")
Back to Journals » Drug Design, Development and Therapy » Volume 18
Propofol and Dexmedetomidine Ameliorate Endotoxemia-Associated Encephalopathy via Inhibiting Ferroptosis
Authors Zhou Y , Yang Y, Yi L, Pan M, Tang W, Duan H
Received 4 January 2024
Accepted for publication 19 April 2024
Published 23 April 2024 Volume 2024:18 Pages 1349—1368
DOI https://doi.org/10.2147/DDDT.S458013
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tuo Deng
Ye Zhou, Yangliang Yang, Liang Yi, Mengzhi Pan, Weiqing Tang, Hongwei Duan
Department of Anesthesiology, Shanghai Pudong Hospital, Fudan University Pudong Medical Center, Shanghai, People’s Republic of China
Correspondence: Hongwei Duan, Email [email protected]
Background: Sepsis is recognized as a multiorgan and systemic damage caused by dysregulated host response to infection. Its acute systemic inflammatory response highly resembles that of lipopolysaccharide (LPS)-induced endotoxemia. Propofol and dexmedetomidine are two commonly used sedatives for mechanical ventilation in critically ill patients and have been reported to alleviate cognitive impairment in many diseases. In this study, we aimed to explore and compare the effects of propofol and dexmedetomidine on the encephalopathy induced by endotoxemia and to investigate whether ferroptosis is involved, finally providing experimental evidence for multi-drug combination in septic sedation.
Methods: A total of 218 C57BL/6J male mice (20– 25 g, 6– 8 weeks) were used. Morris water maze (MWM) tests were performed to evaluate whether propofol and dexmedetomidine attenuated LPS-induced cognitive deficits. Brain injury was evaluated using Nissl and Fluoro-Jade C (FJC) staining. Neuroinflammation was assessed by dihydroethidium (DHE) and DCFH-DA staining and by measuring the levels of three cytokines. The number of Iba1+ and GFAP+ cells was used to detect the activation of microglia and astrocytes. To explore the involvement of ferroptosis, the levels of ptgs2 and chac1; the content of iron, malondialdehyde (MDA), and glutathione (GSH); and the expression of ferroptosis-related proteins were investigated.
Conclusion: The single use of propofol and dexmedetomidine mitigated LPS-induced cognitive impairment, while the combination showed poor performance. In alleviating endotoxemic neural loss and degeneration, the united sedative group exhibited the most potent capability. Both propofol and dexmedetomidine inhibited neuroinflammation, while propofol’s effect was slightly weaker. All sedative groups reduced the neural apoptosis, inhibited the activation of microglia and astrocytes, and relieved neurologic ferroptosis. The combined group was most prominent in combating genetic and biochemical alterations of ferroptosis. Fpn1 may be at the core of endotoxemia-related ferroptosis activation.
Keywords: propofol, dexmedetomidine, endotoxemia, encephalopathy, ferroptosis
Introduction
Over 20 million patients suffer from sepsis worldwide each year, standing as a prominent contributor to fatalities within the Intensive Care Unit (ICU) setting.1,2 During the progression of sepsis, intense inflammatory storm provokes the overactivation of immune system, ultimately causing immunosuppression.3,4 Excessive cytokines can penetrate the blood–brain barrier and aggravate neuroinflammation and secondary neurodegeneration.5 Endotoxemia triggered by LPS administration can closely mimic this acute inflammatory response of sepsis.6
Ferroptosis is a form of non-apoptotic cell debris, featured by the iron-dependent peroxidation of membrane lipids.7 Overaccumulation of iron catalyzes the production of reactive oxygen species (ROS) through Fenton reaction, facilitating the hydroxylation of unsaturated fatty acids (UFA). This process compromises the structure and function of the cell membrane, and activates ferroptosis-related lipid peroxidation.8,9 As research advances, the intricate relationship between ferroptosis and endotoxemia has been progressively unraveled and substantiated. The reinforcement of ferroptosis by ferroportin (Fpn1) knockdown enhances the atrial fibrillation (AF) vulnerability in endotoxemia rat model.10 Meanwhile, resveratrol has been confirmed to protect against LPS-mediated myocardial injury through elevating miR-149, which inhibits ferroptosis.11
Propofol is a short-acting intravenous anesthetic that is widely used for swift induction, stable maintenance, and prompt recovery.12 Dexmedetomidine, a specific α2-adrenoceptor agonist, provides efficient sedation, with a slight analgesic effect.13 The Society of Critical Care Medicine (SCCM) recommends the use of propofol and dexmedetomidine during endotracheal intubation to achieve a low level of sedation.14 Compared with propofol, dexmedetomidine presents distinct benefits, such as anti-inflammatory properties, promotion of biomimetic sleep, and diminishment of acute brain dysfunction risk. However, under light sedation, dexmedetomidine and propofol showed no differential impact on survival outcomes or on the duration of ICU stay for mechanically ventilated adult patients with sepsis.2 Although dexmedetomidine reduces the incidence of delirium in post-cardiac surgery patients undergoing mechanical ventilation, it potentiates the occurrence of bradycardia.15
Balanced general anesthesia, signifying the combination of various anesthetics during surgery, has evolved into the most prevalent management strategy for clinical anesthesia.16 During the perioperative period, dexmedetomidine, often utilized as an adjunct in propofol-dominated anesthesia, can proficiently maintain hemodynamic stability owing to its outstanding anti-central sympathetic effects.17 This conjoint medication is also applicable for sedation in critically ill patients. Selective escalation of the propofol dose with steady dexmedetomidine infusion reduces adjusted 90-day mortality, whereas higher concentrations of dexmedetomidine are associated with increased mortality in patients aged ≤65 years old.18 Simultaneously, dexmedetomidine, as the sole agent, cannot consistently attain intended sedation in all clinical scenarios, necessitating partial supplementation with propofol.19 So far, propofol and dexmedetomidine have been reported to participate in the pathogenesis of several diseases by influencing ferroptotic pathways. For instance, propofol enhances the anti-tumor effects of doxorubicin and paclitaxel in triple-negative breast cancer through the activation of ferroptosis.20 It can also mitigate myocardial ischemia-reperfusion injury (IRI) by repressing AKT/p53-related ferroptosis.12 Apart from the role in cancer and IRI, dexmedetomidine has also been proven to protect against sepsis-associated organ dysfunctions.21 However, research on the effects of propofol and dexmedetomidine on LPS-induced cognitive impairment remains limited.
To date, dexmedetomidine has been proven to alleviate neuroinflammation in sepsis-associated encephalopathy (SAE) mice through α2 adrenergic receptors on astrocytes22 and to improve cognitive performance in aged mice challenged with LPS by sustaining iron homeostasis.23 However, whether propofol owns similar effects has not been explored. Furthermore, whether the combination of dexmedetomidine and propofol in endotoxemia models mimics the potent synergy observed in perioperative anesthesia or offers distinct benefits, as seen in mechanical ventilation sedation, along with the potential involvement of the ferroptosis pathway, is not only intriguing but also holds remarkable value.
Materials and Methods
Animals
A cohort of 218 C57BL/6J male mice (20–25 g, 6–8 weeks) were procured from Beijing Vital River Laboratory Animal Technology. The animals were housed under a 12-hour light–dark cycle (lights on at 8:00 a.m. and off at 8:00 p.m.) and a 25°C temperature environment free of chow and water, conforming to specific pathogen-free (SPF) standards. All animal experimental procedures were approved by the Animal Experimental Ethics Committee of the Fudan University.
Drugs and Treatments
A moderate intraperitoneal LPS injection (5 mg/kg) was used to stir up endotoxemia.24 Propofol was procured from Guorui Pharmaceutical Ltd. (H20143252). LPS was purchased from Sigma-Aldrich (L2880). Dexmedetomidine was obtained from the Yangtze River Pharmaceutical Group (22081331).
In preliminary experiments, mice were randomly assigned to eight groups (n = 6): c (control), lps, l + p50 (lps + p50, propofol 50 mg/kg i.p.), l + p75 (1ps + p75, propofol 75 mg/kg i.p.), l + p100 (lps + p100, propofol 100 mg/kg i.p.), l + d50 (1ps + d50, dexmedetomidine 50 μg/kg i.p.), l + d75 (lps + d75, dexmedetomidine 75 μg/kg i.p.), and l + d100 (lps + d100, dexmedetomidine 100 μg/kg i.p.). The results showed that propofol and dexmedetomidine all had dose-dependent mitigation of LPS-induced neural loss and degeneration, while no significant statistical difference was observed between the medium-dose and high-dose groups (p < 0.05) (Figure S1A–L). Therefore, we chose a relatively more efficient and lower dose of propofol (75 mg/kg i.p.) and dexmedetomidine (75 μg/kg i.p.) to reduce the adverse effects of drug loading. Subsequently, the mice were randomly assigned to five groups: c (control), lps, l + p (lps + p), l + d (lps + d), and l + p + d (lps + p + d). Propofol was diluted to 75 mg/kg with intralipid and administered 1 h before dexmedetomidine administration. Dexmedetomidine was diluted to 75 μg/kg with saline and injected 1 h before LPS treatment. LPS was diluted in saline to a concentration of 5 mg/kg.
