Back to Journals » International Journal of Nanomedicine » Volume 12
Co-delivery nanocarriers targeting folate receptor and encapsulating 2-deoxyglucose and α-tocopheryl succinate enhance anti-tumor effect in vivo
Authors Lei X, Li K, Liu Y, Wang ZY, Ruan BJ, Wang L, Xiang A, Wu D ![]() , Lu Z
, Lu Z
Received 28 February 2017
Accepted for publication 23 May 2017
Published 8 August 2017 Volume 2017:12 Pages 5701—5715
DOI https://doi.org/10.2147/IJN.S135849
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Linlin Sun
Xiaoying Lei,1,* Ke Li,2,3,* Yan Liu,4,* Zhen Yu Wang,1 Ban Jun Ruan,1 Li Wang,1 An Xiang,1 Daocheng Wu,3 Zifan Lu1
1State Key Laboratory of Cancer Biology, Department of Pharmacogenomics, School of Pharmacy, The Fourth Military Medical University, 2Shaanxi Key Laboratory of Ischemic Cardiovascular Disease, Institute of Basic and Translational Medicine, Xi’an Medical University, Xi’an, Shaanxi, 3Key Laboratory of Biomedical Information Engineering of Education Ministry, School of Life Science and Technology, Xi’an Jiaotong University, Xi’an, 4Genetic Engineering Laboratory of PLA, The Eleventh Institute of Academy of Military Medical Sciences of PLA, Changchun, Jilin, People’s Republic of China
*These authors contributed equally to this work
Abstract: A combination administration of chemical agents was highlighted to treat tumors. Recently, tumor cell has been found to be different from normal cell in metabolic manner. Most of cancer cells prefer aerobic glycolysis to mitochondrial oxidative phosphorylation (OXPHOS) to satisfy energy and biomass synthesis requirement to survive, grow and proliferate, which provides novel and potential therapeutic targets for chemotherapy. Here, 2-deoxy-d-glucose (2-DG), a potent inhibitor of glucose metabolism, was used to inhibit glycolysis of tumor cells; α-tocopheryl succinate (α-TOS), a water-insoluble vitamin E derivative, was chosen to suppress OXPHOS. Our data demonstrated that the combination treatment of 2-DG and α-TOS could significantly promote the anti-tumor efficiency in vitro compared with administration of the single drug. In order to maximize therapeutic activity and minimize negative side effects, a co-delivery nanocarrier targeting folate receptor (FR) was developed to encapsulate 2-DG and α-TOS simultaneously based on our previous work. Transmission electron microscope, dynamic light scattering method and UV-visible spectrophotometers were used to investigate morphology, size distribution and loading efficiency of the α-TOS-2-DG-loaded and FR-targeted nanoparticles (TDF NPs). The TDF NPs were found to possess a layer-by-layer shape, and the dynamic size was <100 nm. The final encapsulation efficiencies of α-TOS and 2-DG in TDF NPs were 94.3%±1.3% and 61.7%±7.7% with respect to drug-loading capacities of 8.9%±0.8% and 13.2%±2.6%, respectively. Almost no α-TOS release was found within 80 h, and release of 2-DG was sustained and slow within 72 h. The results of FR binding assay and fluorescence biodistribution revealed that TDF NPs could target FR highly expressed on tumor cell in vitro and in vivo. Further, in vivo anti-tumor experiments showed that TDF NPs had an improved biological function with less toxicity. Thus, our work indicates that the co-delivery TDF NPs have a great potential in tumor therapy.
Keywords: co-delivery nanocarrier, α-tocopheryl succinate, 2-deoxyglucose, anti-tumor
Introduction
The metabolic properties of cancer cells are different from those of normal cells. Most of the malignant cancer cells prefer aerobic glycolysis to mitochondrial oxidative phosphorylation (OXPHOS) to satisfy energy and biomass synthesis requirement to survive, grow and proliferate.1–6 During aerobic glycolysis, an abnormally high rate of glucose uptake was implemented by tumor cells, and most incoming glucose was then metabolized into lactate.7 It was shown that glucose deprivation selectively induces cell injury in transformed human cells via metabolic oxidative stress.8 2-Deoxy-D-glucose (2-DG), an analog of glucose, was phosphorylated by hexokinase to 2-DG-phosphate (2-DG-6P) that cannot undergo further glycolysis and thus was used to mimic glucose deprivation and inhibit glycolysis in vivo.9 Accumulation of 2-DG or 2-DG-6P can cause ATP depletion, cell cycle inhibition and cell death.10
If aerobic glycolysis is inhibited in cancer cells, the function of mitochondrial OXPHOS could be restored to compensate for reduced ATP.11 Thus, dual targeting of mitochondrial and glycolytic pathways was suggested as a promising anti-tumor strategy. At present, a group of compounds targeting mitochondria to induce apoptosis and suppress cancer, mitocans, have been defined.12 As shown as an example, α-tocopheryl succinate (α-TOS), the vitamin E derivative, exerts pro-apoptotic effects in a wide range of tumors and is well tolerated by normal tissues.13 It targets mitochondrial complex II to produce reactive oxygen species (ROS) that then trigger apoptosis.14 More specifically, ROS promote phosphorylation of the Mst1 kinase, resulting in phosphorylation of the transcription factor FoxO1 that translocates into the nucleus. This is followed by increased expression of Noxa that diverts Mcl-1 from Bak, causing the formation of pores in the mitochondrial outer membrane, whereby promoting the apoptotic cascade downstream of mitochondria.15–17 Notably, at the molecular level, α-TOS acts as a Bcl-2 homology domain 3 (BH3) mimetic, effectively sensitizing cancer cells to other drugs. In vitro, α-TOS showed significant anti-tumor effect. Interestingly, in vivo, α-TOS can be easily converted into α-tocopherol (α-TOH) that boosts tumor surveillance in the liver.18 The main obstacle for the successful application of α-TOS-based clinical treatments is the hydrophobic nature of this drug that significantly reduces its bioavailability and therapeutic activity.19 In addition, the intravenous or intraperitoneal administration of α-TOS in an organic phase or oil emulsion can cause important side effects, such as the formation of aggregates as a result of drug mixing with blood plasma, inflammation and embolization processes.20
In recent years, the drug-loaded polymeric nanoparticles (NPs) offer bright promise for cancer therapy.21–24 To potentially maximize therapeutic activity while minimizing negative side effects of 2-DG and α-TOS, an amphiphilic nanocarrier targeting folate receptor (FR) was designed and used in our study to deliver both chemoagents. It is well known that FR was frequently overexpressed in malignant tissue and cells, which provides unique opportunity to specifically target cancer cells by virtue of its high affinity for folic acid and folate analogs.25 Until now, FR targeting has been exemplified using folate conjugates with a wide variety of diagnostic and therapeutic probes.
In this study, we found that mitochondria-targeted drug α-TOS synergizes with 2-DG to trigger various cancer cell death in vitro; the amphiphilic nanocarrier targeting FR on cancer cells and loaded with both showed enhanced anti-tumor effects with less toxicity in vivo. Thus, our study provided a multifunctional nanocarrier with improved anti-tumor therapeutic effects and less toxicity.
