")
Back to Journals » Drug Design, Development and Therapy » Volume 10
Smart nanocrystals of artemether: fabrication, characterization, and comparative in vitro and in vivo antimalarial evaluation
Authors Shah SMH , Ullah F, Khan S , Shah SMM, Matas MD, Hussain Z , Minhas MU , AbdEl-Salam NM, Assi KH, Isreb M
Received 14 June 2016
Accepted for publication 29 August 2016
Published 24 November 2016 Volume 2016:10 Pages 3837—3850
DOI https://doi.org/10.2147/DDDT.S114962
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Wei Duan
Syed Muhammad Hassan Shah,1 Farhat Ullah,2 Shahzeb Khan,2,3 Syed Muhammad Mukarram Shah,4 Marcel de Matas,5 Zahid Hussain,6 Muhammad Usman Minhas,7 Naser M AbdEl-Salam,8 Khaled Hafez Assi,3 Mohammad Isreb3
1Department of Pharmacy, Sarhad University of Science & Information Technology, Peshawar, 2Department of Pharmacy, University of Malakand, Chakdara, Pakistan; 3Institute of Life Sciences Research, School of Pharmacy, University of Bradford, West Yorkshire, 4Department of Pharmacy, University of Swabi, KPK, Pakistan; 5SEDA Pharmaceutical Development Services, The BioHub at Alderley Park, Cheshire, UK; 6Faculty of Pharmacy, Department of Pharmaceutics, Universiti Teknologi MARA, Selangor, Malaysia; 7Faculty of Pharmacy & Alternative Medicine, The Islamia University of Bahawalpur Pakistan, Bahawalpur, Pakistan; 8Riyadh Community College, King Saud University, Riyadh, Saudi Arabia
Abstract: Artemether (ARTM) is a very effective antimalarial drug with poor solubility and consequently low bioavailability. Smart nanocrystals of ARTM with particle size of 161±1.5 nm and polydispersity index of 0.172±0.01 were produced in <1 hour using a wet milling technology, Dena® DM-100. The crystallinity of the processed ARTM was confirmed using differential scanning calorimetry and powder X-ray diffraction. The saturation solubility of the ARTM nanocrystals was substantially increased to 900 µg/mL compared to the raw ARTM in water (145.0±2.3 µg/mL) and stabilizer solution (300.0±2.0 µg/mL). The physical stability studies conducted for 90 days demonstrated that nanocrystals stored at 2°C–8°C and 25°C were very stable compared to the samples stored at 40°C. The nanocrystals were also shown to be stable when processed at acidic pH (2.0). The solubility and dissolution rate of ARTM nanocrystals were significantly increased (P<0.05) compared to those of its bulk powder form. The results of in vitro studies showed significant antimalarial effect (P<0.05) against Plasmodium falciparum and Plasmodium vivax. The IC50 (median lethal oral dose) value of ARTM nanocrystals was 28- and 54-fold lower than the IC50 value of unprocessed drug and 13- and 21-fold lower than the IC50 value of the marketed tablets, respectively. In addition, ARTM nanocrystals at the same dose (2 mg/kg) showed significantly (P<0.05) higher reduction in percent parasitemia (89%) against P. vivax compared to the unprocessed (27%), marketed tablets (45%), and microsuspension (60%). The acute toxicity study demonstrated that the LD50 value of ARTM nanocrystals is between 1,500 mg/kg and 2,000 mg/kg when given orally. This study demonstrated that the wet milling technology (Dena® DM-100) can produce smart nanocrystals of ARTM with enhanced antimalarial activities.
Keywords: artemether, milling, smart nanocrystals, nanosuspension, in vitro dissolution, antimalarial activity
Introduction
Until the middle of the 20th century, malaria was a disease without any effective treatments. Even today, malaria remains endemic in specific regions of the world, with large numbers of deaths related to this disease.1 According to the latest World Health Organization (WHO) report, which was released in October 2015, there were 438,000 deaths due to malaria, with ~214 million cases of malaria also diagnosed between the years 2000 and 2015.
It has become evident from the recent report that the global rates of infection and mortality in the period 2000–2015 have been declining to 37% and 60%, respectively. In 2015, the highest number of malaria cases with >90% of the reported deaths being ascribed to malaria were reported in Sub-Saharan Africa. Among the deaths, ~70% of the victims were children whose ages were <5 years. However, in the period 2000–2015, the death tolls have been decreasing owing to the introduction of new antimalarial therapies.2
Malaria is caused by a parasitic protozoan (a unicellular microorganism) of the genus Plasmodium. Plasmodium falciparum (malaria tropica), Plasmodium vivax (malaria tertian), and Plasmodium malariae and Plasmodium ovale (malaria quaterna) are the species responsible for transmission of the disease in humans.3 The parasite is transmitted into the circulatory system from the bite of an infected female Anopheles mosquito. It then travels to the liver where it matures and reproduces.4–6 According to the recent WHO report, the prevalence of malaria due to P. vivax and P. falciparum is increasing every year, with P. vivax being responsible for transmitting ~75% (previously 64%) of malarial infections. Only 25% (previously 36%) of these infections are caused by P. falciparum, but with both stated species being reported among the most common in Pakistan.7
It has become evident from the current study that 70% of malaria cases in Pakistan are caused by P. vivax. In addition, Khyber Pakhtunkhwa (KP) and Federally Administered Tribal Areas of Pakistan have been reported as the most vulnerable zones for P. vivax and P. falciparum, which has been accentuated by free movement of the Afghan refugees and internally displaced people (WHO report).8 It has also been reported that the local residents of KP and Federally Administered Tribal Areas are immunologically more resistant to malarial infections compared to the migrating Afghani population.9
A number of medications have been used to treat malaria, including quinine, quanidine, chloroquine, primaquine, halofantrine (related quinolone), and pyrimethamine.10 However, the parasite is reported to have evolved to be resistant to most of these drugs. Under these circumstances, the use of artemisinin and its derivatives such as dihydroartemisinin, artesinuate, arteether, artemether (ARTM), and lumefantrine has been suggested by the WHO. A report published by the WHO in 2010 revealed that the numbers of deaths attributed to malaria in 2010 was markedly reduced owing to the use of artemisinin derivatives either individually or in combination therapies.11 Among the artemesinin derivatives, ARTM and its partner drug lumefantrine have been reported to be the most effective drugs to treat malaria.12–15
Artemisinin was originally isolated from the Chinese herb (qinghaosu), which is also known as Artemisia annua. The important part of its structure is the 1,2,4-trioxane ring (Figure 1), which is the active pharmacophore of this compound and responsible for its antimalarial activity.16
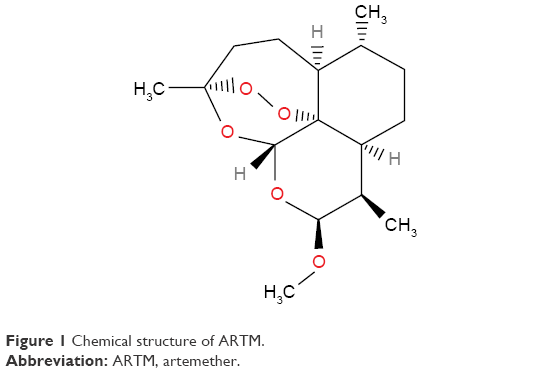 | Figure 1 Chemical structure of ARTM. |
ARTM shows rapid onset of schizontocidal action, and its metabolism in the liver produces the demethylated derivative dihydroartemisinin. It has demonstrated a significant antimalarial activity against both P. falciparum and uncomplicated malaria. It has also been reported to be very effective against cerebral malaria.17 The therapeutic response of this important active pharmaceutical ingredient (API), however, has been reported to be adversely affected by its erratic oral bioavailability due to its poor water solubility.18 Although this compound is a potent and lipid-soluble derivative of artemisinin,19,20 its poor water solubility (133±4 μg/mL) and low bioavailability (<40%) are considered to be major barriers to the development of a commercial dosage form.21
Currently, the commercially available dosage forms of ARTM are tablets, capsules, and injections. In addition to the poor water solubility, another issue associated with the parenteral formulations includes pain on injection, which leads to subsequent poor compliance.22 This pain might be caused by precipitation of the drug at the injection site. It is, therefore, important that a suitable dosage form needs to be designed, which addresses the issues of low aqueous solubility.
