Back to Journals » Journal of Blood Medicine » Volume 11
Disease Burden in Patients with Glanzmann’s Thrombasthenia: Perspectives from the Glanzmann’s Thrombasthenia Patient/Caregiver Questionnaire
Authors Duncan A, Kellum A ![]() , Peltier S, Cooper DL
, Peltier S, Cooper DL ![]() , Saad H
, Saad H
Received 25 April 2020
Accepted for publication 25 August 2020
Published 11 September 2020 Volume 2020:11 Pages 289—295
DOI https://doi.org/10.2147/JBM.S259904
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Martin H Bluth
Alexander Duncan,1 Angela Kellum,2 Skye Peltier,3 David L Cooper,4 Hossam Saad4
1Department of Pathology & Laboratory Medicine, Emory University School of Medicine, Atlanta, GA, USA; 2Louisiana Center for Bleeding and Clotting Disorders, Tulane University, New Orleans, LA, USA; 3Center for Bleeding and Clotting Disorders, University of Minnesota Medical Center – Fairview, Minneapolis, MN, USA; 4Novo Nordisk Inc., Plainsboro, NJ, USA
Correspondence: Alexander Duncan
Emory University School of Medicine, Atlanta, GA, USA
Tel +1 404 712 4637
Fax +1 404 712 4632
Email [email protected]
Purpose: Glanzmann’s thrombasthenia (GT) is a rare bleeding disorder caused by a mutation in the αIIbβ 3 integrin essential for optimal platelet function and hemostasis. The aim of this study was to identify the burden of GT on patients and caregivers through better understanding of the management and psychosocial impact of this disorder.
Patients and Methods: Participants for this online survey were recruited using a rare disease specialty recruiter from Comprehensive Health Education Services. Data were collected from January 31 through March 12, 2019. The questionnaire was designed to collect information regarding demographics, diagnosis, treatment, and psychosocial impact.
Results: Of the 45 respondents (24 patients and 21 caregivers), the majority were female (58%), white (64%), and employed full-time (53%) and had no family history of GT (64%). Many patients reported significant bruising at birth (76%), and the mean age at diagnosis was 2.6 years. About half of the patients experienced 1 bleed per day, and 13% had over 500 bleeds of any severity per year. Most bleeds were skin bruising or mouth bleeds, but patients also reported joint/muscle and gastrointestinal bleeds. Most patients reported receiving a platelet transfusion (82%), and some had developed platelet refractoriness (38%) or antibodies (32%). Common treatments were antifibrinolytics (82%) and recombinant activated factor VII (rFVIIa) (42%), likely due to the presence of antibodies. Many (58%) patients experienced issues with excessive bleeding at school; 38% reported missing school as a result. Female patients struggled to find a gynecologist with knowledge of the management of GT. Most patients were satisfied with the support they receive from their current partner (65%) and their friends (76%).
Conclusion: Most patients with GT are diagnosed early. Patients experience considerable psychosocial impact. Patient and physician education concerning treatment alternatives and the support of the GT community are critical.