Morris Water Maze (MWM) Test
After 72 h from the LPS intervention, when the inflammatory response in the mice had nearly subsided, adaptive training was conducted. Mice were gently placed into water and allowed to swim freely for 60s. Subsequently, spatial navigation trainings were carried out. Each mouse was introduced into the water from an identical location in each of the four quadrants. During spatial exploration time, the platform was removed and the mice were placed in the symmetrical quadrant of the platform. Behavioral trajectories, first escape latency, platform crossing times, time spent in the target quadrant, and swim speed were recorded. All daily experiments were conducted at 8:00 p.m., with the water temperature maintained at 21°C and the environment kept quiet and clean.
Nissl Staining
Twenty-four hours after the final LPS administration, the mice were sacrificed to obtain the brain tissue. After immobilization with 4% paraformaldehyde for 24 h, the brains were dehydrated using a series of alcohol gradients and embedded in paraffin. Dewaxed and rehydrated slices were stained with toluidine blue at 56°C for 2.5 h followed by differentiation. Finally, the slices were rendered transparent in xylene and sealed with neutral gum.
Fluor-Jade C (FJC) Staining
Degenerative neurons were stained using the Fluoro-Jade C (FJC) Staining Kit (G3262, Beijing Solarbio Science & Technology Co., Ltd.), in accordance with the manufacturer’s guidelines. Briefly, the frozen slices underwent a 5-minute immersion in solution A, followed by a 2-minute transfer to 70% ethanol. After a 10-minute bleaching with solution B, the sections were incubated in the dark for 10 min with solution C. Treated slices were snapped using the FITC channel.
Cell Culture and Intervention
HT22 cells (a mouse hippocampal neuron cell line) purchased from Cell Bank, Shanghai, were cultured in DMEM (C11995500BT, Gibco) containing 10% fetal bovine serum (FBS) (F9052, UElandy) and 1% penicillin–streptomycin (15,140–122, Gibco) in 5% CO2 at 37°C. The cells were digested with 0.25% trypsin containing EDTA (25,200–056, Gibco), centrifuged at approximately 80% of the T25 culture bottle, and passaged at a ratio of 1:2. We established a concentration gradient to determine the median lethal dose of LPS and the optimal concentrations of propofol and dexmedetomidine for effectively reversing LPS-induced neuronal toxicity. The groups are as follows: c (control), l1 (1 μg/mL LPS), l2 (2 μg/mL LPS), l5 (5 μg/mL LPS), l10 (10 μg/mL LPS), and l20 (20 μg/mL LPS); lps (20 μg/mL LPS), lp2 (lps + p, 2 μg/mL propofol), lp5 (lps + p, 5 μg/mL propofol), and lp10 (lps + p, 10 μg/mL propofol); ld5 (lps + d, 5 nM dexmedetomidine), ld10 (lps + d, 10 nM dexmedetomidine), and ld20 (lps + d, 20 nM dexmedetomidine). Based on these results, LPS concentration was determined to be 20 μg/mL, with co-incubation concentrations of propofol at 10 μg/mL and dexmedetomidine at 20 nM.
Cell Counting Kit-8 (CCK-8)
Cell viability was assessed using the CCK-8 assay. Suspended cells were seeded in 96-well plates at a density of 5000 cells per well. After incubation with the drugs for 24 h, 10 μL CCK-8 agent with 90 μL basal medium without FBS was added to the cell well for 3-hour reaction. The absorbance in 450 nm wavelength was measured by an enzyme labeling instrument.
Dihydroethidium (DHE) Staining
Dihydroethidium (DHE) is a fluorescent probe commonly used for the detection of ROS, especially superoxide anions. Frozen slices and cell crawling were shielded with 16 μM DHE (50102ES02, Yeasen) for 30 min, followed by a 10-minute DAPI (2 μg/mL, 40728ES03, Yeasen). Images were visualized using an ethidium homodimer-1 channel.
Immunohistochemistry (IHC) Staining
After antigen retrieval in sodium citrate solution (pH 6.0) (G1202, servicebio), the cooled paraffin slices were covered with 3% H2O2 in the dark to inactivate endogenous peroxidase. PBS containing 5% goat serum and 1% Triton X-100 was used to permeabilize the cell membrane for 1 h, followed by blocking for 1 h. The sections were then incubated with primary antibodies (rabbit anti-Iba1, 1:300, 10904-1-AP; Proteintech) at 4°C overnight. The secondary antibody, goat anti-rabbit (HRP) (1:200, A0208, Beyotime), was incubated for 1 h at room temperature. DAB chromogenic solution (PR30010, Proteintech) and hematoxylin were used to visualize microglia and nuclei.
DCFH-DA
DCFH-DA can freely traverse the cell membrane and is hydrolyzed by intracellular ROS, primarily hydrogen peroxide (H2O2), into fluorescent DCFH. Following the manufacturer’s instructions (CA1410, Solarbio), 1 mL of the DCFH-DA probe diluted in serum-free culture medium (1:1000) was added to each well and incubated for 20 min at 37°C. Cell distribution, number, and morphology were determined using bright-field microscopy, while ROS content was assessed using fluorescence intensity in the FITC channel.
Cell Crawling Immunofluorescence Staining
Two hundred microliters of cell suspension with approximately 100,000 cells was tiled on aseptic cover slides (24 × 24 mm) attached to 6-well plates, and 2 mL complete medium was added for cultivation after cell adherence for 6 h. After 24-hour drug incubation, the cells were fixed using 4% paraformaldehyde. The cells were then permeabilized for 20 min and obstructed for 30 min. Primary antibodies, including rabbit anti-CD71 (1:300, 10084-2-AP, Proteintech), rabbit anti-Fpn1 (1:300, 26601-1-AP, Proteintech), and rabbit anti-Nrf2 (1:300, 16396-1-AP, Proteintech), were incubated overnight at 4°C. The treated slides were incubated with Alexa Fluor 594 IgG goat anti-rabbit (1:1000, ab150080, Abcam) for 1 h at room temperature. Nrf2-stained slides experienced with 100 nM phalloidin staining (40735ES75; Yeasen) before 10-minute of DAPI incubation.
Fluorescent Double Labeling with Laser Confocal Microscopy
After antigen retrieval, permeabilization, and blockage, the paraffin slices were incubated with rabbit anti-GFAP (1:300, 16825-1-AP, Proteintech) and mouse anti-Neun (1:300, 66836-1-Ig, Proteintech) overnight at 4°C. Secondary antibodies, including Alexa Fluor 594 IgG goat anti-rabbit and multi-rAb CoraLite Plus 488-goat anti-mouse (1:1000, RGAM002, Proteintech), were added for 1 h at room temperature. After sealing with an anti-fluorescence quencher, scanning was performed using a Zeiss super-resolution laser scanning microscope LSM900 with Airyscan 2. Seven layers of sliced images (1 μm per layer) were scanned in the optimal signal mode of Airyscan using Airyscan 3D processing, and a maximum intensity projection (MIP) was applied. Considering a slice thickness of 8 μm, the projected image captured almost all slice details.
Western Blot
Fresh tissue and cells were disrupted with a high-strength RIPA lysis buffer, followed by 30 min of standing and centrifugation at 12,000 rpm at 4°C for 10 min. The protein content of the supernatant was quantified using a bicinchoninic acid (BCA) standard curve. Ten microliters of samples with equal amounts of total protein were added to 12.5% or 7.5% gels and electrophoresed at a stable voltage of 200 V. The gels were then transferred onto a PVDF membrane at a constant current of 400 mA. After blocking with 5% skimmed milk for 2 h, the membranes were incubated overnight at 4°C with the following primary antibodies: rabbit anti-Bax (1:2000, 50599-2-Ig, Proteintech), rabbit anti-Bcl-2 (1:2000, 26593-1-AP, Proteintech), mouse anti-GPX4 (1:2000, 67763-1-Ig, Proteintech), rabbit anti-Nrf2 (1:3000, A1244, ABclonal), rabbit anti-CD71 (1:1000, A5865, ABclonal), rabbit anti-FTH1 (1:1000, A19544, ABclonal), rabbit anti-DMT1 (1:2000, 20507-1-AP, Proteintech), rabbit anti-Fpn1 (1:1000, 26601-1-AP, Proteintech), and mouse anti-β-actin (1:1000, 66009-1-Ig, Proteintech). Horseradish peroxidase (HRP)-conjugated goat anti-rabbit (1:3000, A0208, Beyotime) and goat anti-mouse (1:1000, A0216, Beyotime) antibodies were added for 1 h, followed by enhanced chemiluminescence (ECL) visualization.
Quantitative-RT PCR (qPCR)
Total RNA was extracted using the SteadyPure Universal RNA Extraction Kit (AG21017, Accurate Biotechnology) after the collection and homogenization of brain tissue and HT22 cells. Total RNA was then converted into cDNA using an Evo M-MLV RT Mix Kit with gDNA clean for qPCR Ver. 2 (AG11728; Accurate Biotechnology). Specific primers, samples, and the SYBR® Premix Pro TaqHS qPCR Kit (Rox Plus) (AG11718, Accurate Biotechnology) were added to a 96-well PCR microplate based on the alignment. The operating program of the qPCR instrument (Applied Biosystems; Thermo Fisher Scientific, Inc.), including temperature and time setting, was conducted in accordance with the manufacturer’s instructions. The primer sets used are listed in Table 1. MRNA expression was calculated using the 2−ΔΔCt method with normalization to β-actin as the reference gene.
