")
Back to Journals » OncoTargets and Therapy » Volume 17
METTL3/MALAT1/ELAVL1 Axis Promotes Tumor Growth in Ovarian Cancer
Authors Xiong J, Lian W, Zhao R, Gao K
Received 25 August 2023
Accepted for publication 24 January 2024
Published 8 February 2024 Volume 2024:17 Pages 85—97
DOI https://doi.org/10.2147/OTT.S431810
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Gaetano Romano
Jian Xiong,1,* Wenqin Lian,2,* Rui Zhao,1 Kefei Gao1
1Department of Obstetrics and Gynecology, Guangzhou Women and Children’s Medical Center, Guangzhou Medical University, Guangzhou, Guangdong, People’s Republic of China; 2Department of Surgery, Guangzhou Women and Children’s Medical Center, Guangzhou Medical University, Guangzhou, Guangdong, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Kefei Gao; Rui Zhao, Department of Obstetrics and Gynaecology, Guangzhou Women and Children’s Medical Center, Guangzhou Medical University, 9 Jinsui Road, Tianhe District, Guangzhou, Guangdong, 510623, People’s Republic of China, Email [email protected]; [email protected]
Background: Studies increasingly recognize the role of N6-methyladenosine (m6A) modification in cancer occurrence and development. METTL3 is a core catalytic subunit of m6A-modified methyltransferases complex, but its regulatory mechanism in ovarian cancer (OC) is not clear.
Methods: In this study, GEPIA 2.0 database was applied for expression analysis, survival analysis and correlation analysis for OC. Additionally, in vitro and in vivo assays were conducted to explore regulatory mechanisms of METTL3 in OC.
Results: We found that METTL3 and MALAT1 were significantly overexpressed in OC tissues and cells compared to normal ovarian tissues and cells. The proliferation rate of OC cells was reduced significantly after knocking down the expression of METTL3 or MALAT1. Subsequently, MALAT1 as oncogene was found to interact with METTL3 and was upregulated in OC tissues and cells. Silencing MALAT1 inhibited OC cell proliferation. Further studies indicated that METTL3 enhanced the stability of MALAT1 by promoting the m6A modification of MALAT1 and that ELAVL1 as a downstream binding protein significantly up-regulated MALAT1 expression.
Conclusion: In conclusion, METTL3 was a carcinogenic molecule that promoted the occurrence of OC. The potential mechanism of the carcinogenic effect of METTL3 was realized by enhancing the m6A modification of MALAT1 mRNA through RNA binding protein ELAVL1.
Keywords: N6-methyladenosine, ovarian cancer, METTL3, MALAT1
Introduction
Ovarian cancer (OC) is the second largest type of cancer in the female reproductive system and accounts for death cases more than any other female reproductive system cancer.1,2 Nearly 90% of OC cases originate from epithelial cells and comprise multiple histologic types, differing in specific molecular changes, clinical behaviours, and treatment outcomes. The remaining 10% are of non-epithelial origins, which mainly include germ cell tumors, sex cord-stromal tumors, and some extremely rare tumors such as small cell carcinomas.3 The treatment of OC is one of the most challenging in gynecological oncology, and the cure rate remains relatively the same over the past 30 years.4 The 5-year survival rate for stage I and II EOC patients is about 76%–91%. Unfortunately, over 75% of OC patients are diagnosed at advanced stage, and only 30% of these patients could survive for more than 5 years due to a lack of specific early clinical symptoms.5 BRCA1/2 germline mutations, which are the most influential genetic risk factors for epithelial OC, are present among 6–15% of women with epithelial OC. The BRCA1/2 status can be used as a therapeutic agent and for estimating prognostic outcomes because BRCA1/2 carriers with epithelial OC respond better than non-carriers to platinum-based chemotherapies. This may improve OC patients’ survival, even though the cancer is generally diagnosed at a later stage and higher grade.6 Clinical treatment outcomes of OC depend on the molecular and biological characteristics of tumor tissues. Epithelial OC develops according to two different carcinogenic pathways, specially, type I includes most endometrioid, clear cells, and mucinous carcinomas, whereas type II is high-grade, biologically aggressive tumors, with a propensity for metastasis from small-volume primary lesions. High-grade serous tumors develop according to the type II pathway and present p53 and BRCA mutations, while low-grade serous tumors are characterized by mutations in KRAS, BRAF, PTEN, PIK3CA.7 In this case, molecular targeted therapy given alone or used in combination with chemotherapy may produce favorable results.8 Genomic alterations in the DNA damage repair pathway are emerging as novel targets for treatment of OC. Platinum compounds and PARP inhibitors are the two main classes of active drugs against cancer cells harboring DNA damage repair alterations. On the other hand, as PD-L1 expression is relatively rare in OC, it is necessary to further investigate potential predictive biomarkers for immune-checkpoint inhibitors.9 Discovery of reliable OC biomarkers and their molecular mechanisms plays an important role in OC management and has a significant impact on the prognosis and molecular targeted therapy for OC patients.10,11 Proteomics, such as mass spectrometry and protein array analysis, have improved the study on underlying molecular signaling events and proteomic characterization of OC. Performing proteomics analysis to explore OC patients’ adaptive responses to therapy can help develop therapeutic choices, suppress drug resistance and potentially improve treatment outcomes.12
N6-methyladenosine (m6A) is a methylation at N6 position of adenosine, that affects the biological processes of cancer by regulating gene expression.13,14 M6A modification is dynamically regulated by methyltransferase, demethylase and RNA binding protein.15 Methyltransferase-like 3 (METTL3) is the only catalytic subunit among at least five methyltransferases that consist of m6A methylase complex.16,17 The expression of METTL3 is maladjusted in cancer via a mechanism dependent on or independent of its methyltransferase activity, and METTL3 acts as an oncogene or tumor suppressor gene to mediate the progression of cancer.18 According to Chen et al, METTL3 could interact with DGCR8 (a processor protein) and positively modulates the pri-miR221/222 process dependent on m6A, which allows it to act as an oncogene for bladder cancer.19 METTL3 in breast cancer accelerates the maturation of pri-miR1246 through m6A modification and further promotes SPRED2/MAPK signal pathway, thereby promoting colorectal cancer metastasis.20 The Warburg effect of cervical cancer could be expedited by METTL3 through enhancing the HK2 stability by YTHDF1-mediated m6A modification.21 However, the precise mechanism of METTL3 in regulating OC is still unclear.
This study explored the effect of METTL3 on the malignant progression of OC and its potential molecular mechanism by performing tissue and cell experiments and confirmed that mRNA m6A modification in MALAT1 was mediated by METTL3 and recognized and bound by ELAVL1, thereby exerting an active role in promoting OC tumorgenesis.
Materials and Methods
Cell Culture
The cell lines SK-OV-3, A2780, IOSE80 were purchased from the Chinese Academy of Sciences Cell Bank (China). All the cells were resuscitated and cultured in RPMI-1640 complete medium (90% RPMI1640 +10% fetal bovine serum) according to the instructions.
