 60 mL/min and TKV ≥ 750 mL). Abbreviations: ACR, albumin–creatinine ratio; ADPKD, autosomal dominant polycystic kidney disease; CKD: chronic kidney disease; CI, confidence interval; CrCl, creatinine clearance; eGFR, estimated glomerular filtration rate; TEMPO, Tolvaptan Efficacy and safety in Management of autosomal dominant Polycystic kidney disease and its Outcomes; TKV, total kidney volume. |
The Tolvaptan Efficacy and Safety in Management of Autosomal Dominant Polycystic Kidney Disease and Its Outcomes (TEMPO) 2:4 phase II clinical trial47 used data from earlier studies that described safety, tolerability, and dosing strategies and applied them to a large population.7 This study was a 3-year, multicenter, open-label study that included 63 (46 North American and 17 Japanese) subjects with ADPKD, randomly matched 1:2 to historical controls from the Consortium of Radiologic Imaging Study of PKD and the Modification of Diet in Renal Disease studies. The primary objective was to assess the long-term safety and tolerability of tolvaptan, and the secondary objective was to acquire pilot efficacy data. The study prespecified primary endpoint was the rate of change in total kidney volume (TKV) over 3 years in subjects and controls, and the secondary endpoint was the rate of change in eGFR. In total, 81% of the subjects (51 patients) completed the 3 years of tolvaptan treatment, and 19% (12 patients) withdrew from the study; 50% of those who withdrew (six patients) did so due to adverse events, which included thirst, polyuria, hyperuricemia, and increased creatinine concentration (acute kidney injury). The results of TEMPO 2:4 showed that the annual TKV growth in tolvaptan-treated subjects was 1.7%, whereas the annual TKV growth in controls was 5.8% (p < 0.001). This represents a 70% slower growth rate per year in tolvaptan-treated patients. Moreover, tolvaptan decreased the rate of decline in eGFR compared with controls (−0.71 vs −2.1 mL/min/1.73 m2 per year, p = 0.01). This study was limited by the small number of patients and unmatched ethnicities between subjects and controls.47
The phase III clinical trial TEMPO 3:4 was a multicenter, double-blinded, placebo-controlled, 3-year trial that aimed to evaluate the safety and efficacy of tolvaptan in slowing the progression of ADPKD. In the study, 1,445 patients with ADPKD were assigned in a 2:1 ratio at 129 sites worldwide to receive a split (morning and night) daily dose (ranging from 90 to 120 mg) of tolvaptan. Patient ages ranged from 18 to 50 years, TKV was 750 mL or more, and estimated creatinine clearance was 60 mL/min or more. The study primary endpoint was the annual rate of change in TKV, and the secondary endpoint was composite and included time to clinical progression (defined as worsening kidney function, kidney pain, hypertension, and albuminuria) and rate of decline in kidney function. Overall, 1,157 patients (80.1%) completed the trial (77% of tolvaptan-treated patients and 86.2% of the placebo group). Findings of this study at 3 years correlated with the results of the phase II study and showed that tolvaptan-treated patients had a lower annual increase in TKV (2.8% per year) in comparison with the placebo group (5.5% per year), and they also had a slower annual decline in kidney function (assessed by means of the reciprocal of the serum creatinine level) with a slope of −2.61 (mg/mL)-1/year in comparison with −3.81 (mg/mL)-1/year in controls. Moreover, the composite endpoint favored tolvaptan over placebo as it showed fewer ADPKD-related events per 100 person-years of follow-up (44 vs 50 events). The discontinuation rate was higher in the tolvaptan group (23%) than in the placebo group (13.8%), and more discontinuations were attributed to adverse events in the tolvaptan group (15.4%) than in the placebo group (5%). Although both groups had similar rates of adverse events, patients treated with tolvaptan had more adverse events related to aquaresis (thirst, polyuria, nocturia, and polydipsia), whereas patients who received placebo had more ADPKD-related adverse events (kidney pain, hematuria, urinary tract infection, and back pain). Moreover, it is important to mention that the tolvaptan group had a greater frequency of elevation of liver-enzyme levels, which caused 12 patients to permanently discontinue the medication. Therefore, liver enzymes levels require monitoring in recipients of tolvaptan, along with uric acid and sodium levels. One important limitation of this study was that all patients were asked to increase hydration and avoid dehydration. This instruction may have led to underestimation of the beneficial effect of tolvaptan due to the suppression of vasopressin levels in the placebo group, resulting in lower rates of kidney growth.48
In light of the previous trials, Boertien et al49 attempted, in a clinical trial, to study the short-term effects of tolvaptan at varying levels of kidney function in patients with ADPKD as well as whether the pharmacodynamic efficacy is dependent on baseline GFR. The authors recruited 29 patients with ADPKD aged between 18 and 70 years and with measured GFRs (mGFR) between 18 and 148 mL/min and gave them increasing dosages of tolvaptan over 3 weeks (60, 90, and 120 mg/day in weeks 1, 2, and 3, respectively). At 3 weeks, 27 patients completed the study, and tolvaptan, as has been described in previous studies, led to an increase in urinary volume and free-water clearance and a decrease in urine osmolality, TKV, and kidney injury marker levels. However, patients with lower baseline mGFR showed decreased responses to tolvaptan for TKV, urinary volume, and osmolality but more distinct responses for fractional free-water clearance, suggesting that they responded more to tolvaptan per functioning nephron. The authors suggested that the lower responses for TKV, urinary volume, and urinary osmolality in those patients were not due to decreased sensitivity to tolvaptan but, rather, due to less functioning kidney mass or structural abnormalities in the kidney. In conclusion, this study reported that ADPKD patients with decreased kidney function may benefit from long-term treatment with tolvaptan, as has been observed for patients with relatively preserved GFRs. Nevertheless, the study was limited by its small sample size and a lack of controls; therefore, the results should be supported by long-term, large-scale, randomized controlled trials.49
Fortunately, 2 years after the Boertien et al study was published, a post hoc analysis of the data from the TEMPO 3:4 study also reported that tolvaptan was equally effective in reducing the rate of TKV growth in patients with ADPKD who had CKD stages 1, 2, and 3 at baseline. Tolvaptan reduced the annual TKV growth by 1.99% for CKD1, 3.12% for CKD2, and 2.61% for CKD3 (all p < 0.001) and reduced eGFR decline by 0.40 in CKD1 (p = 0.23), 1.13 in CKD2 (p < 0.001), and 1.66 mL/min/1.73 m2/year in CKD3 (p < 0.001) patients. This change was not accompanied by an increase in the frequency of adverse events in advanced stages, except for an increased frequency of hypernatremia in CKD3.50 These results are promising for patients with ADPKD.