To date, a range of methods to address the issue of poor water solubility have been explored, including solid dispersions,23 solubilization,24 emulsions,25 microemulsions,26,27 micronization,28 micelles,29 salts, liposomes,30,31 and inclusion complexes using cyclodextrin.32 The literature, however, suggests that none of these approaches were particularly effective at addressing the issues of low aqueous solubility, while having other limitations around manufacturing complexity and physical stability.23,33–35
In this study, we report the use of an alternative approach for delivering drugs with low aqueous solubility. This involves the reduction of particle size of drugs down to submicron sizes. This provides the potential to drive dissolution through increasing the surface area of the drug that is in contact with the media.36–40
In this regard, Freundlich–Ostwald and other researchers have reported that reducing the radius of particles can lead to increased solubility.41–43 This enhanced solubility is more pronounced for particles <1 μm.44 To the best of our knowledge, hitherto nanocrystals of ARTM, which is a very promising drug delivery approach, have not been reported. This article therefore discusses the utility of a novel size reduction system (Dena® DM-100) previously reported by Plakkot et al45 for the rapid production of smart nanocrystals of ARTM with enhanced dissolution, solubility, long-term physical and chemical stability, and enhanced in vitro and in vivo antimalarial performance, against the dominant malaria-causing species in Pakistan, which include P. vivax and P. falciparum.
Materials and methods
Materials
ARTM was kindly gifted by the Shanghai Institute of Material Medica, Shanghai, China (batch no: 110916 from Chengdu Wagott Pharmaceutical Co., Ltd., Chengdu, China), hydroxypropyl methylcellulose (HPMC 6cps, batch no: 8028213) and polyvinylpyrrolidon (PVP-K30, batch no: 08297052G0) were purchased from BASF (Ludwigshafen, Germany), sodium lauryl sulfate (SLS) (batch no: 08421LE) was purchased from Sigma-Aldrich (St Louis, MO, USA). Distilled water was produced in the University of Bradford laboratories.
Preparation of ARTM nanosuspension
ARTM nanosuspensions were produced using the Dena® (DM-100 size reduction machine.46 This machine is composed of fast-moving conical rotor inside a conical sleeve. The grinding media (0.2 μm, yttrium-reinforced zirconium beads) housed inside indentations in the conical rotor and the narrow gap between the rotor and outer sleeve. Turbulence and high shear generated within the narrow gap provides potential for shearing and rupturing the particles, which results in the production of ultrafine powders in the submicron size range. The processed suspension continuously recycles through the apparatus through a stainless steel screen that prevents the contamination of product and retains the grinding media.
ARTM powder was suspended in 250 mL of an aqueous solution consisting of HPMC 6cps (0.5%, w/w), PVP-K30 (0.5%, w/w), and SLS (0.1%, w/w). ARTM suspensions were produced at the concentrations of either 2.5% (w/w) or 10% (w/w). In both suspensions, with solid loads of 2.5% and 10%, the pH was adjusted to 2.0 using HCl. In addition, the impact of milling time on particle size distribution of the ARTM was investigated at both low and high solid loads (2.5% and 10%). The suspensions were processed for 60 minutes by recycling through the size reduction chamber. Samples were collected periodically using the sampling port, and particle size was measured using the Zetasizer Nano-ZS instrument (Malvern Instruments, UK), at the intervals of 5, 10, 15, 30, 45, and 60 minutes.
Characterization of ARTM nanocrystals
Particle size measurement
Particle size of the ARTM nanosuspensions was determined using dynamic light scattering (DLS), Zetasizer Nano-ZS instrument. For 10% (w/w) nanosuspension, 1 mL of the sample was further diluted in water (1:3). Sample analysis of the low feed nanosuspension (2.5%, w/w) was, however, carried out without further dilution. For all measurements, water was selected as dispersant, and temperature was kept as 25°C. Disposable sizing cuvettes were used for the analysis of all samples; all measurements were made in triplicate (n=3), and results were reported as mean ± SD.
Zeta potential measurements
The zeta potential of nanosuspensions was determined using the Zetasizer Nano-ZS instrument. The samples for zeta potential measurements were prepared according to the reported method of Plakkot et al.45 The original samples of the produced nanosuspensions were further diluted in the dispersion medium. One milliliter of the sample was taken for 2.5% nanosuspension, while only 0.2 mL of 10% (w/w) formulation was used, which was further diluted in 10 mL of the dispersion medium. All the samples were analyzed in triplicate (n=3), and results were presented as mean ± SD.
Determination of the loading content of ARTM
The nanosuspensions were evaluated for active content using a Waters 2695, high performance liquid chromatography (HPLC) system connected to the ultraviolet detector. The Ultra™ II (C18 5 μm, 250×4.6 mm column) was used, and the temperature was set at 30°C. The solvent system used as the mobile phase for ARTM assay consisted of acetonitrile and buffer (20:80, v/v; 5 mL of triethylamine diluting to 100 by using water), and the pH was adjusted to 3±0.05 with orthophosphoric acid. The flow rate of the mobile phase was set at 1 mL/min and was analyzed at a wavelength of 216 nm.
Scanning electron microscopy (SEM)
The morphology of unprocessed ARTM was evaluated using the SEM (Quanta 400 SEM; FEI Company, Cambridge, UK). ARTM images were taken at various magnifications. The particles of ARTM were sputter coated with gold prior to the morphological studies.
Transmission electron microscopy (TEM)
TEM (TEM-1200Ex; Japan Electron Optics Laboratory Corporation, Tokyo, Japan) was used to evaluate ARTM nanocrystals at 120 kV. Nanosuspensions of ARTM were deposited on 200 mesh copper grid, which was coated with formvar/carbon (code no: S162), followed by drying the samples at room temperature. The samples were stained negatively with the magnesium uranyl acetate (2%) solution due to the low conductivity of the API.
Differential scanning calorimetry (DSC)
The melting point and heat of fusion of unprocessed and processed ARTM was determined using DSC (Q2000 series; TA Instruments, New Castle, UK). The machine was calibrated with indium 99% (MP at 156.6°C) and zinc (MP at 419.5°C). Under the stream of nitrogen gas, the raw powder and milled samples (nanocrystals) of ARTM were then scanned at a flow rate of 50 mL/min with temperature ranging from 25°C to 200°C at a heating rate of 10°C/min.
Powder X-ray diffraction (PXRD) studies
Crystallinity of the raw and processed ARTM recovered from nanosuspensions was evaluated using the PXRD (D-8, powder diffractometer; Bruker, Kahsruhi, Germany). The samples loaded into silicon wells were scanned in the 5°–50° 2θ range at the rate of 10 2θ/min, at wavelength 1.542 Å with 1 mm slit using Cu Kα as a radiation. The PXRD was calibrated using a corundum standard.
Solubility studies
The solubility studies were also carried out for the ARTM nanocrystals. Samples were isolated from nanosuspensions at the concentration of 10% (w/w) by centrifugation using the method previously reported by Van Eerdenbrugh et al,47 Gao et al,48 and Thakkar et al.49 Approximately 1.5 mL of the ARTM nanosuspension was filled into a centrifugation tube and stored for 24 hours. The sample was then centrifuged using a sigma centrifuge (model: Sigma 0II5982IIII; Scientific Laboratory Supplies Limited, Wilford, Nottingham, UK) at 14,800 rpm for 1 hour. The supernatant layer was taken and filtered through 0.02 μm filter (syringe filter: 20 nm; Whatman International Ltd, Dassel, Germany) to ensure that any undissolved API >0.02 μm (eg, the nanoparticles) was separated from dissolved drug. The supernatant was then analyzed for the ARTM content using the HPLC method as previously described. The solubility study of unprocessed ARTM in pure water and stabilizer solution (HPMC, 0.5% [w/w]; PVP-K30, 0.5% [w/w]; SLS, 0.1% [w/w]) was also carried out to assess the nanocrystals effect on saturation solubility of ARTM. Sufficient quantity of ARTM in pure water and stabilizer solution was placed in vials and sonicated for 2 hours followed by the use the same procedure as mentioned for nanocrystals.
Stability studies
Physical stability of the produced ARTM nanosuspensions was also carried out in the present study and was monitored by measuring particle size and zeta potential with time. Chemical stability of ARTM was assessed by the determination of the active content using HPLC for samples stored for 7 days both in acidic media and in water. In addition, the ARTM nanosuspension with high solids load (10%) was subjected to long-term (90 days) stability studies at 2°C–8°C, 25°C, and 40°C. The purpose of this study was to determine the extent of the particle growth through aggregation and Ostwald ripening. The particle size of stored samples was monitored at regular intervals including 10, 15, 30, 45, 60, 75, and 90 days using DLS.
