")
Back to Journals » Adolescent Health, Medicine and Therapeutics » Volume 15
Cystic Fibrosis in an Adolescent: A “Miranda Warning” Against Blaming TB—A Case-Based Scholarly Update
Authors Adela AY , Kebede AG, Zewdneh D, Kifle M, Dias AB
Received 1 December 2023
Accepted for publication 30 January 2024
Published 2 February 2024 Volume 2024:15 Pages 19—29
DOI https://doi.org/10.2147/AHMT.S451251
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Amanuel Yegnanew Adela,1– 3 Assefa Getachew Kebede,1 Daniel Zewdneh,4 Mahlet Kifle,1 Adriano Basso Dias5
1Radiology Department, Body Imaging Unit, Tikur Anbessa Comprehensive Specialized Referral and Teaching Hospital (TASH), Addis Ababa University, Addis Ababa, Ethiopia; 2Radiology Department, Gondar University Comprehensive Specialized Referral and Teaching Hospital, College of Medical and Health Sciences, University of Gondar, Gondar, Amhara Regional State, Ethiopia; 3Radiology Department, Ethiopian Federal Police Commission Referral Hospital, Addis Ababa, Ethiopia; 4Radiology Department, Pediatric Radiology Unit, Tikur Anbessa Comprehensive Specialized Referral and Teaching Hospital (TASH), Addis Ababa University, Addis Ababa, Ethiopia; 5Department of Medical Imaging, University Medical Imaging Toronto, University of Toronto, Toronto, Ontario, Canada
Correspondence: Amanuel Yegnanew Adela, Tel +251-918-26-1611, Email [email protected]; [email protected]
Abstract: Cystic fibrosis (CF) is a multisystem disorder that occurs as a result of autosomal recessive congenital transmission of CF transmembrane conductance regulator (CFTR) gene mutation on chromosome 7. Because it is considered a disease of the Caucasian pediatric population or due to lack of awareness, it is rarely considered in developing countries like ours. This case report presents the first case of cystic fibrosis ever reported in Ethiopia and possibly East Africa, that of a 17-year-old female diagnosed with the disease following a CT scan of her abdomen and chest. She was initially misdiagnosed and treated for tuberculosis (TB) as she was a chronic cougher. Perhaps due to epidemiological evidence, there is an obstinate tendency of blaming tuberculosis (TB) for almost every case of chronic cough with fibro-bronchiectatic lung parenchymal changes in Ethiopia. Once a diagnosis of TB is posted on such patients, their diagnosis remains in the circle of TB reinfection, relapse or resistance, followed by multiple phases of anti-mycobacterial drugs. This could lead to hazardous implications, including unnecessary prolonged anti-mycobacterial treatments, possibility of developing drug resistance, and mismanagement-related patient morbidity. This patient’s chest and abdominal CT findings, including bronchiectasis, hepatic steatosis, pancreatic lipomatosis, micro-gallbladder and proximal colonic wall thickening, led to the diagnosis of CF. This article, presenting the first documented case of CF in the region, is meant to be a helpful reminder for clinicians and radiologists to also consider presumably “rare” illnesses like CF rather than blaming TB for every chronic cough and highlights the importance of abdominal CT features in the diagnosis of CF.
Keywords: bronchiectasis, cystic fibrosis, CFTR, hepatic steatosis, micro-gallbladder, pancreatic lipomatosis, colonic wall thickening, Ethiopia
Introduction
Cystic fibrosis (CF), also called mucoviscidosis, is a multisystem autosomal recessive congenital disorder secondary to mutation that affects the CF transmembrane conductance regulator (CFTR) gene on chromosome 7.1 As CF is a multisystem disorder, it affects the head and neck, the lungs and airways as well as the gastrointestinal (GI) and the genitourinary (GU) systems. The manifestations of the disease are mainly due to excessive sodium chloride losses in sweat and different epithelial cells of the airways, GI and GU systems as a result of dysfunctional sodium channels in epithelial membranes of patients with the mutated CFTR gene.2 CF patients, therefore, can present with a variety of clinical features, and imaging plays a significant role in the diagnosis and monitoring of CF.
Although CF has been thought of as a Caucasoid disease with variable prevalence in the western community, substantially lower rates of CF have been found in Asia and Africa. To date, in the current literature available, no record of CF could be found in East Africa, particularly Ethiopia. Here, we present a 17-year-old female patient with CF who was being repeatedly treated for tuberculosis (TB) and is the first documented case of (adult) CF in Ethiopia. A discussion of the pulmonary and abdominal manifestations from the imaging perspective follows in the face of the clinical as well as chest and upper abdominal CT features of the patient. Perhaps due to epidemiological evidence, there is an obstinate tendency to incriminate tuberculosis (TB) in almost every case of chronic cough with fibro-bronchiectatic lung parenchymal changes in Ethiopia.3,4 Such misconjecture can have daunting implications, including unnecessarily extended anti-TB treatments, mismanagement-related patient morbidity, and possibility of developing drug resistance. We would like to draw attention to the need to consider CF in a chronic cougher, the importance of abdominal CT features in the diagnosis of CF, and underscore the imperative significance of conducting CF-focused research in a variety of clinical contexts among the scientific community.
