")
Back to Journals » Drug Design, Development and Therapy » Volume 18
Synthesis and Anti-Diabetic Activity of an 8-Purine Derivative as a Novel DPP-4 Inhibitor in Obese Diabetic Zücker Rats
Authors Chayah M, Luque-González A, Gómez-Pérez V, Salagre D , Al-Shdaifat A, Campos JM , Conejo-García A , Agil A
Received 7 December 2023
Accepted for publication 21 March 2024
Published 10 April 2024 Volume 2024:18 Pages 1133—1141
DOI https://doi.org/10.2147/DDDT.S450917
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tuo Deng
Meriem Chayah,1– 3 Angélica Luque-González,1 Verónica Gómez-Pérez,1 Diego Salagre,4,5 Amjad Al-Shdaifat,6 Joaquín María Campos,1,2 Ana Conejo-García,1,2 Ahmad Agil2,4,5
1Department of Medicinal and Organic Chemistry and Excellence Research Unit of Chemistry Applied to Biomedicine and the Environment, Faculty of Pharmacy, University of Granada, Granada, Spain; 2Biosanitary Institute of Granada (Ibs.granada), SAS-University of Granada, Granada, Spain; 3Centre for Genomics and Oncological Research, Pfizer/University of Granada/Andalusian Regional Government (GENYO), Granada, Spain; 4Department of Pharmacology, Faculty of Medicine, University of Granada, Granada, Spain; 5Federico Oloriz Neuroscience Institute, University of Granada, Granada, Spain; 6Department of Medicine and Family Medicine, Faculty of Medicine, Hashemite University, Zarqa, Jordania
Correspondence: Ahmad Agil; Ana Conejo-García, Email [email protected]; [email protected]
Abstract: Type 2 diabetes mellitus (T2DM) is one of the world’s principal metabolic diseases characterized by chronic hyperglycemia. The gut incretin hormones, glucagon-like peptide 1 (GLP-1) and gastric inhibitory polypeptide (GIP), which has been proposed as a new treatment for T2DM, are extensively metabolized by Dipeptidyl peptidase 4 (DPP-4). Inhibitors of DPP-4 block the degradation of GLP-1 and GIP and may increase their natural circulating levels, favoring glycemic control in T2DM. A novel and potent selective inhibitor of DPP-4 with an 8-purine derived structure ( 1) has been developed and tested in vitro and in vivo in Zücker obese diabetic fatty (ZDF) rats, an experimental model of the metabolic syndrome and T2DM to assess the inhibitory activity using vildagliptin as reference standard. ZDF rats were subdivided into three groups (n = 7/group), control (C-ZDF), and those treated with compound 1 (Compound 1-ZDF) and with vildagliptin (V-ZDF), both at 10 mg/kg/d rat body weight, in their drinking water for 12 weeks, and a group of lean littermates (ZL) was used. ZDF rats developed DM (fasting hyperglycemia, 425 ± 14.8 mg/dL; chronic hyperglycemia, HbA1c 8.5 ± 0.4%), compared to ZL rats. Compound 1 and vildagliptin reduced sustained HbAl1c (14% and 10.6%, P < 0.05, respectively) and fasting hyperglycemia values (24% and 19%, P < 0.05, respectively) compared to C-ZDF group (P < 0.001). Compound 1 and vildagliptin have shown a potent activity with an IC50 value of 4.92 and 3.21 μM, respectively. These data demonstrate that oral compound 1 administration improves diabetes in ZDF rats by the inhibitory effect on DPP-4, and the potential to be a novel, efficient and tolerable approach for treating diabetes of obesity-related T2DM, in ZDF rats.
