Back to Journals » International Journal of Nanomedicine » Volume 12
Humanized CD7 nanobody-based immunotoxins exhibit promising anti-T-cell acute lymphoblastic leukemia potential
Authors Yu Y, Li J, Zhu X ![]() , Tang X, Bao Y, Sun X, Huang Y, Tian F
, Tang X, Bao Y, Sun X, Huang Y, Tian F ![]() , Liu X, Yang L
, Liu X, Yang L ![]()
Received 13 November 2016
Accepted for publication 13 February 2017
Published 13 March 2017 Volume 2017:12 Pages 1969—1983
DOI https://doi.org/10.2147/IJN.S127575
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Linlin Sun
Yuan Yu,1–3 Jialu Li,1–3 Xuejun Zhu,4 Xiaowen Tang,2,5 Yangyi Bao,6 Xiang Sun,6 Yuhui Huang,1,2 Fang Tian,4 Xiaomei Liu,1,2 Lin Yang1–3
1The Cyrus Tang Hematology Center, 2Collaborative Innovation Center of Hematology, Soochow University, 3Suzhou Cancer Immunotherapy and Diagnosis Engineering Center, Suzhou, 4Central Laboratory, Department of Hematology, The Affiliated Hospital of Nanjing University of Chinese Medicine, Jiangsu Province Hospital of Traditional Chinese Medicine, Nanjing, 5Department of Hematology, The First Affiliated Hospital of Soochow University, Suzhou, 6Department of Hematology-Oncology, The First People’s Hospital of Hefei, Hefei, People’s Republic of China
Background: Nanobodies, named as VHHs (variable domain of heavy chain of HCAb [heavy-chain antibodies]), are derived from heavy-chain-only antibodies that circulate in sera of camelids. Their exceptional physicochemical properties, possibility of humanization, and unique antigen recognition properties make them excellent candidates for targeted delivery of biologically active components, including immunotoxins. In our previous efforts, we have successfully generated the monovalent and bivalent CD7 nanobody-based immunotoxins, which can effectively trigger the apoptosis of CD7-positive malignant cells. To pursue the possibility of translating those immunotoxins into clinics, we humanized the nanobody sequences (designated as dhuVHH6) as well as further truncated the Pseudomonas exotoxin A (PE)-derived PE38 toxin to produce a more protease-resistant form, which is named as PE-LR, by deleting majority of PE domain II.
Methods and results: Three new types of immunotoxins, dhuVHH6-PE38, dVHH6-PE-LR, and dhuVHH6-PE-LR, were successfully constructed. These recombinant immunotoxins were expressed in Escherichia coli and showed that nanobody immunotoxins have the benefits of easy soluble expression in a prokaryotic expression system. Flow cytometry results revealed that all immunotoxins still maintained the ability to bind specifically to CD7-positive T lymphocyte strains without binding to CD7-negative control cells. Laser scanning confocal microscopy revealed that these proteins can be endocytosed into the cytoplasm after binding with CD7-positive cells and that this phenomenon was not observed in CD7-negative cells. WST-8 experiments showed that all immunotoxins retained the highly effective and specific growth inhibition activity in CD7-positive cell lines and primary T-cell acute lymphoblastic leukemia (T-ALL) cells. Further in vivo animal model experiments showed that humanized dhuVHH6-PE38 immunotoxin can tolerate higher doses and extend the survival of NOD-Prkdcem26Il2rgem26Nju (NCG) mice transplanted with CEM cells without any obvious decrease in body weight. Further studies on NCG mice model with patient-derived T-ALL cells, dhuVHH6-PE38 treatment, significantly prolonged mice survival with ~40% survival improvement. However, it was also noticed that although dhuVHH6-PE-LR showed strong antitumor effect in vitro, its in vivo antitumor efficacy was disappointing.
Conclusion: We have successfully constructed a targeted CD7 molecule-modified nanobody (CD7 molecule-improved nanobody) immunotoxin dhuVHH6-PE38 and demonstrated its potential for treating CD7-positive malignant tumors, especially T-cell acute lymphoblastic leukemia.
Keywords: CD7, humanized nanobody, T-cell acute lymphoblastic leukemia, patient-derived xenograft model, recombinant immunotoxins, Pseudomonas exotoxin A
Introduction
T-cell acute lymphoblastic leukemia (T-ALL) is a highly invasive type of blood cancer that clinically presents primarily as infection, fever, anemia, or abnormal bleeding and often occurs in adults and children. It accounts for 25% of adult acute lymphocyte leukemia cases and 15% of pediatric acute lymphocyte leukemia cases.1 Currently, primary treatment interventions include enhanced chemotherapy,2 allogeneic hematopoietic stem cell transplantation,3 antiviral therapy,4 molecular targeted therapy,5 etc. However, because adult T-ALL patients acquire therapy resistance with elusive mechanisms, treatment effectiveness is limited.6 At the same time, human leukocyte antigen (HLA)-matching difficulties and graft-versus-host reactions present a huge challenge to allogeneic hematopoietic stem cell transplantation.7 In addition, pediatric acute lymphocyte leukemia often recurs, and the lasting remission rate of second-line chemotherapy after recurrence is <25%.8 Therefore, the search for new specific treatment targets for a targeted therapy of T-ALL is particularly urgent.
The molecule CD7 is the most sensitive antigen related to T-cells and is expressed in T-cell precursors, monocytes, and natural killer cells.9 Many research groups have reported that CD7 is highly expressed in T-ALL10 but that it is not expressed in at least one small group of normal T lymphocytes.11 In addition, when CD7 binds to antibodies or antibody derivatives, it is rapidly endocytosed into the cytoplasm.12 Therefore, CD7 is a particularly appropriate target antigen focused on the treatment of T-ALL. In light of these reasons, our laboratory used the CD7 molecule as a target antigen in previous work and screened nanobodies that bind to it with high affinity and specificity. In addition, using this as a foundation, nanobody recombinant immunotoxins were constructed to highly effectively kill T-cell lymphoblastic leukemia cells with CD7 molecular selectivity.