Recently, the phase IIIb, multicenter, randomized-withdrawal, placebo-controlled, double-blinded, parallel-group trial to specifically determine the efficacy and safety of tolvaptan in patients with ADPKD and advanced-stage CKD (late CKD2 to early CKD4) has been published. In this study, 1,370 patients with ADPKD who were either 18 to 55 years old with eGFR of 25 to 65 ml/minute/1.73 m2 or 56 to 65 years old with eGFR of 25 to 44 ml/minute/1.73 m2 were randomly assigned in a 1:1 ratio to receive tolvaptan (683 patients) or placebo (687 patients) for 12 months. The primary end point was the change in eGFR from baseline (before administration of tolvaptan or placebo) to 1 year, which is the trial completion period, and the secondary end point was the slope of the change in the eGFR at 1 year. Hepatic safety of the drug was also monitored during the study. Supporting the evidence of positive effect of tolvaptan from the Boertien et al study,49 results showed that at advanced CKD stages, tolvaptan slowed the progressive renal function loss. The change in eGFR from baseline in tolvaptan group was −2.34 mL/minute/1.73 m2 (95% confidence interval [CI], −2.81 to −1.87), compared to −3.61 mL/min/1.73 m2 (95% CI, −4.08 to −3.14) in the placebo group (difference, 1.27 mL/min/ 1.73 m2; 95% CI, 0.86 to 1.68; P<0.001). At 1 year, the mean slopes of the change in eGFR were −3.16±0.14 mL/minute/1.73 m2 (95% CI, −3.43 to −2.89) in the tolvaptan group, compared to −4.17±0.14 mL/minute/1.73 m2 (95% CI, −4.45 to −3.89) in the placebo group (difference, 1.01 mL/minute/1.73 m2; 95% CI, 0.62 to 1.40; P<0.001). Safety profile in this trial did not differ from that observed in TEMPO 3:4. Elevations in hepatic transaminase 3 times upper the normal limit occurred in 5.6% of tolvaptan group compared to 1.2% of placebo group, however, enzyme levels reversed to normal after discontinuing the treatment. Also, no elevations in bilirubin levels to twice the upper limit of the normal range was noted. Adverse events caused 9.5% of tolvaptan group to discontinue the trial, compared to 2.2% of placebo group. This includes aquaresis-related adverse events which led 2.1% of tolvaptan group to discontinue versus .1% in placebo group and hepatic enzymes-related adverse events which led 1.6% of tolvaptan group to discontinue versus 0.1% of placebo group.51 One limitation of this trial is that it did not study whether the effect of tolvaptan on eGFR was accompanied by a parallel effect on TKV. Also, the effect of treatment at 1-year remains elusive on the long term. Nevertheless, this information can be obtained from Torres et al post hoc analysis.50
Another post hoc analysis of data from TEMPO 3:4 of patients with ADPKD who had baseline information on albuminuria (n = 1375) also delivered promising findings with regard to the efficacy of tolvaptan. The results of the analysis showed that patients with higher baseline albuminuria – expressed as albumin–creatinine ratio (ACR) – had higher blood pressure and TKV and lower eGFR and, during follow-up, those with higher baseline ACR had a more rapid eGFR decline but no association with the rate of TKV growth. Tolvaptan treatment significantly decreased the ACR during the 3-year trial, whereas it increased in the placebo group (−0.40 mg/mmol vs +0.23 mg/mmol). The difference reached a maximum of 24% at the end of the third year (p < 0.001), and this effect remained after the withdrawal of tolvaptan. This finding suggests that tolvaptan provided structural benefits to the kidney. The beneficial effect of tolvaptan on TKV growth and eGFR loss was more readily detected in patients with higher baseline ACR. These findings are contrary to what the original TEMPO 3:4 trial reported – that tolvaptan had no effect compared with placebo on albuminuria – likely because albuminuria was classified categorically as “albuminuria events” instead of on a continuous scale, which may have resulted in a loss of sensitivity to detect treatment-induced changes.52
The series of clinical trials has not ended, as tolvaptan has offered hope for patients with ADPKD for delaying disease progression. Patience is required, however, as ADPKD is a slowly progressive disease, and long-term efficacy and safety will take some time to prove. To this end, a multicenter, open-label, extension study to TEMPO 3:4 (TEMPO 4:4) was recently published. This study was designed to provide an additional 2 years of data on the long-term efficacy and safety of oral tolvaptan tablet regimens in subjects with ADPKD who completed the TEMPO 3:4 trial; 871 (60.3%) of the 1,445 TEMPO 3:4 subjects enrolled in this study, of whom 557 (58.0%) were previously on tolvaptan (early-treatment group) and 314 (64.9%) were previously on prior placebo (delayed-treatment group). Subjects were given daily split-dose regimens of 45/15, 60/30, or 90/30 mg on waking and 9 hours later. The primary endpoint was the change in TKV from TEMPO 3:4 baseline to TEMPO 4:4 at Month 24 in early- vs delayed-treated subjects, and secondary endpoints included changes in eGFR from TEMPO 3:4 baseline to TEMPO 4:4 Month 24 and TKV and eGFR slopes during TEMPO 4:4 in early- and delayed-treatment subjects. Overall, 763 subjects completed the 24-month period of tolvaptan treatment (507 early-treated subjects and 256 delayed-treatment subjects). Results showed that the primary endpoint was not achieved. TEMPO 4:4 did not show preservation of TKV beneficial difference between early vs delayed groups observed at the end of TEMPO 3:4, as TKV increased by 29.9% in early vs 31.6% in delayed-treatment subjects (p = .38). On the contrary, results showed that the key secondary endpoint – that is, change in eGFR from baseline to TEMPO 4:4 Month 24 – was achieved. The effect of tolvaptan in slowing eGFR decline was maintained for 2 more years in TEMPO 4:4 (3.15 mL/min/1.73 m2, p < 0.001). Moreover, eGFR slopes of early-treated subjects were non-inferior to those of delayed-treatment subjects, but TKV growth slopes failed to show such non-inferiority. Generally, the safety profile of tolvaptan in TEMPO 4:4 was similar to that of tolvaptan in TEMPO 3:4; 108 subjects withdrew from the study mostly due to adverse events (n=51; 16 early-treated subjects and 35 delayed-treated subjects). Aquaretic adverse events were the most common and occurred more frequently in the delayed-treatment group. Elevations in transaminase three times upper the normal limit occurred in 3.8% of delayed-treated vs 2.5% of early-treated subjects. This is similar to the rates observed in TEMPO 3:4, where such elevations occurred in 4.2% of tolvaptan-treated vs 1.6% of placebo-treated subjects. While the TKV endpoint did not reach its prespecified threshold for proof of disease-modifying effect, unlike the eGFR endpoint, this may be attributed to unforeseen limitations when the trial was designed, such as loss of randomization and baseline imbalances after TEMPO 3:4. It is important to mention that a post hoc analysis of data from TEMPO 4:4 showed that there is a possible relationship between genotype and response to tolvaptan. Subjects with truncating PKD1 mutation preserved tolvaptan treatment effect on both eGFR and TKV, contrary to a small number of subjects with non-truncating PKD1 mutation or PKD2 mutation who showed no significant effect on TKV at the end of TEMPO 3:4 and TEMPO 4:4. Similarly, subjects with CKD2–3 maintained the tolvaptan effect of eGFR and TKV in TEMPO 4:4, but those with CKD1 did not.53
Side effects
One of the reasons that tolvaptan has not yet been approved by some authorities as a treatment for ADPKD is its side effects. Serious adverse events were reported in the TEMPO trial, including a rise in serum alanine transaminase levels to 2.5 times the upper limit of the normal range, which occurred more frequently in those who received tolvaptan than in those who received placebo (4.9% vs 1.2%, respectively). Two of the patients who received tolvaptan had significant concurrent increases in the alanine aminotransferase or aspartate aminotransferase levels (greater than three times the upper limit of the normal range) and the bilirubin level (greater than two times the upper limit of the normal range), indicating that tolvaptan could lead to serious liver damage. However, for those two patients and for all the others, all abnormalities resolved spontaneously and returned to the baseline after the cessation of tolvaptan.48,54 Although hepatocellular damage following tolvaptan treatment was infrequent and reversible, the risk of potentially irreversible injury remains. For that reason, the US Food and Drug Administration (FDA) issued a statement55 warning about that risk and recommending that healthcare providers conduct liver tests promptly in tolvaptan-receiving patients who complain of symptoms indicating liver injury, and tolvaptan should promptly be discontinued. Other side effects included chest pain (0.8% in tolvaptan group vs 0.4% in placebo) and headache (0.5% in tolvaptan vs none in placebo), and the aquaretic side effects. In addition, certain electrolyte imbalances were documented in the trial, such as an increase in mean serum sodium levels to clinically significant levels (defined as a level greater than 150 mmol/L) in 4% of tolvaptan-treated subjects vs 1.4% in placebo, and an increase in uric acid levels and in frequency of gout of 2.9% in tolvaptan-treated subjects vs 1.4% in placebo.