Transfection of Small Interfering RNAs (siRNA) and qRT-PCR
The siRNA for METTL3 (si-METTL3) was designed and synthesized by GenePharma (China) company. The OC cell lines were transfected with siRNA using an RFect transfection kit (Biogenerating Biotechnologies). The siRNA sequences used in this study were as follows: siMETTL3-1#: 5’-CTGCAAGTATGTTCACTATGA-3’, siMETTL3-2#: 5’-GCCAAGGAACAATCCATTGTT-3’; siMALAT1: 5’-UCUUCAAGAGAGAUAUUUAA-3’. Total RNA was extracted using TRIzol (Invitrogen) reagent according to the protocol. Then, cDNA was prepared by HiScript QRT SuperMix (Vazyme) for qPCR. The qPCR was conducted using SYBR Green PCR Kit (Vazyme). The primers for qPCR in our study were listed as follows: GAPDH-forward:5’-GTCAAGGCTGAGAACGGGAA-3’, GAPDH-reverse:5’-AAATGAGCCCCAGCCTTCTC-3’; METTL3-forward:5’-ACACTGCTTGGTTGGTGTCA-3’, METTL3-reverse:5’-GCGAGTGCCAGGAGATAGTC-3’;
MALAT1-forward: 5’-GCATTAATTGACAGCTGACCCA-3’;
MALAT1-reverse: 5’- GCTTGCTCCTCAGTCCTAGCTT-3’;
MALAT1 for m6A-IP-qPCR forward: 5′- TTCCGGGTGTTGTAGGTTTC-3′;
MALAT1 for m6A-IP-qPCR reverse: 5′- AAAAAACCCACAAACTTGCC-3′.
Western Blot (WB)
The cells were lysed with RIPA (Beyotime, China) buffer to extract total protein. The protein was quantified by BCA protein detection kit (Beyotime, China), and the same amount of protein was added to 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) for separation. After being moved onto polyvinylidene fluoride (PVDF) membranes, the proteins were sealed in 5% skimmed milk. The membrane was washed with PBST and incubated with anti-METTL3 (ab195352) and β-actin (ab8226) and incubated overnight at 4 °C. Then, the membrane was incubated in horseradish peroxidase-coupled goat anti-rabbit (ab6789) for 2 hours (h). After treatment with chemiluminescent Western blot detection kit (GE, USA), the images of the membrane was observed in Odyssey CLx instrument.
Immunohistochemistry (IHC)
IHC analysis for tissue array of OC (Shanghai Outdo Biotech Company, China) was performed strictly according to the instructions provided by GT Vision III kit (Genetech, China). Following the method of Yuan et al,22 the score of immunostaining was evaluated by considering the score of staining intensity and staining area. The staining intensity score (3 represented strong, 2 represented moderate, 1 represented weak, 0 represented negative) was evaluated according to the level of expression. The stained area score (0% represented by 0, 1–25% by 1, 26–50% by 2, and 51–100% by 3) was calculated based on the percentage of positive cells.
Cell Counting Kit-8 (CCK-8) Assay
Cell proliferation assay was performed according to the CCK-8 instructions (Beyotime, China). Cell suspensions were added to the 96-well plate, and 100μL medium was added per well. Ten microliter CCK-8 reagent was added to each well at specified time points (every 24h) after transfection. The absorbance was measured at 450nm.
Colony Formation Assay
Cells with si-METTL3 knockdown were inoculated into 6-well plates at a density of 500 cells per well and cultured at 37°C 1–2 weeks to form visible colonies. The cells were fixed with methanol and then stained with 0.3% crystal violet to count the cell colonies.
5-Ethynyl-2’-Deoxyuridine (EdU) Assay
Click-iT cells EdU cell proliferation imaging kit (Invitrogen, USA) used fluorescence labeling to detect the ability of cell proliferation. The cells (1×104 per well) were placed in a 96-well plate and added with 20 uL EdU reagent and incubated for 2h. After washing with PBS, each well was added with cell fixative and incubated at room temperature for 30 minutes (min), followed by treatment with 0.5%Triton X-100 to increase membrane permeability. Finally, the cells were stained with 4′,6-diamidino-2-phenylindole (DAPI), and the images were obtained under a fluorescence microscope (ZEISS, Oberkochen, Germany).
The m6A Sequencing
Total RNA was extracted and purified by PolyTtract mRNA Isolation System (Promega). After fragmentation, magna methylated RNA immunoprecipitation m6A Kit (Merck Millipore, Germany) was used for m6A RNA immunoprecipitation. In short, total RNA was dispersed into 100nt-long oligonucleotides and then incubated with anti-m6A antibodies or immunoglobulin (IgG). The fragmented RNA containing m6A was eluted and purified with RNA purification kit (Qiagen, Germany). Enriched fragments were analyzed by qPCR. The purified RNA fragments were used to construct libraries using the NEBNext Ultra RNA Library Prep Kit (New England Biolabs, USA) and sequenced using Illumina HiSeq 2000 (Illumina, USA) platform. The level of m6A mRNA was measured by qPCR.
RNA‐binding Protein Immunoprecipitation (RIP)-qPCR
RIP assays were conducted using the Magna RIPTM RNA‐Binding Protein Immunoprecipitation Kit (Millipore). The cells were washed with PBS twice, and the precipitates were collected and re-suspended in the IP lysis buffer. After cracking for 30 min, the lysate was harvested by high-speed centrifugation. Antibodies and protein A/G beads were added to the lysate and incubated together overnight at 4°C. RNA was eluted from magnetic beads with washing buffer, and coprecipitation RNA was extracted by cross-linking and analyzed by qPCR.
In situ Hybridization Staining
Tissue array of OC (Shanghai Outdo Biotech Company, China) was probed for MALAT1 expression using the RNAscope 2.0 HD-Brown Manual Assay (Advanced Cell Diagnostics, USA) according to the manufacturer’s instructions. Briefly, antigen retrieval was performed for 15 min, followed by protease digestion for 30 minutes, probe incubation overnight at 40°C and amplification. The probes used were hs-MALAT1 (400,811). MALAT1 expression was visualized with DAB.
Expression Analysis in GEPIA 2.0 Database
For tumors without normal control tissues, we used the “Expression analysis-Box Plot” module of the GEPIA2 (Gene Expression Profiling Interactive Analysis, version 2) database (http://GEPIA2.cancer-pku.cn/#analysis) to obtain tumor tissue expression and the corresponding expression in normal tissue of the GTEx (Genotype-Tissue Expression) database, under the settings of P-value cutoff = 0.01, log2FC (fold change) cutoff = 1, and “Match TCGA normal and GTEx data”.
Statistical Analysis
The statistical software used in this study included SPSS, GraphPad Prism and R programs. Statistical differences were assessed using Student’s t-test or unpaired two-tailed t-test. Difference of statistical significance was marked * in the figures with more * indicating a greater significant difference, specifically, * represented p < 0.05, ** represented p < 0.01, *** represented p < 0.001, and **** represented p < 0.0001.