Case Presentation
We present the first case of cystic fibrosis ever reported in Ethiopia, a 17-year-old female diagnosed with the disease following a CT scan of her abdomen and chest. She presented with a chronic cough, occasionally productive of whitish sputum, which had started early in her childhood at around 5 years of age. Since then, she was repeatedly visiting health institutions. Except for occasional increments in her white blood cell count, which happened when her symptoms flared up, no striking laboratory abnormality was found among the basic investigations. The search for acid-fast bacilli in her sputum AFB test and for mycobacteria in her sputum PCR (gene expert) examination remained non-revealing. Despite all these, her clinical diagnosis was bouncing between recurrent pneumonia and pulmonary TB. For these diagnoses she had received multiple antimicrobial treatments, including antibiotics and anti-TB medication, on different occasions throughout her life. She, together with her helpless blind father and other family members, resides in a remote area of Gurage zone in the southern part of Ethiopia. At the time of presentation to our facility, she was relatively stable and caring for her father, who was referred from the regional hospital to us for an unrelated medical evaluation. As she was occasionally coughing, a disturbingly dry cough, while attending to her visually impaired father, she attracted the attention of a certain compassionate colleague at our institution. She confided to him the given story and expressed her exhaustion from repeatedly taking the same medications with little improvement, and she subsequently begged if she could get evaluated at our institution. She looked somehow cachectic, but was doing well clinically and had no clubbing of her fingers or any other significant clinical finding. As she had had multiple chest x-rays in the regional hospital at her home town, the recommended line of investigation was a CT scan of the chest and upper abdomen (pre- and post-contrast). The scan revealed bilateral, slightly asymmetric, predominantly perihilar, cylindrical bronchiectasis. There were also a few varicoid forms of bronchiectasis and cavitating and non-cavitating fibrotic changes predominantly in the right lower lobe, while other non-cavitary fibrotic lesions were seen scattered in the other lobes (Figure 1). On CECT, the main pulmonary artery was enlarged compared to the ascending aorta, evidencing pulmonary arterial hypertension (Figure 2). An enlarged liver with severe steatosis (Figure 3) and a small-sized gallbladder suggestive of micro-gallbladder (Figure 3) were also evidenced. A diffuse atrophy and complete fatty replacement of the pancreas with pancreatic duct ectasia, and lipomatous pseudohypertrophy of the tail region (Figure 4), and symmetric concentric smooth thickening and redundancy of the proximal colon (Figure 5) were the features seen on the CT. Only a few differential diagnoses could be made, specifically Shwachman–Diamond syndrome, which can cause pancreatic lipomatosis, and undernutrition, which can additionally cause hepatic steatosis, but all the pulmonary and GI findings in the CT scan of this young adolescent patient pointed to CF.
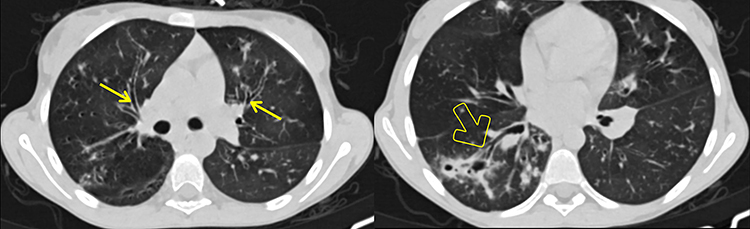 |
Figure 1 Axial NCECT at the level of the right upper lobe bronchus (left) and at the right lower lobe bronchus (right), showing bilateral peri-hilar thick-walled cylindrical bronchiectasis (arrows) and bronchiectatic changes with cylindrical and varicoid components, some with mucus plugs, as well as cavitating and non-cavitating fibrotic changes within the apical segment of the right lower lobe (open arrow). |
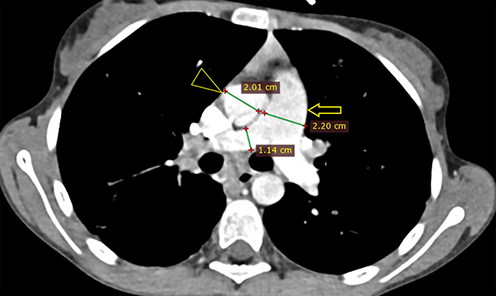 |
Figure 2 Axial CECT at the level of the main pulmonary artery (MPA) showing enlarged MPA (open arrow) compared to the ascending aorta (triangular open arrow head), measuring 22 mm and 20.1 mm, respectively. |
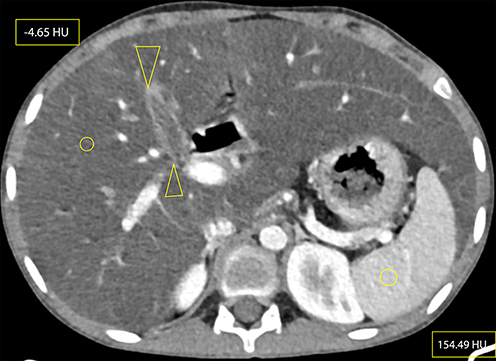 |
Figure 3 Axial CECT at the level of the gallbladder (between open arrow heads) showing micro-gallbladder (here, measuring 3.79 cm long and 1.3 cm wide), hepatomegaly and hepatic steatosis (elliptical areas showing hepatic and splenic attenuation of −4.65 HU and 154.49 HU, respectively). The vascular markings also stand out against the background hepatic parenchyma, demonstrating more evidence of hepatic steatosis . Measurement of hepatic and splenic attenuation at NCECT and CECT at multiple sections showed a range of −14 to −33 HU on NCECT and −2.5 to 22.5 HU on CECT for liver; which was in comparison to the mean splenic attenuations of 45.5–53.5 and 141.61–154.49 HU at NCECT and CECT, respectively. Please also note beaver’s tail, a normal anatomic variant unrelated to CF, where the liver parenchyma of the left hepatic lobe wraps around the spleen. Riedel's lobe, a normal variant of liver parenchymal extension beyond the lower margin of the kidney, was also seen but is not demonstrated here. |