Keywords: diabetes mellitus type 2, DPP-4 inhibitors, purine derivatives, ZDF rats
Introduction
Diabetes is a major global problem nowadays. It currently affects nearly 537 million people worldwide in 2023, and this number will rise to 700 million in 2045.1 Among the latest approaches for the treatment of T2DM, the inhibition of the enzyme DPP-4 emerged as an interesting alternative. This approach is based on the key role that DPP-4 has in the inactivation of the two primary endogenous incretin hormones glucagon-like peptide 1 (GLP-1) and gastric inhibitory polypeptide (GIP), both secreted from the intestine to stimulate insulin secretion from pancreatic β cells.2 The incretin effect accounts for at least 50% of the total insulin secreted after ingestion. In addition to its insulinotropic effects, GLP-1 decreases food intake, inhibits glucagon secretion and gastric emptying, and slows the rate of endogenous glucose production.3 It has also been shown to stimulate β-cell proliferation and to protect them from apoptosis. Both peptides had sparked interest in their therapeutic potential as anti-diabetic agents. However, they are rapidly inactivated by DPP-4 resulting in a half-life of only 1–2 minutes.4 Therefore, the inhibition of DPP-4 prolongs the half-life and increases the levels of endogenous active GLP-1 and thereby greatly enhances its insulinotropic effect.5
Currently, DPP-4 inhibitors are firmly established as a treatment for T2DM proving efficacy and better glycemic control leading to a statistically significant long-term reduction in glycated hemoglobin (HbA1c) without risk of hypoglycemia and cardiovascular side effects.6,7 Thus, several DPP-4 inhibitors, such as vildagliptin, saxagliptin, sitagliptin, alogliptin, or linagliptin (Figure 1), are approved worldwide as monotherapy or combined with other antidiabetic drugs.8
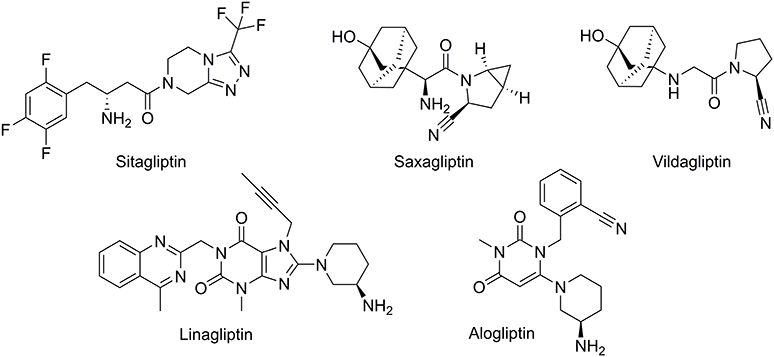 |
Figure 1 Diversity chemical structure of DPP-4 inhibitors. |
The pharmacological landscape for DPP-4 inhibition, as shown in Figure 1, encompasses a diverse array of chemical structures (β-amino acid based, cyanopyrrolidine, pyrimidinedione or xanthine based) with different pharmacokinetic properties. Notably, the concept of tight-binding inhibitors emerges as pivotal in the pharmacological context. The ability of these inhibitors to tightly bind to their target ensures a sustained inhibition of enzyme activity,9 which results in persistent diurnal effects on glucose, β cells and incretin hormones without significant differences between them.9,10
Using computational chemistry in the rational design of drugs and three very common structural modification strategies in pharmaceutical chemistry (bioisosterism, molecular hybridization, and simplification), Semighini et al proposed purine-based structure inhibitors of DPP-4.11 The BindingDB database was used, where out of 447 inhibitors, the most potent one exhibiting a solved complex X-ray structure (PDB code 1RWQ) was chosen, displaying an IC50 of 0.1 nM (lead, Figure 2).12 The replacement of the aminomethypyrimidine by 8-arylmethyl-9H-purin-6-amine maintains the main interactions with DPP-4 and the hybridization with benzomethylenedioxy moiety lead to purine-based compounds. Moreover, additional theoretical toxicity and pharmacokinetic studies suggest no apparent toxicity, good oral availability and pharmacotherapeutic profile.11
 |
Figure 2 Chemical structures of lead and compound 1. |
On the basis of these theoretical studies and in order to validate this scaffold as a DPP-4 inhibitor, we have synthesized one of the proposed structures (1, Figure 2) and assessed its antidiabetic activity in Zücker diabetic fatty (ZDF) rats, an experimental model that best represents the pathogenesis and evolution of human T2DM,13–17 using vildagliptin as a reference standard.
Materials and Methods
Chemistry
General
Reactions were followed via thin-layer chromatography on Merck AL Silicagel 60 F254 TLC layers and UV light. Flash chromatographies were performed with Silicagel Merck 60 with a particle size of 0.040–0.063 mm (230–400 mesh ASTM). Microwave-assisted syntheses were carried out in an Initiator 2.0 system (Biotage AB, Upsala). Reactions were irradiated at 2.450 GHz. 1H NMR spectra were recorded on a Varian Inova Unity (300 MHz) and Varian Direct Drive (400 MHz y 500 MHz) at room temperature in DMSO-d6. Chemical shifts (δ) are given in ppm downfield from tetramethylsilane as the internal standard. Coupling constants (J) are given in Hz. Splitting patterns are labeled as s (singlet), d (doublet), dd (double doublet), t (triplet) and m (multiplet). 13C NMR spectra were performed on a Varian Direct Drive (125 MHz). High-resolution mass-spectrometry (HRMS) analysis was performed on a Waters LCT Premier XE spectrophotometer with electrospray ionization (ESI-TOF). Fusion points were measured in a Stuart Scientific SMP3. Scheme 1 shows the synthesis of compound 1 and Table 1 shows the optimization of the reaction conditions.