Nanobodies are a type of naturally existing single-domain functional proteins. Due to their small molecular weight,13 rapid tissue penetration,14 high solubility and stability,15 high specificity of antigen binding, and ability for large amounts to be synthesized,16 they are often used in cancer treatment. Including the structural recombinant immunotoxins,17 the therapeutic fusion proteins, they are are formed from the fusion of the antibody part that can specifically bind to tumor cell surface antigens and the part with cytotoxic activity.18
In our previous study, we fused an anti-CD7 nanobody that we screened with PE38, a 38 kDa-molecular weight truncated Pseudomonas exotoxin A (PE), to construct the univalent and divalent recombinant immunotoxins VHH6-PE38 and dVHH6-PE38.19 These two recombinant immunotoxins exhibited highly effective CD7 antigen-specific anti-leukemia ability, especially dVHH6-PE38, which could effectively induce apoptosis of patient-initiated T-ALL cells. In NOD/SCID mice transplanted with CEM cells, dVHH6-PE38 arrested the proliferation of tumor cells and significantly extended the survival of the mice. However, dVHH6-PE38 is still partially limited; for example, its antibody part is derived from camel and has not been humanized, and hence there is a risk that it will induce the production of neutralizing antibodies. In addition, because of the presence of lysosome-sensitive sites, its toxin part may be degraded by lysosomes during transport in the cytoplasm, thereby weakening its ability to kill tumor cells. These problems motivated us to further conduct a series of optimizations.
Based on the abovementioned considerations, we designed three modifications of dVHH6-PE38. They include a nanobody humanized version designated dhuVHH6-PE38, a lysosome-tolerant variant in which the lysosome-sensitive sites of domain II in PE38 were deleted designated as dVHH6-PE-LR, and a compatible variant with these two modifications designated as dhuVHH6-PE-LR. In addition, the in vitro and in vivo characteristics of these variants were studied.
Materials and methods
Plasmid, bacterial strain, and cells
Recombinant immunotoxins were inserted into the pET28a (Novagen) plasmid and expressed in BL21 Escherichia coli (DE3; Novagen) cells. Two human acute T-cell leukemia cell lines, CEM and Jurkat, and two human B-cell lymphoma cell lines, Raji and Ramos, purchased from American Type Culture Collection (ATCC), were cultured in RPMI-1640 medium (Hyclone) supplemented with 10% fetal bovine serum (Gibco). 293T-GFP and 293T-CD7-GFP cells, generated in our laboratory by stably transfecting 293T cells with GFP or both GFP and human CD7, respectively, were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) (Hyclone) containing 10% fetal bovine serum. 293T cell line was also purchased from ATCC. T-ALL patient sample cells were provided by the Department of Hematology at Jiangsu Hospital of Traditional Chinese Medicine with the approval of the Ethics Committee of Jiangsu Province Hospital of Traditional Chinese Medicine. Written informed consent was obtained before the experiment. Primary T-ALL cells were cultured in RPMI-1640 medium containing 20% fetal bovine serum. In addition, all cells used in this study were cultured in the presence of 0.1 mg/mL streptomycin and 100 U/mL penicillin (Gibco). All cells were incubated in an incubator maintained at 37°C and 5% CO2. In addition to the abovementioned cell lines or primary cells, other cell lines used in this study were all obtained with the review and approval of the Ethics Committee of Soochow University.
Strategy to humanize nanobody
The sequence of humanized VHH6 was designed based on the strategy suggested by Vincke et al.20 Briefly, the complementarity determining region (CDR) of VHH6 was linked to h-NbBcII10FGLA, a commonly used humanized nanobody scaffold, to create huVHH6, a chimera with specificity to the CD7 antigen. The general humanized nanobody scaffold h-NbBcII10FGLA referenced published research papers.14,20
Construction of immunotoxins
The pET28a-dVHH6-PE38 plasmid was previously constructed successfully in our laboratory.19 huVHH6, humanized VHH6, was synthesized by GENEWIZ Gene Company. Next, the sequence of huVHH6 was substituted for VHH6 to obtain the sequence of the pET28a-dhuVHH6-PE38 plasmid. The PE-LR sequence was synthesized by the same company. Similarly, the PE-LR fragment was substituted for the PE38 fragment of pET28a-dVHH6-PE38 and pET28a-dhuVHH6-PE38, yielding pET28a-dVHH6-PE-LR and pET28a-dhuVHH6-PE-LR. We also constructed a PE38-derived, CD7 unrelated immunotoxin, dVHH22-RIT, that did not bind to the CD7 molecule and served as the negative control. All of these plasmids were transfected into competent BL21 E. coli (DE3; Novagen) cells.