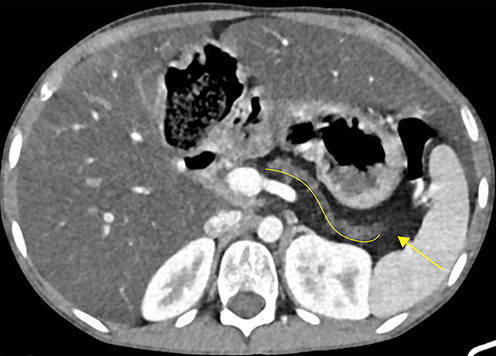 |
Figure 4 CECT at the level of the pancreatic body and tail showing a completely atrophied pancreas, with pancreatic duct ectasia (curved solid line) surrounded by complete fatty replacement (solid arrow). The former two might signify evidence of chronic pancreatitis. |
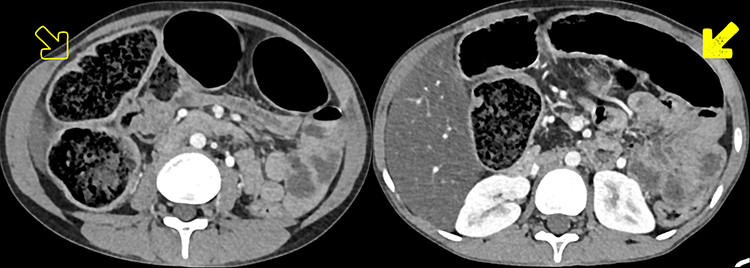 |
Figure 5 Axial CECT showing symmetric concentric thickening and redundancy of the proximal portion of the colon (open arrow): particularly, the ascending colon, hepatic flexure and proximal half of transverse colon, compared to the distal portion of the transverse colon with normal wall thickness (shaded arrow). |
Discussion and Literature Review
Cystic fibrosis (CF) is a multisystem congenital disorder secondary to mutation that affects the CF transmembrane conductance regulator (CFTR) gene on chromosome 7 and is transmitted in an autosomal recessive fashion.1 The manifestations of the disease are mainly due to excessive sodium chloride losses in sweat gland and different epithelial cells of the airways and GI and GU systems as a result of dysfunctional sodium channels in epithelial membranes of patients with mutated CFTR channels.2 Subsequently, intracellular volume reduction, tenacious body fluids, with secondary hyperaldosteronism lead to excessive renal potassium losses and metabolic alkalosis.
CF has been considered a Caucasoid disease, with a reported prevalence ratio of 1:2500–3500 live new births (LNB) in the USA to 1:1400 in Ireland. Lower rates are reported in Latin America (1:1600–14,000) and Mexico (1:8500), while a much lower rate again is reported in Asia and Africa.5,6 Contemporary and available literature shows no record of CF in East Africa, particularly Ethiopia.
CF was previously considered to be lethal in early childhood, mostly from recurrent respiratory tract infections and pancreatic-insufficiency-related malnutrition. However with better understanding of the disease and use of modern therapies, there is a paradigm shift of improvement in life expectancy as well as quality of life in CF patients.7 In Mexico and Latin America, mean survival has improved from 9 years in the early 1990s to about 18 years; and a longer life expectancy, exceeding 40 years, has become a common trend in western nations, with an average survival of 52.1, 47.4 and 47.3 years in Canada, the US and the UK, respectively.8,9 Adult patients account for more than 40–45% of CF patients.10 In spite of her somewhat cachectic appearance, our patient also thrived into adolescence and exhibited good clinical performance. Her antibiotic treatment, unique genetic make-up, or certain dietary or environmental factors might have contributed to her surprising resilience with the disease, despite the fact that she had not received CF-targeted therapy. Whatever the case, as CF is a multisystem condition, it is important to recognize its manifestations in distinct organ systems, which are more pronounced in older patients.11 Here, in light of the case and related research, we aimed to address the systemic manifestations of CF, stressing the role of radiography in the diagnosis of CF in adult and adolescent patients, particularly the lung and abdominal radiologic aspects of CF.
Head and neck manifestations are more common in adults than in children and include chronic sinusitis (in about 100% of cases),12 nasal polyposis in up to 86% of cases2 and chronic otitis media with middle ear effusion. Our patient also had chronic sinusitis manifested by chronic nasal stuffiness accompanied by her chronic cough.
Musculoskeletal manifestations are extremely rare and usually a consequence of chronic illness in most frequently affected organs including lungs and abdomen. Osteopenia, stunting, kyphoscoliosis, cystic fibrosis arthropathy, hypertrophic osteoarthropathy, and associated finger clubbing are the most common musculoskeletal manifestations, which can be detected clinically as well as on imaging.13 The patient, in our case, had no musculoskeletal complaints and no evidence of clubbing of the fingers.