Materials and methods
Materials
2-DG, α-TOS (PHR1029), coumarin-6, bovine serum albumin (BSA), dimethyl sulfoxide (DMSO), 3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide (MTT) and folate acid (FA) were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). Soluble starch, N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC), N-hydroxysuccinimide (NHS), N, N′-dicyclohexylcarbodiimide (DCC), 4-dimethylaminopyridine (DMAP), stearic acid, glycidyltrimethylammonium chloride (GTAC), potassium bromide and DMSO-d6 were purchased from Aladdin (Shanghai, China). DiR dye was purchased from Caliper Life Sciences (Waltham, MA, USA). Annexin V-fluorescein isothiocyanate (FITC)/propidium iodide (PI) was purchased from eBioscience (San Diego, CA, USA). McCoy’s 5A Medium and Dulbecco’s Modified Eagle’s Medium (DMEM) were purchased from Thermo Fisher Scientific (Waltham, MA, USA). Matrigel was purchased from BD (Franklin Lakes, NJ, USA). Besides these, other chemical reagents that were of analytical grade were purchased from Sinopharm Chemical Reagent Co., Ltd (Xi’an, China).
Methods
Cell lines and culture conditions
The research had ethical review board approval of the Fourth Military Medical University. HT29 colon adenocarcinoma cells were purchased from American Type Culture Collection (ATCC); HeLa cervical carcinoma cells and A549 lung adenocarcinoma cells were routinely used in our laboratory. Cells were cultured in McCoy’s 5A Medium or DMEM with 10% fetal bovine serum (FBS) (Thermo Fisher Scientific) following the instructions from ATCC.
Synthesis of encapsulation materials
Synthesis procedure of cationic amphiphilic starch (CStSa, also (+)StSa) was published in Biomaterials.26 Synthesis procedure of BSA-FA followed the literature of Meng et al.27 Briefly, 80 mg of BSA, 40 mg of EDC, 35 mg of NHS and 50 mg of FA were added to DMSO, and the mixture was stirred for 12 h in the dark at 25°C. Then, the sample was centrifuged at 10,000 rpm for 30 min to remove the excess FA and EDC/NHS. In this reaction, EDC and NHS were used to form FA-NHS, which could conjugate with the amino group of BSA. The product was assayed by 1H-nuclear magnetic resonance (1H-NMR).
Preparation of α-TOS-2-DG-loaded and FR-targeted nanoparticles (TDF NPs)
In the first step, we prepared α-TOS-loaded CStSa micelles. A total of 3 mg of α-TOS and 5 mg of CStSa were co-dissolved in 500 μL of DMSO and then dropwise added to 7 mL of distilled water under stirring. After stirring for 10 min, 5 mg of 2-DG was added into the solution and kept under stirring for 10 min. Then, 2 mL of BSA-FA solution (10 mg/mL) was added into the mixture solution under stirring slowly. Finally, TDF NP solution was transferred into a dialysis membrane (3,500 Da molecular weight cut-off [MWCO]) and dialyzed against distilled water for 2 h during which the medium was replaced by clean water thrice, and at the end of the procedure, the volume was set to 10 mL. TDF NPs were further concentrated by treatment with PEG-8000, dialysis and ultrasound redispersion. No drug-loaded NPs with BSA-FA (F NPs) were also synthesized to be a control.
Characterization of TDF NPs
The hydrodynamic diameters and zeta potentials of the TDF NPs were measured by Malvern Instruments (Nano-ZS90, Malvern, UK). The morphology of TDF NPs was observed by a transmission electron microscope (TEM) (H-800; Hitachi, Tokyo, Japan). The solution of TDF NPs was dropped on a copper grid that was coated with ultra-thin carbon support film. When the grid dried, the samples were observed by TEM. The formulas for the calculation of encapsulation efficiency (EE) and drug loading (DL) of TDF NPs are as follows:
|
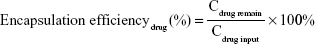
|
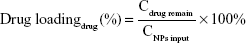
where the Cdrug input was the concentration of α-TOS or 2-DG input before encapsulation, Cdrug remain was the concentration of α-TOS or 2-DG after lyophilization and CNPs input was the total amount of NPs.
In vitro drug release profiles of TDF NPs
Drug release from TDF NPs was measured by a dialysis method; the experiment was conducted using dialysis membrane (MWCO 3500) in phosphate-buffered saline (PBS) (pH 7.4) under stirring at 30 rpm and 37°C for 72 h. The amount of 2-DG in the sample solutions was determined by spectrophotometry. As the control, the same amount of 2-DG- and α-TOS-loaded CStSa micelles (TD NPs without FA-BSA compared with TDF NPs) and free 2-DG were dispersed in PBS (pH 7.4), and drug release profiles were analyzed at the same condition. The amount of α-TOS is water insoluble, and hence it would not release into the PBS.
In vitro anti-tumor activity assay
To demonstrate the anti-tumor activity, MTT assay, colony formation assay and cell death were performed, respectively. For MTT assay, 150 μL of HT29, HeLa and A549 cells were seeded at 6×104/mL density per well in 96-well plates and then treated with 2-DG or α-TOS, or a combination of 2-DG or α-TOS, or TDF NPs. The dose was referred to the previous report.28,29 For colony formation assay, HT29 or HeLa cells were seeded at 300 cells per dish in 6 cm cell culture dishes and treated with 2-DG and α-TOS for 6 h, then grew for 7–14 days, and the number of colonies formed was counted. For cell death, annexin V-FITC/PI was used according to the kit manual and analyzed on a flowcytometer Coulter Epics XL/MCL (Beckman Coulter Inc, Brea, CA, USA).
In vivo biodistribution studies
All the animal experiments were approved by and performed in accordance with the permitted guidelines of the Animal Ethics Committee of the Fourth Military Medical University. FR-positive HeLa cells (5×106 cells in 100 μL of medium) were mixed with 100 μL of Matrigel and subcutaneously injected into the right flank of 8-week-old healthy nude mice. To monitor FR nanocarrier targeting to tumor, the dye DiR, a near-infrared radiation (IR) fluorescent dye, was used to encapsulate DiR-loaded FR-targeted NPs. Free DiR was used as the control. Six tumor-bearing nude mice were randomly divided into two groups, each mouse of one group was injected intravenously with 100 μL of DiR-loaded FR-targeted NPs, while each mouse of another group was injected intravenously with 100 μL of DiR (2 mg/mL). Then, imaging was taken at 0.5 h, 24 h, 48 h and 72 h after injection with IVIS Lumina II In Vivo Imaging System (Caliper Life Sciences).
In vivo tumor suppression experiments
To investigate the anti-tumor efficacy of TPF NPs in vivo, 24 HeLa tumor-bearing mice were randomly divided into four groups (n=6). The administration was started when the tumor volume was ~50 mm3. A total of 100 μL of TDF NPs were intravenously injected at 3-day intervals for six times, an equal volume (100 μL) of normal saline or blank NPs were intravenously administrated as the controls, while the last group was injected with 50 μL of 3 mM free 2-DG intravenously and 50 μL of 30 μM α-TOS in corn oil intraperitoneally, respectively. The body weight of mice and the lengths of the longest tumor axis (a(t), mm) and the vertical axis (b(t), mm) were measured every other day, and the tumor volume (v(t), mm3) was calculated using the following equation: V =0.5× a × b2. The mice were sacrificed on the 21th day after the first administration, and the tumor of each mouse was weighed.