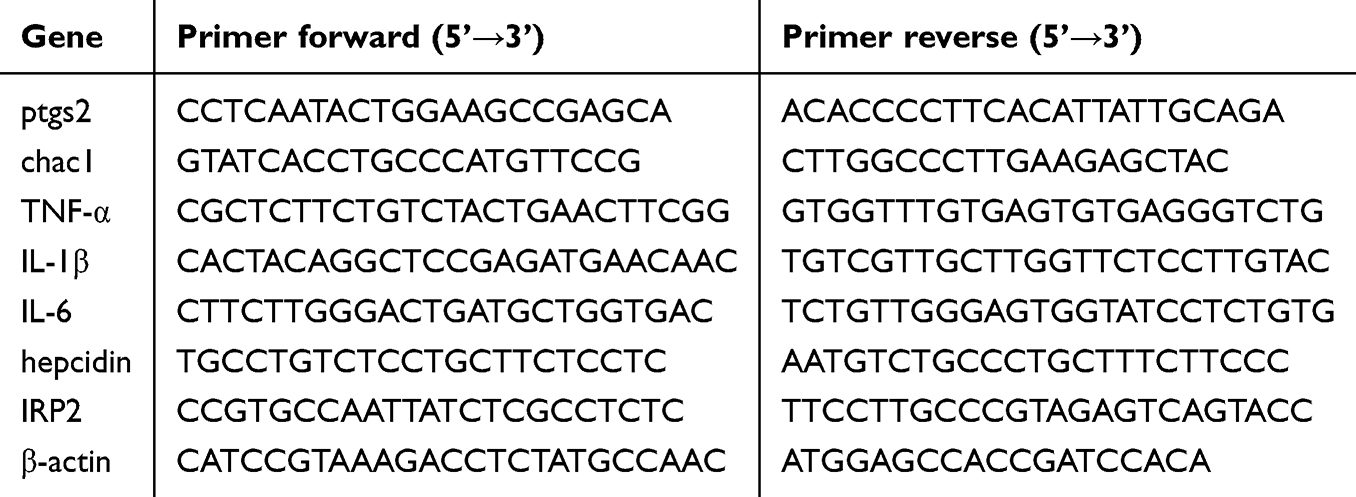 |
Table 1 The Primer Sets Used in qPCR |
Enzyme-Linked Immunosorbent Assay (ELISA)
Supernatant of fresh hippocampal and cortical homogenates after centrifugation was diluted and added to a polystyrene enzyme plate that had been coated, blocked, and washed. After sealing with a plate sealer, the plate was incubated at 37°C for 2 h, followed by the addition of a biotinylated antibody working solution. Enzyme conjugates working solution was then added for 30-minute incubation in the dark. Subsequently, TMB substrate solution was incorporated for another 30 min in darkness. Finally, 2 m sulfuric acid was introduced, resulting in a color change from blue to yellow. The OD value was then measured at 450 nm within 10 min on enzyme labeling instrument.
Iron Content Assay
The iron assay was performed using a Tissue Iron Content Assay Kit (BC4350, Solarbio) and a Cell Iron Content Assay Kit (BC5315, Solarbio). The supernatant from the homogenized brain tissue was mixed with the prepared reagents and heated in boiling water for 5 min. After cooling, chloroform was added for thorough mixing, and the upper organic phase was aspirated for absorbance measurement at 520 nm. After centrifugation, the supernatant of the cell lysate was reacted with the reagents and was allowed to stand at room temperature for 10 min. The absorbance was measured at 510 nm.
MDA Content Assay
Malondialdehyde (MDA) content was evaluated using the lipid peroxidation MDA assay kit (BC0025, Solarbio), following the manufacturer’s instructions. Briefly, tissues and cells were homogenized, centrifuged, and mixed with thiobarbituric acid (TAB) at 100°C for 1 h. The supernatant was collected, and other reagents were added before boiling the water bath. The absorbance of the centrifuged supernatants was measured at 532 nm and 600 nm.
GSH Content Assay
The glutathione (GSH) levels in the brain and HT22 cells were measured using a Reduced GSH Content Assay Kit (BC1175; Solarbio). The supernatant obtained after homogenization was mixed with the reaction reagent, and the absorbance was measured at 412 nm. The final GSH concentration was determined by using a standard curve.
Statistical Analysis
The data are presented as the mean ± standard error of the mean (SEM) and were analyzed using GraphPad Prism 5 (GraphPad Software, Inc., San Diego, CA). The normality and homogeneity of variance of the data were examined using SPSS, version 20.0 (SPSS Inc., Chicago, IL, USA). One-way analysis of variance (ANOVA) was conducted to explore statistical differences among groups, followed by Tukey’s multiple comparison test. Differences were considered significant only when p < 0.05.
Results
Propofol and Dexmedetomidine Ameliorated LPS-Mediated Cognitive Impairment and Neural Deficits as Well as Degeneration
To investigate whether propofol or dexmedetomidine improves behavioral performance after LPS induction, mice were subjected to the MWM test. As shown in Figure 1, the crossing platform times and target quadrant time of endotoxemic mice implied the unfamiliarity of mice to spatial location even after repeated training compared with the mice pretreated with sedatives (p < 0.05) (Figure 1E and F). Single use of either dexmedetomidine or propofol mitigated memory and orientation damage, as evidenced by the gradual reduction in the first escape latency and increased time spent in the platform quadrant, although no significant difference was found in the first escape latency of propofol group (p < 0.05) (Figure 1D and F). Disappointingly, the combination of propofol and dexmedetomidine did not present the expected defensive effects even compared with endotoxemia models, while time spent in the target quadrant supported the protection to some extent (Figure 1D, E and F). Swimming speed demonstrated consistency in motor abilities among all groups (Figure 1G). Despite unsatisfactory results from behavioral experiments, the union of propofol and dexmedetomidine significantly attenuated LPS-induced neuronal loss and degeneration. Nissl-positive neurons were calculated to evaluate functional units, and FJC staining specifically marked degenerative neurons. The joint group had the highest survival and the least degeneration in the hippocampus and cortex compared with the single drug-preconditioned groups (p < 0.05) (Figure 2A–F). In addition, all sedative groups exhibited comparable restoration of declined b-cell lymphoma-2 (bcl-2, an antiapoptotic protein) levels resulting from LPS administration (p < 0.05) (Figure 2G–L). Bcl2-associated x (bax, a proapoptotic protein) remained unchanged (Figure 2G–L). We also constructed an acute endotoxin infection model in vitro, using LPS incubated with HT22 cell lines. In preliminary experiments, we confirmed the median lethal dose of LPS and found that propofol and dexmedetomidine reversed the decline in cell viability in a dose-dependent manner (p < 0.05) (Figure 2M–O). Co-administration of the two drugs provided better therapy to combat cell toxicity than isolated drug usage, in alignment with the histological features (p < 0.05) (Figure 2P).
 |
Figure 1 Propofol and dexmedetomidine ameliorated the endotoxemia-associated cognitive impairment. (A and B) The intervention strategy and experimental procedure for endotoxemia model in vivo and in vitro. (C) Representative trajectory and heatmaps of the MWM. (D) The escape latency for the first platform discovery over five consecutive days. (E) The crossing-platform times on the final day after platform removal. (F) The duration of time spent in the quadrant where the platform was located on the final day. (G) The swim speed on the final day. Data are expressed as the mean ± SEM (*p<0.05 vs c group; #p<0.05 vs lps group; n = 7). |
 |
Figure 2 Propofol and dexmedetomidine mitigated the LPS-induced neuronal loss and degeneration as well as apoptosis. (A–C) Representative images of Nissl staining and the number of Nissl-positive cells (400×), scale bars = 50 μm. (D–F) Representative images of FJC staining and number of FJC-positive cells (400×), scale bars = 50μm. (G–L) The Western blot bands and quantitative analysis of bax and bcl-2. (M–P) Cell viability of treated HT22 cells measured by CCK-8. Data are expressed as the mean ± SEM (*p<0.05 vs lps and l2 group; #p<0.05 vs l+p+d and l5 group; &p<0.05 vs l10 group; n = 6). |
Propofol and Dexmedetomidine Mitigated LPS-Induced Neuroinflammation in vivo and in vitro
DHE staining showed that the single use of propofol hardly restricted neuroinflammation after LPS administration in the hippocampus and cortex (Figure 3A–C). Apart from the notable decrease in TNF-α and IL-6 in the cortex, only-propofol usage exhibited little effect on the transcriptional levels of three inflammatory factors in the hippocampus, as well as cortical IL-1β (p < 0.05) (Figure 3D–E). Contrary to the inert performance of propofol, the anti-inflammatory efficacy of dexmedetomidine has been unequivocally confirmed, regardless of the brain region or the type of inflammatory factor (p < 0.05) (Figure 3A–E). The effectiveness of united sedation did not universally surpass that of individual usage. In certain cases, it demonstrated a superior outcome compared to propofol alone, but fell short of the efficacy achieved with dexmedetomidine alone (p < 0.05) (Figure 3A–E). In vitro experiments, except for the group treated with propofol alone, the others demonstrated excellent capabilities against ROS production, as confirmed by the reversed relative fluorescence intensity of DHE and DCFH-DA (p < 0.05) (Figure 4A–D). To further confirm the relatively limited anti-inflammatory capability of propofol compared to dexmedetomidine, ELISA was used to quantify the content of TNF-α, IL-1β, and IL-6. Results showed that, propofol did indeed partially alleviate LPS-induced neuroinflammation, though not as strong as dexmedetomidine (p < 0.05) (Figure 3F–G). In the hippocampus, the performance of the combined therapy mirrored those of dexmedetomidine alone (p < 0.05) (Figure 3F), while in the cortex, its effects resembled those of sole-propofol application (p < 0.05) (Figure 3G).