Pulmonary Manifestations
A dysfunctional CFTR in chloride channels within the lungs causes intraluminal retention of more chloride ions, with a balancing reabsorption of excess sodium and water from the lumen resulting in scanty, tenacious, thick secretions which tend to dry out. This airway mucosal layer dehydration, with concomitant impaired mucociliary clearance, exposes the affected patient to a chronic bronchopulmonary infection and bronchiectasis.14 The pulmonary manifestations are the most common complaint that lead CF patients to seek health care and they usually present with a chronic cough productive of tenacious sputum, sometimes blood-stained. Likewise, our patient was frequently seeking help for respiratory issues. As the disease progresses, the hemoptysis may intensify in frequency and volume and other complications, including pneumothorax, might follow.2,15
Plain radiography is usually normal and is extremely insensitive in detecting lung findings, particularly in early disease. The chronic CF features that can be detected on chest x-rays include: bronchiectasis, pulmonary hyperinflation, atelectasis, and features of pulmonary arterial hypertension marked by enlarged pulmonary arteries.2 Different severity scores have been used to grade the severity of CF and structural changes on chest x-ray, with all scoring systems liable to inter- and intra-observer variability. The Brasfield (Table 1) and Chrispin–Norman scoring systems show good inter- and intra-observer agreement and appreciable correlation with clinical data and spirometry findings.16 In light of interpretation variability and the lesser sensitivity of chest x-ray as compared to HRCT (high resolution CT), HRCT is being advocated as a gold standard evaluation tool for pulmonary disease in CF patients. The scoring systems using thin section HRCT have been found to be more plausible due to the accuracy in detection of bronchiectasis, and surrogacy of pathological findings with the added bonus of better intra- and inter-observer agreement.17 But the higher radiation exposure and subsequent radiation risks and sedation-related complications still disfavor the use of CT in CF diagnosis and monitoring in children. But once the structural changes of the lung reach their maximum, which usually occurs by school age, the value of chest radiography remains questionable.18
 |
Table 1 Brasfield Scoring System to Evaluate the Evolution of CF Disease Severity on a Plain Radiograph, With a Maximum Possible Score of 25 and a Minimum Score of 3, in Which Points are Subtracted Based on Categorical Score from Each Category Criteria: Higher Overall Score Indicates Greater Disease Severity (slightly modified from reference 16). |
The pulmonary manifestations of CF are well described on CT, which shows lung abnormalities in about 65% of cases when performed using the HRCT protocol.7 In the early stage of the disease, bronchial cuffing with or without peribronchial interstitial thickening, tree-in-bud like centri-lobular nodular branching opacities representative of acute infectious bronchiolitis can be seen.7 As the disease progresses, a bronchiectasis preferentially affecting the upper lobes19 and the apical segments of the lower lobes, and emphysematous bullae and subpleural blebs can be seen.7 This is in contrast to our case, which also showed bronchiectasis but predominantly lower lobe involvement. The bronchiectasis typically undergoes progressive, severity-dependent intensification from cylindrical to varicoid to cystic.2 Air trapping and mucus plugging, two consequences of bronchiectasis, are shown on the CT as mosaic attenuation and finger-in-glove patterns, respectively. Likewise, there were areas of mucus plugging in our case that might echo the severity of bronchiectasis (Figure 1). A study comparing HRCT and magnetic resonance imaging (MRI) showed almost equivalent detection rates of bronchiectasis and mucous plugging, with 80% inter-observer agreement.20 The findings of this study may suggest that, given the high radiation associated with CT, MRI should be utilized for CF diagnosis. However, as CF is a chronic illness, the cost, availability, and technical aspects of MRI, like breathing artefacts, restrict its application in CF patient monitoring.
CECT, particularly CTA, could show pulmonary arterial enlargement, bronchial arterial enlargement and/or aneurysm, and lymphadenopathy. This is consistent with our case study, which showed features of pulmonary artery hypertension marked by enlargement of the pulmonary trunk and its branches (Figure 2).
Abdominal involvement with CF occurs in more than 90–95% of cases and CF can affect the liver, biliary tree and GB, pancreas, GIT, and GUT within the abdomen and pelvis.7,21 The simultaneous presence of abdominal signs could aid in the definitive diagnosis of CF by excluding alternative lung pathology mimics. This is particularly beneficial in developing nations like Ethiopia, where TB is prevalent and frequently incriminated in persistent productive coughs; our case is a classic example.
Pancreatic involvement in CF occurs in 70–100% of cases and is the most common intra-abdominal manifestation of CF.7,11 Pancreatic involvement due to the universal mechanism of CF-related inspissated fluid production primarily affects exocrine function in 80–95% of patients and endocrine function in 30–50% of patients, with 1–2% of the latter developing insulin-dependent CF-related diabetes mellitus.22–24 Subsequently, complete pancreatic atrophy with or without pancreatic fibrofatty replacement, lipomatous pseudohypertrophy, pancreatitis, diffuse fibrosis with microcystic transformation and pancreatic cystosis might follow and be seen on imaging. Clinically, patients may present with a fat malabsorption syndrome owing to exocrine insufficiency, and a unique combined Type I and Type II diabetes mellitus (DM).11 Pancreatic cysts tend to be microscopic (1–3 mm), while the extreme form called pancreatic cystosis could result in cysts in the range of 0.5–12 cm.23 The imaging characteristics of pancreatic fatty replacement are variable with respect to the severity of the disease and the age of the patient, with milder disease at an early age giving a heterogeneous appearance of the pancreas with areas of fatty infiltration.25 Subsequently the pancreas shows partial fatty replacement, with possible pancreatic atrophy and pancreatic duct ectasia. Complete fatty replacement is seen with severe disease and at a mean age of 17 years. Other forms of pancreatic involvement in CF include lipomatous pseudohypertrophy, in which case complete fibrofatty tissue replacement and enlargement of the gland is depicted on imaging. In our case, complete fatty replacement of the pancreas with lipomatous pseudohypertrophy and pancreatic duct ectasia demonstrated on the CT image (Figure 4) might indicate that the pancreatic involvement dates back to the early age of the patient. Shwachman–Diamond syndrome (SDS) is a differential diagnosis that may lead to pancreatic lipomatosis; however, in this case, the pancreatic ductal architecture is typically preserved, and the other findings—hepatic steatosis and micro-gallbladder—that SDS could not account for favored CF.26 Moreover, SDS has other systemic features that are beyond the limits of this article and which our patient did not exhibit.
CF-related acute pancreatitis (in 1.2% of exocrine-insufficient vs 10% of pancreatic-sufficient CF patients) occurs at a mean age of 19.9 years.27 Imaging demonstrates less marked peripancreatic inflammatory changes like fat stranding and fluid collection compared to pancreatitis in non-CF patients,28 and pancreatic calcifications typically along the ducts in 8% of cases.7 Generally, ultrasound, CT and /or MRI can be used to evaluate pancreatic abnormalities, but MRI better delineates fatty replacement versus fibrotic changes compared to the other imaging modalities. Plain radiographs usually have a limited role in pancreatic evaluation and might help depict calcifications which are rare in CF patients.