Dissolution studies
The in vitro drug release of ARTM nanocrystals was compared with micronized ARTM (mean particle size 8.0±2.50 μm) produced using the FPS Spiral Jetmill (FPS, Italy), raw ARTM API powder, marketed tablets (20 mg), and the microsuspension (6.0±3.0 μm) prepared by crushing the tablet using a pestle and mortar followed by sonication in the same dispersion medium used for ARTM nanosuspensions. Dissolution tests were performed using the USP apparatus II (paddle method) at 100 rpm, which has been reported by Umapathi et al.21 The dissolution medium (1,000 mL) chosen for dissolution testing was composed of phosphate buffer at pH 7.2 and 1% sodium dodecyl sulfate, which has previously been reported by Pawar et al.50
For determination of the amount of drug dissolved, 5 mL aliquots were collected from the dissolution bath at specified time intervals (0, 2, 5, 10, 15, 30, 45, and 60 minutes) at 37°C±1°C using the syringe filter (0.2 μm). The dissolution media were then supplemented with 5 mL of fresh dissolution medium to maintain sink conditions. This small-size syringe filter has been previously reported as being effective for use in dissolution studies of nanocrystals.39,51 The drug content of each sample was analyzed using reverse phase HPLC (the method has been described in the “Materials and methods” section).
Microbiological examination and in vitro antimalarial assay
Giemsa-stained smears were examined to identify the Plasmodium species by a skilled microbiologist of Teaching Hospital (Hayat Abad Medical Complex [HMC] Peshawar, Peshawar, Pakistan). In vitro antimalarial assays were undertaken for the ARTM nanosuspension, unprocessed ARTM, and marketed tablets (20 mg) against identified species, ie, P. falciparum and P. vivax. The culture was maintained as described by Trager and Jensen.52 Stock solutions of the ARTM were prepared in solvent at concentrations of 0.1–20 ng/mL for the ARTM nanosuspension, unprocessed ARTM, and marketed tablets with the concentrations of 1–70 ng/mL. The culture (P. vivax 2%–3% and hematocrit 2%–3%) was exposed to various concentrations of the nanosuspensions, unprocessed API, and marketed tablets for 72 hours (37°C, 5% CO2). Lytic buffer comprising Tris pH 7.5 (20 mM), EDTA (5 mM), saponin (0.008%), triton X-100 (0.08%), and syber green-I was added to each well followed by incubation for 3 hours in the dark in incubators at room temperature. Parasites in the thin films were counted against 250 erythrocytes each, with slides being read in triplicate. The mean parasitemia was calculated from the triple-read replicate tests.
The mean parasitemia in the drug-free control wells served as the parameter of optimum and relative growth inhibition in the drug wells and was calculated on the basis of formula as suggested by Fidock et al.53 All the plates were read under fluorescence and IC50 was determined.
|
|
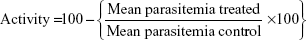
In vivo antimalarial activity
In vivo studies were carried out according to the ethical committee of the University of Malakand and relevant Bye-Laws 2008 (Scientific Procedure Issue-1). In house, male Swiss albino mice aged between 4 weeks and 6 weeks with body weight 30±5 g were used. The protocol for in vivo studies was designed based on the 4 days suppressive studies of Peters.54
P. vivax (HMC-147) collected from the Teaching Hospital was used. Experimental animals were infected by introducing parasitemia intraperitonealy at concentrations of 106 by diluting mice blood in buffer using acid citrate dextrose. The animals were divided into ten groups (n=6 per group). After postinfection starting from day 0 to day 3, ARTM nanosuspensions, microsuspensions, marketed tablets (20 mg), and unprocessed API (raw powder) were given in the form of a suspension by oral gavage at concentrations of 2 mg/kg. Blood was withdrawn from the tail vein on day 4 postinfection. Methanol was used to fix the blood smear followed by staining with Giemsa stain. In the experiment, untreated control mice were used to check any change in the susceptibility of host for infection and for procedural errors. Parasites were counted from each slide as percent paracitemia after counting 250 red blood cells using the formula reported by Fidock et al.53
Acute toxicity studies of ARTM nanosuspension
Acute oral toxicity studies were conducted for ARTM nanosuspensions to estimate the lethal dose (LD50) using male Swiss albino mice (n=8) weighing 35±5 g. ARTM nanosuspensions were given in several doses (500, 1,000, 1,500, and 2,000 mg/kg) by oral gavage. The doses were decided as per the main test specified in the Organization for Economic Cooperation and Development Guideline 420.55 After administering the ARTM nanosuspensions at the above doses, the toxic effects and number of deaths were observed over the period of 24 hours. The protocol for this study was carried out according to the requirements of, and approved by, the Ethical Committee of the University of Malakand and relevant Bye-Laws 2008 (Scientific Procedure Issue-1).
Statistical analysis
Data were presented as mean ± SD. Data were analyzed using either paired t-tests or independent t-test and analysis of variance (ANOVA) followed by Tukey’s post hoc analysis. For particle size, zeta potential, drug content, solubility, stability, and dissolution studies, a P-value of <0.05 was considered to indicate a significant difference between the tested groups. Dissolution data for the different formulations were also analyzed using Statistics 8.1 software (Tallahassee, FL, USA).
Results and discussion
Preparation of ARTM nanocrystals and the impact of processing conditions
The initial size of the unprocessed ARTM was in the size range of 25–30 μm and 120–135 μm (Figure 2A). Figure 2C shows a rapid size reduction of ARTM 800–900 nm within the first 5 minutes of milling at different processing conditions. It became evident that a processing time of 45 minutes was sufficient to achieve maximum attainable size reduction of the particles. The average size of the particles and polydispersity index (PDI) of ARTM for the high concentration suspension (10%, w/w) were found to be 161.3±1.5 nm and 0.172±0.01, respectively, after 60 minutes (Figure 2C). For the low concentration feedstock (2.5%, w/w) prepared both in water and at acidic pH (2.0), the particles were reduced to sizes <250 nm within 60 minutes. The average particle size and PDI values were shown to be 245.5±2.0 nm and 0.150±0.02, respectively (Figure 2C). TEM images clearly show uniform size distribution of the ARTM nanocrystals with size <200 nm (Figure 2B). All the particles shown in TEM images appear to show a defined morphology typically associated with crystalline material with no spherical particles present. The high concentration ARTM suspension (10%, w/w) showed the smallest particle size of 161.3±1.5 nm compared to those from the low concentration feedstock (2.5%, w/w), which had shown an average particle size of 245.5±2.0 nm (Figure 2C).
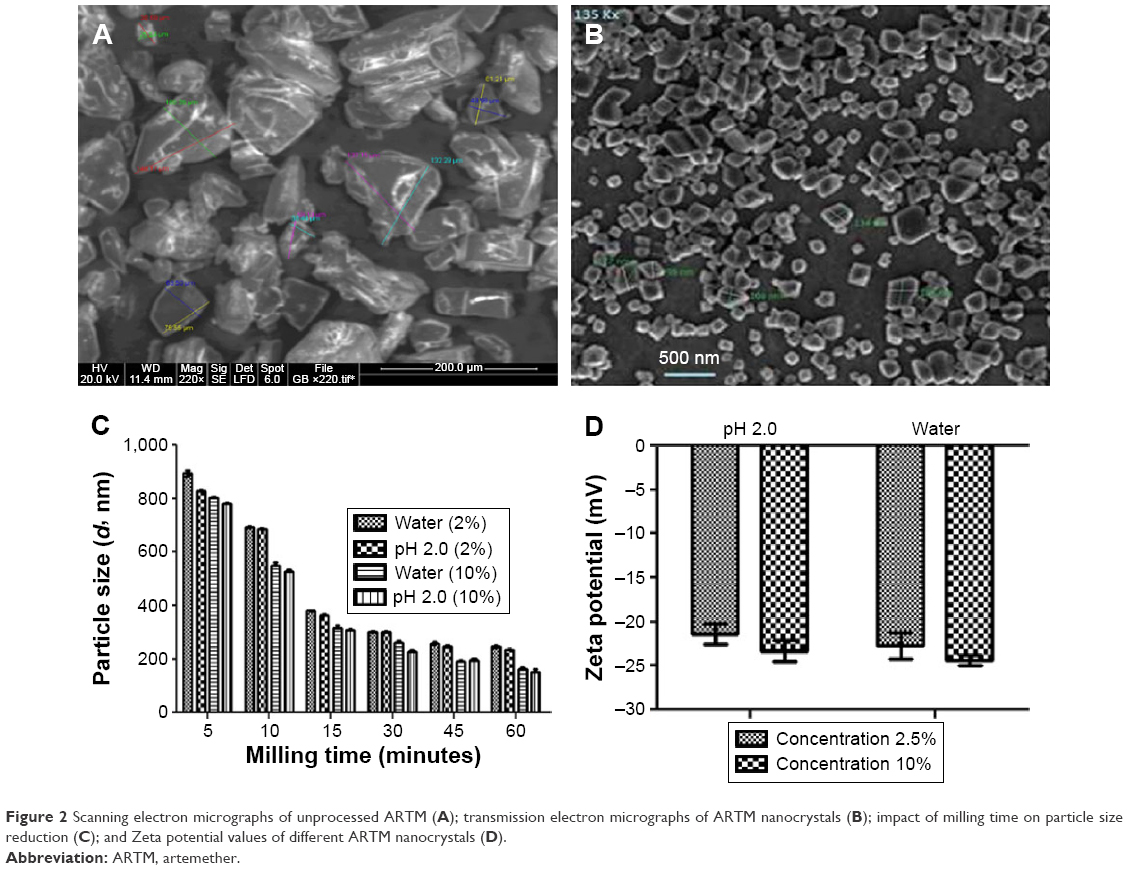 | Figure 2 Scanning electron micrographs of unprocessed ARTM (A); transmission electron micrographs of ARTM nanocrystals (B); impact of milling time on particle size reduction (C); and Zeta potential values of different ARTM nanocrystals (D). |
Stenger and Peukert56 reported that, in wet milling processes, increasing the milling time increases the shear stress, resulting in further reduction in the particle size. In concentrated suspensions, the high stresses and self-attrition forces can potentially cause greater size reduction compared to the dilute suspensions. Furthermore, particles are more likely to collide and interact with the milling medium (beads) resulting in more efficient size reduction. Smaller particles have therefore been achieved for the higher concentration suspension.