Histological assessment
The excised tumor and liver tissues were fixed with 4% paraformaldehyde solution for 1 day and then embedded in paraffin and cut into 5 mm thick sections. Hematoxylin and eosin (HE) staining was performed, and hepatocyte or tumor cell necrosis was observed to assess the liver toxicity and anti-tumor effect under the microscope (OLYMPUS IX71; Olympus Corporation, Tokyo, Japan).
Statistical analysis
The data on in vivo tumor suppression assay were given as mean ± SD of independent experiments. The normality of body weight and tumor size was tested by Kolmogorov–Smirnov test on SPSS Version 16.0 (SPSS Inc., Chicago, IL, USA), then one-way analysis of variance (ANOVA) or Student’s t-test was performed on Prism 5.0 software to determine the difference among the samples (GraphPad Software, Inc., La Jolla, CA, USA). P-values <0.05 were considered as statistically significant.
Results
A synergistic anti-tumor effect of α-TOS and 2-DG in vitro
To determine the anti-tumor effect of α-TOS and 2-DG in combination, we detected the inhibitory effects on cancer cells, including colonic cancer HT29 cell highly favoring glycolytic metabolism, cervix adenocarcinoma HeLa cell preferring to OXPHOS and lung carcinoma A549 cell in a dose- and time-dependent model. MTT assay could reflect the growth of cancer cells by detecting live cells. Considering that the maximum dose of outer encapsulated 2-DG was no >3.5 mM (data not shown), the dose of 3 mM of 2-DG was used in our experiment. Based on 3 mM of 2-DG, 15 μM, 30 μM and 45 μM of α-TOS were combined with 2-DG, respectively. Our results showed that 30 μM, even 15 μM, of α-TOS alone could significantly inhibit the growth of cancer cells in 48 h, and 3 mM of 2-DG by itself also could lead to the inhibition of tumor cells. However, the cell death was no <50% when administrated with single drug. In contrast, a combined treatment of 15 μM of α-TOS and 3 mM of 2-DG could remarkably reduce the survival of cancer cells compared to treatment with α-TOS or 2-DG alone. Expectedly, the survival of cancer cells dramatically decreased with the increased doses of α-TOS to 30 μM or 45 μM (Figure 1A; *P<0.05), which indicated that the combined treatment was dose-dependent. The MTT assay also showed that 45 μM of α-TOS did not significantly reduce the cell variability of HeLa cell compared to 30 μM of α-TOS; meanwhile, the combination treatment of 30 μM of α-TOS and 3 mM of 2-DG induced >50% of cell death in HT29 and A549. Thus, the combination dose of 30 μM of α-TOS and 3 mM of 2-DG was used for drug-loaded dose of nanocarrier next performed.
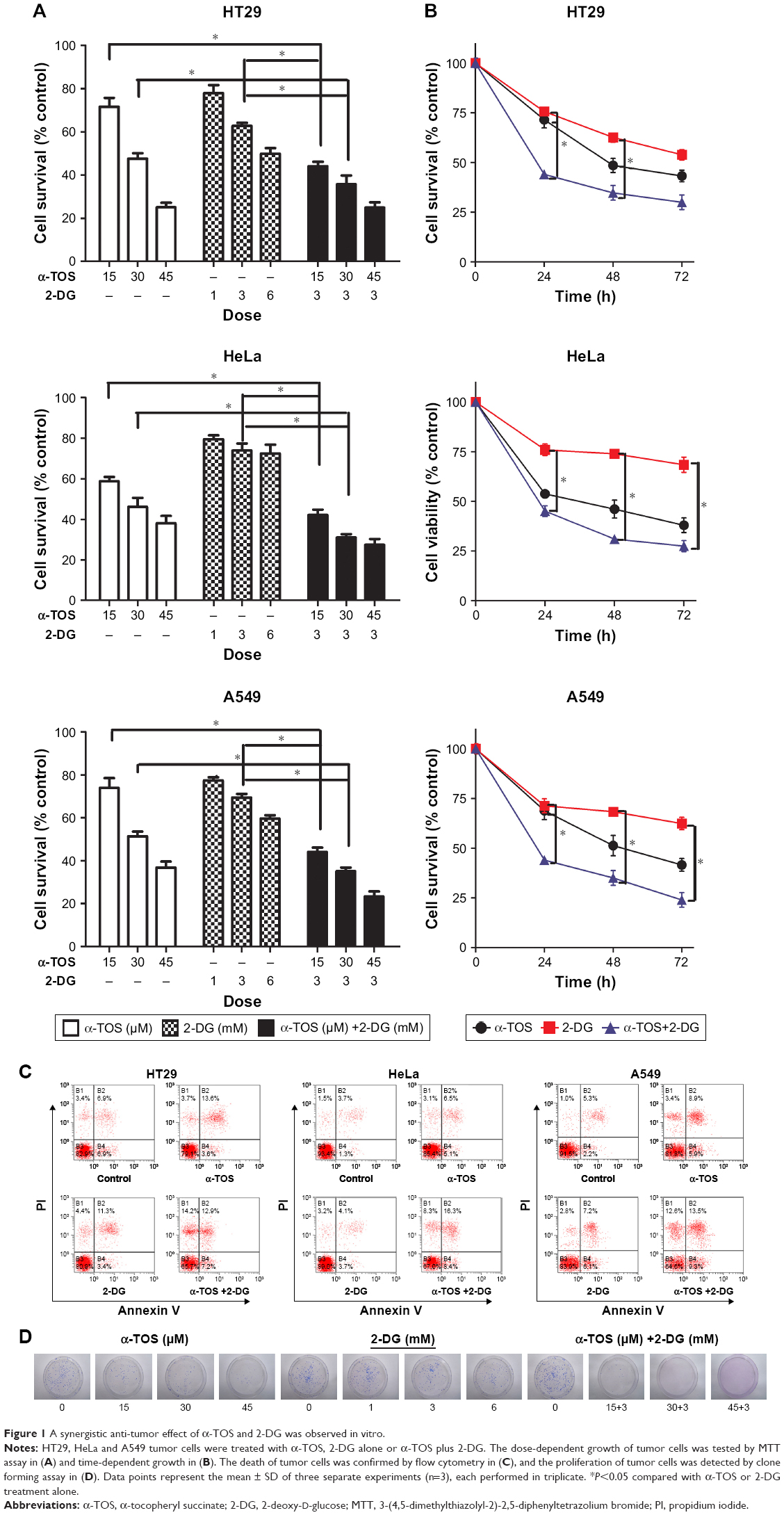 | Figure 1 A synergistic anti-tumor effect of α-TOS and 2-DG was observed in vitro. |
Further, time-dependent effects of the combined treatment were detected in 24 h, 48 h and 72 h after administration. Figure 1B shows that the survival of tumor cells with combined treatment decreased in 24 h; with the treatment prolonged to 48 h, even 72 h, the survival continued to decrease with a mild trend (Figure 1B; *P<0.05), indicating that the anti-tumor effects of combined treatment were superior to that of single drug, and the inhibition to cancer cells was also time-dependent. Even early in 12 h, our data on flow cytometry showed that cell apoptosis and death were induced by combination drug treatment. In HT29, HeLa and A549 cells, the percentage of the early apoptosis and late apoptosis in combination treatment group was higher than that of single drug administration, especially for HeLa cells (Figure 1C).
