")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Hydrogen sulfide-releasing naproxen suppresses colon cancer cell growth and inhibits NF-κB signaling
Authors Kodela R, Nath N, Chattopadhyay M, Nesbitt DE, Velázquez-Martínez CA, Kashfi K
Received 25 June 2015
Accepted for publication 17 July 2015
Published 24 August 2015 Volume 2015:9 Pages 4873—4882
DOI https://doi.org/10.2147/DDDT.S91116
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Shu-Feng Zhou
Ravinder Kodela,1 Niharika Nath,2 Mitali Chattopadhyay,1 Diandra E Nesbitt,1 Carlos A Velázquez-Martínez,3 Khosrow Kashfi1
1Department of Physiology, Pharmacology and Neuroscience, Sophie Davis School of Biomedical Education, City University of New York Medical School, 2Department of Life Sciences, New York Institute of Technology, New York, NY, USA; 3Faculty of Pharmacy and Pharmaceutical Sciences, University of Alberta, Edmonton, AB, Canada
Abstract: Colorectal cancer (CRC) is the second leading cause of death due to cancer and the third most common cancer in men and women in the USA. Nuclear factor kappa B (NF-κB) is known to be activated in CRC and is strongly implicated in its development and progression. Therefore, activated NF-κB constitutes a bona fide target for drug development in this type of malignancy. Many epidemiological and interventional studies have established nonsteroidal anti-inflammatory drugs (NSAIDs) as a viable chemopreventive strategy against CRC. Our previous studies have shown that several novel hydrogen sulfide-releasing NSAIDs are promising anticancer agents and are safer derivatives of NSAIDs. In this study, we examined the growth inhibitory effect of a novel H2S-releasing naproxen (HS-NAP), which has a repertoire as a cardiovascular-safe NSAID, for its effects on cell proliferation, cell cycle phase transitions, and apoptosis using HT-29 human colon cancer cells. We also investigated its effect as a chemopreventive agent in a xenograft mouse model. HS-NAP suppressed the growth of HT-29 cells by induction of G0/G1 arrest and apoptosis and downregulated NF-κB. Tumor xenografts in mice were significantly reduced in volume. The decrease in tumor mass was associated with a reduction of cell proliferation, induction of apoptosis, and decreases in NF-κB levels in vivo. Therefore, HS-NAP demonstrates strong anticancer potential in CRC.
Keywords: nonsteroidal anti-inflammatory drugs, cell cycle, apoptosis, xenograft, NF-κB, thioredoxin reductase, chemoprevention
Introduction
According to a 2015 report by the American Cancer Society, colorectal cancer (CRC) is one of the three top most common cancers in both men and women in the USA. The same report describes that despite a significant decrease in mortality rates in the last 2 decades, there will be an estimated 49,700 deaths from CRC in 2015. In this regard, it is evident that changes in both lifestyle (risk factors) and CRC screening among adults aged 50 years and older have played a significant role in the observed reduction of CRC-related deaths.
Chemoprevention is a general term used to describe the utilization of a chemical compound, administered on a long-term basis, to reduce the incidence of a disease, including cancer. Epidemiological studies illustrate the use of nonsteroidal anti-inflammatory drugs (NSAIDs) as the prototypical drug class to prevent CRC. These observations coincide with scientifically sound evidence generated using a wide variety of in vitro and in vivo drug screening assays. These reports suggest that traditional NSAIDs can reduce the incidence of colon cancer (regress precancerous lesions), and may be promising chemopreventive drugs for CRC.1–5 Nevertheless, long-term administration of NSAIDs is associated with the development of side effects, including gastrointestinal, renal, and cardiovascular toxicity. For example, Bjorkman et al reported that up to 4% of patients using NSAIDs suffer serious gastrointestinal complications, resulting in approximately 8,000 deaths per year.6
Recently, our research group reported on a new type of NSAID, namely hydrogen sulfide (H2S)-releasing NSAIDs, or HS-NSAIDs.7–9 These compounds maintain the anti-inflammatory profile of the parent NSAID without gastrointestinal irritation in animal models. Their chemical structure consists of a conventional NSAID to which a H2S-releasing moiety is covalently attached through an ester linkage.10 This design was based on the observation that H2S is a naturally occurring modulator with cytoprotective and anti-inflammatory effects.11,12 Our research group, along with others, has reported the improved pharmacological profile of different HS-NSAIDs, including HS-diclofenac,13 HS-indomethacin,14 and HS-naproxen.15 An additional example supporting the design of HS-NSAIDs is the paper published by Fiorucci et al who developed an HS-mesalamine derivative with improved efficacy over mesalamine in animal models of inflammation and colitis; this derivative also showed lower toxicity (reduced injury) than the parent compound.16
An interesting observation about HS-NSAIDs is the fact that these compounds also exhibit potentially beneficial anticancer effects. In this regard, our group reported that H2S-releasing derivatives of aspirin, ibuprofen, sulindac, and naproxen inhibit cell proliferation of various human cancer cell lines including those of the breast, prostate, lung, leukemia, pancreas, and colon.8 We have also shown that HS-NSAIDs are effective in vivo. Using a xenograft model of estrogen receptor-negative breast cancer, we observed that HS-aspirin significantly reduced tumor volume and tumor mass through induction of apoptosis and inhibition of cancer cell proliferation.7
Another example of a promising HS-NSAID was published by McGettigan et al who designed an H2S-releasing derivative of naproxen (ATB-346) that suppressed prostaglandin synthesis in colonic tissue as well as thromboxane synthesis in a whole blood assay. These effects were comparable with those observed with naproxen, but the new derivative did not cause significant gastrointestinal injury. In addition, ATB-346 reduced the incidence of precancerous lesions (aberrant crypt foci) in azoxymethane-treated mice.17 Therefore, H2S-releasing derivatives of naproxen are strong chemopreventive candidates.
The transcription factor nuclear factor kappa B (NF-κB) plays an essential role in the normal regulation of cell proliferation, apoptosis, and inflammation; however, this protein can also regulate tumorigenesis, tumor metastasis, and chemoresistance.18–23 NF-κB is activated in human lymphoid malignancies in several tissues, including the prostate, pancreas, colon, rectum, skin (melanoma), liver, and breast.24–29 Using mice models of colitis-associated colon cancer, Greten et al proposed in 2004 a direct contribution of the NF-κB pathway to the development of solid tumors.30 Chemical inhibition of the NF-κB transcriptional pathway results in significant reduction of tumor size and volume, as determined in several mouse xenograft models.20,31,32 Consequently, these results suggest that NF-κB constitutes a suitable drug target.