Hepatobiliary manifestations of CF occur in about 20–72% of adult patients and contribute the third largest share of morbidity and mortality associated with CF.11 The universal pathophysiologic mechanism also applies here as the hepatobiliary complications of CF occur secondary to impaired biliary secretion and inspissated bile accumulation, causing cholangiopathy and subsequent hepatocyte damage.29,30 Focal biliary cirrhosis affects 40–78% and hepatic steatosis 1/3 to 2/3 of patients.31–33 The latter could occur at any age and shows different patterns, including diffuse, focal infiltrating and multi-lobulated pseudomasses of lobulated fatty structures 1–2 cm in size. The involved areas of the liver show increased echo reflectivity on ultrasound, hypo-attenuation on CT, and T1-weighted hyperintensity which is nullified by fat-suppressed sequences on MRI.34
Fibrosis is a result of thick fluid obstructing small intrahepatic biliary ducts and subsequent proliferation and hyperplasia of the ducts with periductal inflammation.30,31 On ultrasound, focal biliary cirrhosis could show periportal and/or diffuse hepatic increased echotexture.35,36 Macronodular cirrhosis, also known as multilobular or multinodular cirrhosis, occurs in 5–12% of cases, 1–2% progressing to portal hypertension which usually manifests as splenomegaly or varices, and ascites representing a poor prognostic sign of decompensated cirrhosis.7,21,30,37 Presence of ascites, unstoppable variceal bleeding, and neoplasia, typically HCC are indications for liver transplantation. Fortunately, HCC in CF is rare, with only 4 cases reported so far. Ultrasound at the multinodular cirrhotic stage shows a small liver with coarse echotexture and irregular surface, an enlarged caudate to right lobe ratio, barely visualized intrahepatic vasculature and, rarely, iso-to-hypoechoic regenerative nodules.38 Features of portal hypertension, including increased portal vein caliber and flow, hepatofugal portal flow, splenomegaly and portosystemic collaterals, can also be demonstrated on ultrasound scans. CECT can also show all these findings except for the spectral parameters.
Studies to determine the correlation between severity of lung involvement and hepatic involvement were unable to reach a consensus; however, severe hepatic and pulmonary disease are rarely found in the same patient.39 In our case as well, a less severe form of lung involvement indicated by a lesser degree of bronchiectasis coexisted with severe hepatic disease, as evidenced by severe hepatic steatosis (Figure 1, Figure 3).
The biliary tree including the gallbladder (GB) and cystic duct are involved in 5–50% of CF patients.25 The biliary complications include biliary sludge, GB atony, cholecystolithiasis (in 12–24% of CF patients), thickened trabeculated GB wall, micro-GB (<2–3 cm long and 0.5–1.5 cm wide, in up to 30% of CF patients), subepithelial cysts, cystic duct atrophy or obstruction, and sclerosing cholangitis.25 Imaging reveals GB abnormalities in 40% of CF patients, with age-matched increases in prevalence reaching 60% in patients between the ages of 15–20. This is consistent with our case because the patient was 17 years old and micro-GB was noted (Figure 3). MRCP detects intrahepatic biliary tree abnormality in 100% of CF patients with known liver disease and in half of patients without known liver disease. CT and ultrasound have lower sensitivity in detecting biliary abnormalities compared to ERCP and MRCP. But considering cost, chronicity of the disease that starts early in age, ultrasound screening of biliary abnormalities has been suggested.
Almost the entire gastrointestinal tract (GIT) can be affected by CF. About 27% of CF patients under the age of five and about 39% of adult patients present with gastroesophageal reflux disease (GERD).40 This could be as a result of multifactorial increased intra-abdominal pressure, and may lead to reflux-related chronic complications, including esophagitis and subsequent strictures and/or Barret metaplasia, especially in adults. The distal esophageal wall and gastric wall thickenings might be seen on imaging and could be due to chronic inflammations as mentioned or primarily due to varices secondary to portal hypertension from biliary cirrhosis. Peptic ulcer diseases are also reported, primarily thought to be due to impaired bicarbonate secretion with subsequent duodenal exposure to unbuffered gastric acid.30 Thickened folds, nodular filling defects, mucosal smudging and dilatation have been reported from barium studies of the duodenum. The duodenum is affected in more than 80% of CF cases.41
One of the most well-described intestinal manifestations of CF is distal intestinal obstruction syndrome (DIOS), previously called adult meconium ileus equivalent (MIE), referring to fecal impaction in the terminal ileum and caecum. It occurs in 10–24% of adult patients with CF, with a slight male preponderance and age-matched increase in incidence. Its clinical features are recurrent abdominal pain, fecal mass in the right lower quadrant, and variable severity of bowel obstruction. Radiographic imaging could typically show fecal loading of the colon with features of distal small bowel obstruction (SBO). The terms DIOS and MIE can be used interchangeably.11,22,23,42,43
Incidence of intussusception in CF patients is 1–2%, ie 10–20 times higher than in the general population. The most common forms are ileo-colo-colic or ileocolic and about 25% of cases are complicated by SBO. A solid mass-like stool in the terminal ileum acts as a lead point in DIOS, while the others can occur without a lead point. Clinically, the patients present with intermittent abdominal pain and plain radiographic findings are usually nonspecific, while other imaging modalities including ultrasound, CT and MRI, can locate the donut (target-like) appearance of the intussusception.