The HPLC results show that no new peaks relating to degradation products were observed on the chromatograms. Furthermore, no substantial aggregation or crystal growth was observed for the nanosuspensions prepared at acidic pH (2.0) when stored for 7 days. This suggests that the nanocrystals will remain stable during residence in the stomach following administration of the nanosuspensions to humans, for which typical stomach residence time (20 minutes on an empty stomach) is far shorter than the period over which stability was monitored (7 days) (Table 1).
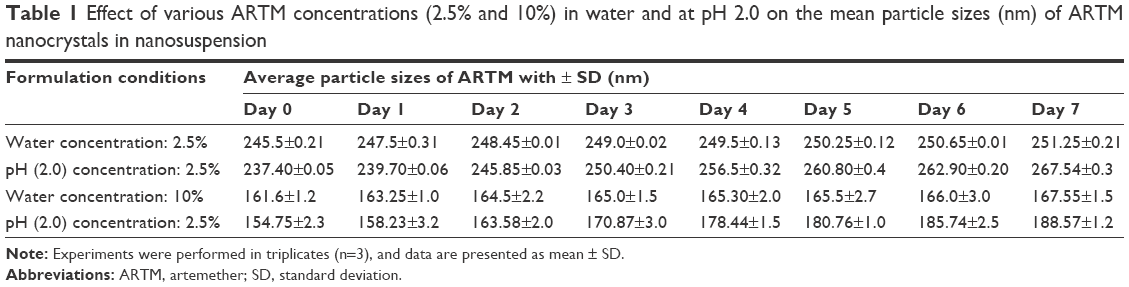 | Table 1 Effect of various ARTM concentrations (2.5% and 10%) in water and at pH 2.0 on the mean particle sizes (nm) of ARTM nanocrystals in nanosuspension |
The percentage of the active agent content of ARTM nanosuspensions (2.5%, w/w) and (10%, w/w) in both water and acidic pH 2.0 was monitored for 7 days. For all four formulations, the percent recovery of the active content did not change significantly (Table 2), which shows that the stability of the drug substance in nanosuspension form is adequate under the conditions tested. In addition, no degradation was observed for both high and low concentration nanosuspensions of ARTM in water and at acidic pH 2.0 over 7 days (Table 2).
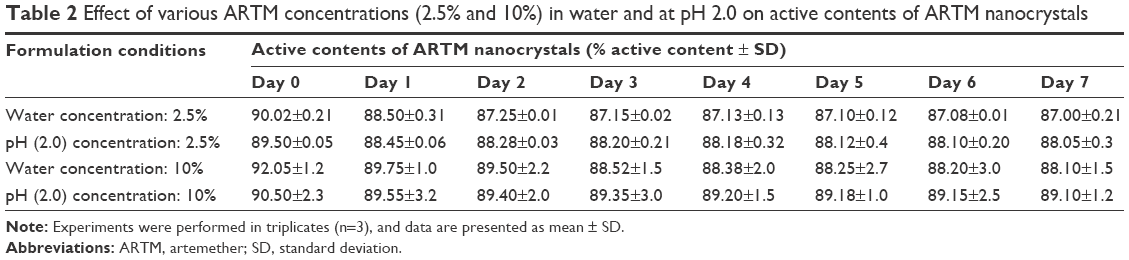 | Table 2 Effect of various ARTM concentrations (2.5% and 10%) in water and at pH 2.0 on active contents of ARTM nanocrystals |
Measurements of zeta potential have also been reported to predict physical stability of formulated nanosuspensions. The zeta potential measurements are dependent on both the surface chemistry of the drug particles and the composition of the stabilizer medium. A range of zeta potential values have previously been reported to give stable nanosuspensions, which include ±30 mV and ±20 mV for electrostatically and sterically stabilized systems, respectively.57,58 In addition, a minimum zeta potential value of ±20 mV has been proposed to deliver adequate stability of nanosuspensions when using both ionic and nonionic polymers and surfactants as stabilizers.59
In this study, the values of zeta potential of ARTM nanosuspensions were found to be within the reported range (±20 mV) (Figure 2D). No significant differences were observed between the tested formulations.
The zeta potential results demonstrated that the combination of HPMC 6cps, PVP, and SLS is sufficient to stabilize nanosuspensions at the selected concentrations. This combination of polymers and surfactants has previously been shown to stabilize drug nanocrystals manufactured using the same top down manufacturing technology.60 In addition, Khan et al reported that the combination of HPMC, PVP, and SLS was very effective at stabilizing nanosuspensions with the excipients being adsorbed onto the surfaces of both crystallized and milled nanoparticles.39
Physical stability studies of ARTM nanocrystals at 2°C–8°C, 25°C, and 40°C for 90 days demonstrated that nanocrystals stored at 2°C–8°C and 25°C (Figure 3A and B) showed improved stability compared to the samples stored at 40°C (Figure 3C). The nanocrystals stored at 2°C–8°C exhibited adequate stability (Figure 3A) with no marked changes in key nanosuspension characteristics. The nanosuspensions stored at 2°C–8°C and 25°C maintained their PDI values, and there was no significant difference (P>0.05, paired t-test, one-way ANOVA) in the mean values of particle size after 90 days storage, which suggests that a homogenous particle size distribution has been achieved after milling, which facilitates the avoidance of Ostwald ripening in the nanosuspensions.61
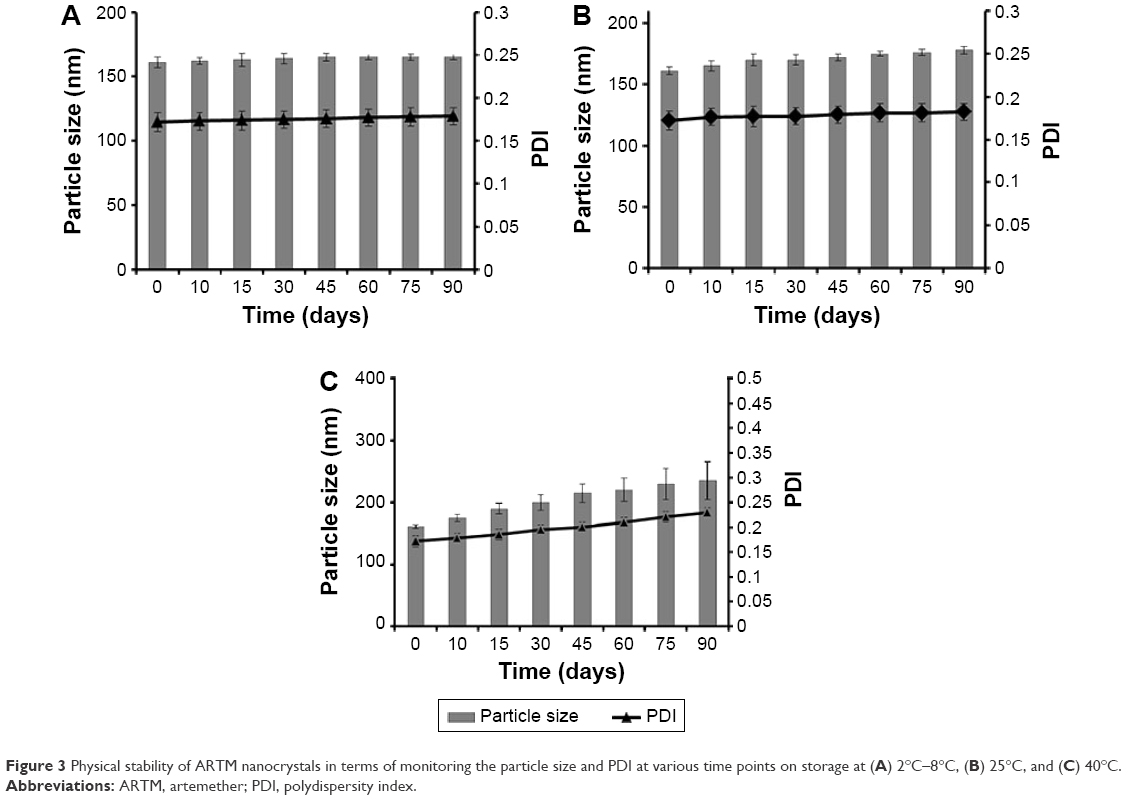 | Figure 3 Physical stability of ARTM nanocrystals in terms of monitoring the particle size and PDI at various time points on storage at (A) 2°C–8°C, (B) 25°C, and (C) 40°C. |
Temperature has previously been reported to demonstrate marked influence on the physical stability of the suspensions, with elevated temperatures increasing the kinetic energy of the suspended particles leading to increased interparticle interactions. The strong van der Waals forces acting between the nanoparticles lead to increased agglomeration and subsequent destabilization of the suspensions.62 Freitas and Müller63 suggested that nanosuspensions should be stored at a temperature range of 2°C–8°C to achieve maximum stability.