Thioredoxin (Trx) is a small redox-active protein responsible for maintaining the reduced state of the enzyme thioredoxin reductase-1 (TrxR).33 One of the many implications of overexpression of the Trx protein is suppression of apoptosis,34 and inactivation of TrxR leads to inhibition of cancer cell proliferation as well as cell cycle arrest and apoptosis.35 Therefore, the Trx/TrxR system is a suitable and complementary target in chemotherapy.
In the present study, we provide evidence demonstrating that H2S-releasing naproxen inhibits proliferation of HT-29 (human colorectal adenocarcinoma) cells, and inhibits tumor growth in a xenograft mouse model, at least in part through modulation of both NF-κB and Trx/TrxR activity.
Materials and methods
Reagents and cell culture
Our group synthesized the H2S-releasing naproxen (chemical name: 4-(5-thioxo-5H-[1,2]-dithiol-3-yl)-phenyl 2-(6-methoxynaphthalen-2-yl)-propionate, or HS-NAP; Figure 1A), using a previously reported procedure.8 We purchased the parent compound naproxen, along with all other chemicals used in this study, from Sigma-Aldrich (St Louis, MO, USA). We obtained the adenocarcinoma cell line (HT-29, ATCC HTB-38) from the American Type Culture Collection (Manassas, VA, USA). No ethics statement was required from the institutional review board for the use of these cell lines. We conducted the cell proliferation inhibition experiments using McCoy’s 5A medium, supplemented with 10% heat-inactivated fetal calf serum, 2 mmol/L l-glutamine, and a mixture of penicillin/streptomycin (100 units/mL), at 37°C and in 5% CO2.
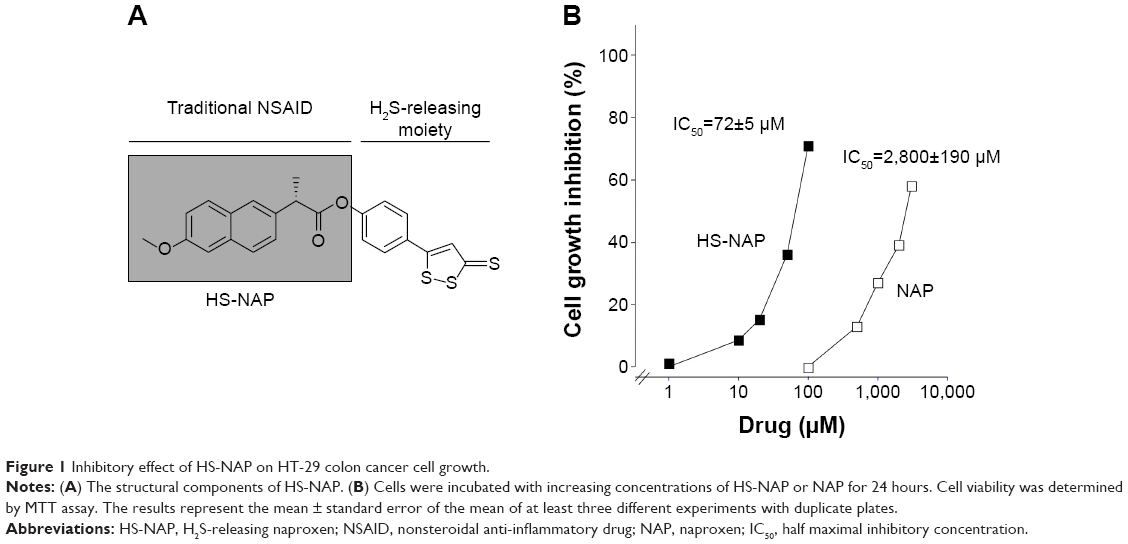 | Figure 1 Inhibitory effect of HS-NAP on HT-29 colon cancer cell growth. |
MTT assay
We determined cell proliferation with and without the HS-NAP test drug using a colorimetric MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide) assay kit (Roche, Indianapolis, IN, USA) as reported previously.7 Briefly, HT-29 cells were plated overnight in 96-well plates at a density of 20,000 cells/well, and then incubated for 24 hours in the presence of different concentrations of HS-NAP. Next, we added to each well 10 μL of 5 mg/mL MTT dye dissolved in phosphate-buffered saline. We then incubated all wells for 2 more hours at 37°C, followed by addition of 100 μL of the solubilization solution. Finally, we measured the absorbance of each plate at a wavelength of 570 nm. We assayed each sample in triplicate on three separate occasions.
Cell proliferation
We determined the proliferating cell nuclear antigen (PCNA) using an enzyme-linked immunosorbent assay kit (Calbiochem, La Jolla, CA, USA), in accordance with the manufacturer’s protocol. Briefly, HT-29 cells (1×106 cells/mL) were incubated with serum-free medium for 24 hours to remove the effect of endogenous growth factors and then incubated for a further 24 hours in the presence of different concentrations of either HS-NAP or the solvent. Briefly, we suspended cells (1×106 cells/mL) in buffer (5 mM ethylenediamine tetraacetic acid, 0.2 mM phenylmethylsulfonyl fluoride, 1 μg/mL pepstatin, 0.5 μg/mL leupeptin, and 50 mM tris-hydroxymethyl aminomethane hydrochloride [Tris-HCl, pH 8.0]). We then transferred aliquots of the cell suspension into the wells of the plate provided with the assay kit; each well contained immobilized antihuman PCNA rabbit polyclonal antibody. We added mouse monoclonal antibody (clone PC10) to each well, and incubated this mixture for 2 hours at room temperature. Subsequently, we washed all wells and added a horseradish peroxidase streptavidin solution, followed by incubation at room temperature for 30 minutes. Finally, we added the substrate tetramethylbenzidine and incubated the plate for 30 minutes, then added quenching solution and measured the absorbance at 450 nm using a plate reader.
Cell cycle analysis
We determined the cell cycle phase distributions of both treated and untreated HT-29 cells using a Coulter Profile XL device equipped with a single argon ion laser, following a previously reported procedure.7
Assay for apoptosis
We treated HT-29 cells (0.5×106 cells/mL) with various concentrations of HS-NAP for 24 hours; we then washed and resuspended the cells with binding buffer containing Annexin V-fluorescein isothiocyanate. We added propidium iodide as a counterstain agent as previously described.7 We determined the percent of cells undergoing apoptosis using BD Biosciences LSR II analyzer with a single argon ion laser. We analyzed approximately 10,000 events for each subset; we collected all parameters in “list mode” files, followed by analysis using Flow Jo software.