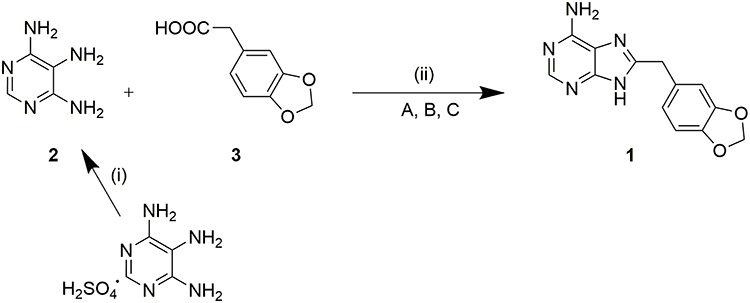 |
Scheme 1 General reaction for one-pot synthesis of purine ring. (i) a. NaOH, H2O, rt - 80 °C - 5 °C, until total dissolution, b. HCl, pH 7. (ii) P(OPh)3, NaOH, anhydrous pyridine, MW 220 °C 15 min. A, B, (C) reaction conditions detailed in Table 1. |
 |
Table 1 Modifications in the Reaction Conditions |
Synthesis
4,5,6-Triaminopyrimidine 2. 4,5,6-Triaminopyrimidine sulfate (1.457g, 6.53mol) and NaOH (0.575 g, 14.37mol) were resuspended in water (15mL) and heated at 80 °C until totally dissolved (approximately 10 minutes). Then, the solution is cooled down to 0~5°C and the pH is adjusted to 7 by adding HCl 1N (approximately 1 mL) allowing the 4,5,6-triaminopyrimidine (2) to crystallize (0.549g, 67%). White needle-shaped solid (0.549 g, 67%); mp: >300 °C fusion with decomposition. 1H NMR (300 MHz, DMSO-d6) δ 7.46 (s, 1H, H-2pyr), 5.53 (s, 4H, NH2-4pyr -and NH2-6pyr), 3.75 (s, 2H, NH2-5pyr).
Benzo[1,3-d]dioxol-5-yl-methyl)-9-H-purin-6-amine 1. Triphenyl phosphite (P(OPh)3) (2.88 mmol), 4,5,6-triaminopyrimidine (2.4 mmol) and 3.4-methylendioxyphenyl acetic acid (2.4 mmol) in anhydrous pyridine (1.5 mL/mmol) were irradiated in a microwave at 220 °C for 75 minutes. Purification was performed by flash chromatography with CH2Cl2/MeOH as eluent. White solid (477mg, 74%); mp: 276 ± 2 °C. 1H NMR (500 MHz, DMSO-d6) δ 11.72 (s, 1H, H-9ad), 8.04 (s, 1H, H-2ad), 7.00 (s, 2H, NH2-6ad), 6.88 (s, 1H, H-4ph), 6.84 (d, J = 7.9, 1H, H-7ph), 6.75 (d, J = 7.8, 1H, H-6ph), 5.96 (s, 2H, O-CH2-O), 4.01 (s, 2H, ad-CH2-ph). 13C NMR (125 MHz, DMSO-d6) δ 155.06 (C-6ad ó C-4 ad), 151.85 (C-2ad), 151.14 (C-4ad or C-6as), 150.43 (C-8ad), 147.27 (C-3aph), 145.89 (C-7aph), 131.00 (C-5ph), 121.65 (C-6ph), 118.60 (C-5ad), 109.08 (C-4ph), 108.20 (C-7ph), 100.83 (C-2ph), 34.57 (ad-CH2-ph). NMR spectra are included in Figures S1–S3. HRMS (m/e): Calculated for C13H11N5O2 269.0913; found 270.0994.
Biological Assays
In vitro DPP-4 Activity Determination
The inhibition of DPP IV activity was performed using the enzyme protocol (Cayman DPP IV screening kit) obtained from Abcam (Barcelona, Spain) with slight modification. Vildagliptin was used as the standard inhibitor. Briefly, 30 µL of the buffer solution, 10 µL of enzyme DPP IV, 10 µL of sample solution (100 ppm), and 50 µL Gly-Pro-AMC as the substrate, were added into the well. The mixture was shaken and incubated for 30 min at 37 °C to have the complete reaction. In control wells, the inhibitor was replaced by aqua bidest. The fluorescence of the free AMC group was measured on an excitation wavelength of 350–360 nm and an emission wavelength of 450–465 nm by using a microplate reader (GloMax® Discover System). The percentage of inhibition was calculated using the formula: % Inhibition = Initial Activity – Inhibitor/Initial Activity ×100. The results are shown in Table 2 and Figure 3.