Keywords: bleeding disorder, psychosocial impact, recombinant activated factor VII, platelet transfusion, platelet antibodies
Introduction
Glanzmann’s thrombasthenia (GT) is an inherited autosomal recessive hemorrhagic syndrome characterized by a qualitative and/or quantitative mutation of the platelet membrane glycoprotein IIb/IIIa (αIIbβ3 integrin) that is essential for effective platelet aggregation and clot formation.1,2 It is a rare bleeding disorder with a general incidence rate of approximately 1 in a million with higher occurrence (approximately 1:200,000) in regions where intracommunity and consanguineous marriages are common.2–4 The disorder is slightly more common among females than males.5 Clinically, acquired platelet disorders, including acquired GT, are relatively more common and can occur with the development of auto-immunization secondary to platelet transfusions, medications, or underlying diseases such as autoimmune disorders, infections, and malignancies.4,6
Clinical presentation is highly variable, ranging from mild bruising to severe life-threatening hemorrhages. Severe bleeding occurs mostly in those with homozygous or compound heterozygous mutations of αIIbβ3, and simple heterozygotes are often asymptomatic.1,2,5 Spontaneous mucocutaneous bleeding is ubiquitously present in GT and commonly includes easy bruising, purpura, epistaxis, mouth bleeds, and menorrhagia in female patients. Severe bleeding can occur within the gastrointestinal system, joints, and central nervous system and often follows trauma, surgery, and childbirth.2,7 Typically, initial symptoms start at birth or early childhood, and most patients with GT are diagnosed before the age of 5 years.2,4 According to data from the international GT registry, the median and mean ages of first bleed are 1 year and 5 years, respectively.2
Laboratory findings include prolonged bleeding time and Platelet Function Analyzer-100 (PFA-100®) closure time, abnormal clot retraction, and no/decreased platelet aggregation in response to all physiologic agonists, including collagen, adenosine diphosphate, epinephrine, thrombin, and arachidonic acid. Aggregation tests with ristocetin are normal in patients with GT. Platelet morphology and count are also often normal in these patients, unlike those with other inherited coagulation disorders.2,8
Management options for localized and minor bleeding in patients with GT include pressure application, fibrin sealants, nasal packing, and topical thrombin gel foam.5,9 Platelet transfusion has been the standard of care for major bleeding episodes. Development of alloantibodies to platelets and refractoriness to future transfusions are common concerns.2 Other treatment options include recombinant activated factor VII (rFVIIa; approved for use in the United States in patients with refractoriness to platelet transfusions and in Europe in patients with past or present refractoriness to platelet transfusions or where platelets are not readily available)10,11 and antifibrinolytics. Allogeneic stem cell transplantation is curative and is considered in selected patients.9,12
Because of the low prevalence of GT, there is a lack of randomized clinical studies that are essential to establish management guidelines.2 Nevertheless, it is important to determine the gaps and areas of unmet needs to effectively tackle disease burden. This article summarizes the results from an online survey that was conducted to better understand the current management strategies of GT and its psychosocial impact in patients and caregivers.
Materials and Methods
Study Design
Participants for this online survey were recruited via a rare disease specialty recruiter from Comprehensive Health Education Services on behalf of Novo Nordisk. Survey respondents included patients with GT or caregivers of pediatric patients with GT in the United States; both sets of respondents received the same version of the questionnaire. Participation was limited to one patient per household. Data were collected from January 31 through March 12, 2019, via a moderator-assisted online survey. The survey consisted of 99 questions; the average time to complete the survey was 45 minutes.
Consent was obtained from all participants. Upon completion of the questionnaire, respondents were given remuneration in appreciation of their time and cooperation. The research survey was conducted in compliance with all national laws protecting personal data and in adherence to relevant guidelines. This survey was considered market research and as such did not require IRB or ethics committee approval.
Study Measures
Questions for patients or caregivers were designed to collect information regarding demographics, symptoms, diagnosis, treatment history, and psychosocial impact. Quantitative assessment of bleeding was done using the International Society on Thrombosis and Haemostasis Bleeding Assessment Tool (ISTH-BAT).13 Responses to questions about symptoms before the diagnosis of GT were used to calculate ISTH-BAT scores. Recently established ISTH-BAT cutoff values were used (≥4, adult males; ≥6, adult females; and ≥3, children) to objectively evaluate the disease state of the individuals.14
Statistical Analysis
Due to the small sample size, only descriptive statistics will be provided.
Results
Patient Demographics
A total of 45 respondents, including 24 patients and 21 caregivers, completed the survey. Patient characteristics are shown in Table 1. Mean (range) age of the 24 patients was 33 years (18–61 years); mean age of children and adolescents whose caregivers completed the survey was 8 years (1–17 years). More than half of survey participants were female (58%), were white (64%), and were employed full-time (53%). Thirty-one percent of participants had a family history of GT, and 64% were the first in their family to be diagnosed with GT.
 |
Table 1 Demographic Characteristics |
Initial Symptoms
The majority of the patients (76%) were born with significant bruising, and many male patients bled extensively during circumcision (47%). Skin bruising was common and was reported in 66% of all patients when first diagnosed. Most patients had a BAT score above the normal range, and the mean (range) BAT score was 9.07 (0–30) (Figure 1). Of note, the reported bleeding scores were calculated based on symptoms prior to GT diagnosis and therefore may not directly predict disease severity; patients with very severe disease are likely to be diagnosed earlier based on their symptoms or family history. Common symptoms (mean BAT score) experienced before diagnosis include bruising (1.89), bleeding from surgery/trauma (1.20), and epistaxis (1.04). Most of the participants were diagnosed before puberty, and female participants did not exhibit signs of excessive bleeding during menstruation. When these female participants who were diagnosed before puberty were removed from the analysis, the estimated range of BAT scores for menstruation was 0.58 to 3.60.