Appendicitis in CF patients is rare compared to the general population (1–2% vs 7%), and its diagnosis should depend on secondary signs as mucoid-impaction-related appendiceal caliber dilatation (>6 mm) without appendicitis is a common finding in CF.11,44
The colon is involved in about 100% of CF patients, radiographically demonstrated as a smooth colonic wall thickening (more than 3 mm) primarily involving the submucosa. The proximal half of the colon, particularly the ascending colon, is almost universally affected while leftside colonic wall thickening is uncommon.44,45 Our case is also consistent with this finding because redundancy and smooth mild thickening of the right half of the colon was evidenced on the CT images (Figure 5). Thickening of the small intestine and mesenteric adenopathies were also reported in 59% and 46% of patients, respectively.46 Fibrosing colonopathy, the extreme form of colonic wall submucosal thickening, was historically considered a disease of children but can also occur in adults. It is a result of progressive submucosal fibrosis and subsequent strictures of the colon, usually associated with high-dose pancreatic enzyme supplements used to treat pancreatic insufficiency in CF.10,30 Diagnostic images can show colonic shortening, nodular thickening, and strictures with obstruction, otherwise diagnosis mainly lies in histopathology. Pneumatosis coli, as a result of dissection of air along connective tissue planes within the abdomen in association with severe obstructive airway disease, are reported in 5% of CF patients at autopsy. Colonic wall thickening with small locules of intramural gas are seen on CT images.30,47
Rectal prolapse, which could occur before the appearance of other manifestations of CF, and is usually related to frequently passing bulky stools, is reported in up to 20% of young CF patients. However, it rarely affects adult patients with CF.30,48
Overall, GI features of CF occur in 85–90% of CF patients and are secondary to excessive viscid mucous accumulation, dysmotility and dysbiosis with intestinal bacterial overgrowth and chronic inflammation, clinically presenting with constipation and/or obstruction and recurrent abdominal pain.49
Renal manifestations of CF include nephrocalcinosis and nephrolithiasis, found in 90% of patients at autopsy series, with symptomatic renal stones occurring in frequencies of more than 1.5–3 fold compared to the general population.50 Renal impairment also occurs in CF and is predominantly caused by glomerulonephritis from immunoglobulin A nephropathy, fueled by chronic infection-related secondary amyloidosis and CF-related diabetes.34 Rarely, renal agenesis and hypochloraemic hypokalaemic metabolic alkalosis without tubulopathy called pseudo Bartter syndrome can also occur.51
Among the genital manifestations of CF, congenital bilateral vas deferens atresia or agenesis is invariably seen in 99% of adult males with CF.52,53 Smaller testicles compared to age-matched controls with microlithiasis, bilateral seminal vesicle agenesis, epididymal tail and body hypoplasia are also reported in males with CF.52
CF: Disease Monitoring and Management
CF is known for viscid epithelial secretions which are responsible for stasis and retention of luminal contents in different hollow structures, including airways, intestines, ducts in the biliary tree, reproductive organs, and pancreatic ducts, etc. This is followed by bacterial overgrowth, recurrent infection, and luminal content hold up, subsequently leading to ectasia and further dysfunction of the hollow structures. CF patients require multidisciplinary team involvement for their well-planned and integrated management and monitoring. This includes pulmonary disease monitoring with lung function tests, radiological imaging and sputum evaluations; gastroenterologic evaluation for malabsorption and obstruction syndromes; endocrinologic screening for DM; ENT evaluation for nasal polyposis and sinusitis; musculoskeletal evaluation for any muscular or bone abnormality, including bone density scans, etc. The treatment of CF patients includes targeted mucoactive agents and physiotherapy support for airway clearance; targeted antibiotic and immunomodulatory therapy via various forms and routes for prevention and treatment of recurrent infections, dietary support, targeted exercise, and gene therapies;54,55 and other specific treatments targeted to various complications to which CF patients might succumb.56 Detailed report of the treatment and monitoring of CF patients is beyond the limits of this article.
Conclusion
This article emphasizes the value of radiography in diagnosing CF in a patient who had a chronic cough and had undergone unsuccessful multiphase anti-mycobacterial TB treatment. But the input of proper clinical data was also indispensable, highlighting the importance of an interdisciplinary approach to CF diagnosis and treatment. Further CF-focused research in the region including genetic screening and developing a clinico-radiologic evaluation protocol in medical assessment of a chronic cougher, inclusive of abdominal ultrasound or CT, might be helpful to better understand and not miss such presumably “uncommon” entities. Future studies should also consider the value of ultrasound and MRI in monitoring of CF patients.
Limitation of the Study
Although there is no other single entity yet known that could describe the constellations of the chest and abdominal CT findings of the patient other than CF, especially in the absence of evidence for TB, this was not backed by sweat chloride and genetic testing. This is mainly due to the economic constraints of the patient and her families, but we are also working on the patient’s condition to let her access a complete evaluation with these tests and CF-targeted therapy.
Institutional Ethical Clearance
Institutional approval is not required at either Addis Ababa University/Tikur Anbessa Comprehensive Specialized Referral and Teaching Hospital or the Federal Police Hospital for a case report if proper patient consent is guaranteed.
Declaration of Patient Consent
The patient has been well informed that we are publishing the case for an academic purpose and she has given consent for her CT images and important clinical data to be included in the case report and literature review. The patient clearly understands that anonymization of her initials and name is guaranteed and due efforts will be made to conceal her identity.
Declaration of Consent from Parents
The plan of publishing the case for academic purposes was discussed with the patient’s parents, especially her father, who duly understood and approved of the publication of the case in compliance with the patient’s wishes.
Acknowledgment
We are grateful to the patient and her father for granting permission to use her anonymized CT scan images and anonymized medical records in this scholarly article. We would like to appreciate Mr. Mitkneh, a radiology technologist and biomedical engineer, for bringing the patient to our attention and for his unwavering work organizing the patient’s medical evaluation records. Ermias Assefa, an engineer and graphic designer, is also owed for his assistance in preparing the patient’s high resolution CT pictures.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Tsui L-C, Buchwald M, Barker D., et al. Cystic fibrosis locus defined by a genetically linked polymorphic DNA marker. Science. 1985;230(4729):1054–1057. doi:10.1126/science.2997931
2. Shaheen NH, Targonska B, Kowal DJ. Imaging spectrum of cystic fibrosis. Contemporary Diagnostic Radiol. 2015;38(26):1–6. doi:10.1097/01.CDR.0000475549.20130.2f
3. Nega B, Ademe Y, Tizazu A. Bronchiectasis: experience of Surgical Management at Tikur Anbessa Specialized Hospital, Addis Ababa, Ethiopia. Ethiopian j Health Sci. 2019;29(4).
