Back to Journals » Journal of Blood Medicine » Volume 9
Genotype–phenotype correlation among beta-thalassemia and beta-thalassemia/HbE disease in Thai children: predictable clinical spectrum using genotypic analysis
Authors Traivaree C ![]() , Monsereenusorn C
, Monsereenusorn C ![]() , Rujkijyanont P
, Rujkijyanont P ![]() , Prasertsin W, Boonyawat B
, Prasertsin W, Boonyawat B ![]()
Received 8 December 2017
Accepted for publication 15 January 2018
Published 10 April 2018 Volume 2018:9 Pages 35—41
DOI https://doi.org/10.2147/JBM.S159295
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Chanchai Traivaree,1 Chalinee Monsereenusorn,1 Piya Rujkijyanont,1 Warakorn Prasertsin,2 Boonchai Boonyawat3
1Division of Hematology/Oncology, Department of Pediatrics, Phramongkutklao Hospital and College of Medicine, Bangkok, Thailand; 2Department of Pediatrics, Queen Savang Vadhana Memorial Hospital, Chonburi, Thailand; 3Division of Genetics, Department of Pediatrics, Phramongkutklao Hospital and College of Medicine, Bangkok, Thailand
Introduction: Beta-thalassemia is a group of inherited hemolytic anemias and one of the most common genetic disorders in Thailand. The clinical spectrum of beta-thalassemia disease ranges from mild to severe clinical symptoms including mild beta-thalassemia intermedia (TI) and severe beta-thalassemia major (TM).
Objective: This study aimed to determine the correlation between beta-globin gene (HBB) mutations and their phenotypic manifestations by evaluating patients’ clinical characteristics, transfusion requirements, growth and hematologic parameters, and hemoglobin typing among pediatric patients treated at Phramongkutklao Hospital.
Materials and methods: Seventy beta-thalassemia patients, including 63 with beta-thalassemia/hemoglobin E (HbE) and 7 with either homozygous or compound heterozygous beta-thalassemia, were enrolled in this study. Their clinical presentation, growth parameters and laboratory findings were reviewed and analyzed. The mean follow-up time was 10.52±5.62 years. Mutation analysis in each individual was performed using multiplex amplification refractory mutation system (M-ARMS), direct DNA sequencing of beta-globin gene and gap PCR for 3.4 kb deletion detection.
Results: All 7 homozygous and compound heterozygous beta-thalassemia patients were classified in TM. Among 63 patients with beta-thalassemia/HbE, 58 were classified in TM and 4 were classified in TI. Mean age at diagnosis was 0.8±0.49 years for homozygous or compound heterozygous beta-thalassemia and 3.43±3.5 years for beta-thalassemia/HbE. The most common HBB mutation was HBB:c.126_129delCTTT [codon 41/42 (-TCTT)] found in 34 alleles (48.6%). The height for age was also lower in homozygous beta-thalassemia patients (<3rd percentile) compared to compound heterozygous beta-thalassemia patients (25–50th percentile).
Conclusion: This study revealed a genotype–phenotype correlation of the most prevalent beta-thalassemia in Thai children using diagnostic capacity in genotypic analysis of HBB mutation. Our findings can provide a better prediction of clinical manifestation and severity by early identification of the type of the HBB mutations.