Results
Overexpression of METTL3 in OC Tissues and Cells
The METTL3 expression in various types of cancer and normal tissues was detected. We found that the METTL3 expression in CHOL, STAD, READ, CESC, BLCA, GBM, LIHC, COAD, LUSC, LUAD, UCEC, HNSC, PRAD and ESCA was significantly higher than that in the normal samples, while the level of METTL3 in PCPG, KIAC and THCA was significantly lower than that in corresponding normal tissues (Figure 1A) in TIMER 2.0 database. Analysis of the differential expression of METTL3 between OC tissues in TCGA and normal tissues in GTEx database from GEPIA 2.0 database showed that METTL3 was significantly up-regulated in OC tumor tissues compared with normal tissues (Figure 1B). According to the median expression of METTL3, there was no significant difference in survival rate between the two groups (Figure 1C) in GEPIA 2.0 database. Compared with adjacent normal tissue, the immunostaining score (H-score) of METTL3 in OC tissues was significantly higher (Figure 1D). The subdivided histological subtypes of OC include serous, borderline mucinous, endometrioid and clear cell and mucinous. The H-score of each histological subtype of METTL3 was evaluated and compared to the adjacent normal tissues. It was found that the H-score difference of METTL3 between endometrioid OC and normal tissues was the most significant (Figure 1E). The results of qRT-PCR and WB in human normal ovarian epithelial cells and OC cells showed that in the two OC cell lines, the expression of METTL3 was significantly higher than human normal ovarian epithelial cells in terms of its mRNA and protein expression (Figure 1F and G).
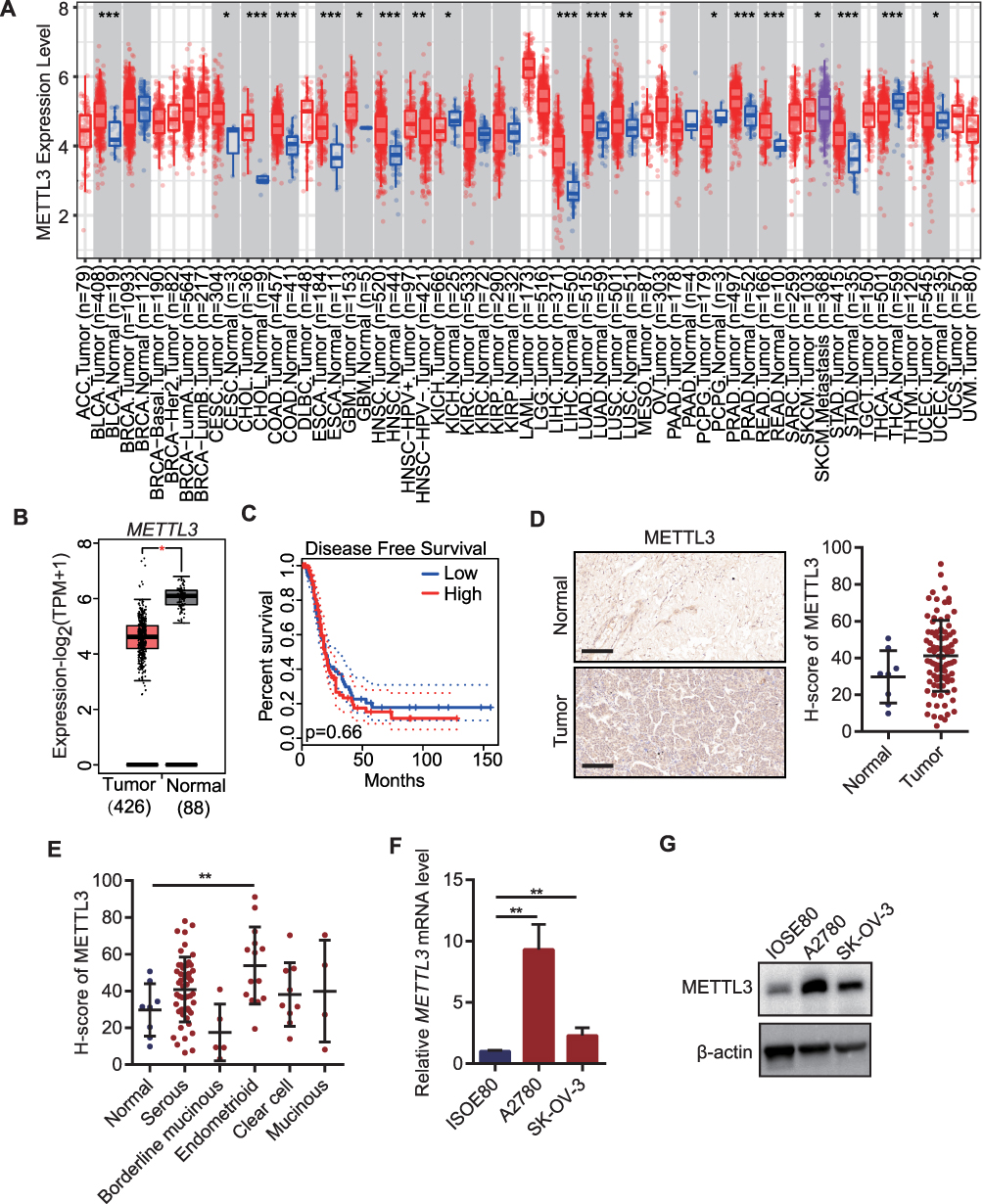 |
Figure 1 Overexpression of METTL3 in ovarian cancer tissues and cells. (A) METTL3 in different tumors in TIMER 2.0 database; (B) METTL3 expression between OC tissues and normal tissues in GEPIA 2.0 database; (C) The Kaplan–Meier curve of OC patients with high METTL3 and low METTL3 in GEPIA 2.0 database; (D) IHC and H-score for METTL3 in OC tissue array; (E) H-score difference of METTL3 between histological subtypes of ovarian cancer; (F) qRT-PCR for METTL3 mRNA in OC cell lines; (G) The protein expression level of METTL3 in ovarian cancer cell line. *p<0.05, **p<0.01, ***p<0.001. |
Knocking Down METTL3 Inhibited the Proliferation of OC Cells
The siRNA of METTL3 was transfected into both OC cell lines, and the effect of knocking down METTL3 was determined by WB (Figure 2A). The effect of knocking down METTL3 on cell proliferation was detected by CCK-8, colony formation and EdU assay. CCK-8 assay results showed that the activity of the two OC cells, with METTL3 knockdown was significantly reduced compared with the control cells (Figure 2B). The number of colonies formed by the cells loaded with si-METTL3 vector was significantly less than that of the cells transferred into the control vector, as shown by the colony formation assay (Figure 2C). From the results of EdU staining, it can be observed that the number of positive EdU staining cells reduced significantly in the group with inhibited expression of METTL3 (Figure 2D). These experiments proved the result that the knockdown of METTL3 significantly weakened the proliferation ability of cells.