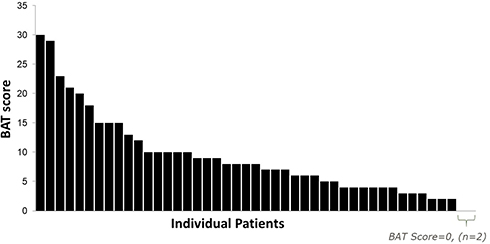 |
Figure 1 Distribution of ISTH-BAT scores. |
Diagnosis
More than 50% of participants were diagnosed with GT prior to their first birthday. For the rest of the participants, the average (range) duration between their initial symptoms and a visit to a specialist was just over 1 year (1–9 years) and to disease diagnosis was just over 2 years (<1–38 years). Mean age (range) at diagnosis was 2.6 years (<1–38 years).
About half of the patients reported 1 bleed daily, and 13% experienced over 500 bleeds in 1 year. These bleeds occurred most frequently as skin bruising and mouth bleeds, but some patients also reported joint or muscle and gastrointestinal bleeds (Figure 2).
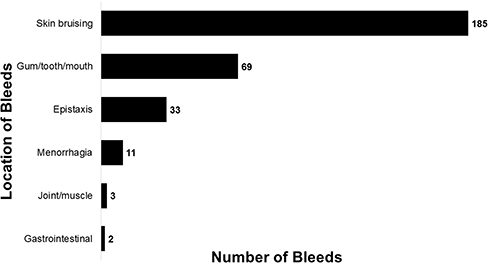 |
Figure 2 Total frequency of bleeds over past 12 months reported by all participants. |
Treatment
Patients were often willing to travel long distances for follow-ups with their preferred hematologist, and the average travel time for these visits was 62 minutes. More than 90% of patients reported regular follow-up visits (71% with a hematologist and 24% with a hemophilia treatment center). The majority (79%) of patients felt confident that most bleeds could be treated at home; 43% reported not requiring any treatment for their recent bleeds. The most common treatments for bleeds during the previous year were antifibrinolytics (82%), followed by rFVIIa (42%) (Figure 3). Only those patients with refractoriness to platelets reported using rFVIIa, primarily for severe bleeding; rFVIIa controlled bleeds with 2 to 3 doses (range, 1–11). Many patients reported taking antifibrinolytics prophylactically (orally, mouth rinses, or nasal sprays).
 |
Figure 3 Treatments used to control recent bleeds (categories were not mutually exclusive). |
The majority (84%) of patients had at least one platelet or blood transfusion, and nearly 25% reported having had more than 20 transfusions in their lifetime. A decrease in efficacy was reported with additional platelet transfusions; 73% of patients reported that platelet transfusions effectively controlled bleeds in the past, and 57% reported that platelet transfusions effectively control bleeds currently. In addition, 8% of patients reported that platelet transfusions did not effectively control bleeds in the past, and 22% of patients reported that platelet transfusions do not currently control bleeds (Figure 4). Many patients reported refractoriness to platelets (38%) or antibodies to platelets (32%). Patients reported concern about uncontrolled bleeding due to refractoriness or antibodies and stated that their physicians ordered platelets without adequately understanding their fears.
 |
Figure 4 Platelet transfusion information. (A) Patient-reported effectiveness of platelet transfusions in the past and the present. (B) Proportion of patients reporting refractoriness to platelets. (C) Proportion of patients tested for antibodies to platelets. (D) Proportion of patients reporting antibodies to platelets. |
Among female patients, controlling menstruation was particularly challenging, and many struggled to find a gynecologist with some knowledge of the management of menstruation, pregnancy, and childbirth in the presence of GT. The majority (74%) reported taking hormonal contraceptives to prevent regular menstruation. Eleven percent of female participants reported menstrual bleeding that required hospitalization and/or emergency treatment.