Keywords: beta-thalassemia disease, hemoglobinopathy, mutation, clinical, children
Introduction
Thalassemia is a group of inherited hemolytic anemias, including alpha-thalassemia and beta-thalassemia, caused by reduced or absent synthesis of the globin chains of hemoglobin (Hb), leading to imbalance of the globin chains. Beta-thalassemia is caused by mutations in the beta-globin gene (HBB) on chromosome 11 which is characterized by reduction of the beta-globin chain. Hemoglobin E (HbE), which is one the most common HBB gene mutations in Southeast Asia, can present with either a hemoglobinopathy or a thalassemic phenotype associated with diverse clinical manifestations. The clinical diversity of beta-thalassemia disease includes chronic hemolytic anemia, hepatosplenomegaly, failure to thrive and other complications that can be lethal during childhood if not treated appropriately.1 The clinical and hematological spectrum of beta-thalassemia disease ranges from mild to clinically manifested conditions including beta-thalassemia major and beta-thalassemia intermedia. Therefore, determination of factors causing such a diverse clinical presentation has clinical significance, and the reason for such diversity is the variety of mutations.2 The molecular basis of beta thalassemia has been studied worldwide. Most beta-thalassemia mutations are caused by point mutations, small deletions or insertions within the coding regions and the exon-intron junctions. In Thailand, the prevalence of beta-thalassemia and HbE carriers were 3%–9% and 13%–50%, respectively.3 At present, more than 30 different mutations have been identified.4 A number of studies revealed genotype–phenotype correlations of beta-thalassemia and beta-thalassemia/HbE in various populations.5,6 The aim of our study was to investigate phenotypic manifestations of these mutations by studying their hematologic parameters, hemoglobin typing, age at presentation, transfusion requirement, age and height for age in both homozygous and compound heterozygous forms of beta-thalassemia. Our data will show the genotype–phenotype correlation of each mutation, including laboratory biomarkers, and genetic profiles in patients with beta-thalassemia and beta-thalassemia/HbE who were followed up in Phramongkutklao Hospital, a tertiary care center for thalassemia patients from all regions in Thailand.
Materials and methods
Patient selection
Seventy beta-thalassemia patients who attended the Hematology Clinic at the Department of Pediatrics, Phramongkutklao Hospital, Bangkok, Thailand were enrolled in our study. Written informed consent and assent forms were obtained from all participants; for children, their parents or guardians provided written informed consent prior to enrollment in the study. The study protocol was approved by the Institutional Review Board of Phramongkutklao Hospital and Phramongkutklao College of Medicine, Bangkok, Thailand, and followed the ethical principles of the Declaration of Helsinki of 1975 and its revision. These patients include 63 with beta-thalassemia/HbE and 7 with homozygous or compound heterozygous beta-thalassemia. All patients were diagnosed at 18 years of age or less. Patients with homozygous beta-thalassemia and beta-thalassemia/HbE were clinically classified into severe transfusion dependent thalassemia major and thalassemia intermedia based on criteria such as age at presentation, average Hb level at the steady state and transfusion frequency history, as previously described.7 There were 45 males and 25 females aged 1.11–25.70 years (median age 15.4 years). A complete clinical history was recorded along with blood transfusion events. Patients were also examined for growth parameters.
Hematological and biochemical parameters
Hematological analyses were carried out using Coulter HMX Automated Hematology Analyzer (Beckman Coulter Corporation, Brea, CA, USA). Hemoglobin profiles and fetal hemoglobin (HbF) concentration were determined using Capillary Electrophoresis (CE, Minicap system, Sebia, Parc Technologique Leonard de Vinci, Evry Cedex, France), as described.
Genetic analysis
After informed consent was obtained, a total of 70 peripheral blood EDTA samples from all individuals were collected. Genomic DNA was extracted from peripheral blood lymphocytes using a standard protocol. First, the HBB gene mutations were characterized by multiplex amplification refractory mutation system (M-ARMS) to detect seven common mutations in Thailand including HBB:c.126_129delCTTT [codon 41/42 (-TCTT)], HBB:c.52A>T [codon 17 (A>T)], HBB:c.-78A>G [nt -28 (A>G)], HBB:c.316-197C>T [IVS-II-654 (C>T)], HBB:c.216_217insA [codon 71/72 (+A)], HBB:c.92+1G>T [IVS-I-1 (G>T)] and HBB:c.92+5G>C [IVS-I-5 (G>C)] as previously described.8 Second, unknown HBB gene mutations were further characterized by direct DNA sequencing of all 3 exons and exon–intron junctions to detect uncommon mutations according to protocols previously described elsewhere.9 Finally, uncharacterized beta-thalassemia alleles by M-ARMS and sequencing methods were subsequently screened by gap-PCR to detect 3.4 kb deletion which was previously reported in Thai populations.10
Statistical analysis
Baseline values of selected variables were calculated as the mean, median, and average according to percentile. Distribution of the quantitative variables was analyzed using Shapiro–Wilk test. Continuous variables were compared between two groups using the unpaired t-test for data with a parametric distribution and the Mann–Whitney U-test for non-parametric distribution. The chi-square test and Fisher exact test were used to analyze the categorical variables for data with a parametric distribution and non-parametric distribution, respectively. Statistical Package for the Social Science (SPSS) version 23 software (IBM Corporation, Armonk, NY, USA) and p-values <0.05 was considered significant.