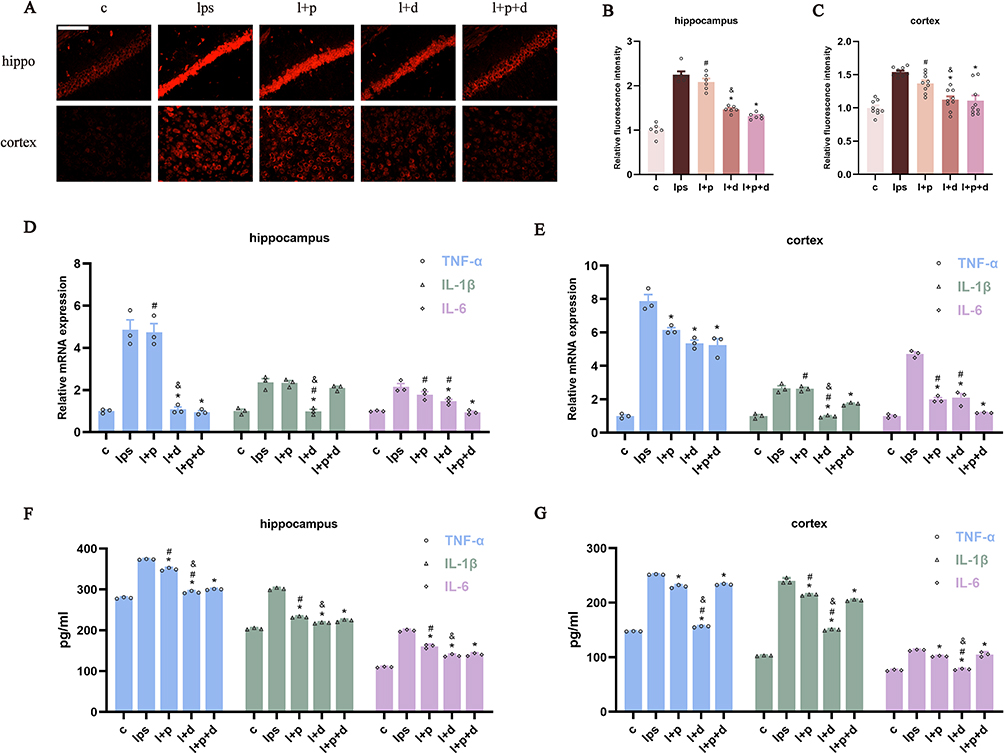 |
Figure 3 Propofol and dexmedetomidine diminished LPS-mediated neuroinflammation in endotoxemic mice. (A–C) DHE staining representative images and analysis of relative fluorescence intensity (400×), scale bar = 50 μm. (D and E) The relative mRNA expression of TNF-α, IL-1β, and IL-6 measured by qPCR; (F and G) The content of TNF-α, IL-1β and IL-6 detected by ELISA. Data are expressed as the mean ± SEM (*p<0.05 vs lps group; #p<0.05 vs l+p+d group; &p<0.05 vs l+p group; n = 6). |
 |
Figure 4 Dexmedetomidine and combination abrogated the ROS production in LPS-incubated HT22 cells. (A and B) The relative fluorescence intensity of DHE staining to evaluate the level of superoxide anions (400×), scale bar = 50 μm. (C and D) The relative fluorescence intensity of DCFH-DA staining to investigate the production of H2O2 (400×), scale bar = 50 μm. Data are expressed as the mean ± SEM (*p<0.05 vs lps group; #p<0.05 vs l+p+d group; &p<0.05 vs l+p group; n = 6). |
Propofol and Dexmedetomidine Inhibited LPS-Activated Microglia and Astrocytes
To explore whether propofol or dexmedetomidine attenuates LPS-initiated microglial and astrocytic activation, we labelled microglia with Iba1 and astrocytes with GFAP. Immunohistochemical results indicated a significant suppression in the number of marked microglia in the hippocampus and cortex after intervention with propofol and dexmedetomidine, either alone or in combination, with no significant difference observed among the three sedative groups (p < 0.05) (Figure 5A–C). Propofol also restricted the augmentation of GFAP-positive astrocytes in the endotoxemic hippocampal CA1 and DG regions, though its suppression on DG did not reach statistical significance. The inhibitory effect was most pronounced with dexmedetomidine alone, whereas the combined drug administration demonstrated an intermediate limiting impact (p < 0.05) (Figure 5D–F).
 |
Figure 5 Propofol and dexmedetomidine reduced LPS-activated microglia and astrocytes. (A–C) The presentative images and the number of Iba1-positive microglia (400×), scale bar = 50 μm. (D–F) The presentative images and the number of GFAP-positive astrocytes (red) co-labelled with Neun-positive neurons (green) in the CA1 and DG of hippocampus (200×), scale bar = 100 μm. Data are expressed as the mean ± SEM (*p<0.05 vs lps group; &p<0.05 vs l+p group; n = 6). |
Propofol and Dexmedetomidine Attenuated LPS-Initiated Neural Ferroptosis in vivo and in vitro
Ferroptosis is well known as the overproduction of iron, which activates the progression of membrane lipid peroxidation. To determine the extent of ferroptosis, we assessed the transcriptional levels of ptgs2 and chac1, which are gene-specific markers of ferroptosis. As shown in Figure 6A, the single use of propofol and dexmedetomidine significantly repressed ferroptosis-related changes in the cortex and the elevation of ptgs2 in the hippocampus. The combined group displayed the best inhibitory effect, though the difference between the joint and single sedative groups was not always statistically significant (p < 0.05) (Figure 6A). Simultaneously, MDA, the end product of lipid metabolism; GSH, an important substrate of the antioxidant barrier; and iron content were detected. The co-administration of propofol and dexmedetomidine exhibited a superior effect in mitigating the elevation of MDA and iron accumulation, as well as the restoration of GSH, compared with their individual use (p < 0.05) (Figure 6B–C). However, the situation varied with respect to the ferroptosis-related proteins. As the central safeguard against ferroptosis, glutathione peroxidase 4 (GPX4) in dexmedetomidine-alone and combined groups in the hippocampus and cortex all showed notable restoration, while only-propofol could hardly reverse (p < 0.05) (Figure 6D, E, K and L). Nrf2 (nuclear factor erythroid 2-related factor 2) is a multifunctional antioxidant transcription factor that regulates several factors in ferroptosis. Single or combined administration of propofol and dexmedetomidine exhibited similar effects on the reversal of LPS-restrained Nrf2 expression in the hippocampus and cortex (p < 0.05) (Figure 6D, F, K and M). Interestingly, the expression of iron metabolism-associated proteins differed in a brain region-specific manner. CD71 (transferrin receptor 1, also known as TfR1) was only recovered in the united group in the hippocampus (p < 0.05) (Figure 6D and G), whereas no changes were observed in the cortex (Figure 6K and N). In contrast, elevated ferritin heavy chain 1 (FTH1) in the cortex showed remarkable constraints in all protective groups, while that in the hippocampus remained unaltered (p < 0.05) (Figure 6D, H, K and O). Notably, the expression of divalent metal transporter 1 (DMT1) showed little changes in any group (Figure 6D, I, K and P), indicating that DMT1 may not be a major factor mediating LPS-induced neural iron overload. Fpn1, the only transporter involved in iron efflux, displayed a significant decline after LPS injection regardless of the brain region. Only the combination of propofol and dexmedetomidine significantly reversed the Fpn1 reduction in the hippocampus, while only propofol-pretreatment was unable to abrogate the decrease in the cortex (p < 0.05) (Figure 6D, J, K and Q). In LPS-incubated cells, the transcriptional levels of ptgs2 and chac1 were not significantly altered in any of the groups (Figure 7A). Nonetheless, excessive MDA and iron were restricted in all pretreated groups and combined usage had the most potent effect (p < 0.05) (Figure 7B). The decrease in GSH levels were also restored in all defensive groups, and the single use of dexmedetomidine appeared to have the weakest protection (p < 0.05) (Figure 7B). The relative quantification of CD71, identified as a biological marker for ferroptosis,25 revealed that only the united group showed effective restoration in HT22 cells (p < 0.05) (Figure 7C and D), consistent with the protein trend in the hippocampus. Meanwhile, only the propofol-preconditioned group failed to enhance the restrained expression of Fpn1 (p < 0.05) (Figure 7E and F). When exposed to stimuli, Nrf2 translocates from cytoplasm to the nucleus. To evaluate the proportion of activated-Nrf2 cells, DAPI was used to localize the nucleus, whereas phalloidin was used to visualize the cytoskeleton. The results demonstrated a potent reversal of activated-Nrf2 cells in all sedative groups, with no group showing outstanding recovery (p < 0.05) (Figure 7G and H).