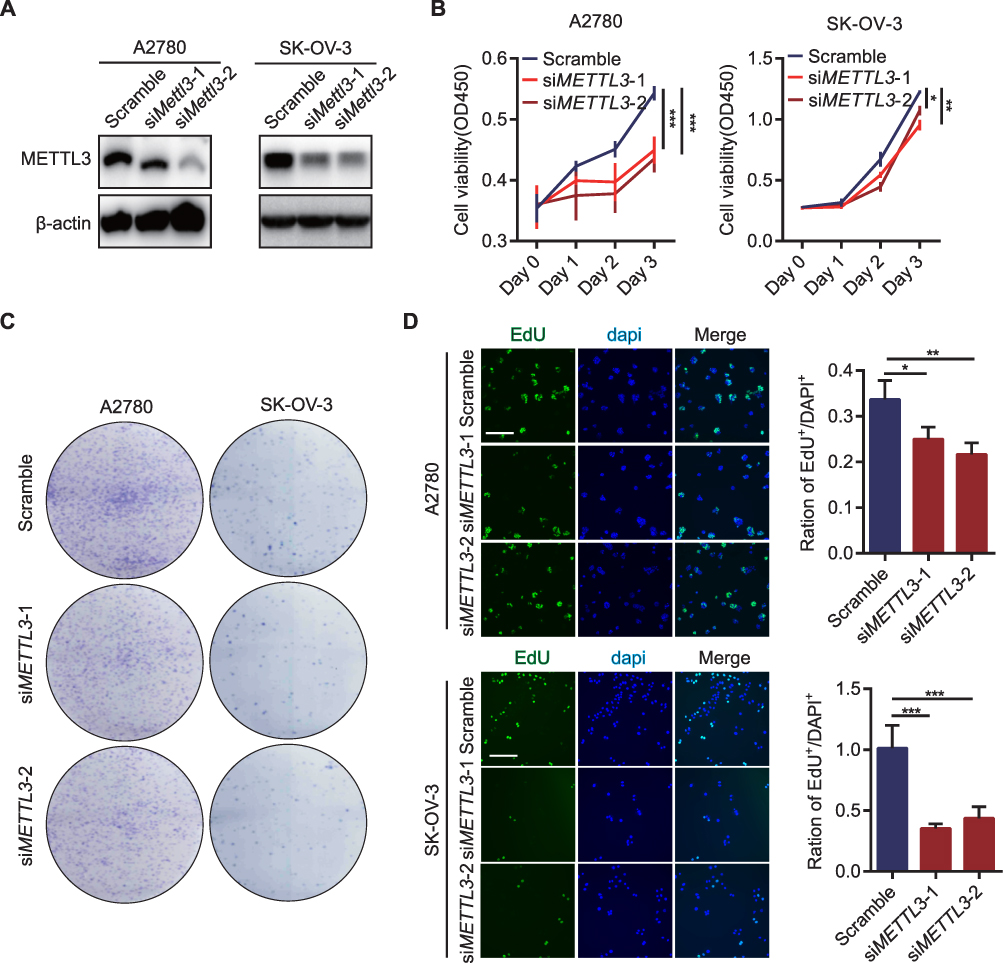 |
Figure 2 Knockdown of METTL3 inhibited the proliferation of OC cells. (A) The knockdown effect of METTL3 in OC cell lines detected by WB assay; (B) Cell proliferation detection by CCK-8 assay in OC cell lines after knocking down METTL3; (C) Clone formation of OC cell lines after knocking down METTL3; (D) EdU staining of OC cell lines after knocking down METTL3. *p<0.05, **p<0.01, ***p<0.001. |
M6A Modification in OC Cells
We compared the m6A modification of human normal ovarian epithelial cells and OC cells, including count distribution of m6A sites, distributed area of various RNA species by m6A modifications and conserved motifs at m6A. The number of m6A loci in human OC cell line A2780 was less than that in human normal ovarian epithelial cell line IOSE80 (Figure 3A). M6A loci in both kinds of cells were mainly distributed on mRNA and enriched in coding sequences (CDSs) and 3 “UTR regions (Figure 3B and C). Specifically, the 3”UTR region of A2780 cells was enriched with about 37.5% of the m6A peak, other exon was enriched with about 38% of the m6A peak, and the 5’UTR region was enriched with about 12% of the m6A peak. The distribution proportion of m6A peak in different regions of IOSE80 cells was not different from that in A2780 cells (Figure 3E). The m6A motif in human OC cells and human normal ovarian epithelial cells was predicted by using the mRNA sequence corresponding to the m6A methylation peak (Figure 3D). The proportion of counts of m6A sites in m6A up group was similar to that in m6A down group, and both of them mainly existed in mRNA, but there were more m6A sites in mRNA in m6A downregulation group, and more m6A sites in lncRNA in m6A upregulation group (Figure 3F). The m6A peak in the m6A downregulation group was mainly concentrated in the 3 “UTR region and other exon, while the m6A peak in the m6A upregulation group distributed more in the 3” UTR region, other exons and 5’UTRs. It was obvious that the enrichment ratio of the m6A peak in the 5’UTR region was significantly higher in m6A upregulation group than that in the m6A downregulation group (Figure 3G). KEGG was employed to determine whether there were different biological pathways between m6A upregulated genes and m6A downregulated genes (Supplementary File 1). We found that the upregulation of m6A was significantly correlated with regulation of RNA biosynthetic process, RNA metabolic process, and cellular metabolic process, and the downregulation of m6A was significantly correlated with rRNA metabolic process, cell differentiation, and TORC2 signaling pathway (Figure 3H).
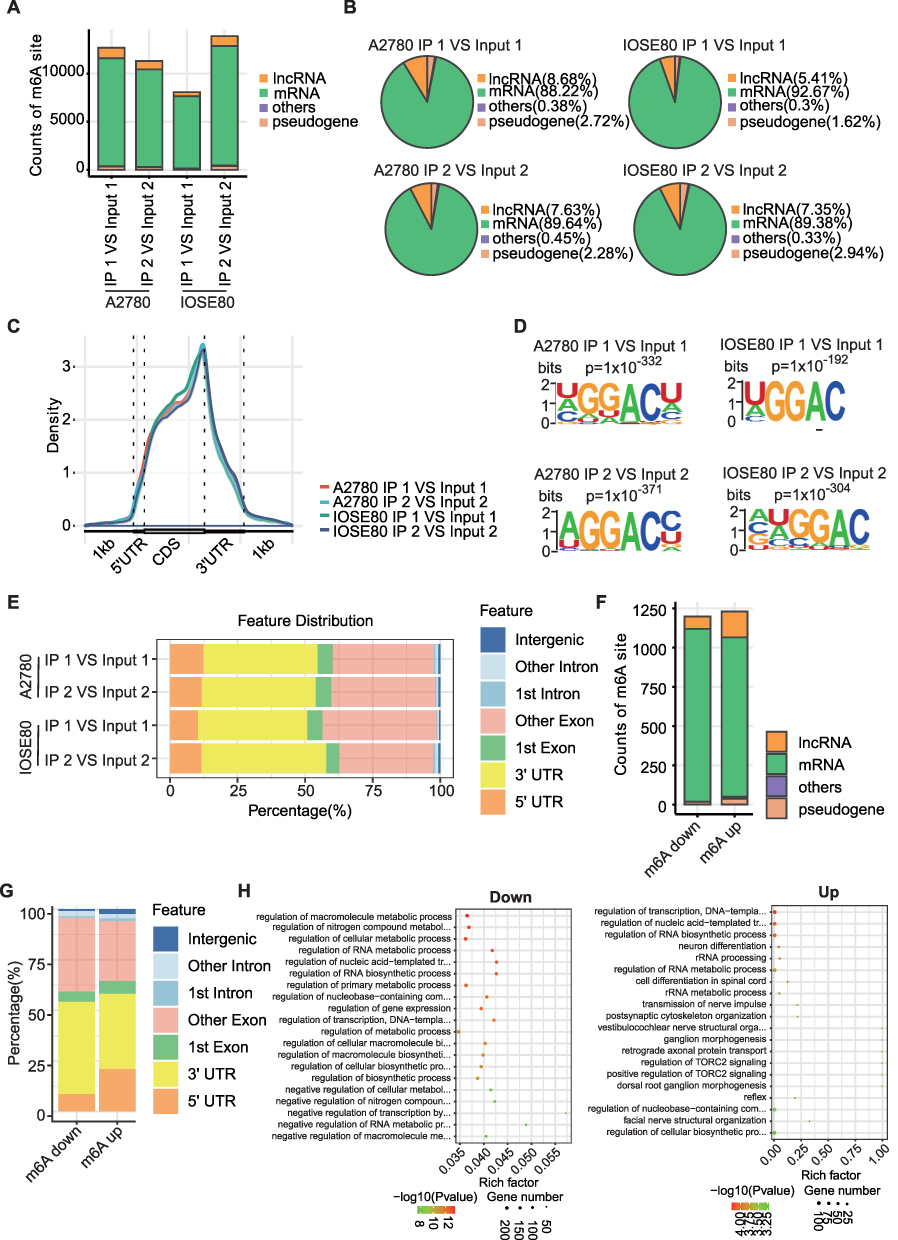 |
Figure 3 m6A modification in OC cells. (A) The number of m6A loci in human OC cell line A2780 and human normal ovarian epithelial cell line IOSE80. (B) Pie chart showed percentages of various RNA types by m6A modifications in A2780 cells and IOSE80 cells. (C) Density of m6A peaks across mRNA transcriptome in A2780 cells and IOSE80 cells. (D) Conserved motif of m6A site in human OC cells and human normal ovarian epithelial cells. (E) The distribution of m6A locus in A2780 cells and IOSE80 cells in different regions of RNA. (F) The proportion of counts of m6A sites among different types of RNA in m6A up group and m6A down group. (G) The proportion of mRNA structure region enriched by m6A peak was in m6A downregulation group and m6A downregulation group. (H) KEGG analysis of m6A up-regulated and down-regulated genes. |
The presence of m6A modification in MALAT1 in OC cells and overexpression and downregulation of MALAT1 level weakened cancer cell proliferation.