Results
Patient characteristics
A total of 70 patients were enrolled in our study, including 63 beta-thalassemia/HbE patients (41 boys and 22 girls) and 7 patients with either homozygous or compound heterozygous beta-thalassemia (5 boys and 2 girls). The average age at diagnosis was 0.8±0.49 years for homozygous or compound heterozygous beta-thalassemia and 3.43±3.5 years for beta-thalassemia/HbE. There were statistical significant between two groups (p-value <0.001). The follow-up time for patients was approximately 10.52±5.62 years with ranges from 0.4–21.70 (median age 9.75 years). The splenectomy was done for 5 beta-thalassemia patients (71.4%) and 15 beta-thalassemia/HbE patients (25%). There were statistical significant between two groups (p-value 0.021). The regular blood transfusion was approximately 11.83±2.19 times/year in beta-thalassemia disease and 9.67±4.03 times/year in beta-thalassemia/HbE disease. There were no statistical significant between two groups (p-value 0.281). The current average age, height, weight and age at splenectomy are shown in Table 1.
 | Table 1 Patient characteristics Notes: Data shown as mean±SD or number (%). p-value from the t-test or chi-square test for data with a parametric distribution and the Mann–Whitney U-test or Fisher exact test for non-parametric distribution. p<0.05 = statistically significant. |
Genotypic data
Excluding HbE mutation, 9 HBB gene mutations were found. The most common HBB gene mutation was HBB:c.126_129delCTTT [codon 41/42 (-TCTT)] found in 34 alleles (48.6%), followed by codon 17 (A>T)] found in 16 alleles (22.8%), HBB:c.316-197C>T [ IVS-II-654 (C>T)] found in 5 alleles (7.1%) and HBB:c.92+5G>C [IVS-I-5 (G>C)] found in 7 alleles (10%). Less common HBB gene mutations included HBB:c.108C>A [codon 35 (C>A)], HBB:c.216_217insA [codon 71/72 (+A)], HBB:c.59A>G [codon 19 (A>G) or Hb Malay], HBB:c.92+1G>T [IVS-I-1 (G>T)], HBB:c.-78A>G [Int -28 (A>G)], and 3.4 kb deletion, accounting for 11.5% of all HBB gene mutations (Table 2). In 7 patients who were diagnosed beta-thalassemia disease, we found 2 homozygous of HBB:c.126_129delCTTT, 2 homozygous of HBB:c.52A>T and 3 compound heterozygous of HBB:c.126_129delCTTT and HBB:c.92+5G>C, HBB:c.216_217insA and HBB:c.108C>A and HBB:c.126_129delCTTT and HBB:c.2T>G [initial codon (ATG>AGG)], respectively.
 | Table 2 The distribution of beta-globin gene (HBB) mutations |
Hematologic and genotypic data
For beta-thalassemia/HbE, the data of 63 patients showed RBC count, hematocrit, and erythrocyte indices (MCV, MCH, MCHC) were decreased at first initial diagnosis, as shown in Table 3. The pattern of fetal hemoglobin was similar for each mutation except for Hb Malay, for which Hb F was 15.9%. For homozygous/compound heterozygous beta-thalassemia diseases, there were no differences in erythrocyte indices and fetal hemoglobin between two groups.