Thioredoxin reductase assay
We treated HT-29 cells with either solvent or HS-NAP at different concentrations, including those equivalent to half the half maximal inhibitory concentration (IC50) (0.5× IC50), the IC50, and twice the IC50 (2× IC50) for 24 hours. We then harvested the cells and washed them with Dulbecco’s phosphate-buffered saline (Invitrogen, Carlsbad, CA, USA), followed by centrifugation. We then resuspended and sonicated the cells in 5 mM potassium phosphate buffer (pH 7.4, 0.5 mM ethylenediaminetetraacetic acid).
We generated a post-microsomal supernatant by ultracentrifugation (105,000× g for 2 hours), and determined the concentrations of protein for all post-microsomal supernatants with a Bradford assay kit (Bio-Rad Laboratories, Hercules, CA, USA) containing bovine serum albumin as the standard. Finally, the activity of the TrxR enzyme was measured using a colorimetric assay (Cayman Chemical Co, Ann Arbor, MI, USA) as described by the manufacturer.
Nuclear protein extraction, NF-κB p65 DNA-binding assay, and immunoblotting
We seeded HT-29 cells (2×106) for approximately 17 hours (overnight) using 10 cm dishes, followed by incubation with various concentrations of HS-NAP at the indicated time intervals. We then extracted nuclear protein using an extraction kit (Cayman Chemical Co); we then determined the protein concentrations using a Bio-Rad reagent (Bio-Rad Laboratories), and the nuclear extracts were stored at -80°C until use. From the nuclear extracts, we determined the DNA binding activity of NF-κB by using the NF-κB (p65) transcription factor assay kit (Cayman Chemical Co), in which the specific DNA sequence containing the NF-κB response element is immobilized in a 96-well plate.
Briefly, we added 50 μg of nuclear proteins to wells containing transcription factor buffer, and then incubated them overnight (total volume of 100 μL at 4°C). We also followed the kit instructions for the use of blank wells, the positive control (tumor necrosis factor alpha-stimulated HeLa cell nuclear extract provided with the kit), and the nonspecific binding samples (provided with the kit). We detected NF-κB binding by adding the NF-κB primary antibody to all wells (except the blank wells), and incubating for 1 hour at room temperature. We then washed the wells and added the secondary antibody (conjugated to horseradish peroxidase), incubating for an additional 1 hour at room temperature; to continue with the procedure, we added 100 μL of developing solution, followed by a 45-minute incubation with gentle shaking; after this time, we added the “stop” solution, and measured the absorbance of all wells at 450 nm. Finally, we calculated the percent of change in activity for each test sample relative to the average of untreated samples.
We used antibodies against IκBα (L35A5), phospho-IκBα (Ser32; 14D4), NF-κB p65 (D14E12), and phospho-IKKα/β (Ser176/180; 16A6) from Cell Signaling Technology (Boston, MA, USA) for the immunodetection of each protein in nuclear extracts or cell lysates.
HT-29 mouse xenograft model
For this prevention study, we used 5-week-old athymic male nude (nu/nu) mice (Charles River Laboratories Inc, Wilmington, MA, USA), housed according to institutional and National Institutes of Health guidelines. Our institutional animal care and research committee approved all experimental procedures. After 1 week of acclimation, we randomly divided the mice into two groups containing six mice each; we started a “pre-initiation” regimen in which we administered vehicle (1% methylcellulose) to the control group and HS-NAP (100 mg/kg body weight) to the treatment group; we dosed both groups daily by gavage. After 1 week of treatment, we inoculated both groups (right hind flank) with HT-29 cells (3×106) suspended in 50% v/v Matrigel (BD Biosciences, San Jose, CA, USA), using a 1 mL syringe and 22-gauge needle. We followed this protocol for a total of 24 days after inoculation, and then euthanized all animals; we identified, isolated, weighed, and stored the tumor tissues in formalin for immunohistochemistry studies.
Additionally, we measured tumor size using electronic calipers at regular intervals, starting at day 6 post-administration until the end of the experiment. We calculated tumor volumes using the formula: (length × width2)/2.
Immunohistochemistry
We fixed tissue specimens in formalin (paraffin-enclosed) for examination, following a previously reported procedure.7 We used the following antibodies: for proliferation, Ki-67 (1:200 dilution) and primary rabbit NF-κB p65 (1:100 dilution); for apoptosis, Promega TUNEL system (Promega Corporation, Madison, WI, USA). For NF-κB scoring, we took five slides from each animal by using the following semiquantitative scoring system.36 The extent of staining was graded as follows: 0, no staining; 1+, ≤25% of cells positive; 2+, 26%–50% of cells positive; 3+, ≥51% of cells positive. The intensity of staining was scored as follows: 0, no staining; 1+, faint; 2+, moderate; and 3+, strong. 1+, 2+, and 3+ were recorded as 1, 2, and 3 points, respectively.
To compare differences in staining, we calculated an expression index (EI) using the following formula: EI = extent of staining × intensity of staining. We evaluated all slides using a light microscope (×200, Carl Zeiss, Oberkochen, Germany). A negative control was done in all cases by omitting the primary antibody.
Statistical analysis
The in vitro data are presented as the mean ± standard error of the mean for at least three different sets of plates done in triplicate. In vivo treatment groups and numbers of animals in each group are indicated in the figure legends. Comparison between treatment groups was done using Student’s t-tests. P<0.05 was considered to be statistically significant.
Results
HS-NAP inhibits growth of HT-29 cells
We evaluated the inhibition of cell proliferation exerted by the test drug (HS-NAP) and the parent compound naproxen using HT-29 human colon cancer cells. This cell line is a commonly used model due to its sensitivity to chemotherapeutic drugs, and it is also commonly used in xenograft tumor models of CRC. Cells that were plated 1 day earlier were treated with vehicle, naproxen, or HS-NAP at various concentrations for 24 hours; growth inhibition was then measured using the MTT assay, and IC50 values were determined. While naproxen inhibited the proliferation of HT-29 cells at very high concentrations (1–5 mM), we observed that HS-NAP exerted a much more potent effect, inhibiting cell proliferation within the 1–100 μM range (Figure 1B). We also observed that the effect exerted by HS-NAP was concentration-dependent, with a calculated IC50 of 72±5 μM (the IC50 of naproxen was 2,800±190 μM). By comparing the two IC50 values, we suggest that there is a 400-fold increase in potency by the test drug HS-NAP.