To further know the proliferation of a single cancer cell, we performed a plate colony formation assay. Our results demonstrated that the colony number was decreased when α-TOS or 2-DG was treated alone, especially α-TOS, while it remarkably reduced, even no colony was present on plate when treatment of α-TOS and 2-DG was combined, which showed that a low dose of α-TOS and 2-DG in combination mostly inhibited the proliferation of HT29 cancer cells (Figure 1D). Taken together, our results showed that the combined treatment of α-TOS and 2-DG could exhibit significantly superior anti-tumor effects on cancer cells to treatment of α-TOS or 2-DG alone, indicating that the treatment in combination by lower doses or shorter time could have a comparable anti-tumor effect.
Preparation and characterization of TDF NPs
In order to overcome the administration of liposoluble agent α-TOS and further reduce the potential toxicity of 2-DG, tumor-targeting nanocarriers were designed. With consideration of the advantages of FR and its ligand as a targeting delivery system, we synthesized BSA-grafted FA and then covered them onto dual drug-loaded micelles, which absorbed 2-DG on surface and encapsulated α-TOS in the core, to prepare targeting dual drug nanocarriers (Scheme 1). FR is frequently overexpressed on the vast majority of cancer tissues such as ovarian, colorectal and breast cancer, while its expression is limited in healthy tissues and organs. The ligand, folic acid, is small, stable over a broad range of temperatures and pH values, inexpensive, and non-immunogenic, and it retains its ability to bind to the FR after conjugation with drugs or diagnostic markers. After folate attaches to its receptors located within caveolae, it is internalized through the endocytotic pathway. As the pH of the endosome approaches five, the BSA-FA was decomposed and the drug released. Figure S1 shows that folate was correctly grafted on BSA. The concentrations of drugs in TDF NPs were adjustable in limited range. Dialysis membrane (3,500 Da MWCO) and PEG-8000 were used in enrichment of drugs. After dewatering, ultrasound was employed for redispersion. Finally, the actual usage dose of α-TOS and 2-DG could match the concentrations of free drugs. Table 1 displays the EE and drug loading capacity (DLC) of TDF NPs. The data were comparable to those drawn from other reports, which were shown to efficiently prevent a burst release of NPs. Indeed, dispersion of free 2-DG and 2-DG- and α-TOS-loaded CStSa micelles without BSA-FA (TD NPs) into the outer dialysis chamber showed different speeds (Figure 2A). Within 5 h, free 2-DG completely dispersed into outer dialysis solutions; within 24 h, 80% of 2-DG was released from TD NPs into outer solutions. While within 48 h, 50% of 2-DG was released from TDF NPs into outer solutions, indicating that TDF NPs covered by BSA-FA have the potential to control drug burst release. For TDF NPs, almost no α-TOS release was found within 80 h, and release of 2-DG was sustained and slow within 72 h.
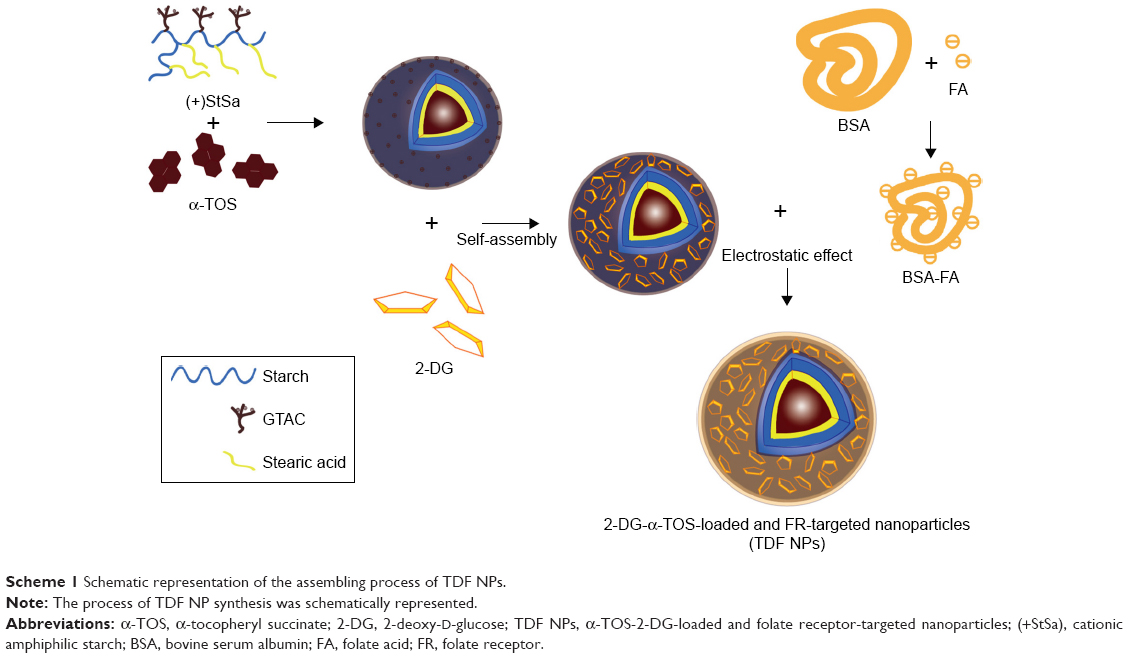 | Scheme 1 Schematic representation of the assembling process of TDF NPs. |
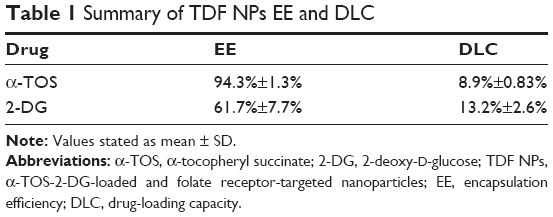 | Table 1 Summary of TDF NPs EE and DLC |
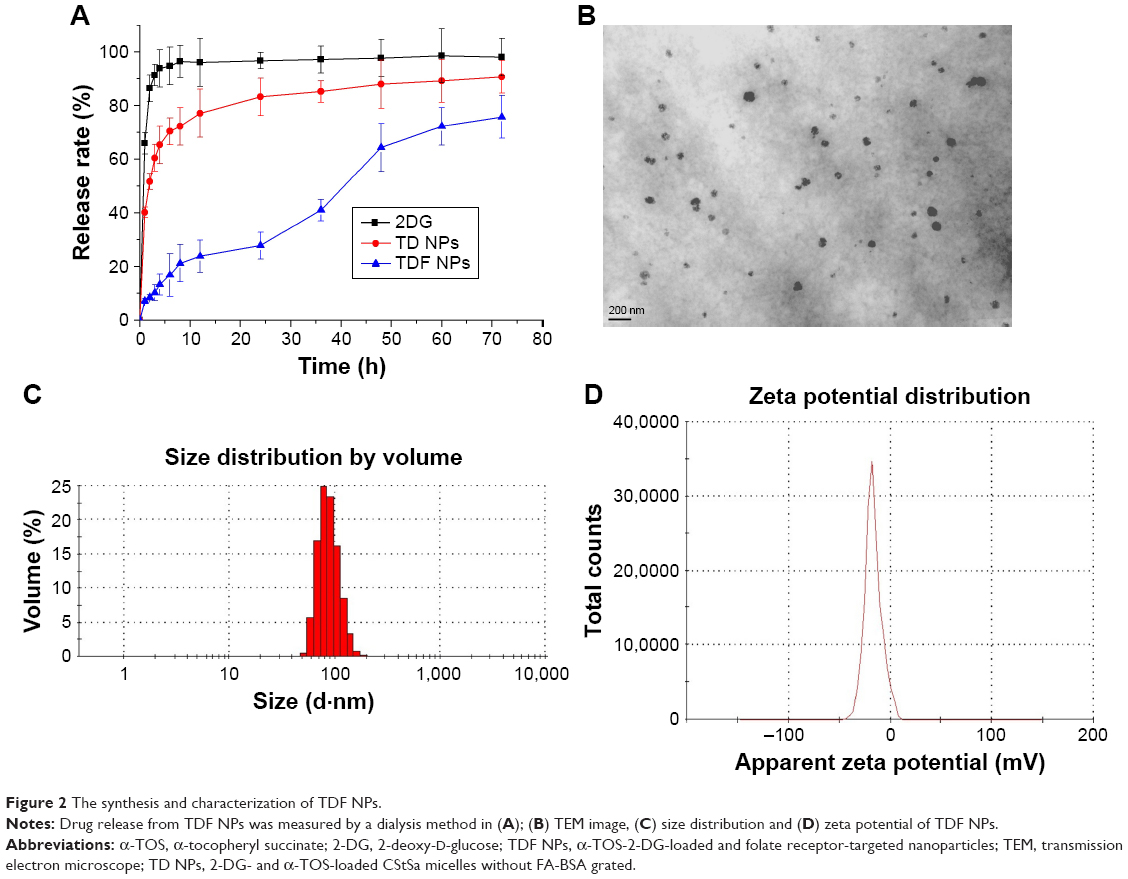 | Figure 2 The synthesis and characterization of TDF NPs. |
Further, we detected the magnifications of TDF NPs through TEM. Figure 2B and C shows that the NPs have a clear spherical shape with a core–shell structure and the size was smaller than 100 nm.