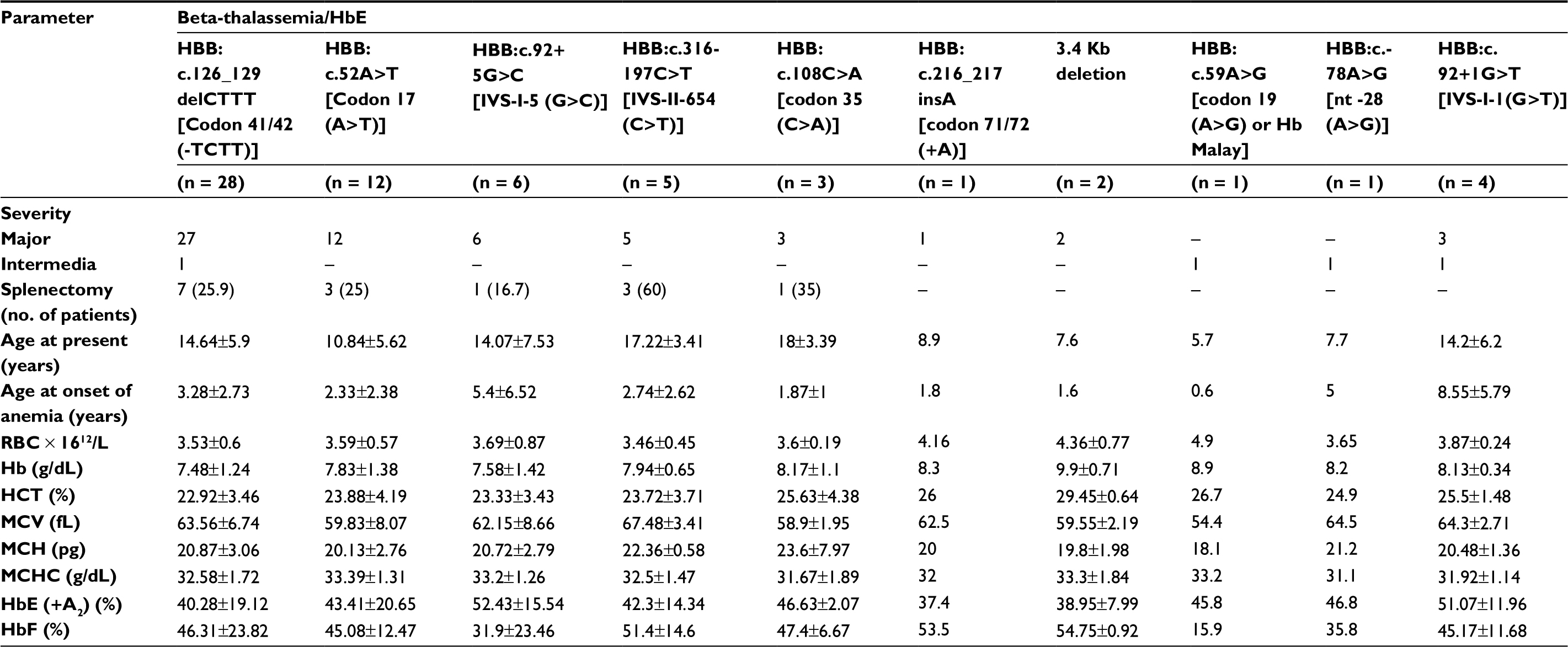 | Table 3 Genotype and hematologic data of beta-thalassemia/HbE patients Note: Data shown as mean±SD or number (%). Abbreviations: Hb, hemoglobin; HbE (+A2), hemoglobin E with hemoglobin A2; HbF, fetal hemoglobin.; HCT, hematocrit; MCH, mean corpuscular hemoglobin; MCHC, mean corpuscular hemoglobin concentration; MCV, mean corpuscular volume; RBC, red blood cells |
Genotypic and phenotypic correlation
The genotype and phenotype correlation was studied in our patients. All homozygous and compound heterozygous beta-thalassemia patients were in thalassemia major. Sixty-three beta-thalassemia/HbE patients were classified in 58 thalassemia major and 4 thalassemia intermedia (Table 3). The HBB:c.126_129delCTTT mutation was noted in 27 beta-thalassemia/HbE patients which was divided into 26 thalassemia major patients and 1 thalassemia intermedia patient. Interestingly, one thalassemia intermedia patient had hemoglobin F 20.8%, which was lower than the average numbers of thalassemia major patients (46.31±23.82%). Further evaluation on this thalassemia intermedia patient also found concomitant alpha-globin gene mutation (Hemoglobin Constant Spring, Hb CS) which could explain the lower hemoglobin F level and less severe clinical symptoms in this patient. The HBB:c.92+1G>T mutation was found in 4 beta-thalassemia/HbE patients: 3 thalassemia major patients and 1 thalassemia intermedia patient. The 3.4 kb deletion was found in 2 beta-thalassemia/Hb E patients and both of them had clinical severity as thalassemia major. The HBB:c.-78A>G and HBB:c.59A>G were identified and both patients were classified as thalassemia intermedia.