Effect of HS-NAP on proliferation, cell cycle progression, and apoptosis
The inhibitory effect on cell proliferation exerted by HS-NAP may occur through effects on basic cellular processes such as modulation of cell cycle progression or induction of cancer cell death. To investigate the effect of HS-NAP on cell proliferation, we treated HT-29 cells with HS-NAP at different concentrations: 0.5× IC50 (36 μM); IC50 (72 μM); and 2× IC50 (144 μM). In all cases, we incubated cells for 24 hours, followed by determination of PCNA expression.
We observed that HS-NAP reduced PCNA expression in HT-29 cells in a dose-dependent manner. In this regard, the magnitude of reduction was 29%±3% at 36 μM, 51%±5% at 72 μM, and 74%±4% at 144 μM (Figure 2A). Likewise, we observed that HS-NAP caused a concentration-dependent accumulation of HT-29 cells in the G0/G1 phase (Figure 2B), while fewer cells were in S and G2/M phases. HS-NAP increased the population of cells in G0/G1 phase from 37.2% (control cells) to 48.8%, 69.7%, and 77.3% at 36, 72, and 144 μM, respectively. The population of cells in the S phase also decreased from 40.9% in control cells to 32.0% (at 36 μM), 17.7% (at 72 μM), and 13.3% (at 144 μM). Finally, we also observed a decrease in the number of cells in the G2/M phase as a result of incubation of HT-29 cells with increasing concentrations of HS-NAP; in this regard, we calculated that approximately 21.9% of cells were in the G2/M phase when no drug was added (control), but this changed to 19.2% (at 36 μM), 12.6% (at 72 μM), and 9.4% (at 144 μM).
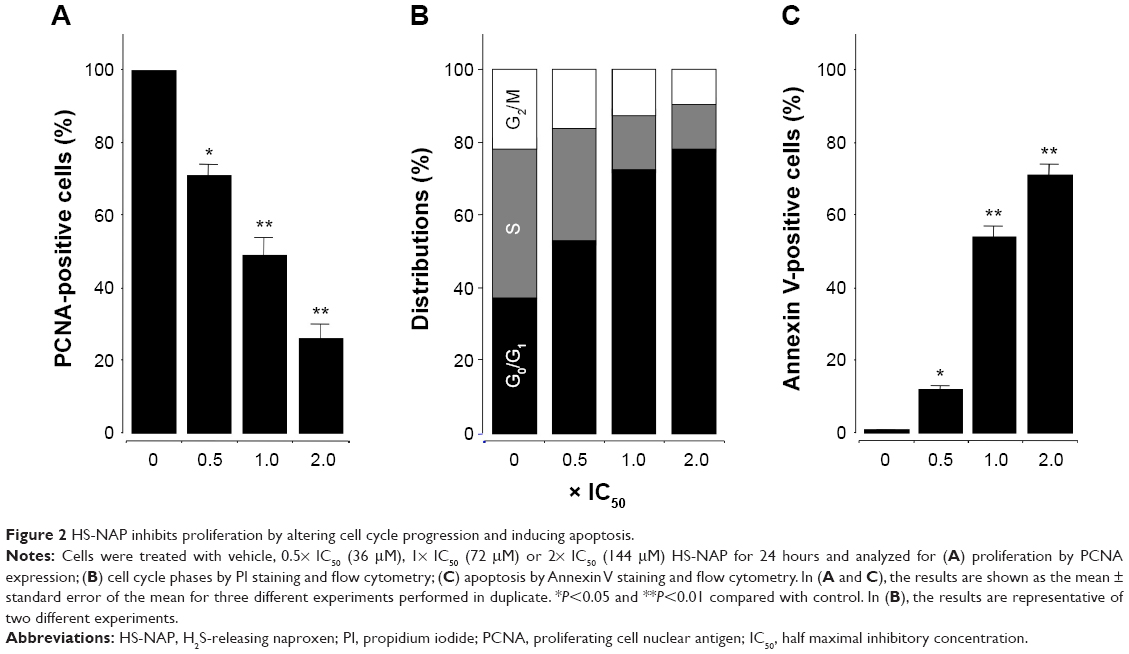 | Figure 2 HS-NAP inhibits proliferation by altering cell cycle progression and inducing apoptosis. |
In this experiment, we observed that HS-NAP significantly increased the number of cells undergoing apoptosis. At 36 μM, 72 μM, and 144 μM, the percentage of apoptotic cells increased to 11.6%±2%, 54.3%±4%, and 71.3%±3%, respectively (Figure 2C). Based on these results, it is likely that HS-NAP inhibits the proliferation of HT-29 cells by G1 arrest and induction of apoptosis.
HS-NAP inhibits the NF-κB DNA signaling pathway in HT-29 cells
Since NF-κB is implicated in cell survival and oncogenesis, we examined the effect of the test drug HS-NAP on NF-κB-DNA binding in HT-29 cells. Nuclear extracts from treated and untreated cells were prepared and examined as described in the Materials and methods section.
Our results suggest that HS-NAP inhibits binding between NF-κB (p65) and DNA in a concentration-dependent manner (Figure 3A). The DNA-NF-κB binding decreased to 22%±3% at 36 μM (0.5× the IC50 value), 53%±2% at 72 μM (IC50), and 59%±4% at 144 μM (2.0× the IC50 value). We also observed a significant decrease in the levels of nuclear p65 (immunoblotting), using lamin as the internal control (Figure 3B). These results suggest that HS-NAP inhibits the nuclear translocation of p65.