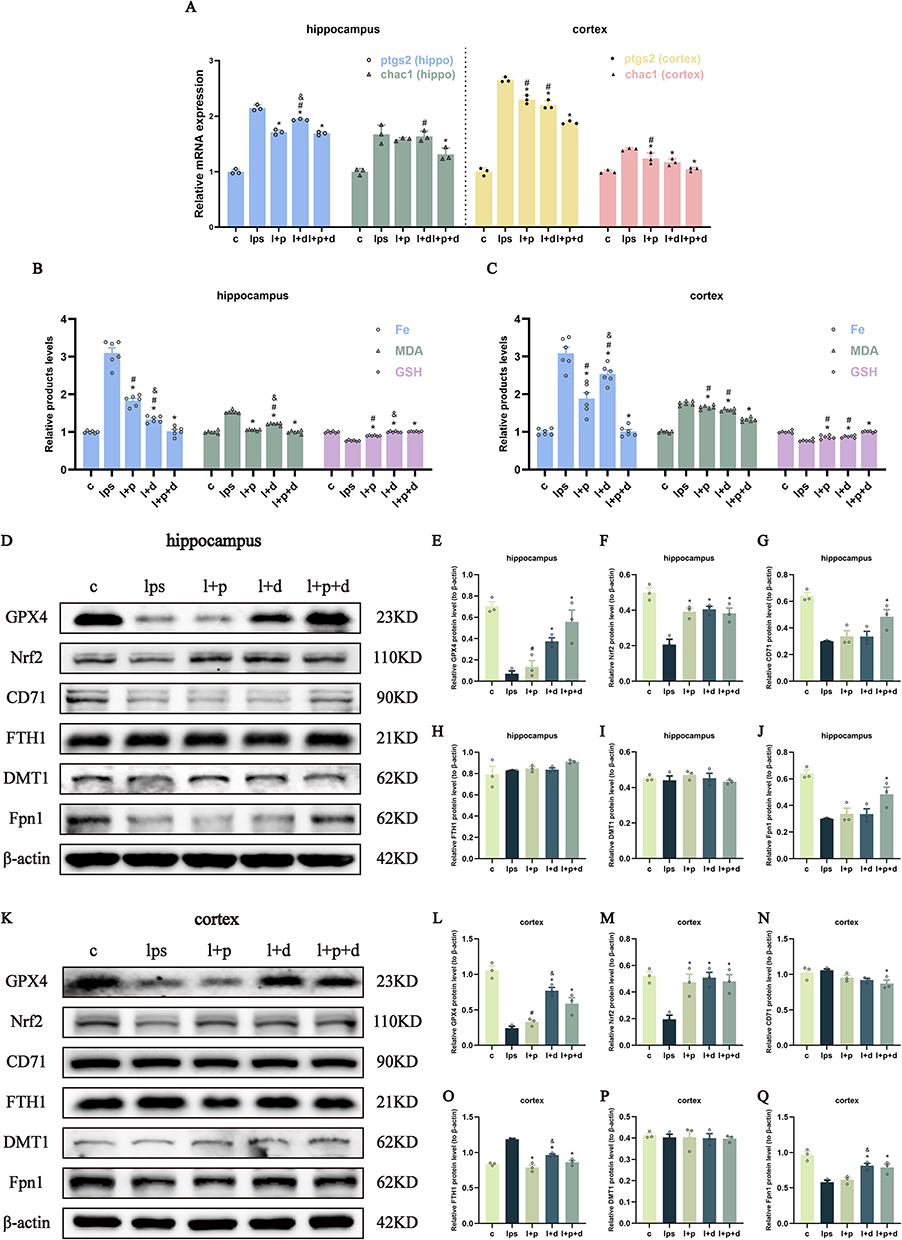 |
Figure 6 Propofol and dexmedetomidine attenuated LPS-initiated ferroptosis in endotoxemic mice. (A) The relative mRNA expression of ptgs2 and chac1. (B and C) The relative products level of Fe, MDA and GSH. (D–Q) The Western blot bands and quantitative analysis of GPX4, Nrf2, CD71, FTH1, DMT1 and Fpn1. Data are expressed as the mean ± SEM (*p<0.05 vs lps group; #p<0.05 vs l+p+d group; &p<0.05 vs l+p group; n = 6). |
 |
Figure 7 Propofol and dexmedetomidine inhibited LPS-induced ferroptosis in HT22 cells. (A) The relative mRNA expression of ptgs2 and chac1. (B) The relative products level of Fe, MDA and GSH. (C–F) The representative images and relative fluorescence intensity of CD71 and Fpn1 (630×), scale bar = 50 μm. (G and H) The representative images and the proportion of activated Nrf2 cells (630×), scale bar = 50 μm. Data are expressed as the mean ± SEM (*p<0.05 vs lps group; #p<0.05 vs l+p+d group; n = 6). |
Propofol and Dexmedetomidine Affected the Transcriptional Upregulation of Hepcidin and IRP2 Caused by LPS Administration
To investigate whether propofol and dexmedetomidine modulate the regulator of iron metabolism, the relative mRNA expression of hepcidin and IRP2 (iron regulatory protein-2) was measured using qPCR. Dexmedetomidine, but not propofol, significantly suppressed hepcidin levels in the hippocampus and cortex of endotoxemic mice (p < 0.05) (Figure 8A and B). All three groups with sedative preconditioning showed a notable decline in LPS-induced IRP2 upregulation, although it was difficult to differentiate the effectiveness among all groups (p < 0.05) (Figure 8C and D).
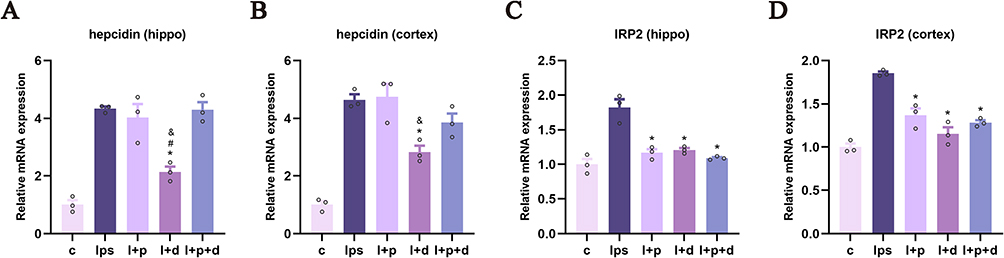 |
Figure 8 Propofol and dexmedetomidine influenced the transcriptional level of hepcidin and IRP2 in endotoxemic mice. (A and B) The relative mRNA expression of hepcidin. (C and D) The relative mRNA expression of IRP2. Data are expressed as the mean ± SEM (*p<0.05 vs lps group; #p<0.05 vs l+p+d group; &p<0.05 vs l+p group; n = 3). |
Discussion
In this study, we compared the ability of propofol and dexmedetomidine to ameliorate LPS-induced cognitive deficits and multiple neuropathic disorders. The following conclusions were drawn: (1) Propofol and dexmedetomidine preconditioning rescued LPS-induced learning and memory deterioration, while the combined medication had little reversal capability. (2) All pretreated groups exhibited alleviation of neural loss and degeneration, with the united group showing the most robust protective effects. (3) All sedative groups repressed neural apoptosis. (4) Dexmedetomidine markedly hindered LPS-induced neuroinflammation, while the anti-inflammatory effect of propofol was somewhat weaker. All sedative pretreatment attenuated the activation of microglia and astrocytes. (5) All sedative therapies diminished ferroptosis, with the combined group exhibiting the most potent protective effects in some cases. The mechanisms involved in propofol and dexmedetomidine against endotoxemia-induced cognitive impairment and brain injury are summarized in Figure 9.