Cost effectiveness
Data on the cost effectiveness of tolvaptan are very limited. Erickson et al,56 however, developed a decision–analytic model to study the cost effectiveness of tolvaptan for slowing the progression of ADPKD. They compared a case in which patients with ADPKD receive tolvaptan for life – assuming its clinical benefit persists for life – until their death, the development of ESRD or liver complications, to a case in which these patients received standard care. Measured outcomes included median age at onset of ESRD, life expectancy, and discounted quality-adjusted life years (QALYs). Results showed that tolvaptan therapy in 40-year-old men and women with ADPKD with an eGFR of 80 mL/min/1.73 m2 would prolong the median age of ESRD onset by 6.3 years in women and 6.8 years in men, increase life expectancy by 2.8 years in women and 2.3 years in men, and yield an increase of 1.2 discounted QALYs in women and 1.1 discounted QALYs in men. This finding suggests a remarkable health benefit if tolvaptan effectiveness can actually be sustained, as it would delay RRT and its huge cost, loss of QOL, and death associated with ESRD. The benefits, however, would come at a very high cost, US$5,760 per month and US$744,100 per QALY gained, compared to standard care, which is seven times the standard willingness-to-pay “WTP” threshold, which ranges from US$50,000–US$100,000 per QALY gained.57 These costs include medication, laboratory, and clinical follow-up. These results clearly showed that tolvaptan was not cost effective.
The cost, furthermore, depended on the eGFR at the time of initiation of tolvaptan and the rate of eGFR decline. The cost in 40-year-olds with an eGFR of 75 mL/min/1.73 m2 would be 16% lower (US$626,938 per QALY) than in 40-year-olds with an eGFR of 80 mL/min/1.73 m2. However, if tolvaptan was given to patients with slower rates of eGFR decline, it would cost US$1,215,200 per QALY gained – meaning that it would be less cost effective. Therefore, for tolvaptan to be cost effective and to meet a US$100,000 WTP threshold per QALY gained, cost would have to be reduced 94% to US$1,155 per month. Moreover, tolvaptan should be used exclusively in patients with ADPKD at a greater risk for a rapid decline in kidney function. Limitations of this study include the assumption of the sustainability of tolvaptan effectiveness observed in the TEMPO trial for life, and not modeling the costs of possible liver injury from tolvaptan or the indirect costs, such as those of caregivers.56,57
Approval of tolvaptan and use in current practice
Based on the results of the TEMPO trials, the regulatory authorities in Japan, Canada, Korea, Switzerland, and the European Medicines Agency (EMA) in Europe approved the use of tolvaptan for the treatment of ADPKD and have granted it marketing authorization. However, in the United States, the FDA has not approved the medication until recently and has requested more efficacy and safety data in a confirmatory trial (REPRISE).49 The approval of tolvaptan calls for the development of widely accepted treatment guidelines for using it in patients with ADPKD, and there are currently none.27,58
In Japan, the current recommendation for starting therapy with tolvaptan is a TKV of more than 750 mL and an annual TKV growth of more than 5% per year.59 In the EU, on behalf of the ERA-EDTA Working Groups of Inherited Kidney Disorders (WGIKD) and European Renal Best Practice (ERBP), Gansevoort et al58 provided guidance for making the decision as to when to start tolvaptan and which patients should be treated with tolvaptan. Tolvaptan – according to the EMA label – is indicated for slowing the progression of cyst growth and renal impairment in adult patients with ADPKD and CKD 1–3 at the time of initiation of treatment and with evidence of rapidly progressing disease. This guideline calls for clarifying the CKD stage and age at which tolvaptan can be started as well as the definition of rapidly progressive disease. Therefore, the statement provided a series of recommendations (Table 2), which resulted in a hierarchical decision algorithm (Figure 4) involving a sequence of the best validated markers for disease progression in descending order of reliability to identify patients with ADPKD with actual or likely rapid disease progression who are eligible for treatment with tolvaptan. Furthermore, this algorithm helps to screen out ineligible patients and identify those who have “possible” rapid disease progression, who should not be treated with tolvaptan, and should be revaluated for indications of treatment after 3–5 years. Gansevoort et al also emphasized the consideration of factors other than eligibility before the initiation of therapy, such as contraindications (Table 3), adverse events, patient motivation and lifestyle factors, and patient’s global risk profile and recommended remaining prudent and allowing some flexibility. This proposed algorithm, we believe, improves the benefit-to-risk ratio and cost effectiveness of this treatment, given its side effects and high costs.