The heights for age were also lower in homozygous beta-thalassemia patients (<3rd percentile) compared to compound heterozygous beta-thalassemia patients (25–50th percentile). Comparisons of height for age in beta-thalassemia/HbE patients among different HBB gene mutations were performed and it was found that 14.3% (4 of 28) of patients with HBB:c.126_129delCTTT; 16.7% (2 of 12) of patients with HBB:c.52A>T; 16.6% (1 of 6) of patients with HBB:c.92+5G>C; and 20% (1 of 5) of patients with HBB:c.316-197C>T had height for age lower than the 3rd percentile (Figure 1).
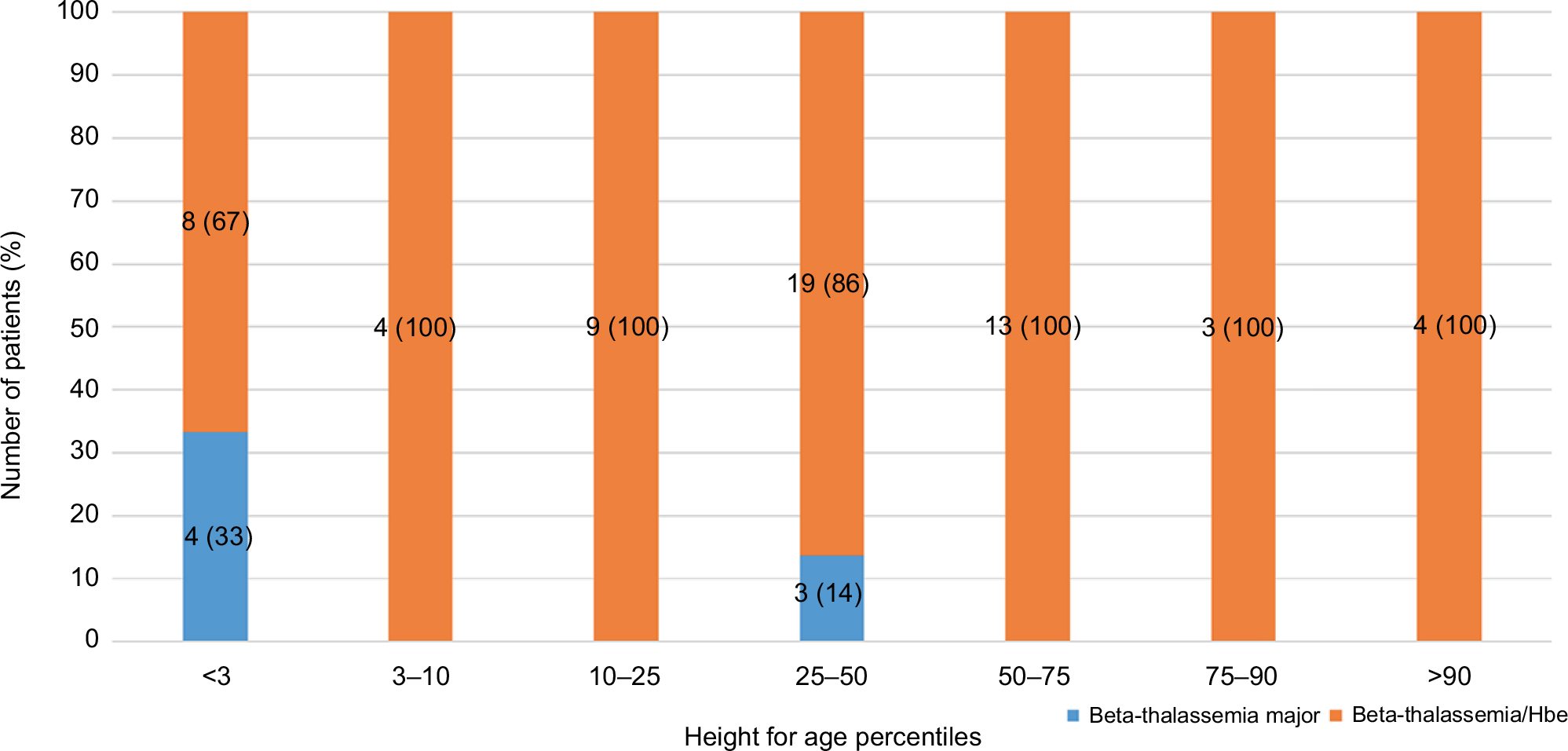 | Figure 1 Comparison of height for age in beta-thalassemia patients. |
Discussion
The present study revealed the distribution of the HBB gene mutations causing beta-thalassemia along with a genotype–phenotype correlation in a Thai pediatrics population. Beta-thalassemia/HbE is the major thalassemia problem in Thailand and can be associated with various clinical phenotypes ranging from thalassemia intermedia to thalassemia major. In our study, the numbers of beta-thalassemia/HbE patients (90%) are higher than the number of homozygous/compound heterozygous beta-thalassemia patients (10%). Knowledge of the molecular basis of the HBB gene mutations elucidates the diversity of clinical manifestations. Beta (zero)-thalassemia apparently has a more severe clinical presentation, causing thalassemia major in homozygous and compound heterozygous states, whereas beta(+)-thalassemia are observed as milder cases of thalassemia intermedia.11
Our findings are mainly in concordance with previous reports.12 Hematologic and clinical presentation correlated with the type of the mutation, confirming the fact that the phenotypic presentation of each mutation is directly related to its molecular basis. Besides the presence of the HbE mutation, the present study demonstrated that the HBB:c.126_129delCTTT