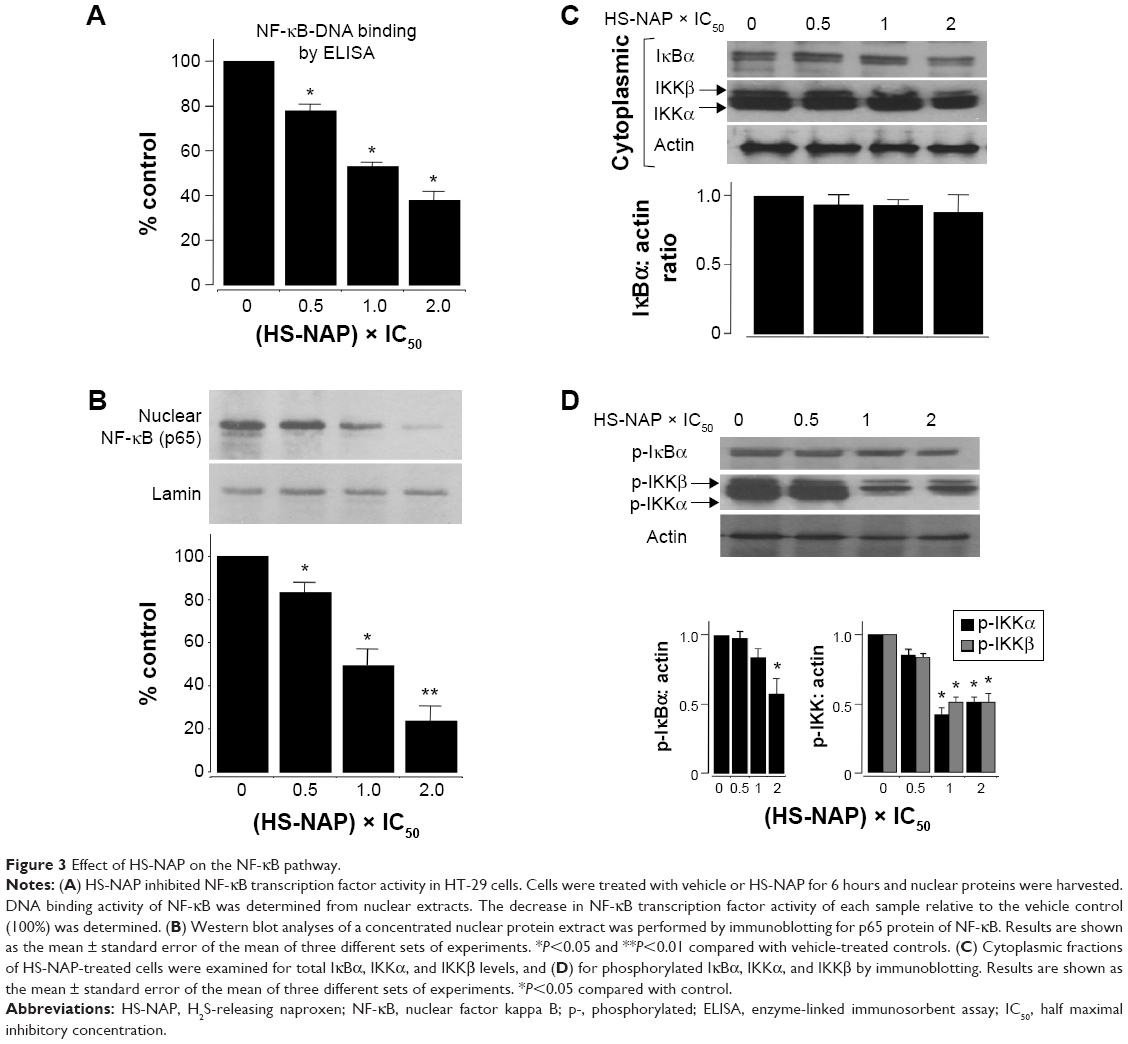 | Figure 3 Effect of HS-NAP on the NF-κB pathway. |
It is well established that integral to NF-κB activation is the dissociation of IκB, which is mediated through phosphorylation and subsequent proteolytic degradation of this inhibitory subunit. Consequently, we also examined if the observed reduction of the nuclear translocation of p65 occurs by suppression of IκBα degradation. We observed that the total amount of cytoplasmic IκBα was not altered by HS-NAP (Figure 3C), whereas the phospho-IκBα was reduced to 60% at 144 μM compared with untreated control (Figure 3D). These results suggest that HS-NAP does not cause degradation of IκBα in HT-29 cells, since phosphorylation IκBα is reduced by HS-NAP, although this response only occurs at the highest concentration (144 μM, 2× IC50 value).
The phosphorylation of IκBα at specific serine residues is mediated by two Iκ kinases (IKKs), namely IKKα and IKKβ.37 Therefore, we examined the levels of both phosphorylated IKKα and phosphorylated IKKβ in the presence of HS-NAP. In this regard, we observed that this compound did not alter the total levels of IKKα and IKKβ; instead, we observed a decreased level of the corresponding phosphorylated IKKα and IKKβ at 1× IC50 (72 μM) and 2× IC50 (144 μM). Therefore, we suggest that HS-NAP prevents the activation of IKKs, inhibits phosphorylation-mediated degradation of IκBα, and prevents the translocation of NF-κB p65 into the nucleus. In repeated experiments for these events, we continued to find reduction of phosphorylation of IκBα to approximately 60% of the control, implying that HS-NAP-mediated reduction of nuclear p65 may occur through additional mechanisms.
HS-NAP inhibits thioredoxin reductase activity
The redox balance of cells via Trx/TrxR may influence the function of NF-κB by regulating its ability to bind to DNA.38–40 The function of Trx is known to be dependent upon the activity of TrxR, so we examined the activity of TrxR in HT-29 colon cancer cells when incubated in the presence of different concentrations of HS-NAP. As shown in Figure 4, HS-NAP strongly inhibited TrxR activity. HS-NAP decreased the enzymatic activity of TrxR from 120 μmol/min/mg protein in the control group to 82.5 μmol/min/mg protein at 0.5× IC50 (69% of control at 36 μM), to 39.7 μmol/min/mg protein at 1× IC50 (33% of control at 72 μM), and to 25.9 μmol/min/mg protein at 2× IC50 (22% of control at 144 μM).
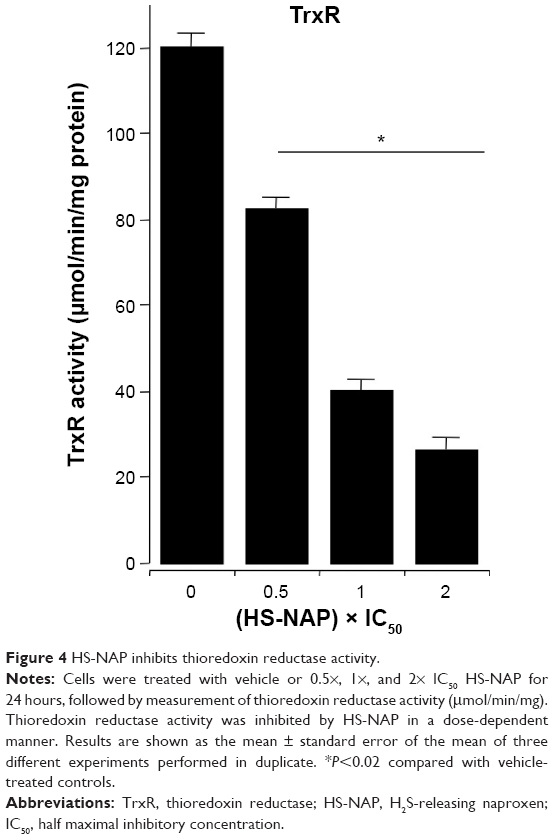 | Figure 4 HS-NAP inhibits thioredoxin reductase activity. |
HS-NAP inhibits tumor growth in a HT-29 xenograft model
Athymic nude male mice were pre-initiated for 7 days via gavage with vehicle or HS-NAP (100 mg/kg) and then injected in the right flank with HT-29 cells for the development of subcutaneous tumors as described in the Materials and methods section. Daily administration of the test agents continued to the end of the experiment (Figure 5A). At the end of the study, HS-NAP-treated mice showed a considerable reduction in tumor volume compared with the control group (Figure 5B and C). The mean tumor volumes at the time of sacrifice for the control group and HS-NAP were 789.6±11 mm3 and 31.1±4 mm3, respectively. This is equivalent to a mean reduction of 96% (P<0.01).