DSC and PXRD studies
DSC and PXRD results for the milled ARTM particles revealed that ARTM maintained its physical form and crystallinity after size reduction. For both nanocrystals and unprocessed ARTM, a single sharp melting endotherm was observed (Figure 4A). However, the melting temperature of the raw ARTM API appeared to show a slightly higher melting temperature (86.56°C) than the nanosuspensions (83.78°C). The endothermic peak of the processed samples was also slightly broadened. These differences can be caused by the particle size difference between the two samples. The DSC profile can be strongly affected by the particle size and packing density.64,65 Additionally, broadening of the DSC peaks can be caused by the presence of impurities or traces of the polymers remaining on the surface of the drug particles.65 The PXRD analysis of both unprocessed and processed ARTM produced sharp X-ray chromatograms, which confirmed that after milling ARTM maintained its crystalline nature (Figure 4B). However, PXRD peaks for the unprocessed samples showed greater intensity and sharper peaks.
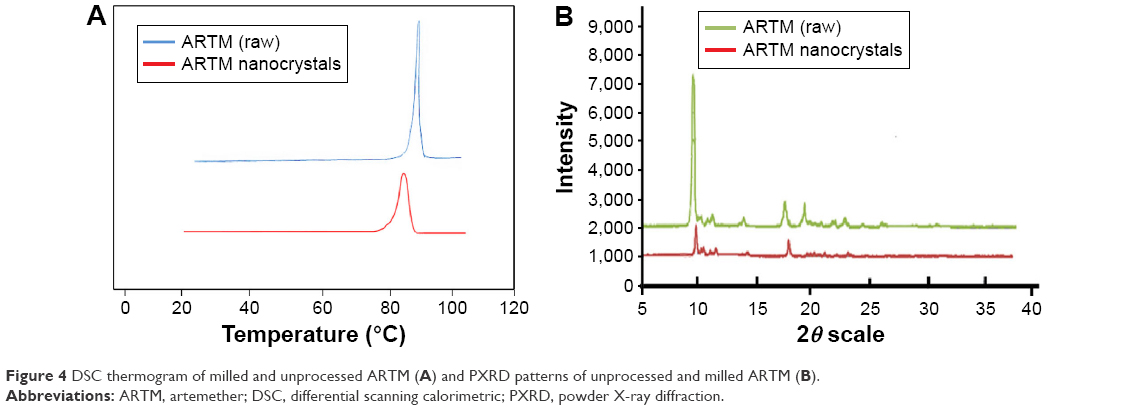 | Figure 4 DSC thermogram of milled and unprocessed ARTM (A) and PXRD patterns of unprocessed and milled ARTM (B). |
These data suggested that both unprocessed ARTM and the particles isolated from the nanosuspension were crystalline in nature. The reduced intensity peaks observed for the milled ARTM are probably related to the smaller particle size of the samples coupled with the presence of traces of the amorphous polymeric stabilizing agent, which contributes to some diffuse scatter.38,39,66 Smaller particles are well known to demonstrate peak broadening with the reduced sharpness of peaks often leading to the disappearance of some peaks.64 In addition, the relatively small sample sizes used for the nanosuspensions raise the risk that fewer particles are exposed to the X-rays, which in turn also leads to reduced peak intensity with lower resolutions of peaks.
Solubility studies
The solubility profile of ARTM nanocrystals and unprocessed ARTM in pure water and stabilizer solution is shown in Figure 5. The solubility of ARTM nanocrystals was found to be 900±2.4 μg/mL, giving an ~3–6-fold increase in solubility, compared to the solubility of unprocessed ARTM in stabilizer solution (321±2.0 μg/mL) and pure water (145±3.0 μg/mL). These differences in solubility were shown to be statistically significant (P<0.05, paired t-test, one-way ANOVA).
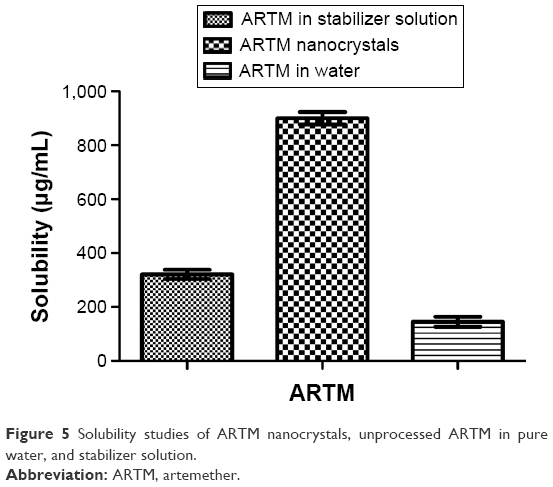 | Figure 5 Solubility studies of ARTM nanocrystals, unprocessed ARTM in pure water, and stabilizer solution. |
Despite water-soluble polymers and surfactants such as HPMC, PVP, and SLS being well known to increase the solubility of drug compounds,67,68 the comparison of the solubility profile of the nanosuspension with that of the unprocessed ARTM dissolved in these same surfactants clearly demonstrates that the nanocrystals provide a substantially increased saturation solubility of ARTM. This phenomenon we believe is ascribed to the marked reduction in particle size (=1,000 nm) and the greatly increased surface area of the particles.
Another possible explanation for the increase in solubility is the production of substantial surface amorphicity following the particle being subject to a high energy comminution process. The results of DSC and X-ray studies, however, show that the ARTM remains predominantly crystalline after size reduction.
The impact of nanonization on the drug and its saturation solubility can be described by the following Freundlich–Ostwald equation:69
|
|
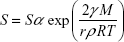
where S is the saturation solubility of nanoparticles, Sα is the saturation solubility of the large crystals, γ is the solid medium interfacial tension, M represents the molecular weight of the substance, r is the radius of particles, ρ represents the density, R is the gas constant, and T represents the temperature. According to this equation, drug solubility will be greater for reduced particle radius. However, Müller et al44 reported that this type of effect is not considerable for larger particles and is more pronounced for the particles in the submicron size range.
Dissolution studies
The results of comparative in vitro dissolution studies of different formulations of ARTM are shown in Figure 6. The figure shows a substantial increase in dissolution rate of the ARTM nanocrystals compared to the marketed tablets (20 mg), unprocessed API (raw drug substance), microsuspension (6.0±2.5 μm), and micronized ARTM powder (8.0±2.5 μm).