The TDF NPs were owing to the electrostatic interaction between negative BSA-FA and positive α-TOS-loaded CStSa micelles. The zeta potential of α-TOS-loaded CStSa micelles and TDF NPs was +28.2±2.9 mV and −22.3±2.1 mV, respectively (Figure 2D). The change of zeta potential in samples showed that TDF NPs comprised TD NPs and BSA-FA, and BSA-FA was covered on the surface of TD NPs factually.
In vitro anti-tumor effect of TDF NPs
To detect the anti-tumor activity of TDF NPs, HT29, HeLa and A549 cells mentioned earlier were tested. The results of MTT assay demonstrated that drug-loaded TDF NPs have remarkable inhibition to the growth of HeLa cells, while no significant suppression to that of HT29 and A549 cells, compared with F NPs without drug loading (Figure 3A). Notably, we found that only F NPs slightly promoted the growth of HeLa cells, which indicated that the nanocarrier was less cytotoxic to tumor cells, as expected low or no cytotoxicity to normal cells. Further detection of FR expression on these tumor cells, we found that HeLa cells expressed highest level of FR, whereas low or undetectable expression in HT29 and A549 cells (Figure S2). The data on FR expression level was consistent with previous reports.30 Our results indicated that α-TOS- and 2-DG-encapsulated TDF NP was effective against tumor cells, and the suppression to tumor cells was dependent on FR-mediated endocytosis. To further confirm the tumor-specific cellular uptake of TDF NPs, HeLa cells were incubated with or without 1% FA after coumarin 6-labeled F NPs were added, then cellular uptake behavior was observed using fluorescent microscopy. Figure 3B exhibits the behavior pattern of HeLa cells after incubating with coumarin 6-labeled F NPs with or without FA for 2 h. Strong green fluorescence was observed in the cytoplasm of HeLa cells incubated without FA, while few fluorescence was seen in the cytoplasm of HeLa cells and most appeared in the proximal outer membrane region when FA was added. It seemed that FA added into medium bound to F NPs, thus preventing the occurrence of endocytosis mediated by the binding of F NPs to FR on the surface of HeLa cells.
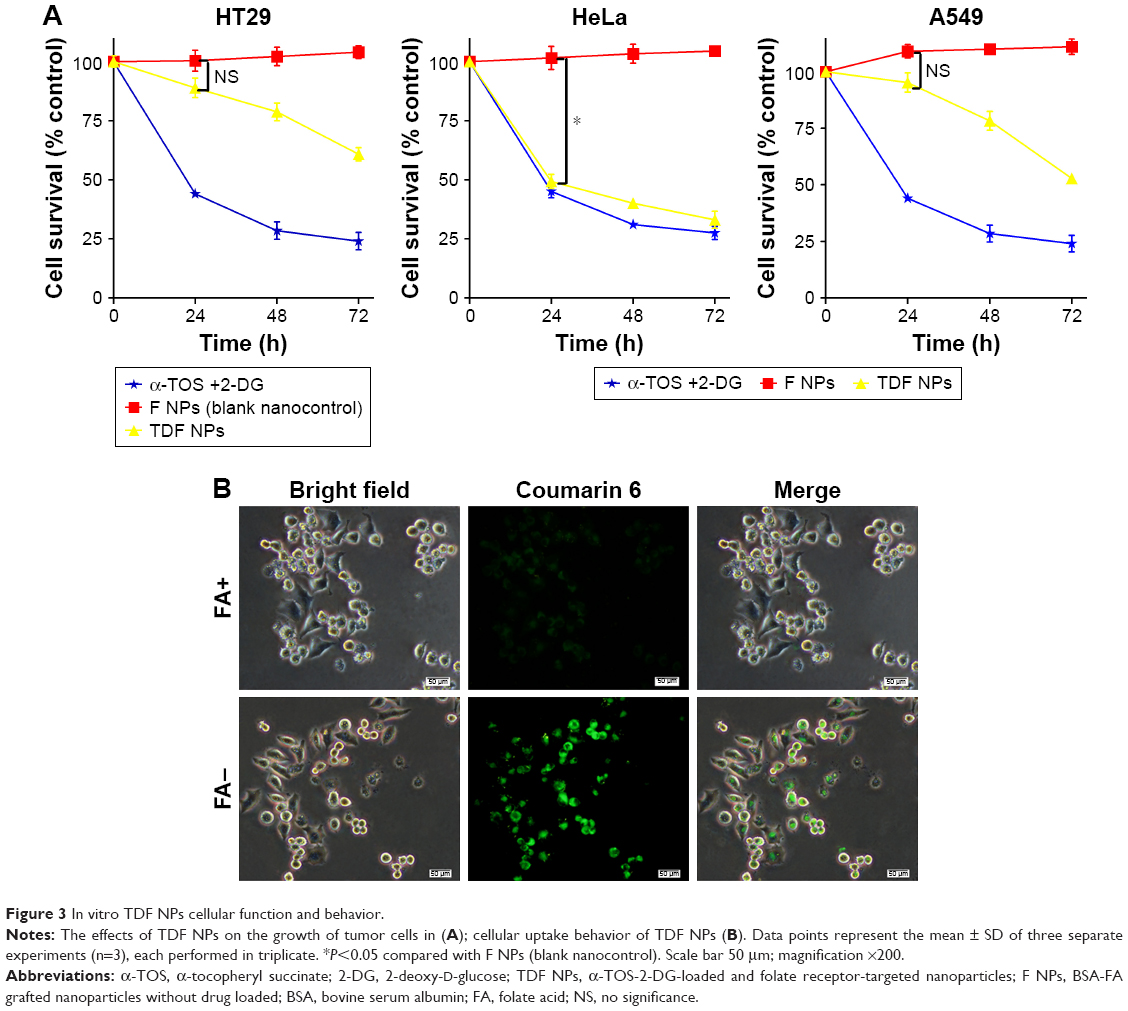 | Figure 3 In vitro TDF NPs cellular function and behavior. |
In vivo distribution of TDF NPs
Further, in vivo the distribution of TDF NPs was studied using near infrared (NIR) fluorescence imaging. In order to facilitate the observation of biodistribution of TDF NPs in vivo, DiR dye was encapsulated by CSaSt to generate DiR-loaded nanoparticles (D-NPs), which comprised CSaSt, BSA-FA and DiR. Then, D-NPs (test group) and free DiR (control group) were injected i.v. into HeLa tumor-bearing nude mice, respectively. The red fluorescence became gradually strong in the tumor-bearing right hindlimb from 0.5 h until 24 h after the injection of D-NPs and was extremely remarkable to 72 h, while no apparent fluorescence accumulated on the tumor of mouse until 24 h after injection with free-DiR (Figure 4), which suggested the tumor-specific distribution of TDF NPs. Besides, considerable fluorescence was only seen around the spleen of mouse test group, which may be caused by phagocytosis of immune cells such as macrophage. Notably, the fluorescence intensity was reduced faster in control group than in test group within 48 h, indicating a slower clearance of TDF NPs in the body. All the earlier results demonstrated that TDF NPs could specifically load drug to FR-positive tumors and prolong the retention in the body, thus improving the anti-tumor efficiency of loaded drugs and reducing the risk of systemic toxicity.