Expression and purification of immunotoxins
After recombinant plasmids were transfected into competent BL21 E. coli (DE3; Novagen) cells, the bacterial broth was incubated overnight in 4 mL lysogeny broth medium containing 100 μg/mL kanamycin at 37°C on a shaking bed. On the second day, the culture medium was transferred at a 1:100 ratio to fresh LB medium supplemented with 100 μg/mL kanamycin and expanded at 37°C on a shaking bed until the maximum absorbance of the culture medium at 600 nm reached 0.6–0.8. Next, the incubation temperature was reduced to 22°C, and expression was induced in the cells by addition of 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG; Calbiochem). Cells were concentrated 6–8 hours after induction. Collected bacterial cells were disrupted by ultrasound and centrifuged at 12,000 rpm for 30 minutes. The supernatant was purified using a nickel chelate affinity chromatography column (GE Healthcare Life Sciences) per the instructions provided by the manufacturer. Afterward, recombinant immunotoxins were placed in phosphate-buffered saline (PBS) overnight at 4°C to remove any remaining imidazole. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and Western blot analysis were performed using standard procedures. Recombinant immunotoxins were detected using Coomassie Brilliant Blue R250 (Amresco) or rabbit anti-His tag antibodies (CST). Horseradish peroxidase (HRP)-conjugated goat anti-rabbit antibodies (Cwbiotech) were used as secondary antibodies. Stop agent and Western chemiluminescence HRP substrate (Merck Millipore) were used for detection.
Flow cytometry
In order to analyze the binding selectivity of the recombinant immunotoxins, cells were incubated with 5 μg/mL of recombinant immunotoxins for 1 hour on ice. After washing twice, they were incubated again with Alexa Fluor 488-labeled anti-His tag antibodies (Qiagen). Finally, cells were analyzed using the FACS Calibur flow cytometer (BD Biosciences).
Internalization analysis
The endocytosis of recombinant immunotoxins was detected using laser scanning confocal microscopy. Coverslips were sterilized and coated with poly-D-lysine (PDL; Beyotime), and the slides were placed in a 24-well plate afterward. 293T-CD7-GFP and 293T-GFP cells were seeded in a 24-well plate and incubated overnight at 37°C. Recombinant immunotoxins were added to cells to a final concentration of 5 μg/mL and incubated for 4 hours at 37°C. Next, cells were washed once with PBS on ice. Recombinant immunotoxins bound to the surfaces of cells were washed away using 0.2 M glycine buffer (pH 2.5) and immediately neutralized using 0.5 M Tris–HCl (pH 7.4). Next, cells were fixed for 1 hour on ice using 4% paraformaldehyde (PFA). After one wash, cells were permeabilized for 5 minutes with 0.2% Triton X-100 and sealed with 2% bovine serum albumin (BSA) for 1 hour. Next, cells were stained overnight at 4°C with anti-His tag antibody labeled with Alexa Fluor 647 (Qiagen). After cells were washed three times, they were stained with 4′,6-diamidino-2-phenylindole (DAPI) for 5 minutes. After one more wash, the coverslips were mounted with 50% glycerin/PBS. Fluorescence was detected using a laser scanning confocal microscope (Olympus).
Cytotoxic activity assay
The 96-well plates were seeded with cells at a density of 1×104 per well. Next, different concentrations of recombinant immunotoxins were added to the cells and incubated for 72 hours. In the next step, 10 μL cell counting reagent (WST-8; Dojindo Molecular Technologies) was added to each sample well. The 96-well plate was incubated at 37°C until the wells got maximum absorbance value at 450 nm. For the blocking experiment, cells were pre-incubated with 100× molar excess of parental dVHH6 nanobody for 1 hour, and then different recombinant immunotoxins were added to a final concentration of 100 ng/mL. Other steps in the procedure were the same as previously described. For cytotoxicity studies in sample cells from one T-ALL patient, cells were seeded in 96-well plates at a density of 2×105 per well and treated with a series of concentrations of recombinant immunotoxins. T-ALL patient sample cells were also treated for 72 hours.
Leukemia cell tumor transplant model
A total of 2.2×106 CEM cells were resuspended in 100 μL PBS and then injected into the tail vein of 6- to 8-week-old female NOD-Prkdcem26Il2rgem26Nju mice (NCG; Laboratory Animal Center of Soochow University). The day on which cells were injected was designated day 0. After 1 week, recombinant immunotoxin was injected into the tail vein at the specified dose every other day for a total of four times. At later time points, the survival status of the mice was assessed. Animal studies were approved by the Institutional Animal Care and Use Committee of Soochow University. All animal experiments were performed following the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH, 8th edition, 2011). Mice were euthanized when their body weight decreased >10%.
Patient-derived xenograft (PDX) model
Primary T-ALL cells were collected and frozen from a 19-year-old T-ALL patient who has signed informed consent to participate in the study, with the approval of the Ethics Committees of Soochow University. The 1×107 sample cells were injected intravenously into NCG mice, and one moribund mouse was euthanized. After a red cells lysis procedure, the peripheral, bone marrow, and spleen T-ALL cells collected from the mouse were immunotyped by flow cytometry assay as nearly 100% CD7+ cells. To test the recombinant immunotoxins effect on the PDX model, we transplanted those T-ALL cells to 12 mice once again at the same dose. After 7 days, drugs were administered as described earlier.
Statistical analysis
Data were analyzed using GraphPad Prism 6. The Student’s t-tests were used for comparisons between groups in animal experiments. Differences with P-values <0.05 were considered statistically significant.
Results
Design and preparation of fusion toxins
With dVHH6-PE38 as a basis, we constructed three new types of nanobody immunotoxins (Figure 1). In dVHH6-PE-LR, the toxin part was replaced with PE-LR, which is a truncated PE38 with region II and region Ib removed, leaving only a furin protein cleavage site in region II and the entire sequence of region III.21 In dhuVHH6-PE38, we replaced the antibody part with a humanized version. In dhuVHH6-PE-LR, both the antibody and toxin parts were modified. In addition, the N terminals of all fusion proteins contained 6X His tags, and the C terminals have the KDEL sequence.