4. Banti AB, Datiko DG, Hinderaker SG, et al. How many of persistent coughers have pulmonary tuberculosis? Population-based cohort study in Ethiopia. BMJ open. 2022;12(5):e058466. doi:10.1136/bmjopen-2021-058466
5. Infante CS, Behar RR, Carpenter LTR, et al. Fibrosis quística en niños y su seguimiento durante 40 años (1977-2017). Revista Cubana de Pediatría. 2019;91(3):1–15.
6. Stephenson AL, Sykes J, Stanojevic S, et al. Survival comparison of patients with cystic fibrosis in Canada and the United States: a population-based cohort study. Ann Internal Med. 2017;166(8):537–546. doi:10.7326/M16-0858
7. Lugo-Olivieri CH, Soyer PA, Fishman EK. Cystic fibrosis: spectrum of thoracic and abdominal CT findings in the adult patient. Clin. Imaging. 1998;22(5):346–354. doi:10.1016/S0899-7071(98)00031-X
8. Elborn JS, Shale D, Britton J. Cystic fibrosis: current survival and population estimates to the year 2000. Thorax. 1991;46(12):881–885. doi:10.1136/thx.46.12.881
9. López-Valdez JA, Aguilar-Alonso LA, Gándara-Quezada V, et al. Cystic fibrosis: current concepts. Boletín médico del Hospital Infantil de México. 2021;78(6):584–596. doi:10.24875/BMHIM.20000372
10. Liong S, Awad D, Jones A, Sukumar S. The adult cystic fibrosis patient with abdominal pain: what the radiologist needs to know. Clin. Radiol. 2011;66(2):132–139. doi:10.1016/j.crad.2010.09.002
11. Fields TM, Michel SJ, Butler CL, Kriss VM, Albers SL. Abdominal manifestations of cystic fibrosis in older children and adults. Am J Roentgenol. 2006;187(5):1199–1203. doi:10.2214/AJR.05.0327
12. Oomen KP, April MM. Sinonasal manifestations in cystic fibrosis. Int j otolaryngol. 2012;2012:1–7. doi:10.1155/2012/789572
13. Cystic fibrosis (musculoskeletal manifestations) | Radiology Reference Article | Radiopaedia.org.
14. El-Feky M, Gaillard F. Cystic fibrosis (pulmonary manifestations). Radiopaediaorg. 2010.
15. Maffessanti M, Dalpiaz G, Antonaglia V. Diffuse lung diseases: clinical features, pathology, HRCT. British Journal of Anaesthesia. 2006;96(4):533–536. doi:10.1093/bja/ael026
16. Banjar H, Khondokar A, Alqarni A, et al. Chest X-Ray Changes in Cystic Fibrosis Patients in A Tertiary Care Center. MAR Pediatrics. 2021;1.
17. de Jong PA, Tiddens HA Cystic fibrosis–specific computed tomography scoring.
18. Terheggen-Lagro S, Arets H, Van der Laag J, Van der Ent C. Radiological and functional changes over 3 years in young children with cystic fibrosis. Eur Respir J. 2007;30(2):279–285. doi:10.1183/09031936.00051406
19. Sequeiros IM, Jarad NA. Radiological features of cystic fibrosis. Cystic Fibrosis. 2012;31–50.
20. Sileo C, Corvol H, Boelle P-Y, Blondiaux E, Clement A, Le Pointe HD. HRCT and MRI of the lung in children with cystic fibrosis: comparison of different scoring systems. J Cyst Fibros. 2014;13(2):198–204. doi:10.1016/j.jcf.2013.09.003
21. Brown J, Mason A, Cooperberg P. Gastrointestinal manifestations of cystic fibrosis in adults: pictorial essay. Canadian Assoc Radiologists j. 1999;50(3):165–169.
22. Robertson MB, Choe KA, Joseph PM. Review of the abdominal manifestations of cystic fibrosis in the adult patient. Radiographics. 2006;26(3):679–690. doi:10.1148/rg.263055101
23. Constantine S, Au V, Slavotinek J. Abdominal manifestations of cystic fibrosis in adults: a review. Australasian Radiol. 2004;48(4):450–458. doi:10.1111/j.1440-1673.2004.01345.x
24. Park RW, Grand RJ. Gastrointestinal manifestations of cystic fibrosis: a review. Gastroenterology. 1981;81(6):1143–1161. doi:10.1016/S0016-5085(81)80027-3
25. King LJ, Scurr ED, Murugan N, Williams SG, Westaby D, Healy JC. Hepatobiliary and pancreatic manifestations of cystic fibrosis: MR imaging appearances. Radiographics. 2000;20(3):767–777. doi:10.1148/radiographics.20.3.g00ma08767
26. Robberecht E, Nachtegaele P, Van Rattinghe R, Afschrift M, Kunnen M, Verhaaren R. Pancreatic lipomatosis in the Shwachman-Diamond syndrome: identification by sonography and CT-scan. Pediatric Radiology. 1985;15(5):348–349. doi:10.1007/BF02386774
27. Gilljam M, Ellis L, Corey M, Zielenski J, Durie P, Tullis DE. Clinical manifestations of cystic fibrosis among patients with diagnosis in adulthood. Chest. 2004;126(4):1215–1224. doi:10.1378/chest.126.4.1215
28. Shanbhogue AKP, Fasih N, Surabhi VR, Doherty GP, Shanbhogue DKP, Sethi SK. A clinical and radiologic review of uncommon types and causes of pancreatitis. Radiographics. 2009;29(4):1003–1026. doi:10.1148/rg.294085748
29. Efrati O, Barak A, Modan-Moses D, et al. Liver cirrhosis and portal hypertension in cystic fibrosis. Eur J Gastroenterol Hepatol. 2003;15(10):1073–1078. doi:10.1097/00042737-200310000-00002
30. Agrons GA, Corse WR, Markowitz RI, Suarez ES, Perry DR. Gastrointestinal manifestations of cystic fibrosis: radiologic-pathologic correlation. Radiographics. 1996;16(4):871–893. doi:10.1148/radiographics.16.4.8835977
31. Akata D, Akhan O, Özcelik U, et al. Hepatobiliary manifestations of cystic fibrosis in children: correlation of CT and US findings. Eur J Radiol. 2002;41(1):26–33. doi:10.1016/S0720-048X(01)00367-9