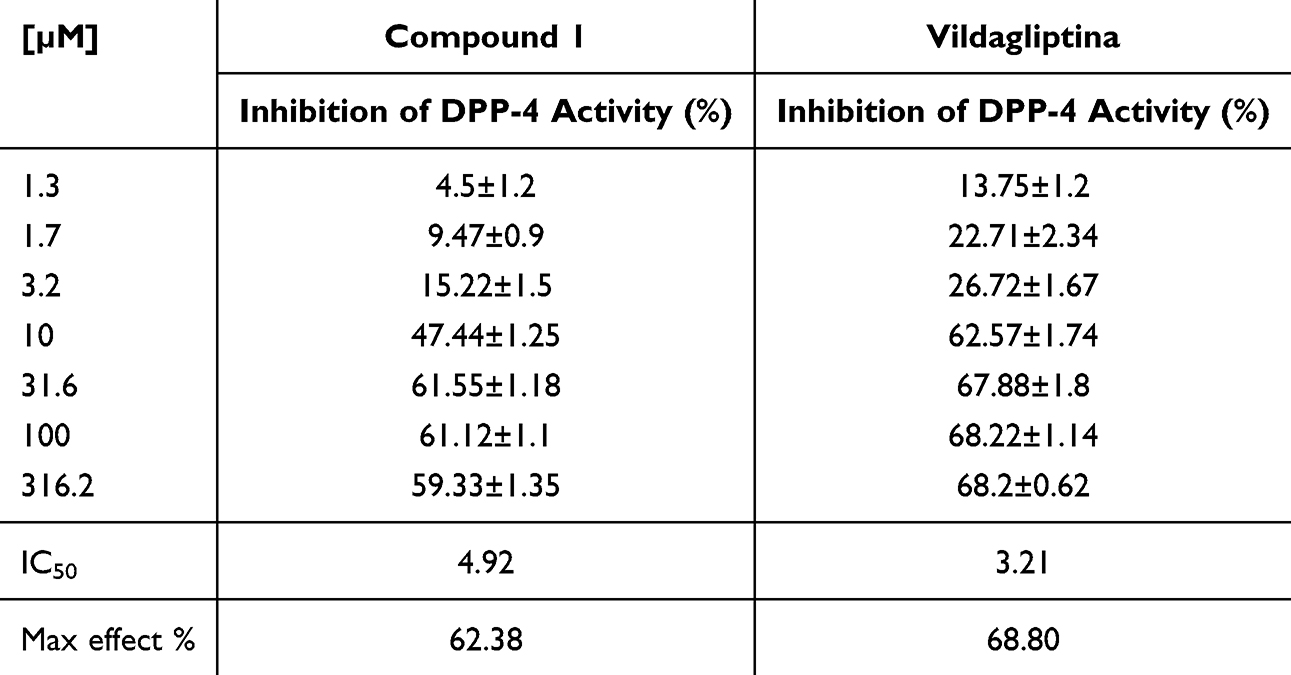 |
Table 2 In vitro Dose-Response Experiment |
 |
Figure 3 Inhibitory effect in vitro of compound 1 and vildagliptin on horse DPP-4 enzyme activity. Each point represents the mean ± S.E.M. of the values from five independent experiments done in triplicate. Statistically significant differences in comparison to the effect of drug solvent: *P < 0.05, **P < 0.01 (two-way followed by Bonferroni test). |
In vivo Biological Evaluation
This study was carried out in accordance with the European Union guidelines for animal care and protection. Male Züker diabetic fat rats (ZDF, fa/fa n = 21) and male Zücker lean littermates (ZL, fa/- n = 7) were obtained at the age of 5 weeks from Charles River (Barcelona, Spain). Animals were maintained on Purina 5008 rat chow (protein 23%, fat 6.5%, carbohydrates 58.5%, fiber 4%, and ash 8%; Charles River) and housed 2 per clear plastic cage in a climate-controlled room at 28–30 °C and 30–40% relative humidity, with a 12 h dark/light cycle (lights on at 7 am). The animals were acclimated to the room conditions in the first week after arrival. Water intake was recorded. ZDF rats were subdivided into three groups treated for 12 weeks: compound1-treated group (compound1-ZDF), vildagliptin-treated group (V-ZDF), and vehicle-treated controls (C-ZDF). A fourth group of lean rats (ZL) was also used. Compound 1 and vildagliptin were separately dissolved in a minimum volume of absolute ethanol and diluted in the drinking water to yield a dose of 10 mg/kg body weight-BW-/day, with a final concentration of 0.066% (w/v) ethanol. Water intake and BW were recorded twice weekly. Freshly compound 1, vildagliptin, and vehicle solutions were prepared daily, and the dose was adjusted to the BW throughout the study period. Water bottles were covered with aluminum foil to protect them from light.