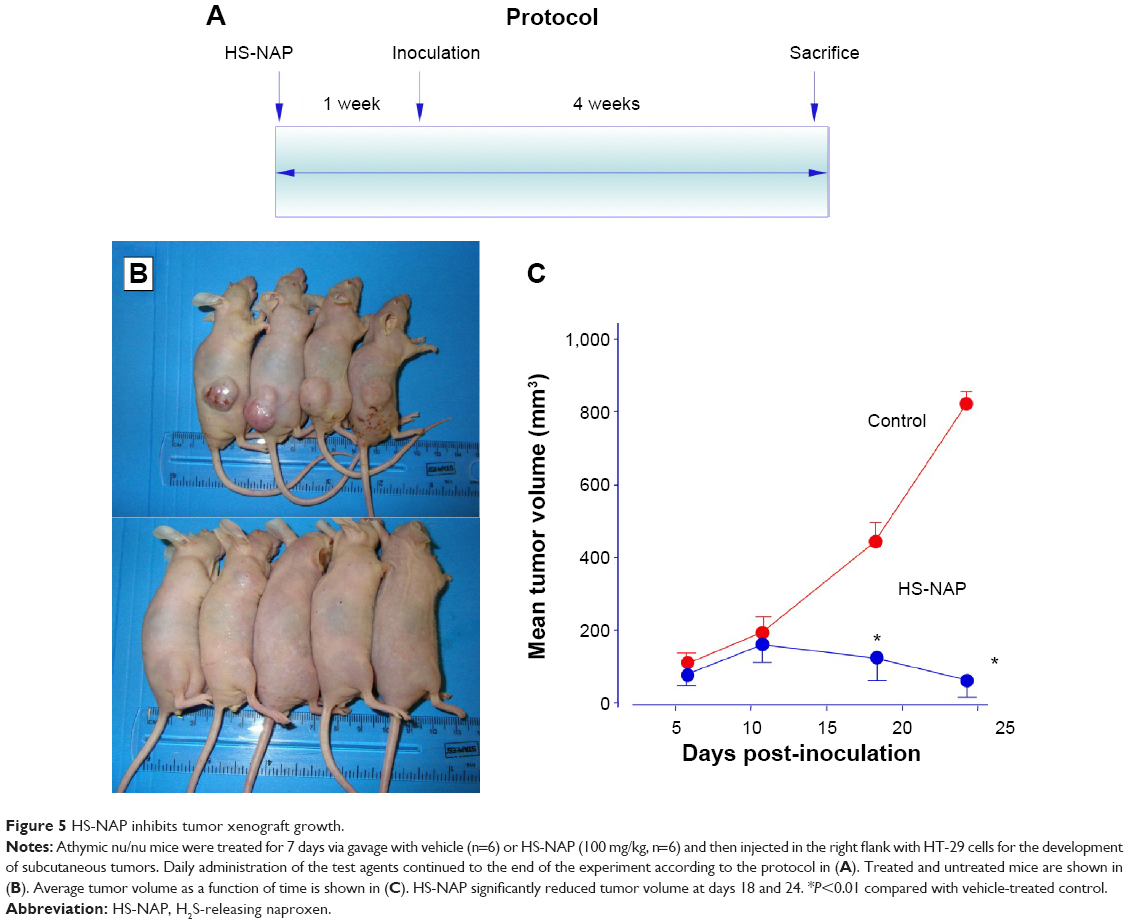 | Figure 5 HS-NAP inhibits tumor xenograft growth. |
Immunohistochemical staining of the tumors were performed for expression of NF-κB p65 and for Ki67, which is a biomarker for tumor cell proliferation. The untreated tumors showed strong expression of Ki67 (85%±3%) and NF-κB (30%±4%) that was reduced in the treated tumors to 38%±2% and 4.5%±2%, respectively (Figure 6A and C). In addition, HS-NAP increased apoptosis in the tumors (19.5%±3%) compared with the control (1.8%±1%) as measured by TUNEL staining for multiple tissue sections (Figure 6B). Therefore, HS-NAP suppressed tumor growth by a combination of increased apoptosis and reduced cell proliferation.