Psychosocial Impact
During childhood, 58% of patients experienced issues with excessive bleeding at school, 38% reported missing school as a result, and 22% reported bullying. Missed work due to GT was reported by 38% of adult patients and 24% of caregivers. Twenty-one percent of adult patients reported that their employer did not take their condition seriously, and only 56% reported that they were satisfied with the support they receive from their employer. Most participants were satisfied with the level of support they receive from their significant other (65%), their family (82%), and their friends (76%) (Figure 5). Overall, patients felt optimistic about their future living with GT. Patients also reported the need for additional resources and education for themselves and their treating physicians, especially regarding menstruation, childbearing, and treatment options.
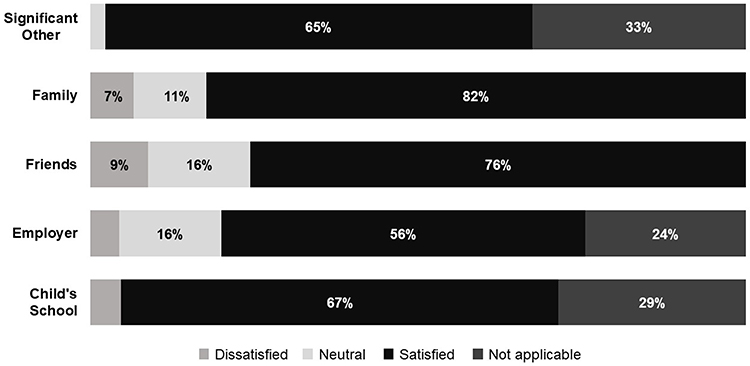 |
Figure 5 Distribution of levels of patient satisfaction with the support they receive. |
Discussion
Although an extremely rare disease, GT is considered a well-understood bleeding disorder with unique clinical and laboratory findings. However, there is a critical need for education and awareness of GT among practicing physicians in order to ensure timely diagnosis and efficient management.2 The goal of this qualitative online survey was to assess the current disease state of GT and its burden as experienced by patients with GT and caregivers of children with GT. Overall, the data presented here bring to light the true impact of this disease on patients, caregivers, and their families and help to provide HCPs with a real-world understanding of the experience of living with this bleeding disorder.
In accordance with previous reports, for most patients, diagnosis occurred within the first year of life and was prompted by the early onset of symptoms.2,5 Although diagnosis of GT can be challenging, expensive, and time-consuming, accurate and prompt diagnosis is essential for successful disease management. In the absence of family history, obtaining a first-time diagnosis of GT can be difficult, especially in regions where confirmatory laboratory tests (eg, platelet aggregometry and flow cytometry) are not available.3,12 Normal coagulation parameters, such as prothrombin time, partial thromboplastin time, and platelet count should be used to rule out other common clotting disorders, including congenital afibrinogenemia and von Willebrand disease.5 As the severity of GT is largely determined by the presence of homozygous vs heterozygous mutations and residual platelet function, prenatal genetic tests and counseling should be considered, especially among susceptible populations and known-carrier parents.4,5 Misdiagnosis and unestablished diagnosis in patients with less severe symptoms has led to an underestimation of the prevalence of GT.12,15
Platelet alloimmunization in inherited GT and refractoriness remain major concerns among patients with GT. Most patients reported receiving platelets or a blood transfusion, and more than half had either antibodies or refractoriness to platelets. About half of those who receive platelet transfusions are likely to develop platelet alloantibodies or refractoriness.12 Although plasmapheresis can be used to remove alloantibodies to allow future platelet transfusion, this procedure may not be always effective or available at all centers. Treatment with rFVIIa has a high success rate and relatively low risk (0.17%) of thromboembolic events.16 rFVIIa acts directly on the platelet surface and enhances thrombin generation and fibrin interaction to control bleeding episodes.4,5,17 Results published in 2019 from the international GT registry suggest that with rFVIIa treatment, with or without antifibrinolytics, for 18 surgical procedures and 205 nonsurgical bleeds, platelets were effective, with a low rate of adverse events in all patients irrespective of their platelet immunization or refractoriness status.7 Alternative therapies to control bleeding episodes, including rFVIIa, should be prudently used, especially in young patients and females of reproductive age to prevent the development of platelet antibodies that can cause potential fetal complications during pregnancy, such as thrombocytopenia and hemorrhage.12 Physicians should encourage their patients with GT to carry a card that highlights the details of their bleeding disorder, including any known antibodies or refractoriness to platelets, and the information for a specialized treatment center in case of an emergency.