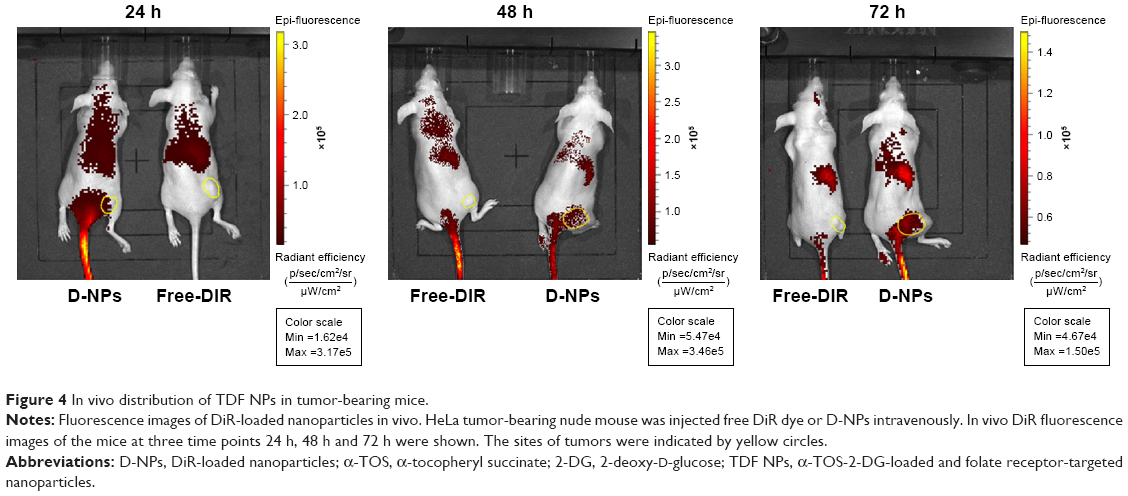 | Figure 4 In vivo distribution of TDF NPs in tumor-bearing mice. |
In vivo tumor suppression effect of TDF NPs
Finally, we studied the tumor suppression effect of TDF NPs in vivo. The weight loss, tumor volume and histologic section of tumor and liver could fully reflect the efficiency and toxicity of treatment to mouse. In Figure 5A, the body weight of TDF NP-treated group grew slowly, suggesting the efficiency of TDF NPs treatment, while that of PBS-treated group lost significantly in the late phase of treatment, indicating the occurrence of cancer cachexia for PBS-treated group. Meanwhile, TDF NP-treated group showed a lowest growth of tumor size compared to groups treated with PBS, F NPs and α-TOS plus 2-DG (Figure 5B). All the tumors were excised and scaled with a ruler (Figure 5C). For PBS group, four mice died before the end point for the cancer cachexia. As expected, the tumor volume of TDF NPs group was remarkably smaller than F NPs, suggesting that the combination treatment took a good anti-tumor effect. Meanwhile, the anti-tumor effect of TDF NPs was better than the non-nano group, indicating that the two drug-loaded NPs had an effective biological function in vivo. HE staining of pathological sections of tumor demonstrated notable necrosis and fewest tumor cells infiltration in TDF NPs group. In addition, the liver tissue of TDF NPs group showed less damage than that of free α-TOS plus 2-DG group (Figure 5D). All these data suggest an effective tumor suppression and less liver cytotoxicity of TDF NPs.