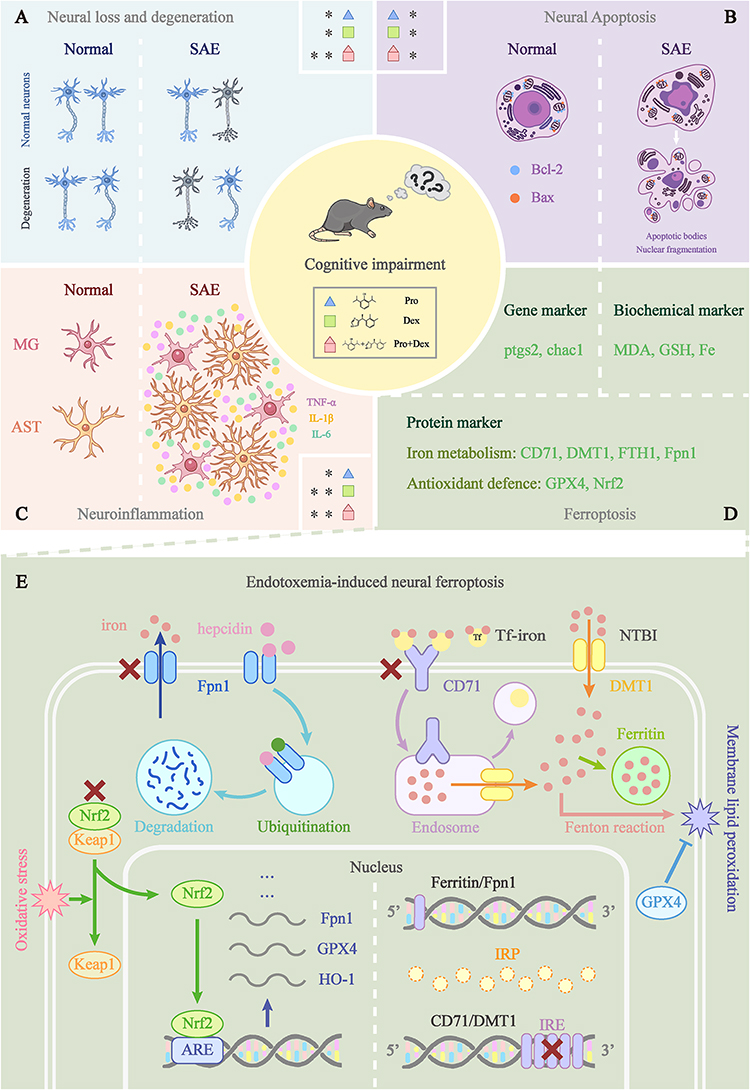 |
Figure 9 Mechanisms of propofol and dexmedetomidine in alleviating endotoxemia-induced encephalopathy. (A-C) The protective effects of propofol, dexmedetomidine and combined usage against LPS-mediated neural loss and degeneration, neural apoptosis and neuroinflammation. Blue triangles, green squares, and red polygons correspond to propofol, dexmedetomidine and combined groups. The asterisk (*) was used to indicate the relative strength of inhibition of the specific pathological conditions by each sedative group. (D) Three aspects of laboratory evidence to evaluate the extent of ferroptosis. (E) The iron metabolism, hepcidin-Fpn1 regulation, IRP/IRE axis, and Nrf2 translocation involved in the LPS-induced neural ferroptosis. |
Propofol and dexmedetomidine are two commonly used anesthetics in the perioperative setting and are among the most frequently used medications in the ICU for critically ill patients undergoing mechanical ventilation.26 Sepsis is a complicated clinical condition with high mortality and morbidity and is often accompanied by multiorgan dysfunction.27 Acute inflammation bursts and subsequent immune constraint finally involve the brain, mediating diffuse brain disorders.28,29 In this study, we utilized in vivo and in vitro models of LPS-induced endotoxemia to simulate the acute inflammatory response in sepsis and investigated whether propofol and dexmedetomidine, either alone or in combination, are involved in LPS-induced cognitive impairment and several neural pathological deficiencies.
According to the MWM behavioral tests, pretreatment with either propofol or dexmedetomidine improved spatial and directional learning and memory deficits mediated by LPS administration. However, the joint group showed disastrous performance, even comparable to that of the endotoxemia group, in stark contradiction to the histological evidence. The highest neural survival and least degeneration indicated the alleviating capability of the combined medication, as evidenced by Nissl and FJC staining in vivo and cell viability in vitro. Similarly, although there was no difference between the three protective groups, neural apoptosis in the hippocampus and cortex was significantly mitigated. The discrepancy between the behavioral and histological results may be due to several factors. Firstly, we conducted MWM tests 72 h after LPS administration, as previous evidence showed that clinical severity scores (CCS) returned to baseline at this time point.24 However, the endurance of mice in response to exercise may not have fully recovered, and continuous swimming-induced exhaustion may reactivate their deteriorating condition. Meanwhile, the compounded metabolic burden from the drug combination may further compromise the tolerance of the mice to high-intensity exercise. In the LPS-induced endotoxemia model, the interval between the last LPS injection and MWM tests varies, ranging from 2 to 10 days.30–35 Secondly, brains of mice which underwent behavioral tests were not harvested. Another portion of the mice was sacrificed with their brains either fixed in 4% paraformaldehyde or rapidly frozen in liquid nitrogen for subsequent experiments after 24 hours of LPS intervention, as widely acknowledged by other researchers.30,34 Thirdly, we selected five mice from the same batch, raised them in identical environments, and housed them in the same cage (seven replicates in total) to minimize intra-group differences in behavioral experiments. However, data from mice that died during MWM were excluded, reducing the sample size. This exclusion may limit the statistical significance of the results and increase the possibility of accidental outcomes. Further experiments are needed to address these problems.
During the early stages of sepsis, excessive inflammatory responses and cytokine storms are crucial factors that drive the progression.36 Therefore, we assessed the effects of propofol and dexmedetomidine on LPS-induced systemic acute inflammation in vivo and in vitro. Results indicated that propofol significantly relieved neuroinflammation, though its capability was relatively restricted compared with the other two pretreated groups. Dexmedetomidine exhibited robust anti-inflammatory properties and its combination with propofol did not interfere with this capability. Interestingly, all sedative groups displayed comparable effects in suppressing the activation of microglia. It is worth noting that although our results indicated that propofol exhibited a weaker anti-inflammatory capacity than dexmedetomidine, this did not imply that propofol had no impact on neuroinflammation. The mechanisms by which propofol modulates neuroinflammation in LPS-induced endotoxemia models require further investigation and validation.
Ferroptosis, a striking form of programmed cell death over the past decade, is involved in the progression of sepsis and associated organ damage.37 The continually discovered ferroptosis inhibitors offer potential prevention and treatment for sepsis. Overfilling with the labile iron pool and deficiency in the anti-lipid peroxidative defense centered around GPX4 both activate the occurrence of ferroptosis.8 Iron homeostasis is maintained by iron influx, storage, and efflux to achieve a dynamic equilibrium. CD71 recognizes and internalizes transferrin (Tf)-bound iron. In the acidic environment of endosomes, iron dissociates from Tf and flows into the cytoplasm through DMT1. Meanwhile, Tf is released into the extracellular space through vesicular transport. DMT1 located on the cell membrane also mediates the influx of non-transferrin-bound iron (NTBI). Ferritin stores excessive iron and Fpn1 facilitates the efflux of surplus iron.38 Our study showed that the combination of propofol and dexmedetomidine exerted an optimal effect on the recovery of genetic and biochemical markers of ferroptosis. However, results were not always consistent in protein changes. The sole use of dexmedetomidine showed the most potent mitigation of GPX4 repression in the cortex, whereas the combination was most effective in the hippocampus. No difference was found in the restoration of Nrf2 among all the sedative groups. In terms of iron metabolism, intriguing region-specificity was observed. In the hippocampus, FTH1 levels did not change even in the LPS group. Only the combination of propofol and dexmedetomidine significantly reversed CD71 expression. In the cortex, CD71 levels displayed unaltered in all groups, while a significant increase in FTH1 levels was seen, which was repressed by both propofol and dexmedetomidine. Interestingly, the trends of DMT1 and Fpn1 were in agreement regardless of the brain region. DMT1 revealed no notable alternation in all groups, suggesting that the role of DMT1 in LPS-mediated neural ferroptosis was relatively insignificant. Meanwhile, the impaired Fpn1 expression was resecured after joint sedation in the hippocampus and cortex. Dexmedetomidine alone also reversed declined Fpn1 in the cortex. The inhibited iron influx by decreased CD71 and enhanced iron storage by increased FTH1 all hindered the intracellular iron overload and may be a compensatory response to high-iron environment. The balance in DMT1 expression may be achieved through the promotion of neuroinflammation and the feedback limitation imposed by excessive iron.39 Therefore, we speculated that the consistent reduction of Fpn1 in the cortex and hippocampus may be the central trigger of ferroptosis. Remarkably, there is still no consensus regarding alterations in iron metabolism factors in sepsis. LPS administration repressed FTH1 levels in LO2 cells (human normal hepatocytes),40 MLE-12 cells (mouse capillary alveolar epithelial cells),41 and H9C2 cells (rat myofibroblasts).42 However, enhanced expression was observed in the myocardium harmed by LPS43–45 and in lungs46 and kidneys injured by cecal ligation and puncture.47 There was also no uniformity in CD71 expression even in a similar LPS-mediated myocardial damage model. Zhou et al resisted the upregulation of CD71,44 whereas Xiao et al reported a remarkable decline.45 However, for Fpn1, the results were highly consistent, showing a decreasing tendency.10,39,45
Many repressors involved in ferroptosis are targets of Nrf2 and are primarily associated with iron, intermediate, and GSH metabolism.48 Under oxidative stress, Nrf2 rapidly dissociates from Kelch-like ECH-associated protein 1 (Keap1) and translocates into the nucleus, where it combines with antioxidant response element (ARE), thereby facilitating transcription of a cascade of cell-protective genes, such as heme oxygenase-1 (HO-1), GPX4, and Fpn1. It is well established how Nrf2 transcriptionally enhances the expression of Fpn1 and ferritin, thus inhibiting ferroptosis.48,49 However, owing to the highly compartmentalized nature of the brain, there is limited research on whether Nrf2 in the central nervous system has a similar regulatory capacity.50 The knockout of Nrf2 impaired the increased ferritin and Fpn1 levels in the striatum of aged mice.51 The specific inhibitor of Nrf2, ML385, significantly inhibited FTH1 and Fpn1 in PC12 cells treated with MPP+ (1-methyl 4-phenyl pyridinium), a classical model of Parkinson’s disease.52 In other words, the modulation of Nrf2 on ferritin and Fpn1 should be synchronous. The disruption of Nrf2 cannot fully explain the sustained FTH1 expression in the hippocampus and the elevated levels in the cortex following LPS treatment. Hence, we suspected that other facilitators were also involved in the decline of Fpn1.