32. Hubbard VS, editor Gastrointestinal complications in cystic fibrosis.
33. Colombo C, Battezzati PM. Liver involvement in cystic fibrosis: primary organ damage or innocent bystander? J Hepatol. 2004;41(6):1041–1044. doi:10.1016/j.jhep.2004.10.002
34. Lavelle LP, McEvoy SH, Ni Mhurchu E, et al. Cystic fibrosis below the diaphragm: abdominal findings in adult patients. Radiographics. 2015;35(3):680–695. doi:10.1148/rg.2015140110
35. Haller J. Sonography of the biliary tract in infants and children. AJR. 1991;157(5):1051–1058. doi:10.2214/ajr.157.5.1927792
36. Quillin S, Siegel M, Rothbaum R. Hepatobiliary sonography in cystic fibrosis. Pediatric Radiology. 1993;23(7):533–535. doi:10.1007/BF02012141
37. Marino CR, Gorelick FS. Scientific advances in cystic fibrosis. Gastroenterology. 1992;103(2):681–693. doi:10.1016/0016-5085(92)90866-W
38. Zwiebel WJ, editor Sonographic diagnosis of diffuse liver disease.
39. Guiu B. Other Diffuse Liver Diseases: Steatosis, Hemochromatosis, Etc. Abdominal Imaging. Hamm B, Ros PR, Eds. Berlin/Heidelberg, Germany: Springer; 2013:1027–1044.
40. Sabati AA, Kempainen RR, Milla CE, et al. Characteristics of gastroesophageal reflux in adults with cystic fibrosis. J Cyst Fibros. 2010;9(5):365–370. doi:10.1016/j.jcf.2010.06.004
41. McKenna E PA, Sweeney LE. Small bowel involvement with cystic fibrosis | eurorad. 2004.
42. Di SA. Cystic fibrosis in adults: 75 cases and a review of 232 cases in the literature. Revista brasileira de odontologia. 1979;26(1):10–16.
43. Koletzko S, Stringer DA, Cleghorn GJ, Durie PR. Lavage treatment of distal intestinal obstruction syndrome in children with cystic fibrosis. Pediatrics. 1989;83(5):727–733. doi:10.1542/peds.83.5.727
44. Haber H, Benda N, Fitzke G, et al. Colonic wall thickness measured by ultrasound: striking differences in patients with cystic fibrosis versus healthy controls. Gut. 1997;40(3):406–411. doi:10.1136/gut.40.3.406
45. Pickhardt PJ, Yagan N, Siegel MJ, Balfe DM, Rothbaum RJ. Cystic fibrosis: CT findings of colonic disease. Radiology. 1998;206(3):725–730. doi:10.1148/radiology.206.3.9494492
46. Uliasz M, Bragoszewska H, Iwanowska B, et al. Sonography of small bowel, colon and appendix in children with cystic fibrosis. Medycyna wieku rozwojowego. 2010;14(1):15–27.
47. Hernanz-Schulman M, Kirkpatrick JJ, Shwachman H, Herman T, Schulman G, Vawter G. Pneumatosis intestinalis in cystic fibrosis. Radiology. 1986;160(2):497–499. doi:10.1148/radiology.160.2.3726132
48. Jm L. Cystic fibrosis: gastrointestinal complications. Br Med Bull. 1992;48(4):847–859. doi:10.1093/oxfordjournals.bmb.a072581
49. Cavalcoli F, Zilli A, Fraquelli M, Conte D, Massironi S. Small bowel ultrasound beyond inflammatory bowel disease: an updated review of the recent literature. Ultrasound Med Biol. 2017;43(9):1741–1752. doi:10.1016/j.ultrasmedbio.2017.04.028
50. Gibney EM, Goldfarb DS. The association of nephrolithiasis with cystic fibrosis. Am J Kidney Dis. 2003;42(1):1–11. doi:10.1016/S0272-6386(03)00403-7
51. Kose M, Pekcan S, Ozcelik U, et al. An epidemic of pseudo-Bartter syndrome in cystic fibrosis patients. Eur J Pediatr. 2008;167:115–116. doi:10.1007/s00431-007-0413-3
52. Blau H, Freud E, Mussaffi H, Werner M, Konen O, Rathaus V. Urogenital abnormalities in male children with cystic fibrosis. Arch Dischildhood. 2002;87(2):135–138. doi:10.1136/adc.87.2.135
53. Yoon JC, Casella JL, Litvin M, Dobs AS. Male reproductive health in cystic fibrosis. J Cyst Fibros. 2019;18:S105–S10.
54. Lomunova MA, Gershovich PM. Gene therapy for cystic fibrosis: recent advances and future prospects. Acta Naturae. 2023;15(2):20. doi:10.32607/actanaturae.11708
55. Pangeni R, Meng T, Poudel S, et al. Airway mucus in pulmonary diseases: muco-adhesive and muco-penetrating particles to overcome the airway mucus barriers. Int J Pharm;2023. 122661. doi:10.1016/j.ijpharm.2023.122661
56. Naehrig S, Chao C-M, Naehrlich L. Cystic fibrosis: diagnosis and treatment. Dtsch Arztebl Int. 2017;114(33–34):564. doi:10.3238/arztebl.2017.0564
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.