 | Figure 1 Design of the recombinant immunotoxins. |
These proteins were induced in E. coli. After ultrasonic disruption of the cultured bacteria, proteins were isolated by centrifugation. After centrifugation, the supernatant and pellet were electrophoresed in SDS-PAGE and dyed with Coomassie Brilliant Blue (Figure 2A). We found that all immunotoxins can be solubly expressed because the target bands can be clearly seen in the supernatant samples. Next, the recombinant immunotoxin in the supernatant was purified using nickel chelate affinity chromatography. Purity was determined by Coomassie Brilliant Blue dye and protein immunoblotting analysis. Figure 2B shows that all proteins had >90% purity. Immunoblotting experiments (Figure 2C) confirmed that each band in Figure 2B stained with Coomassie Brilliant Blue was exactly our target proteins.
 | Figure 2 Characterization of the immunotoxins. |
Testing binding specificity of the new types of immunotoxin
Flow cytometry analysis showed that the modified recombinant immunotoxins maintained the binding specificity of the parent immunotoxin type. In other words, these proteins can bind to CD7-positive CEM and Jurkat cells but cannot bind to CD7-negative Raji and Ramos cells (Figure 3). In addition, dVHH22-RIT, the control immunotoxin that does not recognize the CD7 molecule, does not bind with CD7-positive Jurkat cells (Figure S1), confirming that the immunotoxins we prepared have specificity for recognizing and binding to CD7-positive cells.
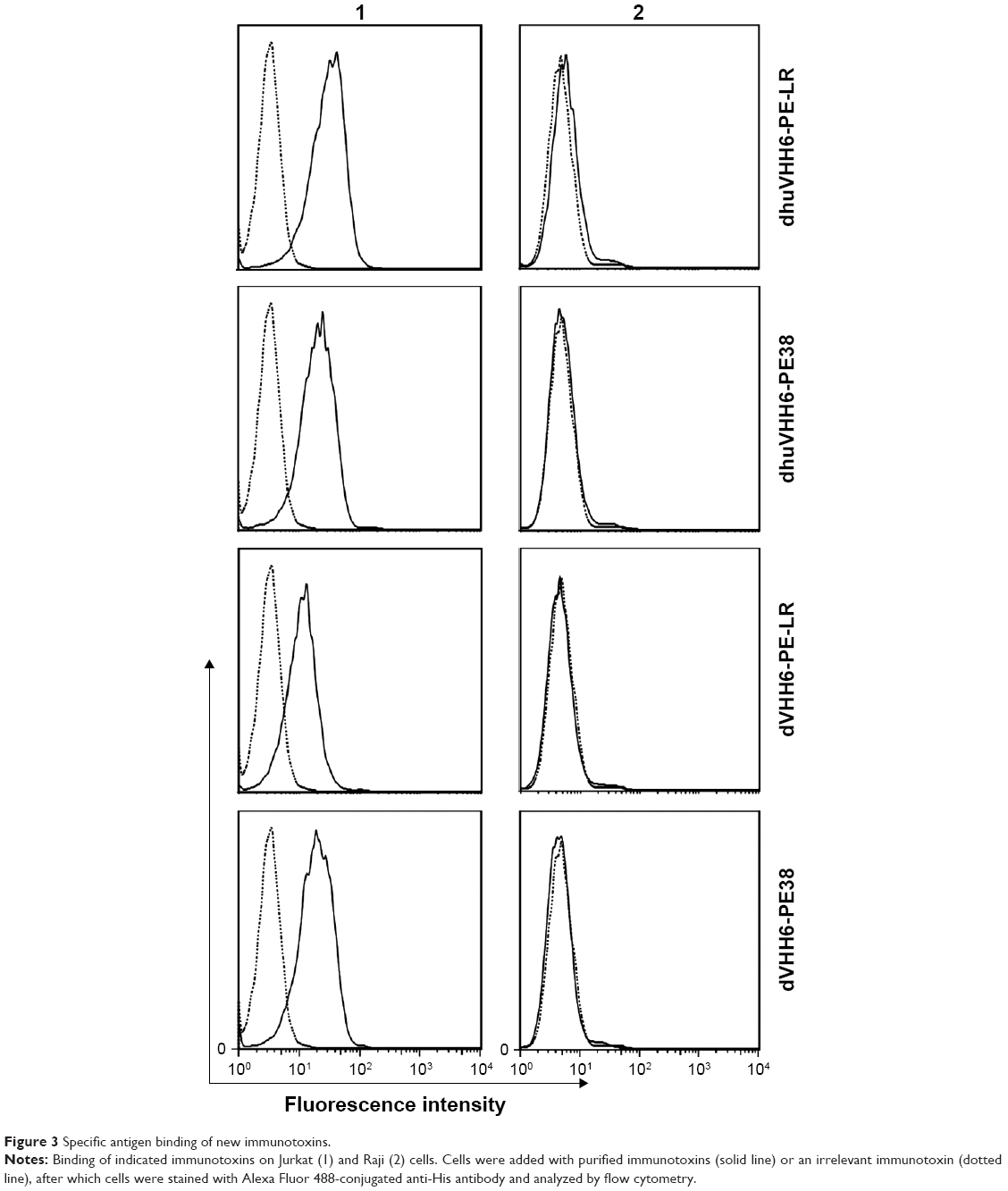 | Figure 3 Specific antigen binding of new immunotoxins. |
Recombinant immunotoxins can be endocytosed by cells
Binding with antigen targets is a necessary but not a sufficient condition for cytotoxicity of immunotoxins, and endocytosis into the target cell is also an important prerequisite for this ability.22 Therefore, in this study, we aimed to confirm that dVHH6-PE38 could be endocytosed into target cells. We used confocal microscopy images to directly observe their endocytosis. 293T-CD7-GFP and 293T-GFP cells were incubated at 37°C with the various recombinant immunotoxins for 4 hours, and the cells were permeabilized and stained with Alexa Fluor 647-labeled anti-His tag antibodies and DAPI dye. Green indicates cell membranes, cell nuclei are stained blue, and red indicates recombinant immunotoxin (antibody part). As shown in Figure 4, upon binding to each of the immunotoxins during incubation at 37°C, clusters of nanobodies (indicated by red arrowheads) appeared in the cytoplasm inside the cell membrane and outside the cell nuclei of CD7-positive 293T-CD7-GFP cells, whereas this phenomenon was not observed in CD7-negative 293T-GFP cells. This result shows that our recombinant immunotoxins were indeed endocytosed into the cytoplasm of CD7-positive cells, fulfilling the prerequisite condition for tumor cell cytotoxicity.