Hepcidin serves as a crucial upstream regulator of Fpn1 by inducing its ubiquitination and degradation, thereby inhibiting the efflux of iron.50 Some researchers contend that the anti-inflammatory effects of hepcidin, along with its increase during inflammation, make it a promising novel biomarker of sepsis.53,54 During the inflammatory phase, microglia encounter LPS signals with toll-like receptor (TLR) 4. Through the secretion of IL-6, microglia stimulate astrocytes to increase hepcidin production. Hepcidin is then secreted paracrinely into neurons, thereby causing neuronal iron overload.55 Based on our findings, we speculated that the restriction on activated microglia and astrocytes and the limitation on IL-6 content from dexmedetomidine may contribute to the hindered hepcidin expression, thus restoring Fpn1 in a manner distinct from Nrf2 regulation. Notably, the inhibitory effect of propofol on microglia and astrocytes was also considerable, while constraint of hepcidin by propofol was rather limited. However, the disability of propofol to repress hepcidin was astonishingly in agreement with its failure to enhance the Fpn1 expression. The IRP/iron-responsive element (IRE) axis is another pathway regulating iron metabolism and is primarily involved in intracellular responses to the iron content.56 IRPs bind to the highly conserved IREs on target mRNAs to form hairpins. Under high-iron environment, IRP dissociates from the 3’-untranslated regions (UTRs) of unstable mRNAs, such as CD71 and DMT1, to initiate mRNA degradation. It also fails to combine with the 5’-UTRs of ferritin and Fpn1 mRNAs, thus promoting the expression. Iron overload also accelerated the degradation of IRP.57 IRP has two subtypes: IRP1 is primarily active in hypoxic tissues like the duodenum and kidneys, while IRP2 is active elsewhere.56 Here, we found that all sedative groups reduced the increase of IRP2. The different regulation on hepcidin and IRP2 provides insights into the distinct anti-ferroptotic mechanisms of propofol and dexmedetomidine. Given that the action of IRP on ferritin and Fpn1 should be analogous, more research is anticipated to determine how propofol hinders ferroptosis.
In this study, LPS was intraperitoneally injected for its strong inducibility of systemic inflammation to mimic the acute inflammatory phase of sepsis. Nonetheless, it hardly fully imitates the progression of sepsis; instead, it is more suitable for studying endotoxemia and systemic inflammatory response syndrome (SIRS).24 Furthermore, as discussed above, the time interval between behavioral tests and LPS administration remained controversial. The unexpectedly poor cognitive performance in combined sedative group reminded us to contemplate and solve this issue. Additionally, after determining the severity of ferroptosis, we cast light on the iron homeostasis disorder critical to ferroptosis. Research on factors involved in lipid peroxidation and antioxidant defense disruption is also indispensable for a comprehensive assessment of the intervention of propofol and dexmedetomidine in LPS-induced ferroptosis. Despite the limitations mentioned above, our study still holds clinical prospects. We compared the protection of propofol and dexmedetomidine, alone or in combination, against LPS-induced cognitive deficiencies, neurotoxicity and degeneration, neural apoptosis, neuroinflammation, and ferroptosis. Dexmedetomidine pretreatment exhibited potent anti-inflammatory abilities. Besides, combined drug therapy showed remarkable synergic effects on neuronal survival and partial resistance to ferroptosis, laying a solid foundation for clinical multi-drug sedation therapy. To better translate the synergistic effects of this combined sedation into clinical strategies, it is important to figure out the optimal dosage ratio, administration route, and infusion time for propofol and dexmedetomidine union. Moreover, as previously hypothesized, the reduction of Fpn1 is likely to be the key trigger for LPS-mediated neural ferroptosis. Elucidating the pivotal role of Fpn1 and exploring specific drugs targeted at restoring Fpn1 function may potentially pioneer remedies against sepsis.
Conclusion
The scarcity of treatment options and persistently high incidence of sepsis pose a significant threat to the survival of ICU patients, imposing a heavy burden on the healthcare system. Propofol and dexmedetomidine are two commonly used sedatives in critically ill patients experiencing mechanical ventilation. Investigating their potential advantages in alleviating symptoms and improving the outcomes of sepsis has substantial clinical value.
Data Sharing Statement
Enquiries about data availability should be directed to the first author.
Ethics Approval
All animal studies were approved by the Animal Ethics Committee of Fudan University Pudong Medical Center and conducted in accordance with the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals.
Acknowledgment
This project was supported by the Key Specialty Construction Project of Pudong Health and Family Planning Commission of Shanghai (PWZzk2017-06).
Disclosure
The authors confirm that there are no conflicts of interest in this work.
References
1. Singer M, Deutschman CS, Seymour CW, et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA. 2016;315(8):801–810. doi:10.1001/jama.2016.0287
2. Hughes CG, Mailloux PT, Devlin JW, et al. Dexmedetomidine or propofol for sedation in mechanically ventilated adults with sepsis. N Engl J Med. 2021;384(15):1424–1436. doi:10.1056/NEJMoa2024922
3. van der Poll T, Shankar-Hari M, Wiersinga WJ. The immunology of sepsis. Immunity. 2021;54(11):2450–2464. doi:10.1016/j.immuni.2021.10.012
4. Rajendrakumar SK, Revuri V, Samidurai M, et al. Peroxidase-mimicking nanoassembly mitigates lipopolysaccharide-induced endotoxemia and cognitive damage in the brain by impeding inflammatory signaling in macrophages. Nano Lett. 2018;18(10):6417–6426. doi:10.1021/acs.nanolett.8b02785
5. Xin Y, Tian M, Deng S, et al. The key drivers of brain injury by systemic inflammatory responses after sepsis: microglia and neuroinflammation. Molecular Neurobiology. 2022;60(3):1369–1390. doi:10.1007/s12035-022-03148-z
6. Dickson K, Lehmann C. Inflammatory response to different toxins in experimental sepsis models. Int J Mol Sci. 2019;20(18):4341. doi:10.3390/ijms20184341
7. Chen X, Li J, Kang R, Klionsky DJ, Tang D. Ferroptosis: machinery and regulation. Autophagy. 2021;17(9):2054–2081. doi:10.1080/15548627.2020.1810918
8. Tang D, Chen X, Kang R, Kroemer G. Ferroptosis: molecular mechanisms and health implications. Cell Res. 2021;31(2):107–125. doi:10.1038/s41422-020-00441-1
9. Yang K, Zeng L, Yuan X, et al. The mechanism of ferroptosis regulating oxidative stress in ischemic stroke and the regulation mechanism of natural pharmacological active components. Biomed Pharmacother. 2022;154:113611. doi:10.1016/j.biopha.2022.113611
10. Fang J, Kong B, Shuai W, et al. Ferroportin-mediated ferroptosis involved in new-onset atrial fibrillation with LPS-induced endotoxemia. Eur J Pharmacol. 2021;913:174622. doi:10.1016/j.ejphar.2021.174622
11. Wang X, Simayi A, Fu J, Zhao X, Xu G. Resveratrol mediates the miR-149/HMGB1 axis and regulates the ferroptosis pathway to protect myocardium in endotoxemia mice. Am J Physiol Endocrinol Metab. 2022;323(1):E21–E32. doi:10.1152/ajpendo.00227.2021
12. Li S, Lei Z, Yang X, et al. Propofol protects myocardium from ischemia/reperfusion injury by inhibiting ferroptosis through the AKT/p53 signaling pathway. Front Pharmacol. 2022;13:841410. doi:10.3389/fphar.2022.841410
13. Wei Q, Chen J, Xiao F, Tu Y, Zhong Y, Xie Y. High-dose dexmedetomidine promotes apoptosis in fetal rat hippocampal neurons. Drug Des Devel Ther. 2021;15:2433–2444. doi:10.2147/DDDT.S300247
14. Devlin JW, Skrobik Y, Gelinas C, et al. Clinical practice guidelines for the prevention and management of pain, agitation/sedation, delirium, immobility, and sleep disruption in adult patients in the ICU. Crit Care Med. 2018;46(9):e825–e873. doi:10.1097/CCM.0000000000003299
15. Heybati K, Zhou F, Ali S, et al. Outcomes of dexmedetomidine versus propofol sedation in critically ill adults requiring mechanical ventilation: a systematic review and meta-analysis of randomised controlled trials. Br J Anaesth. 2022;129(4):515–526. doi:10.1016/j.bja.2022.06.020
16. Brown EN, Pavone KJ, Naranjo M. Multimodal General Anesthesia: theory and Practice. Anesth Analg. 2018;127(5):1246–1258. doi:10.1213/ANE.0000000000003668
17. Tang CL, Li J, Zhang ZT, et al. Neuroprotective effect of bispectral index-guided fast-track anesthesia using sevoflurane combined with dexmedetomidine for intracranial aneurysm embolization. Neural Regen Res. 2018;13(2):280–288. doi:10.4103/1673-5374.226399
18. Shehabi Y, Serpa Neto A, Bellomo R, et al. Dexmedetomidine and propofol sedation in critically Ill patients and dose-associated 90-day mortality: a secondary cohort analysis of a randomized controlled trial (SPICE III). Am J Respir Crit Care Med. 2023;207(7):876–886. doi:10.1164/rccm.202206-1208OC
19. Shehabi Y, Howe BD, Bellomo R, et al. Early sedation with dexmedetomidine in Critically Ill Patients. N Engl J Med. 2019;380(26):2506–2517. doi:10.1056/NEJMoa1904710
20. Sun C, Liu P, Pei L, Zhao M, Huang Y. Propofol inhibits proliferation and augments the anti-tumor effect of doxorubicin and paclitaxel partly through promoting ferroptosis in triple-negative breast cancer cells. Front Oncol. 2022;12:837974. doi:10.3389/fonc.2022.837974
21. She H, Hu Y, Zhou Y, et al. Protective effects of dexmedetomidine on sepsis-induced vascular leakage by alleviating ferroptosis via regulating metabolic reprogramming. J Inflamm Res. 2021;14:6765–6782. doi:10.2147/JIR.S340420
22. Mei B, Li J, Zuo Z. Dexmedetomidine attenuates sepsis-associated inflammation and encephalopathy via central alpha2A adrenoceptor. Brain Behav Immun. 2021;91:296–314. doi:10.1016/j.bbi.2020.10.008
23. Guo F, Kang J, Tan J, Wang Y, Jia L, Xu H. Dexmedetomidine pretreatment improves lipopolysaccharide-induced iron homeostasis disorder in aged mice. Curr Neurovasc Res. 2020;17(2):164–170. doi:10.2174/1567202617666200217105109
24. Seemann S, Zohles F, Lupp A. Comprehensive comparison of three different animal models for systemic inflammation. J Biomed Sci. 2017;24(1). doi:10.1186/s12929-017-0370-8
25. Feng H, Schorpp K, Jin J, et al. Transferrin receptor is a specific ferroptosis marker. Cell Rep. 2020;30(10):3411–3423 e7. doi:10.1016/j.celrep.2020.02.049
26. Moore JPR, Shehabi Y, Reade MC, et al. Stress response during early sedation with dexmedetomidine compared with usual-care in ventilated critically ill patients. Crit Care. 2022;26(1):359. doi:10.1186/s13054-022-04237-0
27. Hurtado-Navarro L, García-Palenciano C, Pelegrín P. Inflammasomes in sepsis. Inflammasome Biology. 2023;2023:369–382.