 | Figure 4 Algorithm to assess indications for initiation of treatment in ADPKD. Notes: aIn our opinion, the indication ‘CKD stages 1–3 at initiation of treatment’ is not sufficiently specific as eGFR should be indexed for age. ADPKD patients with a high eGFR for age are unlikely to show rapid disease progression. There is currently no published evidence for the effect of tolvaptan in patients below the age of 18 or above the age of 50 years. beGFR may vary over time in individual patients, especially when close to the normal range. To confidently define ‘rapid disease pro- gression’, the rate of eGFR decline should be supported by multiple measurements that reliably indicate a rate of decline in eGFR. For this reason, this criterion should also be defined more strictly when historical data are available for only a short period compared with when available for a longer period. cWhen ‘evidence of rapid disease progression’ is based on historical eGFR data, the decline in renal function should be due to ADPKD and not related to other diseases, medications or factors that may contribute (reversibly or irreversibly) to a decline in renal function (e.g. diabetes mellitus, NSAIDs, calcineurin inhibitors, dehydration or contrast agents). dThe criterion decline in eGFR ≥5 mL/min/1.73 m2 in 1 year is adopted from the KDIGO CKD Guideline. eThe criterion decline in eGFR ≥2.5 mL/min/1.73 m2 per year over a period of 5 years is comparable to class 1C patients in the Mayo classification of ADPKD. fIn young ADPKD patients with CKD stage 1, the observation of ‘no change in eGFR’ in general is not considered a sensitive marker of slow disease progression, as eGFR often remains fairly stable during a prolonged period of time, whereas TKV increases steadily, suggesting disease progression. In such patients, changes in TKV and/or prediction models should be applied to assess historical or predicted disease progression. gThe criterion of increase in TKV ≥5% per year is likely to be conservative. It is based on the threshold defining the Mayo class 1C patients. This criterion has also been advocated by the Japanese regulatory authorities. The average rate of TKV growth in placebo-treated patients in the TEMPO 3:4 trial was 5.5% per year. hThe ellipsoid equation estimates TKV reliably when compared with classical volumetry. iThe Mayo classification of ADPKD is based on height-adjusted TKV indexed for age. It predicts that patients with class 1C, 1D and 1E have more rapid disease progression. A kidney length ≥16.5 cm, as assessed by ultrasound (or MRI), can be used in patients younger than 45 years to indicate a high likelihood of rapid disease progression. jThe PRO-PKD score suggests that patients with a truncating PKD1 mutation and early onset of clinical signs (i.e. hypertension, macroscopic hematuria, cyst infection or flank pain before the age of 35 years) have rapid disease progression with start of RRT at a relatively young age. kAlthough there is significant variability in the age of reaching ESRD within families that share the same mutation, clinical experience as well as observational studies have shown that a detailed family history can provide important information for risk prediction. Reproduced with permission from Gansevoort RT, Arici M, Benzing T, et al. Recommendations for the use of tolvaptan in autosomal dominant polycystic kidney disease: a position statement on behalf of the ERA-EDTA working groups on inherited kidney disorders and European renal best practice. Nephrol Dial Transplant. 2016;31(3):337–348. ©2016 Oxford University Press.58 Abbreviations: ADPKD, autosomal dominant polycystic kidney disease; CKD, chronic kidney disease; CT, computed tomography; eGFR, estimated glomerular filtration rate; ESRD, end-stage renal disease; PKD, polycystic kidney disease; MRI, magnetic resonance imaging; TKV, total kidney volume; (ht)TKV, height-adjusted total kidney volume. |
 | Table 2 Recommendations for the use of tolvaptan in ADPKD as retrieved from the Gansevoort et al statement58 Note: Reproduced with permission from Gansevoort RT, Arici M, Benzing T, et al. Recommendations for the use of tolvaptan in autosomal dominant polycystic kidney disease: a position statement on behalf of the ERA-EDTA working groups on inherited kidney disorders and European renal best practice. Nephrol Dial Transplant. 2016;31(3):337–348. ©2016 Oxford University Press.58 Abbreviations: ADPKD, autosomal dominant polycystic kidney disease; CKD, chronic kidney disease; eGFR, estimated glomerular filtration rate; ESRD, end-stage renal disease; PKD, polycystic kidney disease; MRI, magnetic resonance imaging; TKV, total kidney volume. |
 | Table 3 Contraindications for tolvaptan as retrieved from the Gansevoort et al statement derived from the EMA-approved label58 Note: Reproduced with permission from Gansevoort RT, Arici M, Benzing T, et al. Recommendations for the use of tolvaptan in autosomal dominant polycystic kidney disease: a position statement on behalf of the ERA-EDTA working groups on inherited kidney disorders and European renal best practice. Nephrol Dial Transplant. 2016;31(3):337–348. ©2016 Oxford University Press.58 Abbreviations: ADPKD, autosomal dominant polycystic kidney disease; EMA, European Medicines Agency. |
Considerations for future trials
There are a number of questions that can be raised concerning the benefit of tolvaptan that have not been answered yet. Does tolvaptan have different efficacy in different races? This question can be raised after reviewing the report of a subgroup analysis of the Japanese patient subset from the TEMPO 3:4 trial.60 Although tolvaptan exhibited the same efficacy in the Japanese population as in the total population in the TEMPO 3:4 trial in reducing the rate of ADPKD progression, there is no explanation for the difference of 1.5% in the annual rate of TKV growth between the total population and this subpopulation. The annual rate of TKV growth in tolvaptan-treated subjects was 2.8% in the total TEMPO 3:4 trial population, whereas it was 1.3% in the Japanese subpopulation. It is notable that Japanese patients had a lower baseline than that of the total patient population, and they also had a smaller body build (thinner and shorter) but received the same dose of tolvaptan as the total population in TEMPO 3:4; therefore, they might have been exposed to higher concentrations of the medication. In addition, the results may have been influenced by the sample size of the Japanese subpopulation, which was too small to approach statistical significance. The possibility of racial differences in the efficacy of tolvaptan should be considered in future trials to acquire more data concerning this matter.