Many of the participants in this survey agreed that additional education on GT is needed for both patients and health-care professionals. Female patients have struggled to find a gynecologist with knowledge of GT. Among pregnant females, a thorough maternal history, appropriate prophylaxis, and a multidisciplinary approach can optimize maternal and fetal outcomes.18,19 However, well-controlled clinical trials are needed to standardize management protocols for these patients.18 Avoidance of unnecessary trauma that can increase the risk of bleeding is important; therefore, many patients with GT are unable to participate in sports or physical education classes. These patients also often have many visible bruises, and this can have a negative effect on their self-esteem and social life. There is a need for multidisciplinary comprehensive care to alleviate anxiety, address psychosocial issues, and improve patients’ overall health and quality of life.9,12
It should be noted that this is a qualitative research survey, and the results cannot be generalized to the overall population due to sample selection, interviewing methods, and small sample size. There is also potential recruitment bias, as the organization used to recruit patients has offered disease-specific educational programs; however, another study found very similar results despite different recruiting methods.20 In addition, although the survey did ask basic questions about surgical procedures, data on surgical outcomes and adverse events were not collected.
Conclusion
GT is an extremely rare disorder, and patients can feel isolated. Connecting with other patients and receiving additional support from family and organizations can be helpful. Prompt diagnosis, with comprehensive workup including platelet aggregation and flow cytometry testing, and a multidisciplinary approach to treatment are essential for good prognosis. Optimal patient, caregiver, and physician education are lacking but are of utmost importance for efficient management of GT. Efforts should be undertaken to enhance the availability of resources and information regarding treatment alternatives.
Abbreviations
GT, Glanzmann’s thrombasthenia; ISTH-BAT, International Society on Thrombosis and Haemostasis Bleeding Assessment Tool; rFVIIa, recombinant activated factor VII.
Acknowledgments
The survey was sponsored by Novo Nordisk. Writing and editorial support were provided by PRECISIONscientia, Yardley, PA, which were in accordance with Good Publication Practice (GPP3) guidelines and funded by Novo Nordisk. The authors would like to acknowledge Helen Smith and Dr Shilpa Jain for their assistance with the survey data analysis. Select data from this paper was presented at the 61st American Society of Hematology Annual Meeting & Exposition as a poster presentation. The poster’s abstract was published in Blood. (2019);134 (Supplement 1):3456, https://doi.org/10.1182/blood-2019-128408.
Disclosure
AD is a consultant for Novo Nordisk. AK is a member of advisory boards for Novo Nordisk and Takeda and is a member of the speakers bureau for Takeda. SP is a consultant for Novo Nordisk. DLC and HS were employees of Novo Nordisk at the time of survey conception, drafting, and data collection and analysis. HS disclosed that Novo Nordisk has a product that is being used to treat bleeding in patients with Glanzmann thrombasthenia (rFVIIa). DLC is currently an employee of uniQure, Lexington, MA, USA. HS is currently an employee of Amgen, Thousand Oaks, CA, USA. The authors report no other conflicts of interest in this work.