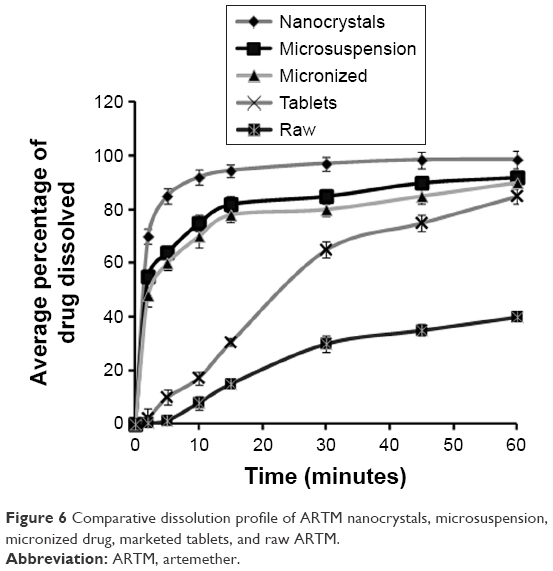 | Figure 6 Comparative dissolution profile of ARTM nanocrystals, microsuspension, micronized drug, marketed tablets, and raw ARTM. |
Figure 6 also shows that ~>70% of drug nanocrystals of ARTM are dissolved in the first 2 minutes compared to the unprocessed API (0.5%) and the commercial tablets (2.2%). Increases in the dissolution rate of 32- and 140-fold were observed for nanocrystals compared to the commercial tablets (20 mg) and unprocessed ARTM powder, respectively. This significant (P<0.05, paired t-test, one-way ANOVA) increase in the dissolution rate of ARTM nanocrystals demonstrated that ARTM nanocrystals maintained their surface area with no marked agglomeration occurring after completion of the size reduction process.
The relationship of surface curvature, vapor pressure, and the particle size can be illustrated by the following Kelvin equation:
|
|
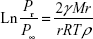
where Pr is the vapor pressure of a particles having the radius r. P∞ is the vapor pressure of an infinitely large particle, γ is the surface tension, R is the gas constant, T is the absolute temperature, M represents the molecular weight, and ρ is the density of the particle. Based on this mathematical expression, nanosized particles having larger surface curvature demonstrate noticeably higher vapor pressure compared to the bigger micronized particles. Molecular transfer from liquid to gas is similar to the transfer of molecules from solid phase to liquid medium. In this case, the dissolution pressure can be converted to vapor pressure that is expected to be enhanced for smaller submicron particles.70
In addition to the enhanced solubility of nanosized particles, the following Prandtl equation demonstrates an important relationship between particle size and hydrodynamic boundary layer (hH);
|
|
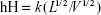
where hH is the hydrodynamic boundary layer, k is a constant, L is the length of the surface in the direction of the fluid flow, and V is the relative velocity of the liquid surrounding the particles.71 When used in combination with the following Noyes–Whitney equation, it is clear that dissolution velocity will have a tendency to increase with reducing the diffusion boundary thickness:
|
|
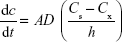
where dc/dt is the rate of drug dissolution, A is the drug surface area accessible for dissolution, D is the drug diffusion coefficient, Cs is the saturation solubility of the drug, Cx is the bulk concentration, and h is the thickness of the boundary layer adjacent to the dissolving drug surface.
In vitro antimalarial activity
P. falciparum and P. vivax were confirmed using optical microscopy. The in vitro antimalarial activity of ARTM nanocrystals showed significant (P<0.05) activity against P. falciparum and P. vivax at the very low concentrations of 0.7 ng/mL and 0.3 ng/mL, respectively (amount of ARTM in nanosuspension was 10%, w/w). However, the vehicle/control comprising aqueous stabilizer (HPMC 6cps, PVP-K30, and SLS) solution showed no activity against the tested strains. The IC50 values of the ARTM nanosuspension against P. falciparum and P. vivax were 28- and 54-fold lower than those of the unprocessed ARTM (20.0 ng/mL and 16.3 ng/mL) and 13- and 21-fold lower than those of the marketed ARTM tablets (9.0 ng/mL and 6.3 ng/mL), respectively. The enhanced in vitro performance of nanoformulation against the selected strains (P. falciparum and P. vivax) is encouraging, and it is because of the smaller particle size, which provides immense surface area resulting in an enhanced dissolution rate and consequently marked absorption.72
In vivo antimalarial activity
The in vivo antimalarial potential of ARTM with respect to antimalarial activity and reduction in the total parasitemia for a range of different pharmaceutical presentations including nanocrystals, unprocessed API, and marketed tablets is shown in Figure 7A and B. As shown in Figure 7A, the highest parasitemia was observed in control group on day 4 (receiving vehicle). The highest parasitemia observed in the control group confirmed that the chosen animal model for the current study was valid and accurate.
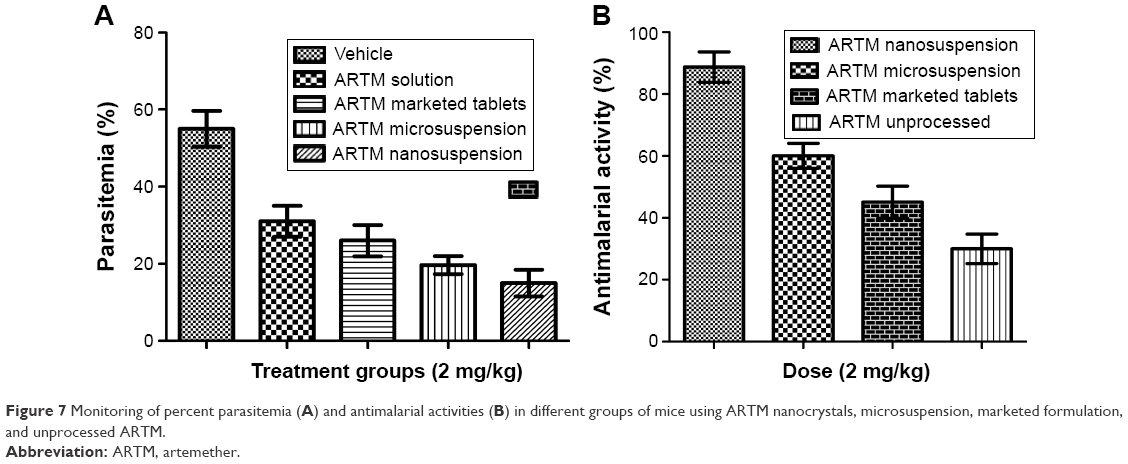 | Figure 7 Monitoring of percent parasitemia (A) and antimalarial activities (B) in different groups of mice using ARTM nanocrystals, microsuspension, marketed formulation, and unprocessed ARTM. |
The different ARTM presentations demonstrated significant antimalarial performance compared to the control group. ARTM nanocrystals showed (P<0.05) the highest reduction in the percent parasitemia (89%) in the treated animals compared to the microsuspension, unprocessed API, and marketed ARTM formulation. The unprocessed API, marketed tablets, and microsuspension were, however, only able to reduce the paracitemia by 27%, 45%, and 60%, respectively, at same dose (2 mg/kg). The antimalarial activity of ARTM nanocrystals was 3.2-, 2-, and 1.5-fold higher than that of the unprocessed API, marketed formulation, and microsuspension with no significant difference in the survival of experimental animals receiving ARTM nanocrystals, unprocessed API, and marketed ARTM formulations (Table 3). These data suggest that the therapeutic window of this drug is reasonably high.
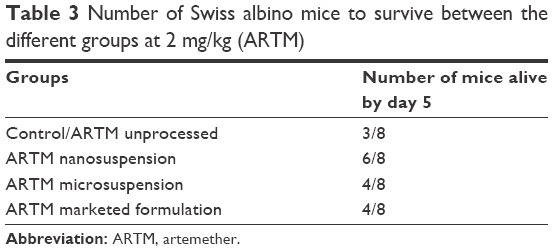 | Table 3 Number of Swiss albino mice to survive between the different groups at 2 mg/kg (ARTM) |
The enhanced antimalarial efficacy of ARTM nanocrystals is probably related to the rapid dissolution of the high surface area of nanoparticles in vivo, with the oral bioavailability likely to be highest for this system in line with hypotheses described in the literature.72 In this regard, the dose of ARTM in nanocrystals form has the potential to be reduced which in turn could potentially reduce the dose-related toxicity of this drug, with the additional opportunity to ameliorate the dose-related resistance problems, which are well known for this very potent drug.
Acute toxicity
LD50 (median lethal oral dose) is a statistically derived single dose of a substance that can be expected to cause death in 50% of treated animals when administered by the oral route. The LD50 value is expressed in terms of weight of test substance per unit weight of test animal (milligram per kilogram). In the acute toxicity studies, no mortality was observed in the groups receiving 500, 1,000, and 1,500 mg/kg dose of both ARTM nanosuspensions after 24 hours. However, animals of the group receiving 2,000 mg/kg were shown to be deceased. It can therefore be concluded that the LD50 value of ARTM nanocrystals is between 1,500 mg/kg and 2,000 mg/kg when given orally. The LD50 value of ARTM was previously reported by Zhou et al,73 which was found to be >2,000 mg/kg. The high LD50 value of ARTM conventional dosage form could be due to its low solubility and hence low bioavailability as compared to nanoformulation, which has also become evident in our solubility and dissolution studies.