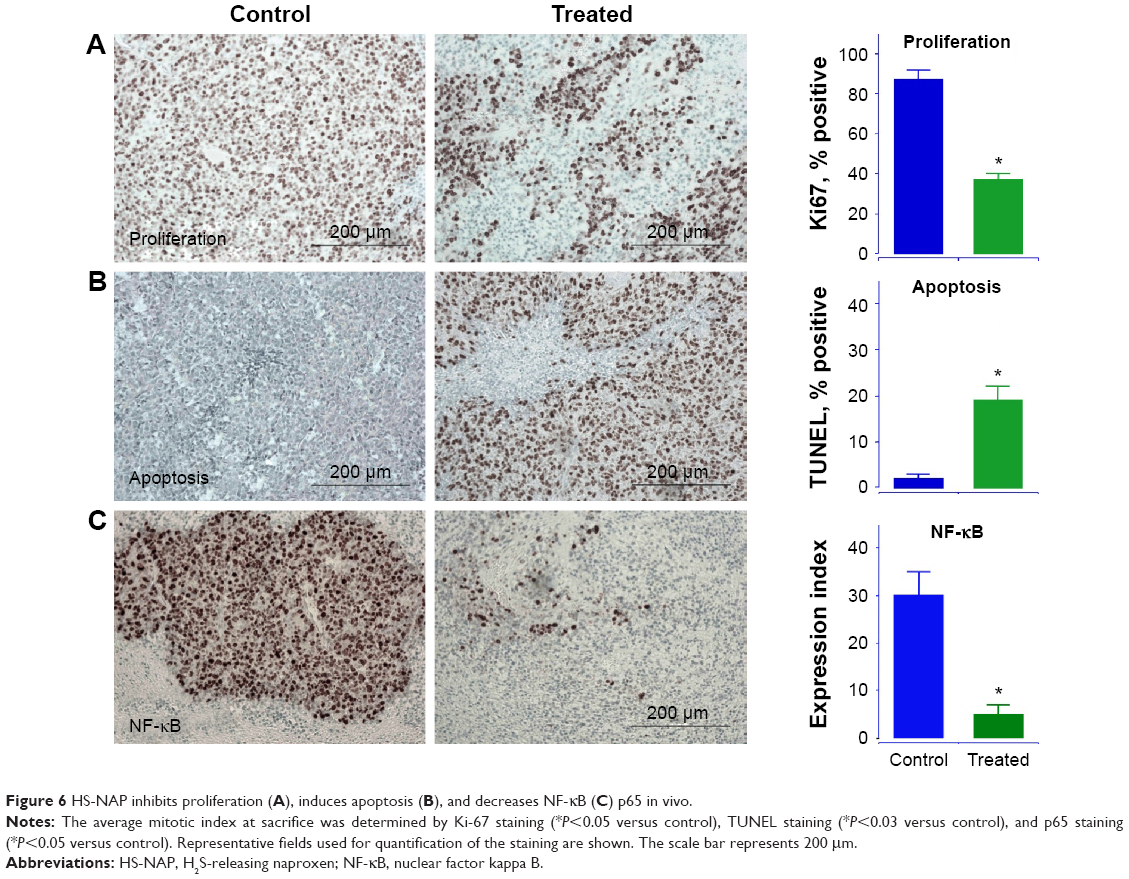 | Figure 6 HS-NAP inhibits proliferation (A), induces apoptosis (B), and decreases NF-κB (C) p65 in vivo. |
Discussion
The overarching milestone of this investigation is the significant decrease in tumor growth (by ~96%), observed in animals dosed orally with the test drug HS-NAP, which represents a remarkable effect, and suggests a potential cancer chemoprevention use for this H2S-releasing naproxen derivative. Taken together with previous published reports showing that HS-NAP essentially produced no toxicity in the gastrointestinal tract,15,41 a feature that was also observed in the present study but not formally evaluated, sets the pharmacological properties of HS-NAP apart from that of the much more toxic parent compound naproxen.
At the cellular level, we determined that HS-NAP inhibited cell proliferation, altered the cell cycle progression, and induced apoptosis of cancer cells. These events were associated with inhibition of NF-κB signaling, both by reducing the nuclear translocation of NF-κB and decreasing NF-κB-DNA binding. These results are further supported by the enzyme inhibitory effect exerted by HS-NAP on the TrxR enzyme, which according to some literature reports, may be related to decreased NF-κB-DNA binding.39 Besides, the inhibition of TrxR activity may also cause cancer cell apoptosis via caspase-dependent and caspase-independent pathways.35 Consequently, we submit that the mechanism of action exerted by HS-NAP is the result of a combination of all the effects described earlier, and collectively contribute to the significant reduction in tumor volume.
Our results also suggest that HS-NAP inhibits the translocation of p65 to the nucleus by inhibiting IKK phosphorylation; this is encouraging considering that inactivation of NF-κB in different cancer cells has been shown to stop the ability of cancer cells to grow.19 However, HS-NAP reduced the phospho-IκBα to only approximately 50%–60% at high concentration (2× IC50) and did not completely inhibit IκBα phosphorylation, although nuclear translocation of NF-κB was strongly reduced. This discrepancy cannot be explained at the current time. In previous studies, we reported the effects of a similar hybrid compound, namely a H2S-releasing aspirin. In that report, we observed a marked reduction of phosphorylation of IκBα and a concomitant inactivation of IKKs.7 For HS-NAP, it may be envisaged that the observed reduction of p65 (and its subsequent nuclear translocation), could be due to NF-κB-independent pathways, such as the likelihood of a modification of NF-κB itself by sulfuration via H2S released from the HS-NAP. At the level of biochemical events, some known molecular targets of H2S are intracellular signaling proteins and transcription factors (reviewed in Kashfi and Olson10). For this, further studies of HS-NAP and its metabolic kinetics will be explored. In conclusion, HS-NAP suppressed the proliferation of CRC HT-29 cells, inhibited xenograft tumor progression, and suppressed NF-κB activity. Therefore, HS-NAP is a potential cancer chemopreventive agent and merits further study.
Acknowledgment
This research was supported in part by a National Institutes of Health grant (R24 DA018055). The funding agency had no role in the study design, collection, analysis, or interpretation of data, in the writing of the manuscript; or in the decision to submit the manuscript for publication.
Author contributions
MC, NN, RK, CAV-M, and KK participated in the design of this research. MC, RK, and DEN conducted the experiments. MC, NN, CAV-M, and KK performed the data analysis. MC, NN, CV-M, and KK wrote or contributed to the writing of the manuscript. All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no potential conflicts of interest in this work.