RNA analysis of human OC cell line A2780 and m6A antibody precipitation of human normal ovarian epithelial cell-line IOSE80 revealed peaks mapping to MALAT1 mRNA, indicating the presence of m6A site on MALAT1 (Figure 4A). M6A RNA immunoprecipitation showed a significant increase in m6A levels in two OC cell lines compared with normal ovarian epithelial cell lines (Figure 4B). Considering the effect of m6A modification, MALAT1 expression was analyzed in different OC sources. By comparing the MALAT1 expression between the OC tissues in the TCGA database and the normal tissues in the GTEx database, no significant difference in MALAT1 expression between the two tissues was found, and the MALAT1 expression did not significantly cause the survival difference of OC in the TCGA (Figure 4C and D). We performed IHC in the collected tissues and evaluated H-score. It was found that the H-score of MALAT1 was significantly higher in OC tissue than normal adjacent tissue (Figure 4E). The five histological subtypes of ovarian carcinoma also showed significantly higher in H-score of MALAT1 (Figure 4F) than normal tissues. Cell experiments also demonstrated significant overexpression of MALAT1 levels (Figure 4G) in two OC cell lines compared to normal ovarian epithelial cells. MALAT1 levels (Figure 4H) were inhibited by the transfection of MALAT1 siRNAs in two OC cell lines, and the effect of MALAT1 inhibition on cell proliferation was observed. According to the increasing trend of absorbance at 450nm measured by CCK-8, the absorbance in OC cells transfected si-MALAT1 were significantly weaker than those in the control group (Figure 4I). Colony density and EdU-positive rate were also significantly lower than in the control group (Figure 4J and K).
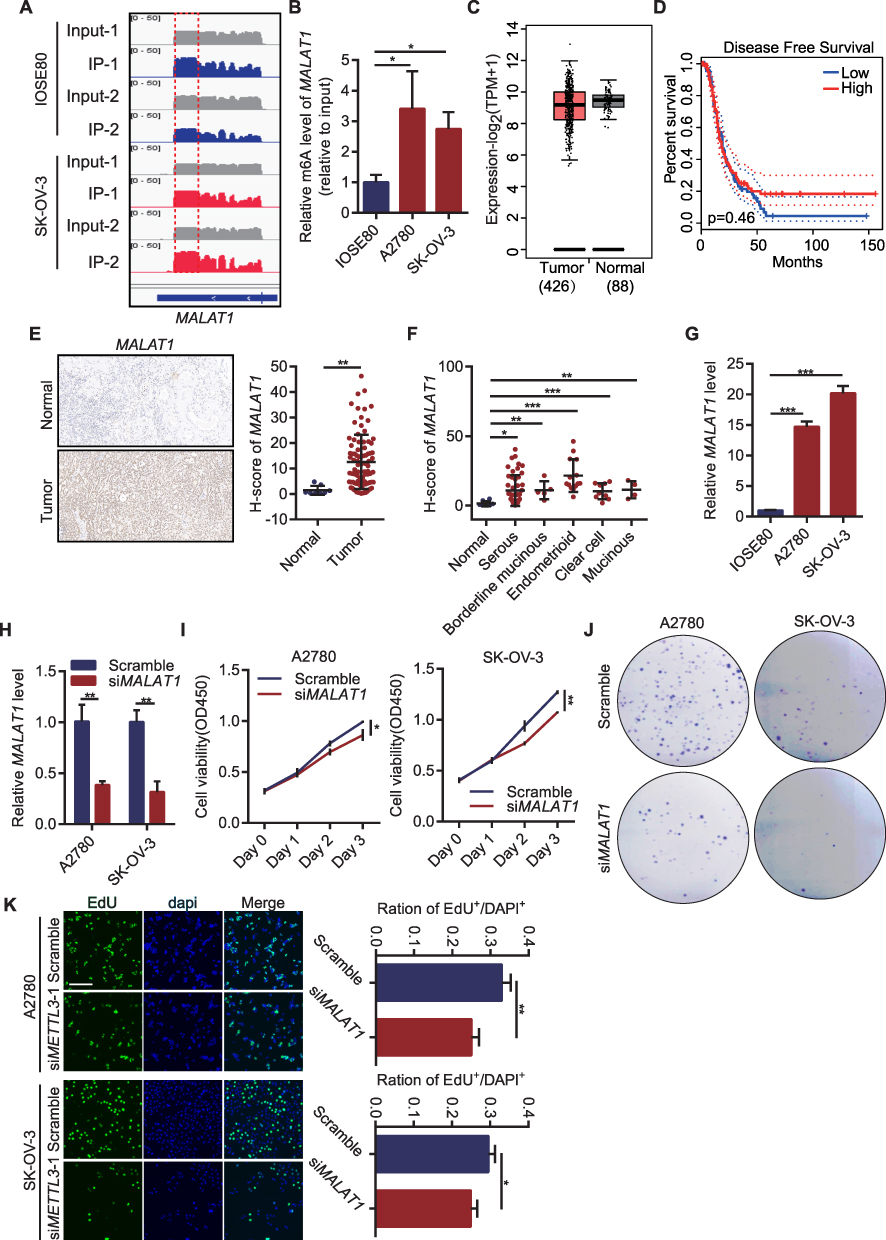 |
Figure 4 MALAT1 with m6A modification promoted OC proliferation. (A) IgV showed m6A level of MALAT1 in the human OC cell line A2780 and human normal ovarian epithelial cell line IOSE80; (B) m6A-IP-qPCR detected m6A levels of MALAT1 in human OC cell lines and human normal ovarian epithelial cell lines; (C) MALAT1 expression in OC tissues and normal tissues in GEPIA 2.0 database; (D) The Kaplan–Meier curve of OC patients with high MALAT1 and low MALAT1 in GEPIA 2.0 database; (E) MALAT1 expression in OC tissue array by in situ hybridization; (F) Expression of MALAT1 in different pathological types of OC; (G) MALAT1 level in two OC cell lines and in human normal ovarian epithelial cells; (H). Knock down efficiency after knocking down MALAT1 in two OC cell lines; (I) CCK-8 assays showed changes in the viability of OC cells transfected with siMALAT1. (J) Colony formation of OC cells transfected with siMALAT1; (K) EdU-positive rate of OC cells transfected with siMALAT1. *p<0.05, **p<0.01,***p<0.001. |
METTL3 Enhanced MALAT1 Stability via ELAVL1
The m6A modification of MALAT1 was likely to be catalyzed primarily by MALAT1 because of the obvious modification of m6A in the METTL3, which was the core enzyme that catalyzed the modification of m6A. To verify the accuracy of this hypothesis, the relationship between METTL3 and MALAT1 was analyzed. In the OC dataset of TCGA, both METTL3 and MALAT1 were positively correlated at mRNA level and expression level (Figure 5A and B). By analyzing the results of RIP-qPCR, we found that a large number of co-precipitation complexes of METTL3 and MALAT1 (Figure 5C) formed in the OC cell line. After transfecting si-METTL3 into two OC cell lines, compared with control OC cells, the m6A level in MALAT1 