At the end of the treatment period, animals fasted overnight, and on the next day, they were anesthetized with sodium thiobarbital (thiopental) and sacrificed between 9 and 11 pm. Blood was collected via heart puncture into EDTA Vacutainer tubes, centrifuged, and the plasma was aliquoted and frozen at −80°C for subsequent determinations.
The determination of biochemical parameters of glycemia and HbA1c was measured using commercial kits in an automatic analyzer (Roche Laboratory Systems, Mannheim, Germany). The results are shown in Figures 4 and 5.
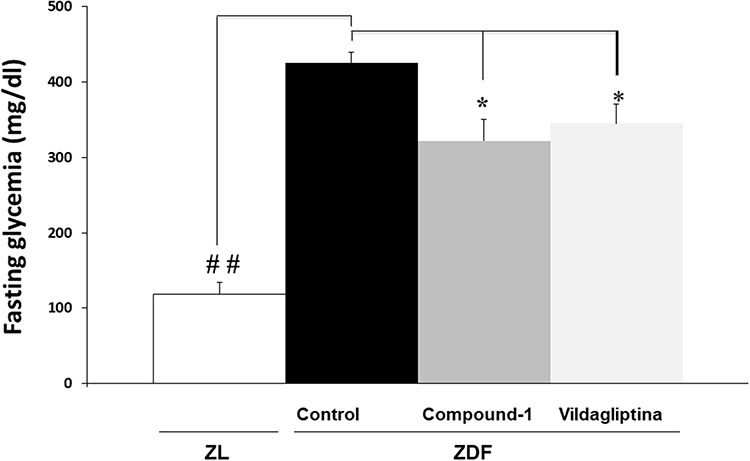 |
Figure 4 Hyperglycemia values measured in control, compound 1 and vildagliptin-treated animals. Values are the means ± S.E.M. of 7 animals/group. Values not sharing a common superscript letter are significantly different by two-way ANOVA/Tukey. *P < 0.05 compound 1-Zucker diabetic fatty [Compound 1-ZDF] versus Control vehicle Zücker diabetic fatty [C-ZDF] rats, *P < 0.05 Vildagliptin-Zücker diabetic fatty [Vildagliptin-ZDF] versus C-ZDF rats, ##P < 0.01 C-ZDF versus Zücker lean [ZL] rats. |
 |
Figure 5 HbA1c values measured in control, compound 1 and vildagliptin-treated animals. Values are the means ± S.E.M. of 7 animals/group. Values not sharing a common superscript letter are significantly different by two-way ANOVA/Tukey (*P < 0.05 compound 1-Zücker diabetic fatty [Compound 1-ZDF] versus Control vehicle Zücker diabetic fatty [C-ZDF] rats, *P < 0.05 Vildagliptin-Zücker diabetic fatty [V-ZDF] versus C-ZDF rats; ##P < 0.01 C-ZDF versus Zücker lean [ZL] rats. |
The animal experimentation conducted in this study adhered to the protocols approved by the Ethical Committee at the University of Granada (Granada, Spain), under the reference number 4–09-2016-CEEA. Furthermore, the study was carried out in accordance with the animal care and protection guidelines of the European Union in accordance with the principles outlined in the Declaration of Helsinki.
Statistical Analysis
All results are expressed as mean ± standard error of the mean (S.E.M) values. The % inhibition was calculated using the formula, control- test/control x 100. IC50 (concentration of drug that produced half of the maximal diminishment of DPP-4 activity) and Emax (minimum decrease in DPP-4 activity) were calculated from the concentration–response curves using a non-linear regression analysis with the Graph-Pad Inplot computer program (GraphPad Software Inc., USA). Statistical analysis was performed using two-way ANOVA followed by Bonferroni post-tests, and the differences were considered significant when P < 0.05. Levels of significance were labeled on the figures as follows: ** P < 0.01, * P < 0.05 and ##P < 0.01.
Results and Discussion
Chemistry
The hit compound 1 with a general structure based on 8-arylmethyl-9H-purin-6-amine (Scheme 1) was synthesized. It has an adenine-like structure with two oxygen atoms linked via a methylene bridge.