28. Marques A, Torre C, Pinto R, Sepodes B, Rocha J. Treatment advances in sepsis and septic shock: modulating pro- and anti-inflammatory mechanisms. J Clin Med. 2023;12:8.
29. Ferlini L, Gaspard N. What’s new on septic encephalopathy? Ten things you need to know. Minerva Anestesiol. 2023;89(3):217–225. doi:10.23736/S0375-9393.22.16689-7
30. Huang X, Ye C, Zhao X, et al. TRIM45 aggravates microglia pyroptosis via Atg5/NLRP3 axis in septic encephalopathy. J Neuroinflammation. 2023;20(1):284. doi:10.1186/s12974-023-02959-8
31. Liu K, Wan G, Jiang R, et al. Astragalus injection ameliorates lipopolysaccharide-induced cognitive decline via relieving acute neuroinflammation and BBB damage and upregulating the BDNF-CREB pathway in mice. Pharm Biol. 2022;60(1):825–839. doi:10.1080/13880209.2022.2062005
32. Xue W, Li Y, Zhang M. Pristimerin inhibits neuronal inflammation and protects cognitive function in mice with sepsis-induced brain injuries by regulating PI3K/Akt signalling. Pharm Biol. 2021;59(1):1351–1358. doi:10.1080/13880209.2021.1981399
33. Wang K, Sun M, Juan Z, et al. The Improvement of Sepsis-Associated Encephalopathy by P2X7R Inhibitor through Inhibiting the Omi/HtrA2 apoptotic signaling pathway. Behav Neurol. 2022;2022:3777351. doi:10.1155/2022/3777351
34. Han YG, Qin X, Zhang T, et al. Electroacupuncture prevents cognitive impairment induced by lipopolysaccharide via inhibition of oxidative stress and neuroinflammation. Neurosci Lett. 2018;683:190–195. doi:10.1016/j.neulet.2018.06.003
35. Li Y, Ma J, Diao J, Chen W, Wang Z. Esmolol inhibits cognitive impairment and neuronal inflammation in mice with sepsis-induced brain injury. Transl Neurosci. 2023;14(1):20220297. doi:10.1515/tnsci-2022-0297
36. Huang M, Cai S, Su J. The pathogenesis of sepsis and potential therapeutic targets. Int J Mol Sci. 2019;20:21.
37. Huo L, Liu C, Yuan Y, Liu X, Cao Q. Pharmacological inhibition of ferroptosis as a therapeutic target for sepsis-associated organ damage. Eur J Med Chem. 2023;257:115438. doi:10.1016/j.ejmech.2023.115438
38. Qian ZM, Ke Y. Brain iron transport. Biol Rev Camb Philos Soc. 2019;94(5):1672–1684. doi:10.1111/brv.12521
39. Urrutia P, Aguirre P, Esparza A, et al. Inflammation alters the expression of DMT1, FPN1 and hepcidin, and it causes iron accumulation in central nervous system cells. J Neurochem. 2013;126(4):541–549. doi:10.1111/jnc.12244
40. Wang J, Zhu Q, Li R, Zhang J, Ye X, Li X. YAP1 protects against septic liver injury via ferroptosis resistance. Cell Biosci. 2022;12(1):163. doi:10.1186/s13578-022-00902-7
41. Zhang J, Zheng Y, Wang Y, et al. YAP1 alleviates sepsis-induced acute lung injury via inhibiting ferritinophagy-mediated ferroptosis. Front Immunol. 2022;13:884362. doi:10.3389/fimmu.2022.884362
42. Li N, Wang W, Zhou H, et al. Ferritinophagy-mediated ferroptosis is involved in sepsis-induced cardiac injury. Free Radic Biol Med. 2020;160:303–318. doi:10.1016/j.freeradbiomed.2020.08.009
43. Chen Z, Cao Z, Gui F, et al. TMEM43 protects against sepsis-induced cardiac injury via inhibiting ferroptosis in mice. Cells. 2022;11:19.
44. Zhou B, Zhang J, Chen Y, et al. Puerarin protects against sepsis-induced myocardial injury through AMPK-mediated ferroptosis signaling. Aging. 2022;14(8):3617–3632. doi:10.18632/aging.204033
45. Xiao Z, Kong B, Fang J, et al. Ferrostatin-1 alleviates lipopolysaccharide-induced cardiac dysfunction. Bioengineered. 2021;12(2):9367–9376. doi:10.1080/21655979.2021.2001913
46. Li J, Li M, Li L, Ma J, Yao C, Yao S. Hydrogen sulfide attenuates ferroptosis and stimulates autophagy by blocking mTOR signaling in sepsis-induced acute lung injury. Mol Immunol. 2022;141:318–327. doi:10.1016/j.molimm.2021.12.003
47. Guo J, Wang R, Min F. Ginsenoside Rg1 ameliorates sepsis-induced acute kidney injury by inhibiting ferroptosis in renal tubular epithelial cells. J Leukoc Biol. 2022;112(5):1065–1077. doi:10.1002/JLB.1A0422-211R
48. Dodson M, Castro-Portuguez R, Zhang DD. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 2019;23:101107. doi:10.1016/j.redox.2019.101107
49. Kerins MJ, Ooi A. The Roles of NRF2 in modulating cellular iron homeostasis. Antioxid Redox Signal. 2018;29(17):1756–1773. doi:10.1089/ars.2017.7176
50. Galy B, Conrad M, Muckenthaler M. Mechanisms controlling cellular and systemic iron homeostasis. Nat Rev Mol Cell Biol. 2023;25:1. doi:10.1038/s41580-023-00648-1.
51. Han K, Jin X, Guo X, et al. Nrf2 knockout altered brain iron deposition and mitigated age-related motor dysfunction in aging mice. Free Radic Biol Med. 2021;162:592–602. doi:10.1016/j.freeradbiomed.2020.11.019
52. Li M, Zhang J, Jiang L, et al. Neuroprotective effects of morroniside from Cornus officinalis sieb. Et zucc against Parkinson’s disease via inhibiting oxidative stress and ferroptosis. BMC Complement Med Ther. 2023;23(1):218. doi:10.1186/s12906-023-03967-0
53. Olinder J, Borjesson A, Norrman J, et al. Hepcidin discriminates sepsis from other critical illness at admission to intensive care. Sci Rep. 2022;12(1):14857. doi:10.1038/s41598-022-18826-0
54. Scindia Y, Wlazlo E, Leeds J, et al. protective role of hepcidin in polymicrobial sepsis and acute kidney injury. Front Pharmacol. 2019;10:615. doi:10.3389/fphar.2019.00615
55. You LH, Yan CZ, Zheng BJ, et al. Astrocyte hepcidin is a key factor in LPS-induced neuronal apoptosis. Cell Death Dis. 2017;8(3):e2676. doi:10.1038/cddis.2017.93
56. Camaschella C, Nai A, Silvestri L. Iron metabolism and iron disorders revisited in the hepcidin era. Haematologica. 2020;105(2):260–272. doi:10.3324/haematol.2019.232124
57. Peng Y, Chang X, Lang M. Iron homeostasis disorder and alzheimer’s disease. Int J Mol Sci. 2021;22(22):12442. doi:10.3390/ijms222212442
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.