 | Figure 4 Internalization of immunotoxins by confocal fluorescence microscopy. |
Evaluation of recombinant immunotoxin cytotoxicity in cell lines
After confirming the specific binding and uptake capacity, WST-8 assay was used to determine the cytotoxic activity of immunotoxins. Experimental results showed that all the three modified recombinant immunotoxins can effectively restrain the proliferation of CD7-positive tumor cell lines in a dose-dependent manner (Figure 5A and B; Table 1), appearing that the half maximal effective inhibitory concentrations (EC50s) of them ranged from 3.311 to 8.374 ng/mL in the CEM cell line. In the Jurkat cell line, the EC50s were ranging from 5.998 to 17.750 ng/mL. Among these, the cytotoxicity of dhuVHH6-PE38 was the highest, with an EC50, respectively, 3.311 (CEM) and 5.998 ng/mL (Jurkat). We also assessed the in vitro cytotoxicity of dVHH6-PE38, which had an EC50 with 0.644 (CEM) and 2.977 ng/mL (Jurkat) as reported in our previous publication.19 Figure 5A and B also shows that the inhibition rate against positive cells of the two PE-LR forms of immunotoxin was generally slightly lower than that of the two PE38-type toxins. In addition, all the immunotoxins could not inhibit the growth of CD7-negative Raji and Ramos cells even at high concentrations (Figure 5C and D). After pre-incubation with an excess of the parental nanobody dVHH6, which was expected to block CD7 molecule on the cells, the cytotoxic activity of all of these immunotoxins against CEM cells was blocked (Figure S2). This further corroborates the finding that their cytotoxic activity is selective to the CD7 molecule.
 | Figure 5 Dose-dependent cytotoxicity of the fusion toxins in cell culture. |
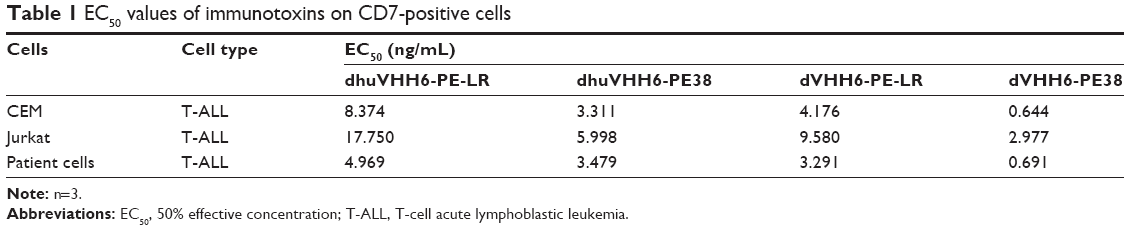 | Table 1 EC50 values of immunotoxins on CD7-positive cells |
Efficacy of modified recombinant immunotoxins in the NCG tumor transplant model
Considering the excellent in vitro cytotoxic activity of dhuVHH6-PE38 on CEM cells, we assessed its anti-leukemia activity in NCG mice transplanted with CEM cells (Figure 6). In the meantime, the in vivo antitumor effect of dhuVHH6-PE-LR was also evaluated. NCG mice were divided into four groups: a group injected with PBS, a group injected with dhuVHH6-PE38, a group injected with dhuVHH6-PE-LR, and a group injected with dVHH22-RIT (CD7 unrelated nanobody immunotoxin). The doses were adjusted so that mice were injected with 50 μg each time for a total of four times. The 2.2×106 CEM cells were injected into the mice via the tail vein on day 0, and mice were injected with PBS or protein drugs 7 days later. The reagents were injected via the tail vein every other day for a total of four times (Figure 6A).
 | Figure 6 In vivo evaluation of dhuVHH6-PE38 in CEM xenograft tumor model. |
Experimental results showed that the PBS-treated group of mice only survived an average of 12 days, the dhuVHH6-PE-LR-treated group had a moderately increased survival extension with a median survival of 16 days, whereas mice that received dhuVHH6-PE38 treatment survived significantly longer (median survival of 19 days). Not surprisingly, mice in the dVHH22-RIT group did not exhibit a clear increase in survival, similar to the PBS group (Figure 6B). This result shows that dhuVHH6-PE38 antitumor activity was effective in vivo and exhibited specificity. And comparing with dhuVHH6-PE-LR, the better antitumor effect of dhuVHH6-PE38 makes it deserving of further study in second animal model, such as a patient tumor-derived xenograft model.