Conclusion
A novel top down technology (Dena® DM-100) was shown to be very effective at producing ARTM nanocrystals (<300±2.5 nm) over a short duration (<30 min), with high drug concentrations and improved saturation solubility achieved for both the high concentration (10%, w/w) and low concentration (2.5%, w/w) nanosuspensions.
The smallest average particle size of ARTM (<170±2 nm) was achieved in 60 minutes for the high concentration suspensions (10%, w/w) when processed both in water and at pH 2.0. The combination of HPMC 6cps (0.5%, w/w), PVP-K30 (0.5%, w/w), and SLS (0.1%, w/w) was sufficient to stabilize the nanocrystals in suspension. The nanocrystals showed much increased dissolution rate with ~32- and ~140-fold enhancement in the dissolution rate compared to the marketed tablets and the raw unprocessed ARTM.
In both the in vitro and in vivo studies, the antiplasmodial activity of the produced ARTM nanocrystals is compared favorably with that of the raw unprocessed API and the marketed tablet product. The nanocrystals of ARTM demonstrated 3.2-, 2-, and 1.5-fold higher activity against the selected strains (P. vivax) compared to those of the unprocessed API, marketed tablets, and microsuspension, respectively. Moreover, the nanocrystals of ARTM also demonstrated 28- and 54-fold lower IC50 values compared to those of the unprocessed API and marketed ARTM tablets, respectively, against P. falciparum, while also demonstrating 13- and 21-fold improvements for the same presentations against P. vivax.
These data clearly demonstrate that the nanocrystals of ARTM produced using a top down size reduction technology have superior physicochemical characteristics, which lead to improved in vivo antimalarial performance, compared to those of unprocessed API, micronized drug, and commercially available ARTM tablets. The produced ARTM nanocrystals demonstrated rapid in vitro dissolution, which provided benefits in vivo drug absorption and resultant improved in vivo antiplasmodial activity.
This study clearly demonstrated that the Dena® DM-100 system is able to rapidly and efficiently reduce the particle size to the level at which the dissolution rate and bioavailability of ARTM are enhanced markedly. These results suggest that the ARTM nanocrystal preparation could have the potential to demonstrate improved therapeutic performance in humans; hence, future work will be focused on completing the experimental studies, which could provide a line of sight to the clinical evaluation of the nanocrystal system.
Acknowledgments
The authors gratefully acknowledge the Department of Pharmacy, University of Malakand, Chakdara, Dir (L), KPK, Pakistan; Faculty of Pharmacy, Sarhad University Peshawar, Pakistan; Institute of Life Sciences Research, School of Pharmacy, University of Bradford, West Yorkshire, UK; and Universiti Teknologi MARA for support and providing resources to execute this research work. The authors are also thankful to the Deanship of Scientific Research, King Saud University, for funding the work through the research group project no RGP-210.
Disclosure
The authors report no conflicts of interest in this work.
References
Meshnick SR, Dobson MJ. The history of antimalarial drugs. In: Rosenthal PJ, editor. Antimalarial Chemotherapy. Springer; 2001:15–25. | ||
Bhatt S, Weiss D, Cameron E, et al. The effect of malaria control on Plasmodium falciparum in Africa between 2000 and 2015. Nature. 2015;526(7572):207–211. | ||
Snounou G, Viriyakosol S, Jarra W, Thaithong S, Brown KN. Identification of the four human malaria parasite species in field samples by the polymerase chain reaction and detection of a high prevalence of mixed infections. Mol Biochem Parasitol. 1993;58(2):283–292. | ||
Wiesner J, Ortmann R, Mitsch A, et al. Inhibitors of farnesyltransferase: 5-arylacryloyl-aminobenzophenones show antimalarial activity. Int J Pharm Sci. 2003;58(4):289–290. | ||
Wiesner J, Kettler K, Sakowski J, et al. Farnesyltransferase inhibitors inhibit the growth of malaria parasites in vitro and in vivo. Angew Chem Int Ed. 2004;43(2):251–254. | ||
Wiesner J, Ortmann R, Jomaa H, Schlitzer M. Double ester prodrugs of FR900098 display enhanced in-vitro antimalarial activity. Arch Pharm (Weinheim). 2007;340(12):667–669. | ||
Khatoon L, Baliraine FN, Bonizzoni M, Malik SA, Yan G. Genetic structure of Plasmodium vivax and Plasmodium falciparum in the Bannu district of Pakistan. Malar J. 2010;9(1):112. | ||
WHO. World Malaria Report 2011. Geneva: World Health Organization; 2011. | ||
Basseri H, Raeisi A, Holakouie K, Shanadeh K. Malaria prevention among afghan refugees in a malarious area, southeastern Iran. Bull Soc Pathol Exot. 2010;103(5):340–345. | ||
Panisko DM, Keystone JS. Treatment of malaria – 1990. Drugs. 1990;39(2):160–189. | ||
Okiro EA, Alegana VA, Noor AM, Snow RW. Changing malaria intervention coverage, transmission and hospitalization in Kenya. Malar J. 2010;9(285). | ||
Nayyar GM, Breman JG, Newton PN, Herrington J. Poor-quality antimalarial drugs in southeast Asia and sub-Saharan Africa. Lancet Infect Dis. 2012;12(6):488–496. | ||
Falade C, Makanga M, Premji Z, Ortmann C-E, Stockmeyer M, de Palacios PI. Efficacy and safety of artemether–lumefantrine (Coartem®) tablets (six-dose regimen) in African infants and children with acute, uncomplicated falciparum malaria. Trans R Soc Trop Med Hyg. 2005;99(6):459–467. | ||
Ridley RG. Medical need, scientific opportunity and the drive for antimalarial drugs. Nature. 2002;415(6872):686–693. | ||
Bunnag D, Viravan C, Looareesuwan S, Karbwang J, Harinasuta T. Clinical trial of artesunate and artemether on multidrug resistant falciparum malaria in Thailand. A preliminary report. Southeast Asian J Trop Med Public Health. 1991;22(3):380–385. | ||
El-Dakdoky M. Evaluation of the developmental toxicity of artemether during different phases of rat pregnancy. Food Chem Toxicol. 2009;47(7):1437–1441. | ||
Klayman DL. Qinghaosu (artemisinin): an antimalarial drug from China. Science. 1985;228(4703):1049–1055. | ||
Fule RA, Meer TS, Sav AR, Amin PD. Artemether-soluplus hot-melt extrudate solid dispersion systems for solubility and dissolution rate enhancement with amorphous state characteristics. J Pharm (Cairo). 2013;2013:151432. | ||
Sinha S, Medhi B, Sehgal R. Challenges of drug-resistant malaria. Parasite. 2014;21:61. | ||
Tayade N, Nagarsenker MS. Development and evaluation of artemether parenteral microemulsion. Indian J Pharm Sci. 2010;72(5):637. | ||
Umapathi P, Ayyappan J, Quine SD. Development and validation of a dissolution test method for artemether and lumefantrine in tablets. Trop J Pharm Res. 2011;10(5):643–653. | ||
Mandawgade SD, Sharma S, Pathak S, Patravale VB. Development of SMEDDS using natural lipophile: application to β-artemether delivery. Int J Pharm. 2008;362(1):179–183. | ||
Serajuddin A. Solid dispersion of poorly water-soluble drugs: early promises, subsequent problems, and recent breakthroughs. J Pharm Sci. 1999;88(10):1058–1066. | ||
Aungst BJ. Novel formulation strategies for improving oral bioavailability of drugs with poor membrane permeation or presystemic metabolism. J Pharm Sci. 1993;82(10):979–987. | ||
Jang D-J, Jeong EJ, Lee H-M, Kim B-C, Lim S-J, Kim C-K. Improvement of bioavailability and photostability of amlodipine using redispersible dry emulsion. Eur J Pharm Sci. 2006;28(5):405–411. | ||
Lawrence MJ, Rees GD. Microemulsion-based media as novel drug delivery systems. Adv Drug Deliv Rev. 2000;45(1):89–121. | ||
Sahu G, Sharma H, Gupta A, Kaur C. Advancements in microemulsion based drug delivery systems for better therapeutic effects. Int J Pharm Sci Dev Res. 2015;1(1):008–015. | ||