The microwave-assisted synthesis of benzimidazole and other heterocycles through the condensation of a diamine and a carboxylic acid has been described.18 This method has been adapted for the synthesis of more complex aminopyrimidin structures in one pot.19
Scheme 1 shows the general synthesis procedure for compound 1. The purine backbone is synthesized by the condensation between the amino groups of position 4 and 5 of the pyrimidine and the carboxylic acid group of reactant 3 which carries the substituent at position 8 of the final purine ring.
Compound 1 has been synthesized following the conditions described by the group of Chiosis et al to form purine rings.19 4,5,6-triaminopyrimidine sulfate (2), 3.4-methylendioxyphenyl acetic acid (3), triphenyl phosphite and sodium hydroxide are mixed and solved in anhydrous pyridine (Scheme 1). The reaction mixture is irradiated on the microwave at 220 °C for 15 minutes, leading to compound 1 with a 2% yield.
The main difference between the reaction performed and the one described by Chiosis group19 is the amount used. In our case, we scaled it up which might explain the low yield obtained, making it difficult for the triamino salt derivative to become in situ into the free base form. Therefore, to improve the yield, the triamino salt derivative was previously converted into the free base form 4,5,6-triaminopyrimidine (2) (Scheme 1). Thus, an aqueous suspension of 4,5,6-triaminopyrimidine sulfate was heated with sodium hydroxide until totally dissolved.20
When using the free base form of the pyrimidine derivative to form the purine ring, compound 1 is obtained with an 18% yield (Table 1, method a). Subsequent changes in molar ratio, solvent volume and reaction time were carried out to study their influence on the yield of the formation of the purine ring (Table 1). The best yield, 74%, was achieved when 1 eq. of 2 and 3 in 1.5 mL of solvent were irradiated on the microwave at 220 °C for 75 minutes.
Biological Evaluation
The incubation in vitro of DPP-IV with different concentrations (0.0313 to 2 µM) of compound 1 (Table 2) inhibited DPP-IV activity in a concentration-dependent manner (Figure 3). By non-linear regression analysis, the data were fitted to a sigmoid curve, enabling the Emax to be calculated as 62 ± 1.04% of inhibition and the IC50 as 4.9 µM. Vildagliptin (0.0313 to 2 µM) also concentration-dependently inhibited DPP-IV activity (Table 2 and Figure 3), but it was more efficacious (Emax = 69 ± 1.10% of inhibition) and more potent (IC50 = 3.2 µM) than compound 1. The solvent did not significantly modify DPP-IV activity.
Regarding the in vivo assay, as expected, the C-ZDF group developed severe fasting hyperglycemia (425 ± 14.8 mg/dl) which was significantly reduced by compound 1 and vildagliptin treatment to 322 ± 29 and 345 ± 26 mg/dl, respectively (P < 0.05) (Figure 4).
Moreover, chronic long-day hyperglycemia, as indicated by HbA1c levels, were significantly decreased from 8.5 ± 0.5% in the C-ZDF group to 7.3 ± 0.2 and 7.6 ± 0.2% in compound 1 and vildagliptin treated groups, respectively (P < 0.05) (Figure 5).
Discussion
Vildagliptin’s absorption is unaffected by food, its oral bioavailability is approximately 85% and it reaches maximal plasma concentrations within a few hours (tmax 1.7 hours).21 In addition, this drug is one of the most common DPP-4 inhibitors used in clinical because it is currently approved to be taken simultaneously with other antidiabetics such as metformin, sulphonylureas, thiazolidinediones, or even insulin becoming a reference drug.22 In humans, the most used dose is 100 mg, corresponding to about 10 mg/kg in animals.22 This dose was also previously used and was found to be the most effective.23 We use the same dose of 10 mg/kg in vivo for both compounds to compare their effects at the same dose and to be bioequivalent.