The antitumor effect of immunotoxins against primary T-ALL sample
To assess the anti-leukemia potential of these therapeutic drugs on primary T-ALL samples, T-ALL patient sample cells with high expression of CD7 (CD7-positive rates shown in Figure S3A) were treated with increasing concentrations of each recombinant immunotoxin, including dhuVHH6-PE38, dhuVHH6-PE-LR, dVHH6-PE38, and dVHH6-PE-LR. Cytotoxicity was detected using the WST-8 reagent kit. Results showed that all four of these immunotoxins could effectively inhibit the proliferation of T-ALL cells from patients, with an inhibition rate as high as ~70%–80% (Figure 7). This inspiring discovery triggered us to investigate the in vivo activity of those immunotoxins against patient-derived T-ALL mouse model (Figure 8), especially for dhuVHH6-PE38 that has shown potent in vivo activities (Figure 6). The animal experiment scheme was the same as the design described earlier. Finally, in dhuVHH6-PE38-treated group, the last mouse was alive until 50 days after tumor inoculation (Figure 8B). Meanwhile, all the PBS-treated mice died by day 34 and all the dVHH22-RIT-injected mice died by day 35 after tumor transplantation. Slight weight loss was transiently observed for majority of mice, which fully recovered after the last injection of immunotoxin (Figure 8C). We also detected the CD7 expression rate of dhuVHH6-PE38-treated group and -untreated group at the end of treatment (Figure S3).
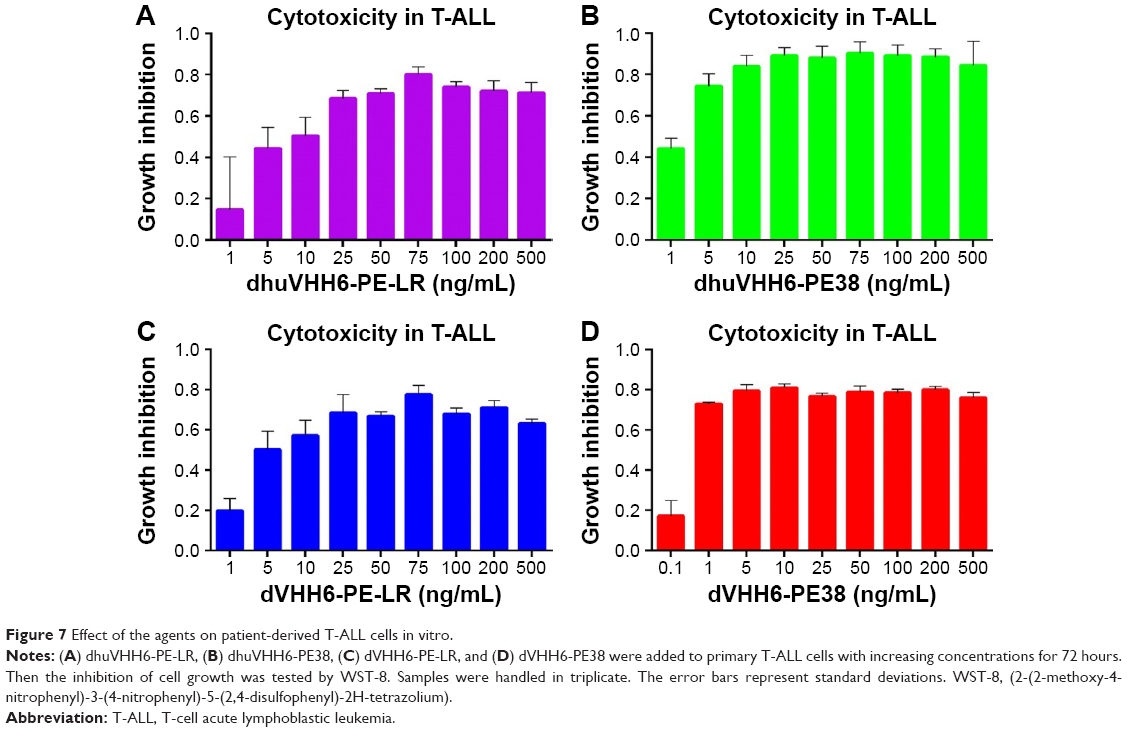 | Figure 7 Effect of the agents on patient-derived T-ALL cells in vitro. |
 | Figure 8 Antitumor activity of dhuVHH6-PE38 in PDX model. |
Discussion
The human CD7 molecule is a 40 kDa-molecular weight cell surface glycoprotein that belongs to the immunoglobulin superfamily.23 It is highly expressed in many human T-cell lymphoblastic leukemias and is negative for expression in ~9% of normal peripheral T-cells.24 There have already been many attempts to use the CD7 molecule as a target antigen as a research focus for the treatment of T-cell lymphoblastic leukemia, but many experiments have not obtained clinical approval because of the lack of sufficient efficacy or safety.
Our laboratory’s previous research on dVHH6-PE38 demonstrated its highly effective, CD7 molecule-specific antitumor activity both in vitro and in vivo, but because its antibody part is derived from camel and its toxin part has lysosome-sensitive sites, its clinical application is associated with some risks and limitations. In this study, we constructed a series of modified immunotoxins to reduce their immunogenic and nonspecific toxicity and tried to avoid lysosomal degradation during cytoplasm transport. In this way, the cytotoxicity of the protein toxin against tumor cells is maximized, and its cytotoxic efficacy is increased as well. Prokaryotic expression results showed that they can be solubly expressed in E. coli at excellent levels; 1 L of bacterial broth can yield ~5 mg of purified product. This greatly decreases the cost of our immunotoxin preparations, meaning that they have some commercial value as well as clinical application potential.
Flow cytometry results showed that these modifications still retained the binding specificity for the CD7 molecule and that the specificity was not lost due to humanization or lysosome tolerance modifications. In addition, one of the keys for successful immunotoxins is delivery into the cytoplasm by endocytosis after the part that targets the antigen binds to surface antigens of the tumor cell. After a series of catalytic reactions, the toxin (for example, PE), and the antibody part are separated, activated PE may be degraded by the lysosome or transported from the Golgi apparatus to the endoplasmic reticulum where it inhibits protein synthesis, resulting in cell death.25 Therefore, entry of the immunotoxin into the tumor cell by endocytosis is an important prerequisite for its cytotoxic function. Laser scanning confocal microscopy showed that our immunotoxins can be endocytosed into CD7-positive cells, fulfilling this prerequisite condition for cytotoxic activity against tumor cells. In addition, the immunotoxins cannot be endocytosed into CD7-negative cells, again confirming its specificity to the CD7 molecule.