References
Smalley W, Ray WA, Daugherty J, Griffin MR. Use of nonsteroidal anti-inflammatory drugs and incidence of colorectal cancer: a population-based study. Arch Intern Med. 1999;159:(2):161–166. | ||
Chan TA. Nonsteroidal anti-inflammatory drugs, apoptosis, and colon-cancer chemoprevention. Lancet Oncol. 2002;3(3):166–174. | ||
Rao CV, Reddy BS. NSAIDs and chemoprevention. Curr Cancer Drug Targets. 2004;4(1):29–42. | ||
Bertagnolli MM, Eagle CJ, Zauber AG, et al. Celecoxib for the prevention of sporadic colorectal adenomas. N Engl J Med. 2006;355(9):873–884. | ||
Cooper K, Squires H, Carroll C, et al. Chemoprevention of colorectal cancer: systematic review and economic evaluation. Health Technol Assess. 2010;14(32):1–206. | ||
Bjorkman DJ. Current status of nonsteroidal anti-inflammatory drug (NSAID) use in the United States: risk factors and frequency of complications. Am J Med. 1999;107(6A):3S–8S. | ||
Chattopadhyay M, Kodela R, Nath N, Barsegian A, Boring D, Kashfi K. Hydrogen sulfide-releasing aspirin suppresses NF-kappaB signaling in estrogen receptor negative breast cancer cells in vitro and in vivo. Biochem Pharmacol. 2012;83(6):723–732. | ||
Chattopadhyay M, Kodela R, Nath N, et al. Hydrogen sulfide-releasing NSAIDs inhibit the growth of human cancer cells: a general property and evidence of a tissue type-independent effect. Biochem Pharmacol. 2012;83(6):715–722. | ||
Chattopadhyay M, Kodela R, Nath N, et al. Hydrogen sulfide-releasing aspirin modulates xenobiotic metabolizing enzymes in vitro and in vivo. Biochem Pharmacol. 2012;83(6):733–740. | ||
Kashfi K, Olson KR. Biology and therapeutic potential of hydrogen sulfide and hydrogen sulfide-releasing chimeras. Biochem Pharmacol. 2013;85(5):689–703. | ||
Wallace JL, Dicay M, McKnight W, Martin GR. Hydrogen sulfide enhances ulcer healing in rats. FASEB J. 2007;21(14):4070–4076. | ||
Wallace JL, Ferraz JG, Muscara MN. Hydrogen sulfide: an endogenous mediator of resolution of inflammation and injury. Antioxid Redox Signal. 2012;17(1):58–67. | ||
Li L, Rossoni G, Sparatore A, Lee LC, Del Soldato P, Moore PK. Anti-inflammatory and gastrointestinal effects of a novel diclofenac derivative. Free Radic Biol Med. 2007;42(5):706–719. | ||
Wallace JL. Hydrogen sulfide-releasing anti-inflammatory drugs. Trends Pharmacol Sci. 2007;28(10):501–505. | ||
Wallace JL, Caliendo G, Santagada V, Cirino G. Markedly reduced toxicity of a hydrogen sulphide-releasing derivative of naproxen (ATB-346). Br J Pharmacol. 2010;159(6):1236–1246. | ||
Fiorucci S, Orlandi S, Mencarelli A, et al. Enhanced activity of a hydrogen sulphide-releasing derivative of mesalamine (ATB-429) in a mouse model of colitis. Br J Pharmacol. 2007;150(8):996–1002. | ||
Elsheikh W, Blackler RW, Flannigan KL, Wallace JL. Enhanced chemopreventive effects of a hydrogen sulfide-releasing anti-inflammatory drug (ATB-346) in experimental colorectal cancer. Nitric Oxide. 2014;41:131–137. | ||
Chen F, Castranova V, Shi X. New insights into the role of nuclear factor-kappaB in cell growth regulation. Am J Pathol. 2001;159(2):387–397. | ||
Luo JL, Kamata H, Karin M. IKK/NF-kappaB signaling: balancing life and death – a new approach to cancer therapy. J Clin Invest. 2005;115(10):2625–2632. | ||
Karin M, Cao Y, Greten FR, Li ZW. NF-kappaB in cancer: from innocent bystander to major culprit. Nat Rev Cancer. 2002;2(4):301–310. | ||
Mantovani A. Molecular pathways linking inflammation and cancer. Curr Mol Med. 2010;10(4):369–373. | ||
Ben-Neriah Y, Karin M. Inflammation meets cancer, with NF-kappaB as the matchmaker. Nat Immunol. 2011;12(8):715–723. | ||
Baker RG, Hayden MS, Ghosh S. NF-kappaB, inflammation, and metabolic disease. Cell Metab. 2011;13(1):11–22. | ||
Hoesel B, Schmid JA. The complexity of NF-kappaB signaling in inflammation and cancer. Mol Cancer. 2013;12:86. | ||
Nakshatri H, Bhat-Nakshatri P, Martin DA, Goulet RJ Jr, Sledge GW Jr. Constitutive activation of NF-kappaB during progression of breast cancer to hormone-independent growth. Mol Cell Biol. 1997;17(7):3629–3639. | ||
Sovak MA, Bellas RE, Kim DW, et al. Aberrant nuclear factor-kappaB/Rel expression and the pathogenesis of breast cancer. J Clin Invest. 1997;100(12):2952–2960. | ||
Biswas DK, Cruz AP, Gansberger E, Pardee AB. Epidermal growth factor-induced nuclear factor kappa B activation: a major pathway of cell-cycle progression in estrogen-receptor negative breast cancer cells. Proc Natl Acad Sci U S A. 2000;97(15):8542–8547. | ||
Romieu-Mourez R, Landesman-Bollag E, Seldin DC, Traish AM, Mercurio F, Sonenshein GE. Roles of IKK kinases and protein kinase CK2 in activation of nuclear factor-kappaB in breast cancer. Cancer Res. 2001;61(9):3810–3818. | ||
Weldon CB, Burow ME, Rolfe KW, Clayton JL, Jaffe BM, Beckman BS. NF-kappa B-mediated chemoresistance in breast cancer cells. Surgery. 2001;130(2):143–150. | ||
Greten FR, Eckmann L, Greten TF, et al. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. 2004;118(3):285–296. | ||
Reuther JY, Reuther GW, Cortez D, Pendergast AM, Baldwin AS Jr. A requirement for NF-kappaB activation in Bcr-Abl-mediated transformation. Genes Dev. 1998;12(7):968–981. | ||
Baldwin AS. Control of oncogenesis and cancer therapy resistance by the transcription factor NF-kappaB. J Clin Invest. 2001;107(3):241–246. | ||
Mustacich D, Powis G. Thioredoxin reductase. Biochem J. 2000;346 Pt 1:1–8. | ||
Arner ES, Holmgren A. The thioredoxin system in cancer. Semin Cancer Biol. 2006;16(6):420–426. | ||
Zhao F, Yan J, Deng S, et al. A thioredoxin reductase inhibitor induces growth inhibition and apoptosis in five cultured human carcinoma cell lines. Cancer Lett. 2006;236(1):46–53. | ||
Ouyang N, Williams JL, Tsioulias GJ, et al. Nitric oxide-donating aspirin prevents pancreatic cancer in a hamster tumor model. Cancer Res. 2006;66(8):4503–4511. | ||
Bonizzi G, Karin M. The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 2004;25(6):280–288. | ||
Ueno M, Masutani H, Arai RJ, et al. Thioredoxin-dependent redox regulation of p53-mediated p21 activation. J Biol Chem. 1999;274(50):35809–35815. | ||
Matthews JR, Wakasugi N, Virelizier JL, Yodoi J, Hay RT. Thioredoxin regulates the DNA binding activity of NF-kappa B by reduction of a disulphide bond involving cysteine 62. Nucleic Acids Res. 1992;20(15):3821–3830. | ||
Bannister AJ, Cook A, Kouzarides T. In vitro DNA binding activity of Fos/Jun and BZLF1 but not C/EBP is affected by redox changes. Oncogene. 1991;6(7):1243–1250. | ||
Blackler R, Syer S, Bolla M, Ongini E, Wallace JL. Gastrointestinal-sparing effects of novel NSAIDs in rats with compromised mucosal defence. PLoS One. 2012;7(4):e35196. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.