As discussed earlier, ADPKD and ESRD have substantial impacts on patient QOL. Information about the actual magnitude of the benefits of tolvaptan (in terms of kidney pain) and information about the impact of adverse events on patient QOL is lacking. Therefore, future studies should include information about the effect of tolvaptan therapy on QOL in patients with ADPKD.
Additional studies should also be directed toward investigating the cost effectiveness of tolvaptan via modeling the cost that would meet the WTP threshold based on the estimated outcome benefit in large clinical intervention trials such as TEMPO.57
Other points that remain elusive and necessitate additional research include drug–drug interactions, the ideal dosing regimen that achieves maximal efficacy and minimal adverse events, whether the effect of tolvaptan differs between the two genotypes, and the possible synergistic effect when tolvaptan is co-administered with other medications such as mTOR inhibitors.
Conclusion
ADPKD is a slowly progressive structural cystic kidney disease that often leads to ESRD requiring RRT. The progressiveness of this disease and its complications cause debilitating effects on health of patients and impose a significant burden, both economically and on the QOL, of patients. Thus, patients should be treated holistically, paying particular attention to their psychosocial status. Recently, tolvaptan, which is a vasopressin V2 receptor antagonist, has been suggested by several studies as a promising agent. Patients who were treated with tolvaptan had a lower annual increase in total kidney volume, a slower rate of decline of kidney function, and prolonged life expectancy. However, there have been several side effects associated with the administration of tolvaptan, including elevated liver enzymes, headache, chest pain, and electrolyte imbalances. Data concerning the cost effectiveness of tolvaptan are limited, but one study regarding this matter concluded that it was not cost effective. Therefore, cost effectiveness, along with other points that remain elusive such as drug–drug interactions, the ideal dosing regimen, and the possible variation in efficacy among races, should be studied in large clinical trials such as TEMPO. As tolvaptan has currently been approved for use in several countries, there are different recommendations as to when to start treatment with tolvaptan. Therefore, widely accepted universal guidelines are needed.
Disclosure
The authors report no conflicts of interest in this work.
References
Walker BR, Colledge NR, Ralston S, Penman ID. Davidson’s Principles and Practice of Medicine. Edinburgh: Churchill Livingstone; 2014. | ||
Hogan MC, Torres V. Polycystic kidney disease. BMJ Best Practice. 2015. Available from: http://bestpractice.bmj.com/best-practice/monograph/481.html. Accessed March 20, 2016. | ||
Higgins JC, Fitzgerald JM. Evaluation of incidental renal and adrenal masses. Am Fam Phys. 2001;63(2):288–295. | ||
National Institute of Diabetes and Digestive and Kidney Diseases. Simple Kidney Cysts. Available from: http://www.niddk.nih.gov/health-information/health-topics/kidney-disease/kidney-cysts/Pages/facts.aspx#sup1. Accessed March 20, 2016. | ||
Torres VE, Bennett WM. Diagnosis of and screening for autosomal dominant polycystic kidney disease. Available from: http://www.uptodate.com/contents/diagnosis-of-and-screening-for-autosomal-dominant-polycystic-kidney-disease?source=see_link. Accessed March 14, 2016. | ||
Srivastava A, Patel N. Autosomal dominant polycystic kidney disease. Am Fam Phys. 2014;90(5):303–307. | ||
Baur BP, Meaney CJ. Review of tolvaptan for autosomal dominant polycystic kidney disease. Pharmacotherapy. 2014;34(6):605–616. | ||
Spithoven EM, Kramer A, Meijer E, et al. Renal replacement therapy for autosomal dominant polycystic kidney disease (ADPKD) in Europe: prevalence and survival—an analysis of data from the ERA-EDTA Registry. Nephrol Dial Transplant. 2014;29(Suppl 4):iv15–iv25. | ||
Chapman AB, Rahbari-Oskoui FF, Bennett WM. Renal manifestations of autosomal dominant polycystic kidney disease. Available from: http://www.uptodate.com/contents/renal-manifestations-of-autosomal-dominant-polycystic-kidney-disease#H9123853. Accessed March 14, 2016. | ||
Grantham JJ, Torres VE, Chapman AB, et al; CRISP Investigators. Volume progression in polycystic kidney disease. N Engl J Med. 2006;354(20):2122–2130. | ||
Torres VE, Harris PC. Autosomal dominant polycystic kidney disease: the last 3 years. Kidney Int. 2009;76(2):149–168. | ||
Torres VE, Harris PC, Pirson Y. Autosomal dominant polycystic kidney disease. Lancet. 2007;369(9569):1287–1301. | ||
Braun WE. Autosomal dominant polycystic kidney disease: emerging concepts of pathogenesis and new treatments. Cleve Clin J Med. 2009;76(2):97–104. | ||