With respect to cytotoxicity results of the immunotoxins to cell lines in vitro, the cytotoxic activity of dhuVHH6-PE38, dVHH6-PE-LR, and dhuVHH6-PE-LR against tumor cell lines was slightly lower compared to that of dVHH6-PE38. This means that each modification of the recombinant immunotoxins reduces cytotoxicity activity to some extent. Considering that the increase in cytotoxic activity of the recombinant immunotoxin was related to increased affinity,26 we propose that the increased EC50 of humanized recombinant immunotoxin is related to decreased affinity of its antibody part after humanization. In our previous work, we confirmed that the affinity of the divalent antibody was much higher than that of the monovalent antibody.19 We could also try to construct variant with higher valence, such as trivalent humanized nanobody immunotoxins, to solve the problem of affinity reduction due to humanization. Interestingly, the inhibition rate of the two PE-LR types of immunotoxins on positive cells is slightly lower than those of the two PE38 types of toxins. However, in previous studies, HA22-LR was found to have greater cytotoxic efficacy against B-cell tumor lines compared to HA22.21 If we assume that the PE toxin sensitivity sites of some T-cell lymphoblastic leukemia are in region II of the PE toxin, the removal of region II in PE-LR, along with the drawbacks of these sensitive sites, would ultimately result in PE-LR toxin having weakened cytotoxic activity again T-cell leukemia cells, though the actual reasons still require further investigation. However, in view of the results of this study, despite PE-LR being more tolerant to lysosomes, the improved cytotoxic efficacy of PE38 toxin against T-cell lymphoblastic tumors compared to PE-LR makes it more suitable for being the cytotoxic part of the recombinant immunotoxin. In addition, the specificity of the cytotoxic activity of these immunotoxins is limited to CD7-positive cells. This way, the CD7-negative T lymphocytes in the body avoid CD7 nanobody immunotoxins clearing all CD7-positive cells and causing a loss of immunity, and normal immune function is maintained in the body.
In vivo studies of the modified recombinant immunotoxins showed that consistent with the in vitro study, dhuVHH6-PE38 has more effective antitumor activity than dhuVHH6-PE-LR in vivo. Interestingly, during the entire course of dosing, dhuVHH6-PE38 did not result in any significant decrease in body weight in the mice, meaning that it is well tolerated by animals and representing an important step in clinical transformation of anti-CD7 nanobody recombinant immunotoxins. However, our recombinant immunotoxin dhuVHH6-PE38 still has some room for improvement; for example, human sFasL or sTRAIL27,28 or endogenous proteins to which T-cell leukemias are sensitive may be used as the toxin part, thereby constructing a completely humanized recombinant protein immunotoxin and removing immunogenicity as much as possible and increasing safety, especially when we observed a disappointing in vivo activity by dhuVHH6-PE-LR. In addition, as discussed previously, we can try to construct trivalent humanized nanobody immunotoxins, or increase the dose or frequency to achieve greater antitumor efficacy.
Previous drugs for antitumor therapy often exhibited significant efficacy against cell lines, but poor or no efficacy against cancer cells derived from patients. After treating primary T-ALL cells with a 99.43% CD7-positive rate with our immunotoxins in vitro, we achieved a growth inhibition rate of ~70%–80%. The in vivo anti-leukemic evaluation of dhuVHH6-PE38 in NCG mice xenotransplanted with primary T-ALL cells was also performed. The group treated with dhuVHH6-PE38 exhibited a dramatic overall survival increase compared with the PBS and another negative control dVHH22-RIT-administered group, demonstrating great value for future clinical application.
This inspiring result motivated us to gather more patient samples with different CD7 expression levels and study the efficacy of treatment with our nanobody immunotoxins, laying the foundation for clinical transformation of this new immunotoxin.
Conclusion
In this study, we constructed three modifications of a nanobody recombinant immunotoxin and studied their functional characteristics. All of these modified immunotoxins maintained binding specificity for the CD7 molecule and the ability to be endocytosed into the cytoplasm after binding, and could significantly inhibit the growth of the CD7-positive cell lines Jurkat and CEM as well as T-ALL patient sample cells. Especially with the humanized nanobody immunotoxin dhuVHH6-PE38, the survival of leukemia model mice treated with this immunotoxin was extended effectively without any significant decrease in body weight. Therefore, dhuVHH6-PE38 may be a promising choice for future pre-clinical and clinical research for the treatment of CD7-positive T-ALL.
Acknowledgments
We would like to give thanks for the contributions of Dr Jingle Tang for screening and identifying CD7 nanobodies, Huiming Meng and Gangli An for providing the CEM and Jurkat cell lines, Pengjun Jiang for preparing the primary T-ALL cells, Xichen Zheng for managing the animal models, and Fengtao You and Bozhen Zhang for proof reading and modifying the manuscript. This work was supported by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), the National Natural Science Foundation of China (Grant No 31471283), and the Collaborative Innovation Major Project (Grant No XYXT2015304).
Disclosure
Lin Yang is a co-founder of PersonGen BioTherapeutics Co., Ltd. which focuses on R&D of CAR-T and CAR-NK technologies and translating them into clinics. The other authors report no conflicts of interest in this work.