This study demonstrates that, orally, a new potent and selective DPP-4 inhibitor with an 8-purine-derived structure enhances glycemic control in ZDF rats. Indeed, this is the first demonstration that this new DPP-4 inhibitor reduces both fasting hyperglycemia and chronic maintained hyperglycemia, as evidenced by a −1% decrease in absolute HbA1c values. As expected, the in vivo oral administration of vildagliptin also decreases fasting as well as chronically maintained hyperglycemia, as shown by a reduction of −1% in absolute HbA1c values. In effect, this is a mild antihyperglycemic effect. However, it approaches the efficacy of the potent oral antihyperglycemic agents such as sitagliptin and metformin in the same animal model at the same age and for similar treatment periods.24 However, in this same animal model, vildagliptin did not show a positive effect on antihyperglycemic, likely due to using a lower dose (3g/kg),25 less treatment time period (5 weeks), and treatment time onset (12 and 10 weeks).25,26 In the present study, vildagliptin (10mg/kg/d) improved glycemic parameters, an effect consistent with other studies at the same dose in experimental Type 2 Diabetes rat.27 Moreover, in several monotherapies, vildagliptin 100 mg daily studies in T2DM human, vildagliptin significantly reduced mean HbA1c.28,29
Incubation in vitro of DPP-4 with compound 1 and vildagliptin concentration-dependently inhibited DPP-4 enzyme activity. The IC50 obtained for vildagliptin (3,21 µM) 0.88 nM agrees with previous studies that reported inhibition of DPP-4 in Caco-2 cells with EC50 = 2.286 μM.29 Obviously, the effect of compound 1 on DPP-4 activity had not previously been explored and we found that it showed lesser efficacy but greater potency than vildagliptin in the inhibition of DPPT-4 activity. The in vitro results show that compound 1 is slightly less active than vildagliptin, 7% less for Emax and 1.7 µM less for IC50. However, in vivo assays assessing the effects on hyperglycemia and HbA1c, a trend of improvement in the efficacy of compound 1 with respect to vildagliptin can be observed, though not significant due probably to pharmacokinetic processes that occur in vivo but not in vitro.
Conclusion
In the present study, a new 8-purine derivative 1 was obtained through an optimized synthesis pathway and unequivocally characterized. In vitro studies demonstrated that compound 1 inhibits DPP-IV in a dose-dependent manner. Moreover, chronic oral treatment in an experimental model of T2DM with obese diabetic ZDF rats demonstrated an interesting anti-diabetic effect that could make it an efficient and tolerable approach for treating diabetes. Therefore, further studies are needed to determine the pharmacotherapeutic profile and potential toxicity effects of this inhibitor.
Acknowledgments
A.A. would like to thank the support of the visitant professor Dr Amjad Al-Shdaifat.
Funding
This research was supported by the Spanish Ministry of Science and Innovation through (grants PID2021.128109OB.I00 and PID2021-125900OB-I00) by European Regional Development Fund (ERDF) as a way of making Europe.
Disclosure
The authors report no conflicts of interest in this work.
References
1. International Diabetes Federation. IDF Diabetes Atlas (10th e). Available from: http://www.diabetes.atlas.org.
2. Holst JJ, Deacon CF. Inhibition of the activity of dipeptidyl-peptidase IV as a treatment for type 2 diabetes. Diabetes. 1998;47:1663–1670. doi:10.2337/diabetes.47.11.1663
3. Kreymann B, Williams G, Ghatei MA, Bloom SR. Glucagon-like peptide-1: a physiological incretin in man. Lancet. 1987;2:1300–1304. doi:10.1016/s0140-6736(87)91194-9
4. Baggio LL, Drucker DJ. Biology of incretins: GLP-1 and GIP. Gastroenterology. 2007;132:2131–2157. doi:10.1053/j.gastro.2007.03.054
5. Deacon CF, Hughes TE, Holst JJ. Dipeptidyl peptidase IV inhibition potentiates the insulinotropic effect of glucagon-like peptide 1 in the anesthetized pig. Diabetes. 1998;47:764–769. doi:10.2337/diabetes.47.5.764
6. Ahrén B, Simonsson E, Larsson H, et al. Inhibition of dipeptidyl peptidase IV improves metabolic control over a 4-week study period in type 2 diabetes. Diabetes Care. 2002;25:869–875. doi:10.2337/diacare.25.5.869
7. Scheen AJ. The safety of gliptins: updated data in 2018. Expert Opin Drug Saf. 2018;17:387–405. doi:10.1080/14740338.2018.1444027