in the cells transfected with si-METTL3 was significantly reduced (Figure 5D) and the inhibition of METTL3 also lowered the MALAT1 level (Figure 5E). M6A modification was co-regulated by modifier enzymes and RNA-binding proteins. ELAVL1 was an important RNA-binding protein. By analyzing the expression of ELAVL1 in the OC tissues from TCGA and the normal tissues from the GTEx database, we found that the ELAVL1 was overexpressed in the OC tissues, but there was no significant difference between the OC tissues and normal tissues in GEPIA 2.0 database (Figure 5F); moreover, the ELAVL1 was also positively correlated with the MALAT1 expression (Figure 5G) in GEPIA 2.0 database. In the two OC cell lines, ELAVL1 showed enrichment of MALAT1 with significantly upregulated mRNA (Figure 5H) in comparison to IgG. In OC cells with METTL3 knockdown, there was a significant increase of MALAT1 in the RNA of MALAT1 antibody precipitation (Figure 5I). After inhibiting the level of ELAVL1, the expression of MALAT1 was also significantly inhibited (Figure 5J). Also, in the OC cells with inhibited ELAVL1, the stability was greatly weakened (Figure 5K).
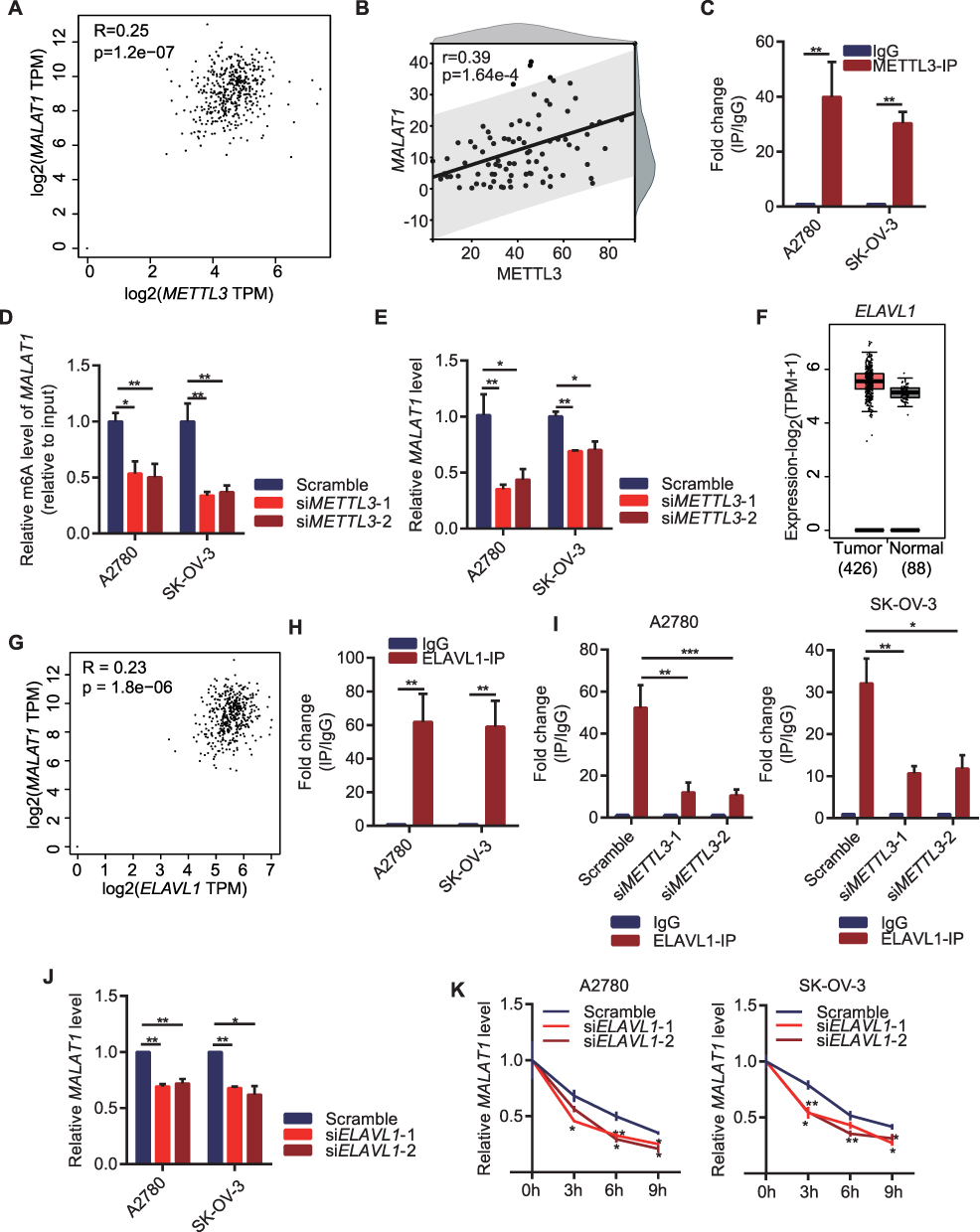 |
Figure 5 METTL3/MALAT1/ELAVL1 axis promotes ovarian cancer cell proliferation via m6A modification. (A) Correlation between METTL3 and MALAT1 in OC in GEPIA 2.0 database; (B) Correlation of METTL3 and MALAT1 in OC tissue array; (C) Interaction between METTL3 and MALAT1 according to RIP-qPCR in OC cell lines; (D) The m6A level in MALAT1 after knocking down METTL3 in OC cell lines; (E) MALAT1 level of OC cell lines after knocking down METTL3; (F) ELAVL1 level between OC tissues and normal ovarian tissue in GEPIA 2.0 database; (G) Correlation between ELAVL1 and MALAT1 in OC in GEPIA 2.0 database; (H) Interaction between ELAVL1 and MALAT1 according to RIP-qPCR in OC cell lines; (I) Interaction between ELAVL1 and MALAT1 after knocking down METTL3 in OC cell lines; (J) The expression of MALAT1 after knocking down ELAVL1 in OC cell lines; (K) The stability of MALAT1 after inhibiting ELAVL1 in OC cell lines. *p<0.05, **p<0.01. |
Discussion
The development of METTL3 targeted drugs in recent years relied on the studies of regulation, function and mechanisms of METTL3 in cancers.15 METTL3 involvement and several regulatory mechanisms have also been reported in OC. Bi et al performed in vitro and in vivo experiments and observed that METTL3 silencing inhibits PTEN through impairing on miR-126-5p targeting, thereby blocking PI3K/Akt/mTOR pathways and hindering OC progression and tumorigenesis.23 METTL3 accelerates cisplatin resistance in OC through RHPN1-AS1 regulated by m6A modification via activating PI3K/AKT pathway.24 According to Hua et al, METTL3 has a very strong carcinogenic effect on OC by stimulating AXL translation and EMT.25 Moreover, previous experimental findings showed that METTL3 activates the AKT pathway in OC cells. A low expression of METTL3 may facilitate OC cell apoptosis through the mitochondrial apoptotic pathway and inhibit cancer cell invasion by reducing the activation of the AKT signaling pathway and the expression of the downstream effector Cyclin D1.26 Although these studies analyzed the tumor-causing mechanisms of METTL3 mediated by m6A modification in OC, m6A modification is co-regulated by modifying enzymes and RNA-binding proteins, therefore we cannot rule out the potential involvement and involvement of molecular mechanisms underlying the interaction between METTL3 and RNA-binding proteins in OC.