mutation was the predominant genotype (48.6%) in our population, which was compatible with earlier findings.13,14 The homozygous frameshift HBB:c.126_129delCTTT mutation and the compound heterozygosity of HBB:c.126_129delCTTT with either beta (zero)-thalassemia (initiation codon mutation) or severe beta (+)-thalassemia (HBB:c.92+5G>C) produces severe clini- cal symptoms leading to thalassemia major. The remaining beta-thalassemia patients were homozygous/compound heterozygous for the beta (zero)-thalassemia mutation, including HBB:c.52A>T, HBB:c.216_217insA and HBB:c.108C>A which also presented with severe thalassemia major phenotype. Patients with beta-thalassemia/HbE whose beta-globin gene mutations were HBB:c.126_129delCTTT and HBB:c.52A>T had the same genotypic pattern as beta (zero)-thalassemia, and had severe clinical symptoms. In a previous study, beta-thalassemia/HbE patients with HBB:c.316-197C>T and HBB:c.92+5G>C had the same genotypic pattern as beta(+)-thalassemia.15 However, this study revealed that there were severe clinical symptoms classified as thalassemia major, e.g. continued regular blood transfusion and growth failure (height for age lower than 3rd percentile). Both HBB:c.92+5G>C and HBB:c.316-197C>T were previously reported by transcriptional analysis which revealed that these mutations caused alternative cryptic splicing, resulting in severe beta(+)-thalassemia with only low levels of normal beta-thalassemia mRNA.16,17 In addition, one of our beta-thalassemia/HbE patients who carries the HBB:c.126_129delCTTT mutation presents with thalassemia intermedia with concomitant alpha-globin gene mutation (Hb CS), which was identified only by molecular analysis for the Hb CS mutation. This could explain the less severe clinical symptoms in this patient.