Bennett WM, Torre VE. Extrarenal manifestations of autosomal dominant polycystic kidney disease. Available from: http://www.uptodate.com/contents/extrarenal-manifestations-of-autosomal-dominant-polycystic-kidney-disease#H9. Accessed March 14, 2016. | ||
Igarashi P, Somlo S. Genetics and pathogenesis of polycystic kidney disease. J Am Soc Nephrol. 2002;13(9):2384–2398. | ||
Pirson Y. Extrarenal manifestations of autosomal dominant polycystic kidney disease. Adv Chronic Kidney Dis. 2010;17(2):173–180. | ||
Rossetti S, Strmecki L, Gamble V, et al. Mutation analysis of the entire PKD1 gene: genetic and diagnostic implications. Am J Hum Genet. 2001;68(1):46–63. | ||
Magistroni R, He N, Wang K, et al. Genotype-renal function correlation in type 2 autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2003;14(5):1164–1174. | ||
Rossetti S, Chauveau D, Kubly V, et al. Association of mutation position in polycystic kidney disease 1 (PKD1) gene and development of a vascular phenotype. Lancet. 2003;361(9376):2196–2201. | ||
Chebib FT, Jung Y, Heyer CM, et al. Effect of genotype on the severity and volume progression of polycystic liver disease in autosomal dominant polycystic kidney disease. Nephrol Dial Transplant. 2016;31(6):952–960. | ||
Porath B, Gainullin VG, Cornec-Le GE, et al; Genkyst Study Group, HALT Progression of Polycystic Kidney Disease Group; Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease. Mutations in GANAB, encoding the glucosidase IIα subunit, cause autosomal-dominant polycystic kidney and liver disease. Am J Hum Genet. 2016;98(6):1193–1207. | ||
Ong AC, Harris PC. A polycystin-centric view of cyst formation and disease: the polycystins revisited. Kidney Int. 2015;88(4):699–710. | ||
Ferreira FM, Watanabe EH, Onuchic LF. Polycystins and molecular basis of autosomal dominant polycystic kidney disease. In: Li X, editor. Polycystic Kidney Disease [Internet]. Brisbane (AU): Codon Publications; 2015. Chapter 7. Available from: https://www.ncbi.nlm.nih.gov/books/NBK373394/. Accessed July 9, 2016. | ||
Pei Y, Watnick T, Bennett WM. Genetics of autosomal dominant polycystic kidney disease and mechanisms of cyst growth. Available from: http://www.uptodate.com/contents/genetics-of-autosomal-dominant-polycystic-kidney-disease-and-mechanisms-of-cyst-growth?source=see_lin. Accessed March 21, 2016. | ||
Eccles MR, Stayner CA. Polycystic kidney disease—where gene dosage counts. F1000Prime Rep. 2014;6:24. | ||
Chang MY, Ong AC. New treatments for autosomal dominant polycystic kidney disease. Br J Clin Pharmacol. 2013;76(4):524–535. | ||
Chapman AB, Rabari-Oskoui FF, Bennett WM. Course and treatment of autosomal dominant polycystic kidney disease. Available from: http://www.uptodate.com/contents/course-and-treatment-of-autosomal-dominant-polycystic-kidney-disease?source=search_result. Accessed March 25, 2016. | ||
Thong KM, Ong AC. The natural history of autosomal dominant polycystic kidney disease: 30-year experience from a single centre. QJM. 2013;106(7):639–646. | ||
Lee H, Manns B, Taub K, et al. Cost analysis of ongoing care of patients with end-stage renal disease: the impact of dialysis modality and dialysis access. Am J Kidney Dis. 2002;40(3):611–622. | ||
Glassock RJ, Winearls C. The global burden of chronic kidney disease: how valid are the estimates? Nephron Clin Pract. 2008;110(1):c39–c46. | ||
Smith DH, Gullion CM, Nichols G, Keith DS, Brown JB. Cost of medical care for chronic kidney disease and comorbidity among enrollees in a large HMO population. J Am Soc Nephrol. 2004;15(5):1300–1306. | ||
Zelmer JL. The economic burden of end-stage renal disease in Canada. Kidney Int. 2007;72(9):1122–1129. | ||
Kimmel PL, Patel SS. Quality of life in patients with chronic kidney disease: focus on end-stage renal disease treated with hemodialysis. Semin Nephrol. 2006;26(1):68–79. | ||
Valderrábano F, Jofre R, López-Gómez JM. Quality of life in end-stage renal disease patients. Am J Kidney Dis. 2001;38(3):443–464. | ||
Kimmel PL, Peterson RA. Depression in end-stage renal disease patients treated with hemodialysis: tools, correlates, outcomes, and needs. Semin Dial. 2005;18(2):91–97. | ||
Ong AC, Devuyst O, Knebelmann B, Walz G; ERA-EDTA Working Group for Inherited Kidney Diseases. Autosomal dominant polycystic kidney disease: the changing face of clinical management. Lancet. 2015;385(9981):1993–2002. | ||
Brunelli SM, Blanchette CM, Claxton AJ, Roy D, Rossetti S, Gutierrez B. End-stage renal disease in autosomal dominant polycystic kidney disease: a comparison of dialysis-related utilization and costs with other chronic kidney diseases. Clinicoecon Outcomes Res. 2015;7:65–72. | ||
Knight T, Schaefer C, Krasa H, Oberdhan D, Chapman A, Perrone RD. Medical resource utilization and costs associated with autosomal dominant polycystic kidney disease in the USA: a retrospective matched cohort analysis of private insurer data. Clinicoecon Outcomes Res. 2015;7:123–132. | ||