References
1. Nurden AT, Fiore M, Nurden P, Pillois X. Glanzmann thrombasthenia: a review of ITGA2B and ITGB3 defects with emphasis on variants, phenotypic variability, and mouse models. Blood. 2011;118(23):5996–6005. doi:10.1182/blood-2011-07-365635
2. Poon MC, Di Minno G, d’Oiron R, Zotz R. New insights into the treatment of Glanzmann thrombasthenia. Transfus Med Rev. 2016;30(2):92–99. doi:10.1016/j.tmrv.2016.01.001
3. Iqbal I, Farhan S, Ahmed N. Glanzmann thrombasthenia: a clinicopathological profile. J Coll Physicians Surg Pak. 2016;26(8):647–650. doi:2396
4. Solh T, Botsford A, Solh M. Glanzmann’s thrombasthenia: pathogenesis, diagnosis, and current and emerging treatment options. J Blood Med. 2015;6:219–227. doi:10.2147/JBM.S71319
5. Nurden AT. Glanzmann thrombasthenia. Orphanet J Rare Dis. 2006;1:10. doi:10.1186/1750-1172-1-10
6. Nurden AT. Acquired Glanzmann thrombasthenia: from antibodies to anti-platelet drugs. Blood Rev. 2019;36:10–22. doi:10.1016/j.blre.2019.03.004
7. Zotz RB, Poon MC, Di Minno G, D’Oiron R. The international prospective Glanzmann thrombasthenia registry: pediatric treatment and outcomes. TH Open. 2019;3(3):e286–e294. doi:10.1055/s-0039-1696657
8. Diz-Kucukkaya R. Inherited platelet disorders including Glanzmann thrombasthenia and Bernard-Soulier syndrome. Hematology Am Soc Hematol Educ Program. 2013;2013:268–275. doi:10.1182/asheducation-2013.1.268
9. Krause KA, Graham BC. Glanzmann thrombasthenia. StatPearls. Treasure Island (FL): StatPearls Publishing LLC.; 2019.
10. Novo Nordisk Inc. NovoSeven®RT Package Insert. 2019.
11. Novo Nordisk A/S. NovoSeven®RT Package Leaflet. 2019.
12. Lee A, Poon MC. Inherited platelet functional disorders: general principles and practical aspects of management. Transfus Apher Sci. 2018;57(4):494–501. doi:10.1016/j.transci.2018.07.010
13. Rodeghiero F, Tosetto A, Abshire T, et al; on behalf of the ISTH/SSC Joint VWF and Perinatal/Pediatric Hemostasis Subcommittees Working Group. ISTH/SSC Bleeding Assessment Tool: a standardized questionnaire and a proposal for a new bleeding score for inherited bleeding disorders. J Thromb Haemost. 2010;8:2036–2065.
14. Elbatarny M, Mollah S, Grabell J, et al. Normal range of bleeding scores for the ISTH-BAT: adult and pediatric data from the merging project. Haemophilia. 2014;20(6):831–835. doi:10.1111/hae.12503
15. Nava T, Rivard GE, Bonnefoy A. Challenges on the diagnostic approach of inherited platelet function disorders: is a paradigm change necessary? Platelets. 2018;29(2):148–155. doi:10.1080/09537104.2017.1356918
16. Rajpurkar M, Croteau SE, Boggio L, Cooper DL. Thrombotic events with recombinant activated factor VII (rFVIIa) in approved indications are rare and associated with older age, cardiovascular disease, and concomitant use of activated prothrombin complex concentrates (aPCC). J Blood Med. 2019;10:335–340. doi:10.2147/JBM.S219573
17. Di Minno G, Coppola A, Di Minno MN, Poon MC. Glanzmann’s thrombasthenia (defective platelet integrin alphaIIb-beta3): proposals for management between evidence and open issues. Thromb Haemost. 2009;102(6):1157–1164. doi:10.1160/TH09-04-0225
18. Bannow BS, Konkle BA. Inherited bleeding disorders in the obstetric patient. Transfus Med Rev. 2018;32(4):237–243. doi:10.1016/j.tmrv.2018.06.003
19. Wijemanne A, Watt-Coote I, Austin S. Glanzmann thrombasthenia in pregnancy: optimising maternal and fetal outcomes. Obstetric Med. 2016;9(4):169–170. doi:10.1177/1753495X16655021
20. Chitlur M, Recht M, Rajpurkar M, et al. Unmet needs in diagnosis and treatment of Glanzmann’s Thrombasthenia (GT): perceptions of US hematologists and nurses. Blood. 2014;124(21):2179. doi:10.1182/blood.V124.21.2179.2179
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.