The clinical severity of beta-thalassemia can be explained by at least 3 factors. The first factor is the type of HBB gene mutations in which 97% (68 out of 70 patients) of our patients had mutation-correlated clinical severity. Only 2 patients with beta-thalassemia/HbE who carried beta(zero)-thalassemia mutations, including 1 patient with the HBB:c.126_129delCTTT mutation and 1 patient with the HBB:c.92+1G>T mutation, presented with thalassemia intermedia. The milder clinical phenotype in one of our patients was explained by the second factor, which is the co-inheritance of alpha-thalassemia. The alpha-globin gene mutation reduces the synthesis of alpha-globin chains, causing less imbalance of alpha/beta globin chains, which is the main pathogenesis of thalassemia severity. The third factor is the existence of other genetic modifiers which can compensate for the lack of beta-globin chains. Further study needs to define the genetic modification which interacts with beta-thalassemia, to provide better understanding of the process of the disease. Several methods can determine beta-thalassemia mutations. This study revealed that using M-ARMS to detect 7 common mutations in Chinese and Southeast Asian populations, it was possible to detect 85.2% of the alleles, as in previous reports.18 Since M-ARMS can detect only a given set of mutations specific to the primers employed, direct DNA sequencing is the next step to identify various point mutations and small rearrangements in the beta-globin gene; however, the disadvantage of DNA sequencing is that large deletions of the gene are undetectable. Thus, gap-PCR is the final step to detect the 3.4 kb deletion previously reported in Thai populations.10 A combination of these techniques could identify beta-thalassemia mutations in all 140 alleles (100%) of pediatric patients in our study. The 3.4 kb deletion was detected in only 2 patients in our study. Those patients’ phenotype (3.4 kb deletion) was beta-thalassemia major major, diagnosed since 1.6 years of age. This phenomenon can be explained by several genetic factors, which may play roles in determining the variability of the disease. Our study shed some light on a genotype–phenotype correlation and therefore on the diagnostic capacity of genotypes of beta-thalassemia mutations, especially in pediatric patients (Tables 2 and 3).This may serve as a tool to predict clinical severity if detected in early life. The correlation can help to broaden our understanding of genotypic and phenotypic diversity and varied clinical presentation. This will help early intervention in children at high risk and promote prevention by genetic counseling. We believe that a prenatal diagnosis plan, or at least newborn screening for molecular markers, can lead to better outcomes in developing countries with thalassemia populations.
In conclusion, the present study revealed genotype and phenotype correlations of the most prevalent beta-thalassemia types in Thai children. According to our data, hematological parameters and consequently the clinical presentation are closely related to the type of the mutation. Our findings can provide a better prediction of clinical manifestation and severity by early identification of the type of the beta-thalassemia mutations.
Acknowledgment
This study was approved and supported by funding from the Phramongkutklao College of Medicine.
Disclosure
The authors report no conflicts of interest in this work.