8. Deacon CF. Dipeptidyl peptidase 4 inhibitors in the treatment of type 2 diabetes. Nat Rev Endocrinol. 2020;16:642–653. doi:10.1038/s41574-020-0399-8
9. Saini K, Sharma S, Khan Y. DPP4 inhibitors for treating T2DM - hype or hope? An analysis based on the current literature. Front Mol Biosci. 2023;10:1130625. doi:10.3389/fmolb.2023.1130625
10. Yin R, Xu Y, Wang X, et al. Role of dipeptidyl peptidase 4 inhibitors in antidiabetic treatment. Molecules. 2022;27(10):3055. doi:10.3390/molecules27103055
11. Semighini EP, Resende JA, de Andrade P, et al. Using computer-aided drug design and medicinal chemistry strategies in the fight against diabetes. J Biomol Struct Dyn. 2011;28:787–796. doi:10.1080/07391102.2011.10508606
12. Peters JW, Weber S, Kritter S, et al. Aminomethylpyrimidines as novel DPP-IV inhibitors: a 105-fold activity increase by optimization of aromatic substituents. Bioorg Med Chem Lett. 2004;14:1491–1493. doi:10.1016/j.bmcl.2004.01.019
13. Peterson R, Shaw W, Neel M, et al. Zücker diabetic fatty rat as a model for non-insulin-dependent diabetes mellitus. ILARNews. 1990;32:16–27. doi:10.1093/ilar.32.3.16
14. Kasiske BL, O’Donnell MP, Keane WF. The Zücker rat model of obesity, insulin resistance, hyperlipidemia, and renal injury. Hypertension. 1992;19:110–115. doi:10.1161/01.hyp.19.1_suppl.i110
15. Kojima K, Hama T, Kato T, Nagatsu T. Rapid chromatographic purification of dipeptidyl peptidase IV in human submaxillary gland. J Chromatogr. 1980;189(2):233–240. doi:10.1016/s0021-9673(00)81523-x
16. Shafrir E. Animal models of non-insulin-dependent diabetes. Diabetes Metab Rev. 1992;8:179–208. doi:10.1002/dmr.5610080302
17. Aleixandre De Artinano A, Miguel Castro M. Experimental rat models to study the metabolic syndrome. Br J Nutr. 2009;102:1246–1253. doi:10.1017/S0007114509990729
18. Lin SY, Isome Y, Stewart E, Liu J-F, Yohannes D, Yu L. Microwave-assisted one step high-throughput synthesis of benzimidazoles. Tetrahedron Lett. 2006;47:2883–2886. doi:10.1016/j.tetlet.2006.02.127
19. Tao H, Kang Y, Taldone T, Chiosis G. Microwave-assisted one step synthesis of 8-arylmethyl-9H-purin-6-amines. Bioorg Med Chem Lett. 2009;19:415–417. doi:10.1016/j.bmcl.2008.11.057
20. Biamonte MA, Shi J, Hurst D, Hong K, Boehm MF, Kasibhatla SR. Preparation of 8-(arylsulfanyl)adenines with diazonium salts under mild, aerobic conditions. J Org Chem. 2005;70(2):717–720. doi:10.1021/jo048522a
21. Davis H, Jones Briscoe V, Dumbadze S, Davis SN. Using DPP-4 inhibitors to modulate beta cell function in type 1 diabetes and in the treatment of diabetic kidney disease. Expert Opin Investig Drugs. 2019;28(4):377–388. doi:10.1080/13543784.2019.1592156
22. Nair AB, Jacob S. A simple practice guide for dose conversion between animals and human. J Basic Clin Pharm. 2016;7(2):27–31. doi:10.4103/0976-0105.177703
23. Aldakinah AA, Al-Shorbagy MY, Abdallah DM, El-Abhar HS. Trigonelline and vildagliptin antidiabetic effect: improvement of insulin signalling pathway. J Pharm Pharmacol. 2017;69(7):856–864. doi:10.1111/jphp.12713
24. Han SJ, Choi S-E, Kang Y, et al. Effect of sitagliptin plus metformin on b-cell function, islet integrity and islet gene expression in Zücker diabetic fatty rats. Diabet Res Clin Pract. 2011;92:213–222. doi:10.1016/j.diabres.2011.01.016
25. Wang Y, Landheer S, van Gilst WH, et al. Attenuation of renovascular damage in Zücker diabetic fatty rat by NWT-03, an egg protein hydrolysate with ACE- and DPP4-inhibitory Activity. PLoS One. 2012;7(10):e46781. doi:10.1371/journal.pone.0046781
26. Eom YS, Gwon AR, Kwak KM, et al. Protective effects of vildagliptin against pioglitazone-induced bone loss in type 2 diabetic rats. PLoS One. 2016;20(12):e0168569. doi:10.1371/journal.pone.0168569
27. Refaat R, Sakr A, Salama M, El Sarha A. Combination of vildagliptin and pioglitazone in experimental type 2 diabetes in male rats. Drug Dev Res. 2016;77(6):300–309. doi:10.1002/ddr.21324
28. Keating GM. Vildagliptin: a review of its use in type 2 diabetes mellitus. Drugs. 2014;74(5):587–610. doi:10.1007/s40265-014-0199-3
29. Gupta A, Jacobson GA, Burgess JR, et al. Citrus bioflavonoids dipeptidyl peptidase-4 inhibition compared with gliptin antidiabetic medications. Biochem Biophys Res Commun. 2018;3(1):21–25. doi:10.1016/j.bbrc.2018.04.156
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.