Charoenchaitrakool M, Dehghani F, Foster N, Chan H. Micronization by rapid expansion of supercritical solutions to enhance the dissolution rates of poorly water-soluble pharmaceuticals. Ind Eng Chem Res. 2000;39(12):4794–4802. | ||
Vo CL-N, Park C, Lee B-J. Current trends and future perspectives of solid dispersions containing poorly water-soluble drugs. Eur J Pharm Biopharm. 2013;85(3):799–813. | ||
Schwarz C, Mehnert W, Lucks J, Müller R. Solid lipid nanoparticles (SLN) for controlled drug delivery. I. Production, characterization and sterilization. J Control Release. 1994;30(1):83–96. | ||
Pattni BS, Chupin VV, Torchilin VP. New developments in liposomal drug delivery. Chem Rev. 2015;115(19):10938–10966. | ||
Loftsson T, Brewster ME. Pharmaceutical applications of cyclodextrins. 1. Drug solubilization and stabilization. J Pharm Sci. 1996;85(10):1017–1025. | ||
Jones M-C, Leroux J-C. Polymeric micelles – a new generation of colloidal drug carriers. Eur J Pharm Biopharm. 1999;48(2):101–111. | ||
Huang Y, Dai W-G. Fundamental aspects of solid dispersion technology for poorly soluble drugs. Acta Pharm Sin B. 2014;4(1):18–25. | ||
Narang AS, Delmarre D, Gao D. Stable drug encapsulation in micelles and microemulsions. Int J Pharm. 2007;345(1):9–25. | ||
Merisko-Liversidge E, Liversidge GG, Cooper ER. Nanosizing: a formulation approach for poorly water-soluble compounds. Eur J Pharm Sci. 2003;18(2):113–120. | ||
Khadka P, Ro J, Kim H, et al. Pharmaceutical particle technologies: an approach to improve drug solubility, dissolution and bioavailability. Asian J Pharm Sci. 2014;9(6):304–316. | ||
O’Mahony M, Leung AK, Ferguson S, Trout BL, Myerson AS. A process for the formation of nanocrystals of active pharmaceutical ingredients with poor aqueous solubility in a nanoporous substrate. Org Process Res Dev. 2015;19(9):1109–1118. | ||
Khan S, Matas MD, Zhang J, Anwar J. Nanocrystal preparation: low-energy precipitation method revisited. Cryst Growth Des. 2013;13(7):2766–2777. | ||
Li M, Azad M, Davé R, Bilgili E. Nanomilling of drugs for bioavailability enhancement: a holistic formulation-process perspective. Pharmaceutics. 2016;8(2):17. | ||
Schade H. Kolloidchemie und Balneologie. Colloid Polym Sci. 1909;5(1):2–8. | ||
Freundlich H. Neuere Fortschritte der Kolloidchemie und ihre biologische Bedeutung. Protoplasma. 1927;2(1):278–299. | ||
Wu W, Nancollas GH. A new understanding of the relationship between solubility and particle size. J Solution Chem. 1998;27(6):521–531. | ||
Müller R, Jacobs C, Kayser O. Nanosuspensions as particulate drug formulations in therapy: rationale for development and what we can expect for the future. Adv Drug Deliv Rev. 2001;47(1):3–19. | ||
Plakkot S, De Matas M, York P, Saunders M, Sulaiman B. Comminution of ibuprofen to produce nano-particles for rapid dissolution. Int J Pharm. 2011;415(1–2):307–314. | ||
Sulaiman B, Inventor. “The Milling System”. Patent No: WO/2007/0204072007. 2007. | ||
Van Eerdenbrugh B, Vermant J, Martens JA, et al. Solubility increases associated with crystalline drug nanoparticles: methodologies and significance. Mol Pharm. 2010;7(5):1858–1870. | ||
Gao L, Zhang D, Chen M. Drug nanocrystals for the formulation of poorly soluble drugs and its application as a potential drug delivery system. J Nanopart Res. 2008;10(5):845–862. | ||
Thakkar H, Nangesh J, Parmar M, Patel D. Formulation and characterization of lipid-based drug delivery system of raloxifene-microemulsion and self-microemulsifying drug delivery system. J Pharm Bioallied Sci. 2011;3(3):442. | ||
Pawar JN, Shete RT, Gangurde AB, et al. Development of amorphous dispersions of artemether with hydrophilic polymers via spray drying: physicochemical and in silico studies. Asian J Pharm Sci. 2016;11(3):385–395. | ||
Shekunov BY, Chattopadhyay P, Seitzinger J, Huff R. Nanoparticles of poorly water-soluble drugs prepared by supercritical fluid extraction of emulsions. Pharm Res. 2006;23(1):196–204. | ||
Trager W, Jensen JB. Human malaria parasites in continuous culture. Science. 1976;193(4254):673–675. | ||
Fidock DA, Rosenthal PJ, Croft SL, Brun R, Nwaka S. Antimalarial drug discovery: efficacy models for compound screening. Nat Rev Drug Discov. 2004;3(6):509–520. | ||
Peters W. The chemotherapy of rodent malaria, XXII. The value of drug-resistant strains of P. berghei in screening for blood schizontocidal activity. Ann Trop Med Parasitol. 1975;69(2):155–171. | ||
OECD. Acute Inhalation Toxicity – Fixed Concentration Procedure. Available from: http://www.oecd.org/dataoecd/63/7/32035886.pdf. Accessed February 2, 2008. | ||
Stenger F, Peukert W. The role of particle interactions on suspension rheology–application to submicron grinding in stirred ball mills. Chem Eng Tech. 2003;26(2):177–183. | ||
Yang JZ, Young AL, Chiang PC, Thurston A, Pretzer DK. Fluticasone and budesonide nanosuspensions for pulmonary delivery: preparation, characterization, and pharmacokinetic studies. J Pharm Sci. 2008;97(11):4869–4878. | ||
Jacobs C, Müller RH. Production and characterization of a budesonide nanosuspension for pulmonary administration. Pharm Res. 2002;19(2):189–194. | ||
Ali HS, York P, Blagden N. Preparation of hydrocortisone nanosuspension through a bottom-up nanoprecipitation technique using microfluidic reactors. Int J Pharm. 2009;375(1):107–113. | ||
Plakkot S, de Matas M, York P, Saunders M, Sulaiman B. Comminution of ibuprofen to produce nano-particles for rapid dissolution. Int J Pharm. 2011;415(1–2):307–314. | ||
Deng J, Huang L, Liu F. Understanding the structure and stability of paclitaxel nanocrystals. Int J Pharm. 2010;390(2):242–249. | ||
Wu L, Zhang J, Watanabe W. Physical and chemical stability of drug nanoparticles. Adv Drug Deliv Rev. 2011;63(6):456–469. | ||
Freitas C, Müller RH. Effect of light and temperature on zeta potential and physical stability in solid lipid nanoparticle (SLN™) dispersions. Int J Pharm. 1998;168(2):221–229. | ||
Bunjes H, Koch MH, Westesen K. Effect of particle size on colloidal solid triglycerides. Langmuir. 2000;16(12):5234–5241. | ||
Valleri M, Mura P, Maestrelli F, Cirri M, Ballerini R. Development and evaluation of glyburide fast dissolving tablets using solid dispersion technique. Drug Dev Ind Pharm. 2004;30(5):525–534. | ||
Ali HS, York P, Ali AM, Blagden N. Hydrocortisone nanosuspensions for ophthalmic delivery: a comparative study between microfluidic nanoprecipitation and wet milling. J Control Release. 2011;149(2):175–181. | ||
Leuner C, Dressman J. Improving drug solubility for oral delivery using solid dispersions. Eur J Pharm Biopharm. 2000;50(1):47–60. | ||
Aziz HA, Tan YTF, Peh KK. Solubility of drugs in aqueous polymeric solution: effect of ovalbumin on microencapsulation process. AAPS PharmSciTech. 2012;13(1):35–45. | ||
Kesisoglou F, Panmai S, Wu Y. Nanosizing – oral formulation development and biopharmaceutical evaluation. Adv Drug Del Rev. 2007;59(7):631–644. | ||
Junghanns J-UAH, Mueller RH. Nanocrystal technology, drug delivery and clinical applications. Int J Nanomedicine. 2008;3(3):295–309. | ||
Kayaert P, Van den Mooter G. Is the amorphous fraction of a dried nanosuspension caused by milling or by drying? A case study with naproxen and cinnarizine. Eur J Pharm Biopharm. 2012;81(3):650–656. | ||
Patravale V, Date AA, Kulkarni R. Nanosuspensions: a promising drug delivery strategy. J Pharm Pharmacol. 2004;56(7):827–840. | ||
Zhou Y, Ning D, Wang S, et al. Antimalarial compositions: Google Patents; 1997. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.