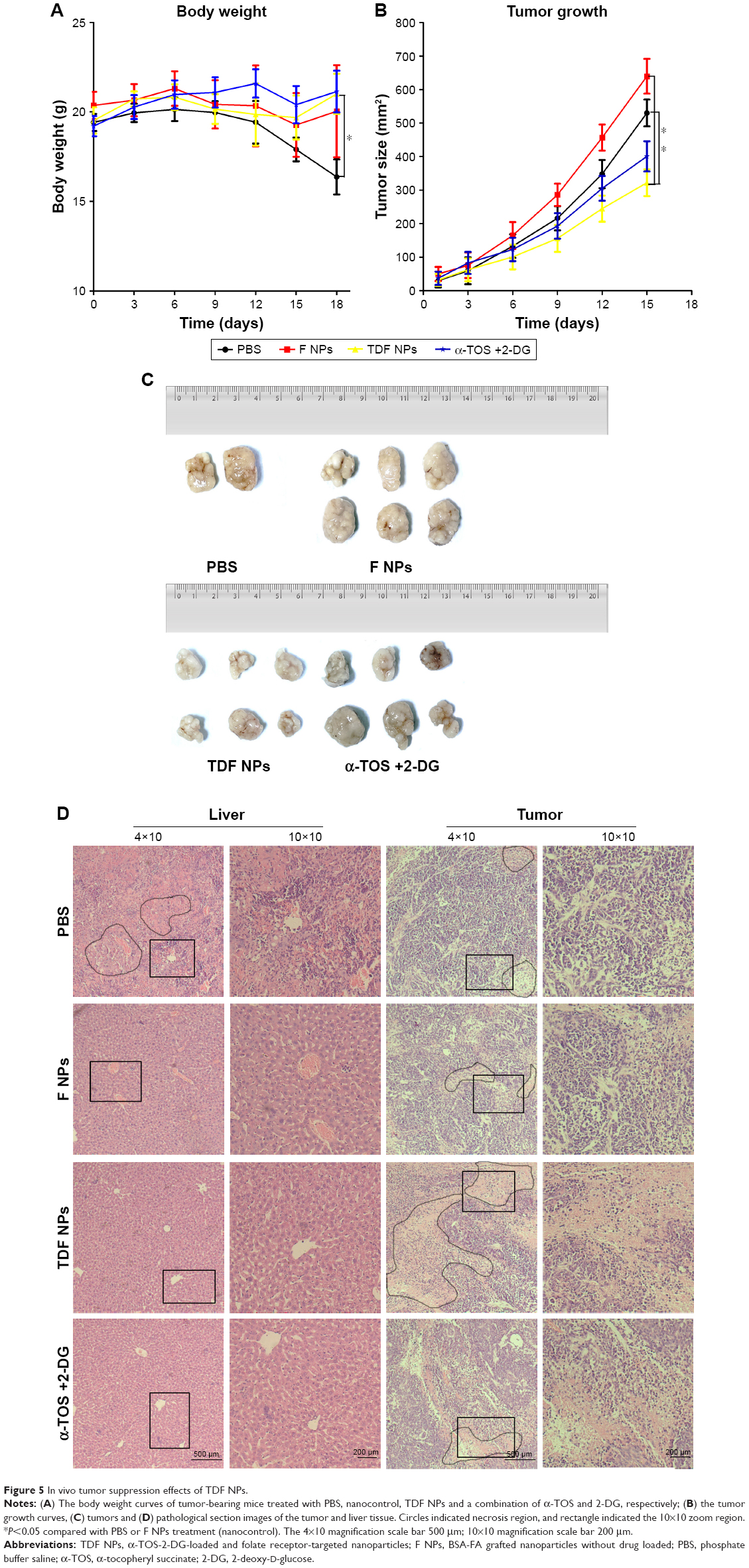 | Figure 5 In vivo tumor suppression effects of TDF NPs. |
Discussion
In this study, we synthesized a novel anti-tumor nano-medicine, TDF NPs, which was proved to be effective and less cytotoxic compared to free drug 2-DG or α-TOS, thus providing a promising cancer treatment. Several reports showed that metabolic profiles of cancer cells divert significantly from those of non-neoplastic cells; thus, tumors rely on these metabolic alterations for growth, metastasis and survival, the atypical pathways might be potential targets of antineoplastic drugs. The glucose metabolism in cancerous cells is remarkably different from that in their normal counterparts. Proliferating cancer cells prefer to obtain ATP via aerobic glycolysis rather than OXPHOS even in the presence of ample oxygen. We thus use 2-DG, non-metabolizable glucose analog, which targets glucose metabolism to deplete cancer cells of energy. Our results showed that 3 mM of 2-DG alone could inhibit the growth and proliferation of cancer cells in vitro. Previous work showed that the ratio of 3 mM of 2-DG to glucose in the culture media was 1:3.3 and the ratio proved to inhibit glycolysis. Our results were consistent with previous cell culture studies.28 However, the suppression effect of 2-DG by itself was limited since 6 mM of 2-DG did not significantly increase the inhibition ratio. Previous studies showed that 2-DG combined with other therapeutic agents or radiotherapy could exhibit a synergistic anticancer effect.
Since OXPHOS is another pathway to contribute to energy production in cancers, targeting mitochondrial metabolism has been proposed for cancer therapy. α-TOS, the vitamin E analogs, showed great promise for future clinical applications as a potential anticancer agent. However, the low solubility in physiological media limited the use of α-TOS. By designing an amphiphilic nanocarrier encapsulating α-TOS and 2-DG, we overcome poor bioavailability of α-TOS, thus dramatically improving the anti-tumor effect of 2-DG treatment alone. In our study, both α-TOS and 2-DG, when administered as single agents, were limited to inhibit the proliferation of HT29, HeLa and A549 tumor cells, and when administered in combination, their effect on such tumor growth was synergistic, thus resulting in significant cell death, as demonstrated in vitro by MTT and Annexin V/PI staining. The advantage of combined administration was proved by other such studies. Goldberg et al31 combined Ras inhibitor farnesylthiosalicylic acid (FTS) and 2-DG to treat pancreatic tumors, Cheong et al32 used 2-DG and metformin to treat breast tumor, Zhang et al33 combined α-TOS and doxorubicin to treat gastric cancer and so on.
Co-delivery nanocarrier means two or more therapeutic drugs are co-encapsulated into one nanocarrier. Many reports have proved that it is an efficient tool to simultaneous administration of multiple therapeutics, including cytotoxic agents, chemosensitizers, small interfering RNA (siRNA) and anti-angiogenic agents.26 In our previous work, we have designed and synthesized PgS and CSaSt micelles.26,34 For CSaSt micelles, we successfully encapsulated insoluble apogossypolone and soluble doxorubicin into one nanocarrier targeting hyaluronic acid receptor CD44. Further, the EE and DL were measured; the drug-loaded NP was characterized by IR spectra, TEM and drug release assay. As expected, the dual drug nanocarriers showed remarkable accumulation in tumor region and promoted the anti-tumor efficacy. Notably, in vivo acute toxicity experiment showed that LD50 of drug-loaded NP was remarkably lower compared with the combination group (apogossypolone and doxorubicin administrated simultaneously without encapsulation into one nanocarrier). All these work indicated that we have established a safe and effective nanocarrier system. For α-TOS and 2-DG, the main problem is to set a proper dose ratio of α-TOS and 2-DG for FR-positive HeLa cells. Dong et al35 tested for the sensitivity of several tumor cells to α-TOS and found IC50 value varied greatly for various tumor cells. We also found the difference of live cell percentage among HT29, HeLa and A549 cells when treated with α-TOS. Compared with HT29 cells, HeLa cells had a slightly lower IC50 value (Table S1). Based on the data of our experiment, a combination dose of 30 μM of α-TOS and 3 mM of 2-DG was determined, which was suitable for HeLa cells. For breast carcinomas and Ras overexpression-induced tumor, the in vivo dose of α-TOS varied from 6 μM to 15 μM.29,35 Thus, for different tumors, an optimized in vivo dose of TDF NPs should be explored.
For FR highly expressed HeLa cells, the TDF NPs showed a significant anti-tumor effect compared with the combination of the two free drugs (Figure 5C and D). Although the combination of the two free drugs did not show significant toxicity to mice (Figure 5A and D), it did not lead to a parallel anti-tumor effect to the TDF NPs. In turn, the combination of the two free drugs needs to increase dose to show the notable anti-tumor effect, which may cause significant toxicity in vivo. Thus, the TDF NPs showed a great potential in tumor therapy.
Conclusion
In this study, we targeted the tumor cell metabolism pathway to inhibit their growth and proliferation with the combination of two chemical agents α-TOS and 2-DG. Our results showed that in vitro α-TOS and 2-DG had a synergistic anti-tumor effect on tumor cells, especially on HeLa cells; TDF NPs encapsulating α-TOS and 2-DG also had a significant inhibition to the growth of FR highly expressed tumor cells. In vivo TDF NPs could target to FR-positive tumor and displayed a remarkable anti-tumor effect with less toxicity to liver.