According to a previous study, m6A modification is catalyzed by METTL3 and enriched in the 3’ untranslated region of a large subset of mRNAs at the sites close to the stop codon.27 In this study, we analyzed m6A modification in OC cells and found that m6A sites were mainly distributed on mRNAs and enriched in coding sequences (CDSs) and 3’UTR regions, which was in accordance with the previous findings. Past research28 also showed that METTL3 expression in OC cells and tissues is significantly higher than that in normal cells and tissues. Hence, knocking down METTL3 could greatly weakened the proliferation capacity of OC cells.
Another novel molecular mechanism of METTL3 in promoting OC has been reported.29 Regulatory effects of METTL3 on MALAT1 expression have been observed in non-small cell lung30 and breast cancer.31 METTL3 increases the stability of MALAT1 and enhances glioma progression.32 In this study, METTL3 promoted MALAT1 m6A modification of MALAT1 in OC but different from the above studies, we found that such a regulation of MALAT1 by METTL3 in OC required the involvement of RNA-binding protein ELAVL1. According to a study, the recruitment of ELAVL1 to m6A-modified RNA is necessary for METTL3 to stabilize SOX2 mRNA.33 These findings supported that ELAVL1 is one of the key factors in m6A modification mediated by METTL3.
ELAVL1 was found to regulate RNA stability in an m6A-dependent manner. The total amount of m6A in RNA can be detected by a variety of methods. METTL3 plays a crucial role in the development of gastrointestinal cancer, but there are still many challenges that require further analysis. Future research could focus on the role of METTL3 in the tumor microenvironment, the molecular mechanisms that regulate METTL3 expression and activity, and the screening of specific inhibitors for clinical application.34
The underlying cause-effect mechanism between METTL3 and OC development should be clarified.35 METTL3 enhances IGF2BP3 axis for inhibiting tumor immune surveillance by upregulating m6A modification of PD-L1 mRNA in breast cancer. Recent studies have revealed the roles and molecular mechanisms associated with METTL3 in many cancers.16 In most cases, METTL3 is a carcinogenic gene that promotes the occurrence and development of multiple cancers by depositing m6A modifications on key transcripts.15 Full-length METTL3 has 580 amino acids, and m6A is enriched in 3′ untranslated regions (3′UTRs), around stop codons and within long internal exons.36
Although the present study provides compelling evidence for the overexpression of METTL3 and MALAT1 in OC and their role in promoting OC cell proliferation through the m6A modification of MALAT1 mRNA, it still has some limitations. Firstly, the study mainly relied on in vitro experiments using cell lines and did not provide any data from in vivo studies or patient samples. Therefore, our findings should be validated in a larger cohort of patient samples and animal models before direct applying METTL3 and MALAT1 in OC treatment. Additionally, we did not investigate the downstream effects of the METTL3/MALAT1 axis on other cellular processes involved in OC, such as migration, invasion, and metastasis. Which requires further studies to explore the complete role of the METTL3/MALAT1 axis in OC and its impact on overall OC progression.
In conclusion, our study showed that METTL3 promoted OC progression by mediating m6A modification via which ELAVL1 further interacted with MALAT1 to regulate MALAT1 stability.
Data Sharing Statement
The original contributions presented in the study are included in the article/Supplementary File 1. The raw m6A-seq data presented in this study have been deposited in the Genome Sequence Archive in BIG Data Center, Beijing Institute of Genomics (BIG), Chinese Academy of Sciences, under accession number HRA003854 that is accessible at https://bigd.big.ac.cn. Further inquiries can be directed to the first author and corresponding authors.
Ethics Approval and Consent to Participate
The studies involving human ovarian cancer samples were reviewed and approved by the Ethics Committee of Shanghai Outdo Biotech Company (SHYJS-CP-1907005). Our study was also approved by the Ethics Committee of Guangzhou Women and Children’s Medical Center (2022-082B00). The present study was conducted with written informed consent from all of the patients.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was funded by the Research foundation of Guangzhou Women and Children’s Medical Center for Clinical Doctor (2021BS044, 2020RC004), and the Science and Technology Program of Guangzhou, China (2024A03J0807, 2024A03J0956).
Disclosure
The authors disclose no conflicts of interest in this work.
References
1. Siegel RL, Miller KD, Fuchs HE, Jemal A, Cancer statistics, 2022. CA Cancer J Clin. 2022;72(1):7–33.
2. Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–249. doi:10.3322/caac.21660
3. Saani I, Raj N, Sood R, et al. Clinical challenges in the management of malignant ovarian germ cell tumours. Int J Environ Res Public Health. 2023;20(12):6089. doi:10.3390/ijerph20126089
4. Sehouli J, Grabowski J. Surgery in recurrent ovarian cancer. Cancer. 2019;125(24):4598–4601. doi:10.1002/cncr.32511
5. Arend R, Martinez A, Szul T, et al. Biomarkers in ovarian cancer: to be or not to be. Cancer. 2019;125(24):4563–4572. doi:10.1002/cncr.32595
6. Shah S, Cheung A, Kutka M, et al. Epithelial ovarian cancer: providing evidence of predisposition genes. Int J Environ Res Public Health. 2022;19(13):8113. doi:10.3390/ijerph19138113
7. Pavlidis N, Rassy E, Vermorken JB, et al. The outcome of patients with serous papillary peritoneal cancer, fallopian tube cancer, and epithelial ovarian cancer by treatment eras: 27 years data from the SEER registry. Cancer Epidemiol. 2021;75:102045. doi:10.1016/j.canep.2021.102045
8. Luvero D, Plotti F, Aloisia A, et al. Ovarian cancer relapse: from the latest scientific evidence to the best practice. Crit Rev Oncol Hematol. 2019;140:28–38. doi:10.1016/j.critrevonc.2019.05.014
9. Revythis A, Limbu A, Mikropoulos C, et al. Recent insights into PARP and immuno-checkpoint inhibitors in epithelial ovarian cancer. Int J Environ Res Public Health. 2022;19(14):8577. doi:10.3390/ijerph19148577