References
Taher AT, Weatherall DJ, Cappellini MD. Thalassaemia. Lancet 2018;391(10116):155–167. | ||
Sankaran VG, Lettre G, Orkin SH, Hirschhorn JN. Modifier genes in Mendelian disorders: the example of hemoglobin disorders. Ann N Y Acad Sci. 2010;1214:47–56. | ||
Wasi P, Pootrakul S, Pootrakul P, Pravatmuang P, Winichagoon P, Fucharoen S. Thalassemia in Thailand. Ann N Y Acad Sci. 1980;344:352–363. | ||
Thein SL, Winichagoon P, Hesketh C, et al. The molecular basis of beta-thalassemia in Thailand: application to prenatal diagnosis. Am J Hum Genet. 1990;47(3):369–375. | ||
Maryami F, Azarkeivan A, Fallah MS, Zeinali S. A large cohort study of genotype and phenotype correlations of beta-thalassemia in Iranian population. Int J Hematol Oncol Stem Cell Res. 2015;9(4):198–202. | ||
Sahu PK, Pati SS, Mishra SK. Genotype–phenotype correlation of beta-thalassemia spectrum of mutations in an Indian population. Hematol Rep. 2012;4(2):e9. | ||
Ho PJ, Hall GW, Luo LY, Weatherall DJ, Thein SL. Beta-thalassaemia intermedia: is it possible consistently to predict phenotype from genotype? Br J Haematol. 1998;100(1):70–8. | ||
Bhardwaj U, Zhang YH, Lorey F, McCabe LL, McCabe ER. Molecular genetic confirmatory testing from newborn screening samples for the common African-American, Asian Indian, Southeast Asian, and Chinese beta-thalassemia mutations. Am J Hematol. 2005;78(4):249–255. | ||
Sirichotiyakul S, Saetung R, Sanguansermsri T. Analysis of beta-thalassemia mutations in northern Thailand using an automated fluorescence DNA sequencing technique. Hemoglobin. 2003;27(2):89–95. | ||
Sanguansermsri T, Pape M, Laig M, Hundrieser J, Flatz G. Beta zero-thalassemia in a Thai family is caused by a 3.4 kb deletion including the entire beta-globin gene. Hemoglobin. 1990;14(2):157–168. | ||
Asadov C, Abdulalimov E, Mammadova T, Gafarova S, Guliyeva Y, Aliyeva G. Genotype–phenotype correlations of beta-thalassemia mutations in an Azerbaijani population. Turk J Haematol. 2017;34(3):258–263. | ||
Weatherall DJ. The challenge of haemoglobinopathies in resource-poor countries. Br J Haematol. 2011;154(6):736–744. | ||
Boonyawat B, Monsereenusorn C, Traivaree C. Molecular analysis of beta-globin gene mutations among Thai beta-thalassemia children: results from a single center study. Appl Clin Genet. 2014;7:253–258. | ||
Laosombat V, Fucharoen SP, Panich V, et al. Molecular basis of beta thalassemia in the south of Thailand. Am J Hematol. 1992;41(3):194–198. | ||
Laosombat V, Wongchanchailert M, Sattayasevana B, Wiriyasateinkul A, Fucharoen S. Clinical and hematological features of beta(+)-thalassemia (IVS-1 nt 5, G-C mutation) in Thai patients. Eur J Haematol. 2001;67(2):100–104. | ||
Cheng TC, Orkin SH, Antonarakis SE, et al. beta-Thalassemia in Chinese: use of in vivo RNA analysis and oligonucleotide hybridization in systematic characterization of molecular defects. Proc Natl Acad Sci U S A. 1984;81(9):2821–2825. | ||
Treisman R, Orkin SH, Maniatis T. Specific transcription and RNA splicing defects in five cloned beta-thalassaemia genes. Nature. 1983;302(5909):591–596. | ||
Old JM, Khan SN, Verma I, et al. A multi-center study in order to further define the molecular basis of beta-thalassemia in Thailand, Pakistan, Sri Lanka, Mauritius, Syria, and India, and to develop a simple molecular diagnostic strategy by amplification refractory mutation system-polymerase chain reaction. Hemoglobin. 2001;25(4):397–407. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.