Lentine KL, Xiao H, Machnicki G, Gheorghian A, Schnitzler MA. Renal function and healthcare costs in patients with polycystic kidney disease. Clin J Am Soc Nephrol. 2010;5(8):1471–1479. | ||
de Barros BP, Nishiura JL, Heilberg IP, Kirsztajn GM. Anxiety, depression, and quality of life in patients with familial glomerulonephritis or autosomal dominant polycystic kidney disease. J Bras Nefrol. 2011;33(2):120–128. [Article in English, Portuguese] | ||
Tong A, Rangan GK, Ruospo M, et al. A painful inheritance-patient perspectives on living with polycystic kidney disease: thematic synthesis of qualitative research. Nephrol Dial Transplant. 2015;30(5):790–800. | ||
Simms RJ, Thong KM, Dworschak GC, Ong AC. Increased psychosocial risk, depression and reduced quality of life living with autosomal dominant polycystic kidney disease. Nephrol Dial Transplant. 2016;31(7):1130–1140. | ||
Miskulin DC, Abebe KZ, Chapman AB, et al; HALT-PKD Study. Health-related quality of life in patients with autosomal dominant polycystic kidney disease and CKD stages 1–4: a cross-sectional study. Am J Kidney Dis. 2014;63(2):214–226. | ||
Suwabe T, Ubara Y, Mise K, et al. Quality of life of patients with ADPKD-Toranomon PKD QOL study: cross-sectional study. BMC Nephrol. 2013;14:179. | ||
Halvorson CR, Bremmer MS, Jacobs SC. Polycystic kidney disease: inheritance, pathophysiology, prognosis, and treatment. Int J Nephrol Renovasc Dis. 2010;3:69–83. | ||
Torres VE, Harris PC. Strategies targeting cAMP signaling in the treatment of polycystic kidney disease. J Am Soc Nephrol. 2014;25(1):18–32. | ||
Higashihara E, Torres VE, Chapman AB, et al; TEMPOFormula and 156-05-002 Study Investigators. Tolvaptan in autosomal dominant polycystic kidney disease: three years’ experience. Clin J Am Soc Nephrol. 2011;6(10):2499–2507. | ||
Torres VE, Chapman AB, Devuyst O, et al; TEMPO 3:4 Trial Investigators. Tolvaptan in patients with autosomal dominant polycystic kidney disease. N Engl J Med. 2013;367(25):2407–2418. | ||
Boertien WE, Meijer E, de Jong PE, et al. Short-term effects of tolvaptan in individuals with autosomal dominant polycystic kidney disease at various levels of kidney function. Am J Kidney Dis. 2015;65(6):833–841. | ||
Torres VE, Higashihara E, Devuyst O, et al; TEMPO 3:4 Trial Investigators. Effect of tolvaptan in autosomal dominant polycystic kidney disease by CKD stage: results from the TEMPO 3:4 trial. Clin J Am Soc Nephrol. 2016;11(5):803–811. | ||
Torres VE, Chapman AB, Devuyst O, et al. Tolvaptan in later-stage autosomal dominant polycystic kidney disease. N Engl J Med. 2017;377(20):1930–1942. | ||
Gansevoort RT, Meijer E, Chapman AB, et al; TEMPO 3:4 Investigators. Albuminuria and tolvaptan in autosomal-dominant polycystic kidney disease: results of the TEMPO 3:4 Trial. Nephrol Dial Transplant. 2016;31(11):1887–1894. | ||
Torres VE, Chapman AB, Devuyst O, et al; TEMPO 4:4 Trial Investigators. Multicenter, open-label, extension trial to evaluate the long-term efficacy and safety of early versus delayed treatment with tolvaptan in autosomal dominant polycystic kidney disease: the TEMPO 4:4 Trial. Nephrol Dial Transplant. Epub 2017 Mar 31. | ||
Watkins PB, Lewis JH, Kaplowitz N, et al. Clinical pattern of tolvaptan-associated liver injury in subjects with autosomal dominant polycystic kidney disease: analysis of clinical trials database. Drug Saf. 2015;38(11):1103–1113. | ||
U.S. Food and Drug Administration. Samsca (tolvaptan): drug warning-potential risk of liver injury. [cited January 25, 2013]. Available from: http://www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm336669.htm?source=govdelivery. Accessed April 19, 2016. | ||
Erickson KF, Chertow GM, Goldhaber-Fiebert JD. Cost-effectiveness of tolvaptan in autosomal dominant polycystic kidney disease. Ann Intern Med. 2013;159(6):382–389. | ||
Nee R, Yuan CM, Abbott KC. Isn’t it ironic? Cost-effectiveness and willingness to pay for tolvaptan in the prevention of kidney failure in autosomal dominant polycystic kidney disease. Am J Kidney Dis. 2014;63(4):552–554. | ||
Gansevoort RT, Arici M, Benzing T, et al. Recommendations for the use of tolvaptan in autosomal dominant polycystic kidney disease: a position statement on behalf of the ERA-EDTA working groups on inherited kidney disorders and European renal best practice. Nephrol Dial Transplant. 2016;31(3):337–348. | ||
Horie S. Will introduction of tolvaptan change clinical practice in autosomal dominant polycystic kidney disease? Kidney Int. 2015;88(1):14–16. | ||
Muto S, Kawano H, Higashihara E, et al. The effect of tolvaptan on autosomal dominant polycystic kidney disease patients: a subgroup analysis of the Japanese patient subset from TEMPO 3:4 trial. Clin Exp Nephrol. 2015;19(5):867–877. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.