10. Bonifácio VDB. Ovarian cancer biomarkers: moving forward in early detection. Adv Exp Med Biol. 2020;1219:355–363.
11. Zheng Y, Wang Y, Zou C, et al. Tumor-associated macrophages facilitate the proliferation and migration of cervical cancer cells. Oncologie. 2022;24(1):147–161. doi:10.32604/oncologie.2022.019236
12. Ghose A, Gullapalli SVN, Chohan N, et al. Applications of proteomics in ovarian cancer: dawn of a New Era. Proteomes. 2022;10(2):16. doi:10.3390/proteomes10020016
13. He L, Li H, Wu A, et al. Functions of N6-methyladenosine and its role in cancer. Mol Cancer. 2019;18(1):176. doi:10.1186/s12943-019-1109-9
14. Yao W, Bin Q, Shen H, Deng H, Tang F. Serum level of tumor-specific growth factor in patients with cervical cancer and its potential prognostic role. Oncologie. 2022;24(3):499–512. doi:10.32604/oncologie.2022.024951
15. Jiang X, Liu B, Nie Z, et al. The role of m6A modification in the biological functions and diseases. Signal Transduct Target Ther. 2021;6(1):74. doi:10.1038/s41392-020-00450-x
16. Chen J, Guo B, Liu X, et al. Roles of N(6)-methyladenosine (m(6)A) modifications in gynecologic cancers: mechanisms and therapeutic targeting. Exp Hematol Oncol. 2022;11(1):98. doi:10.1186/s40164-022-00357-z
17. Zeng C, Huang W, Li Y, et al. Roles of METTL3 in cancer: mechanisms and therapeutic targeting. J Hematol Oncol. 2020;13(1):117. doi:10.1186/s13045-020-00951-w
18. Liu S, Zhuo L, Wang J, et al. METTL3 plays multiple functions in biological processes. Am J Cancer Res. 2020;10(6):1631–1646.
19. Han J, Wang J-Z, Yang X, et al. METTL3 promote tumor proliferation of bladder cancer by accelerating pri-miR221/222 maturation in m6A-dependent manner. Mol Cancer. 2019;18(1):110. doi:10.1186/s12943-019-1036-9
20. Peng W, Li J, Chen R, et al. Upregulated METTL3 promotes metastasis of colorectal Cancer via miR-1246/SPRED2/MAPK signaling pathway. J Exp Clin Cancer Res. 2019;38(1):393. doi:10.1186/s13046-019-1408-4
21. Wang Q, Guo X, Li L, et al. N6-methyladenosine METTL3 promotes cervical cancer tumorigenesis and Warburg effect through YTHDF1/HK2 modification. Cell Death Dis. 2020;11(10):911. doi:10.1038/s41419-020-03071-y
22. Yuan C, Zhu X, Han Y, et al. Elevated HOXA1 expression correlates with accelerated tumor cell proliferation and poor prognosis in gastric cancer partly via cyclin D1. J Exp Clin Cancer Res. 2016;35:15. doi:10.1186/s13046-016-0294-2
23. Bi X, Lv X, Liu D, et al. METTL3-mediated maturation of miR-126-5p promotes ovarian cancer progression via PTEN-mediated PI3K/Akt/mTOR pathway. Cancer Gene Ther. 2021;28(3–4):335–349. doi:10.1038/s41417-020-00222-3
24. Cui S. METTL3-mediated m6A modification of lnc RNA RHPN1-AS1 enhances cisplatin resistance in ovarian cancer by activating PI3K/AKT pathway. J Clin Lab Anal. 2022;36(12):e24761. doi:10.1002/jcla.24761
25. Aliyuda F, Moschetta M, Ghose A, et al. Advances in ovarian cancer treatment beyond PARP inhibitors. Curr Cancer Drug Targets. 2023;23(6):433–446. doi:10.2174/1568009623666230209121732
26. Hua W, Zhao Y, Jin X, et al. METTL3 promotes ovarian carcinoma growth and invasion through the regulation of AXL translation and epithelial to mesenchymal transition. Gynecol Oncol. 2018;151(2):356–365. doi:10.1016/j.ygyno.2018.09.015
27. Choe J, Lin S, Zhang W, et al. mRNA circularization by METTL3-eIF3h enhances translation and promotes oncogenesis. Nature. 2018;561(7724):556–560. doi:10.1038/s41586-018-0538-8
28. Liang S, Guan H, Lin X, et al. METTL3 serves an oncogenic role in human ovarian cancer cells partially via the AKT signaling pathway. Oncol Lett. 2020;19(4):3197–3204. doi:10.3892/ol.2020.11425
29. Jin D, Guo J, Wu Y, et al. m(6)A mRNA methylation initiated by METTL3 directly promotes YAP translation and increases YAP activity by regulating the MALAT1-miR-1914-3p-YAP axis to induce NSCLC drug resistance and metastasis. J Hematol Oncol. 2019;12(1):135. doi:10.1186/s13045-019-0830-6
30. Li S, Jiang F, Chen F, et al. Effect of m6A methyltransferase METTL3 -mediated MALAT1/E2F1/AGR2 axis on Adriamycin resistance in breast cancer. J Biochem Mol Toxicol. 2022;36(1):e22922. doi:10.1002/jbt.22922
31. Visvanathan A, Patil V, Arora A, et al. Essential role of METTL3-mediated m(6)A modification in glioma stem-like cells maintenance and radioresistance. Oncogene. 2018;37(4):522–533. doi:10.1038/onc.2017.351
32. Linares CA, Varghese A, Ghose A, et al. Hallmarks of the tumour microenvironment of gliomas and its interaction with emerging immunotherapy modalities. Int J Mol Sci. 2023;24(17):13215. doi:10.3390/ijms241713215
33. Wang Q, Geng W, Guo H, et al. Emerging role of RNA methyltransferase METTL3 in gastrointestinal cancer. J Hematol Oncol. 2020;13(1):57. doi:10.1186/s13045-020-00895-1
34. Meyer KD, Saletore Y, Zumbo P, et al. Comprehensive analysis of mRNA methylation reveals enrichment in 3’ UTRs and near stop codons. Cell. 2012;149(7):1635–1646. doi:10.1016/j.cell.2012.05.003
35. Yang H. Comprehensive analysis of the expression and clinical significance of a ferroptosis-related genome in ovarian serous cystadenocarcinoma: a study based on TCGA data. Oncologie. 2022;24(4):835–863. doi:10.32604/oncologie.2022.026447
36. Sun T, Wu R, Ming L. The role of m6A RNA methylation in cancer. Biomed Pharmacother. 2019;112:108613. doi:10.1016/j.biopha.2019.108613
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.