References
Durinck K, Goossens S, Peirs S, et al. Novel biological insights in T-cell acute lymphoblastic leukemia. Exp Hematol. 2015;43(8):625–639. | ||
Utsunomiya A, Choi I, Chihara D, Seto M. Recent advances in the treatment of adult T-cell leukemia-lymphomas. Cancer Sci. 2015;106(4):344–351. | ||
Lin HN, Liu CY, Pai JT, et al. How to predict the outcome in mature T and NK cell lymphoma by currently used prognostic models? Blood Cancer J. 2012;2:e93. | ||
Kchour G, Tarhini M, Kooshyar MM, et al. Phase 2 study of the efficacy and safety of the combination of arsenic trioxide, interferon alpha, and zidovudine in newly diagnosed chronic adult T-cell leukemia/lymphoma (ATL). Blood. 2009;113(26):6528–6532. | ||
Zain JM, O’Connor O. Targeted treatment and new agents in peripheral T-cell lymphoma. Int J Hematol. 2010;92(1):33–44. | ||
Habiel DM, Krepostman N, Lilly M, et al. Senescent stromal cell-induced divergence and therapeutic resistance in T cell acute lymphoblastic leukemia/lymphoma. Oncotarget. 2016;7(50):83514–83529. | ||
Yonekura K, Utsunomiya A, Takatsuka Y, et al. Graft-versus-adult T-cell leukemia/lymphoma effect following allogeneic hematopoietic stem cell transplantation. Bone Marrow Transplant. 2008;41(12):1029–1035. | ||
Yadav BD, Samuels AL, Wells JE, et al. Heterogeneity in mechanisms of emergent resistance in pediatric T-cell acute lymphoblastic leukemia. Oncotarget. 2016;7(37):58728–58742. | ||
Eto T, Akashi K, Harada M, et al. Biological characteristics of CD7 positive acute myelogenous leukaemia. Br J Haematol. 1992;82(3):508–514. | ||
Venditti A, Del Poeta G, Buccisano F, et al. Prognostic relevance of the expression of Tdt and CD7 in 335 cases of acute myeloid leukemia. Leukemia. 1998;12(7):1056–1063. | ||
Reinhold U, Abken H, Kukel S, et al. CD7-T cells represent a subset of normal human blood lymphocytes. J Immunol. 1993;150(5):2081–2089. | ||
Pauza ME, Doumbia SO, Pennell CA. Construction and characterization of human CD7-specific single-chain Fv immunotoxins. J Immunol. 1997;158(7):3259–3269. | ||
Muyldermans S. Nanobodies: natural single-domain antibodies. Annu Rev Biochem. 2013;82:775–797. | ||
Vaneycken I, Govaert J, Vincke C, et al. In vitro analysis and in vivo tumor targeting of a humanized, grafted nanobody in mice using pinhole SPECT/micro-CT. J Nucl Med. 2010;51(7):1099–1106. | ||
Dumoulin M, Conrath K, Van Meirhaeghe A, et al. Single-domain antibody fragments with high conformational stability. Protein Sci. 2002;11(3):500–515. | ||
Revets H, De Baetselier P, Muyldermans S. Nanobodies as novel agents for cancer therapy. Expert Opin Biol Ther. 2005;5(1):111–124. | ||
Li T, Qi S, Unger M, et al. Immuno-targeting the multifunctional CD38 using nanobody. Sci Rep. 2016;6:27055. | ||
Liu W, Onda M, Kim C, et al. A recombinant immunotoxin engineered for increased stability by adding a disulfide bond has decreased immunogenicity. Protein Eng Des Sel. 2012;25(1):1–6. | ||
Tang J, Li J, Zhu X, et al. Novel CD7-specific nanobody-based immunotoxins potently enhanced apoptosis of CD7-positive malignant cells. Oncotarget. 2016;7(23):34070–34083. | ||
Vincke C, Loris R, Saerens D, Martinez-Rodriguez S, Muyldermans S, Conrath K. General strategy to humanize a camelid single-domain antibody and identification of a universal humanized nanobody scaffold. J Biol Chem. 2009;284(5):3273–3284. | ||
Weldon JE, Xiang L, Chertov O, et al. A protease-resistant immunotoxin against CD22 with greatly increased activity against CLL and diminished animal toxicity. Blood. 2009;113(16):3792–3800. | ||
Fuchs H, Weng A, Gilabert-Oriol R. Augmenting the efficacy of immunotoxins and other targeted protein toxins by endosomal escape enhancers. Toxins. 2016;8(7):E200. | ||
Aruffo A, Seed B. Molecular cloning of two CD7 (T-cell leukemia antigen) cDNAs by a COS cell expression system. EMBO J. 1987;6(11):3313–3316. | ||
Moll M, Reinhold U, Kukel S, et al. CD7-negative helper T cells accumulate in inflammatory skin lesions. J Invest Dermatol. 1994;102(3):328–332. | ||
Alewine C, Hassan R, Pastan I. Advances in anticancer immunotoxin therapy. Oncologist. 2015;20(2):176–185. | ||
Bera TK, Onda M, Brinkmann U, Pastan I. A bivalent disulfide-stabilized Fv with improved antigen binding to erbB2. J Mol Biol. 1998;281(3):475–483. | ||
Bremer E, ten Cate B, Samplonius DF, de Leij LF, Helfrich W. CD7-restricted activation of Fas-mediated apoptosis: a novel therapeutic approach for acute T-cell leukemia. Blood. 2006;107(7):2863–2870. | ||
Bremer E, Samplonius DF, Peipp M, et al. Target cell-restricted apoptosis induction of acute leukemic T cells by a recombinant tumor necrosis factor-related apoptosis-inducing ligand fusion protein with specificity for human CD7. Cancer Res. 2005;65(8):3380–3388. |
Supplementary materials
 | Figure S1 Characterization of dVHH22-RIT. |
 | Figure S2 A block experiment by dVHH6 using WST8 assays. |
 | Figure S3 CD7-positive rate of primary T-ALL. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.