Acknowledgments
This study was supported by the Innovation Project of Shaanxi Science and Technology (2015KTCL03-13) and National Natural Science Foundation of China (NSFC No 31571215). We acknowledge the technician Jintao Hu for flow cytometry analysis.
Disclosure
The authors report no conflicts of interest in this work.
References
Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029–1033. | ||
Kim JW, Dang CV. Cancer’s molecular sweet tooth and the Warburg effect. Cancer Res. 2006;66(18):8927–8930. | ||
Chen Z, Lu W, Garcia-Prieto C, Huang P. The Warburg effect and its cancer therapeutic implications. J Bioenerg Biomembr. 2007;39(3):267–274. | ||
Gatenby RA, Gillies RJ. Glycolysis in cancer: a potential target for therapy. Int J Biochem Cell Biol. 2007;39(7–8):1358–1366. | ||
Kroemer G, Pouyssegur J. Tumor cell metabolism: cancer’s Achilles’ heel. Cancer Cell. 2008;13(6):472–482. | ||
DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7(1):11–20. | ||
Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer. 2011;11(2):85–95. | ||
Lin X, Zhang F, Bradbury CM, et al. 2-Deoxy-D-glucose-induced cytotoxicity and radiosensitization in tumor cells is mediated via disruptions in thiol metabolism. Cancer Res. 2003;63(12):3413–3417. | ||
Pelicano H, Martin DS, Xu RH, Huang P. Glycolysis inhibition for anticancer treatment. Oncogene. 2006;25(34):4633–4646. | ||
Zhao Y, Butler EB, Tan M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis. 2013;4:e532. | ||
Zheng J. Energy metabolism of cancer: glycolysis versus oxidative phosphorylation. Oncol Lett. 2012;4(6):1151–1157. | ||
Rohlena J, Dong LF, Ralph SJ, Neuzil J. Anticancer drugs targeting the mitochondrial electron transport chain. Antioxid Redox Signal. 2011;15(12):2951–2974. | ||
dos Santos GA, Abreu e Lima RS, Pestana CR, et al. (+)α-Tocopheryl succinate inhibits the mitochondrial respiratory chain complex I and is as effective as arsenic trioxide or ATRA against acute promyelocytic leukemia in vivo. Leukemia. 2012;26(3):451–460. | ||
Kluckova K, Bezawork-Geleta A, Rohlena J, Dong L, Neuzil J. Mitochondrial complex II, a novel target for anti-cancer agents. Biochim Biophys Acta. 2013;1827(5):552–564. | ||
Prochazka L, Dong LF, Valis K, et al. Alpha-Tocopheryl succinate causes mitochondrial permeabilization by preferential formation of Bak channels. Apoptosis. 2010;15(7):782–794. | ||
Valis K, Prochazka L, Boura E, et al. Hippo/Mst1 stimulates transcription of the proapoptotic mediator NOXA in a FoxO1-dependent manner. Cancer Res. 2011;71(3):946–954. | ||
Kruspig B, Nilchian A, Bejarano I, Orrenius S, Zhivotovsky B, Gogvadze V. Targeting mitochondria by α-tocopheryl succinate kills neuroblastoma cells irrespective of MycN oncogene expression. Cell Mol Life Sci. 2012;69(12):2091–2099. | ||
Neuzil J, Massa H. Hepatic processing determines dual activity of alpha-tocopheryl succinate: a novel paradigm for a shift in biological activity due to pro-vitamin-to-vitamin conversion. Biochem Biophys Res Commun. 2005;327(4):1024–1027. | ||
Palao-Suay R, Rodrigáñez L, Aguilar MR, et al. Mitochondrially targeted nanoparticles based on α-TOS for the selective cancer treatment. Macromol Biosci. 2016;16(3):395–411. | ||
Koudelka S, Turanek Knotigova P, Masek J, et al. Liposomal delivery systems for anti-cancer analogues of vitamin E. J Control Release. 2015;207:59–69. | ||
Chen MC, Sonaje K, Chen KJ, Sung HW. A review of the prospects for polymeric nanoparticle platforms in oral insulin delivery. Biomaterials. 2011;32(36):9826–9838. | ||
Park S, Kang S, Chen X, et al. Tumor suppression via paclitaxel-loaded drug carriers that target inflammation marker upregulated in tumor vasculature and macrophages. Biomaterials. 2013;34(2):598–605. | ||
Zeng X, Tao W, Mei L, Huang L, Tan C, Feng SS. Cholic acid-functionalized nanoparticles of star-shaped PLGA-vitamin E TPGS copolymer for docetaxel delivery to cervical cancer. Biomaterials. 2013;34(25):6058–6067. | ||
Zhang H, Liu G, Zeng X, et al. Fabrication of genistein-loaded biodegradable TPGS-b-PCL nanoparticles for improved therapeutic effects in cervical cancer cells. Int J Nanomedicine. 2015;10:2461–2473. | ||
Low PS, Henne WA, Doorneweerd DD. Discovery and development of folic-acid-based receptor targeting for imaging and therapy of cancer and inflammatory diseases. Acc Chem Res. 2008;41(1):120–129. | ||
Li K, Liu H, Gao W, et al. Mulberry-like dual-drug complicated nanocarriers assembled with apogossypolone amphiphilic starch micelles and doxorubicin hyaluronic acid nanoparticles for tumor combination and targeted therapy. Biomaterials. 2015;39:131–144. | ||
Meng H, Chen JY, Mi L, et al. Conjugates of folic acids with BSA-coated quantum dots for cancer cell targeting and imaging by single-photon and two-photon excitation. J Biol Inorg Chem. 2011;16(1):117–123. | ||
Aft RL, Zhang FW, Gius D. Evaluation of 2-deoxy-D-glucose as a chemotherapeutic agent: mechanism of cell death. Br J Cancer. 2002;87(7):805–812. | ||
Dong LF, Jameson VJ, Tilly D, et al. Mitochondrial targeting of α-tocopheryl succinate enhances its pro-apoptotic efficacy: a new paradigm for effective cancer therapy. Free Radic Biol Med. 2011;50(11):1546–1555. | ||
Müller C, Schubiger PA, Schibli R. In vitro and in vivo targeting of different folate receptor-positive cancer cell lines with a novel 99mTc-radiofolate tracer. Eur J Nucl Med Mol Imaging. 2006;33(10):1162–1170. | ||
Goldberg L, Israeli R, Kloog Y. FTS and 2-DG induce pancreatic cancer cell death and tumor shrinkage in mice. Cell Death Dis. 2012;3:e284. | ||
Cheong JH, Park ES, Liang J, et al. Dual inhibition of tumor energy pathway by 2-deoxyglucose and metformin is effective against a broad spectrum of preclinical cancer models. Mol Cancer Ther. 2011;10(12):2350–2362. | ||
Zhang X, Peng X, Yu W, et al. Alpha-tocopheryl succinate enhances doxorubicin-induced apoptosis in human gastric cancer cells via promotion of doxorubicin influx and suppression of doxorubicin efflux. Cancer Lett. 2011;307(2):174–181. | ||
Liu H, Li K, Lan L, et al. Double-layered hyaluronic acid/stearic acid-modified polyethyleneimine nanoparticles encapsulating (-)-gossypol: a nanocarrier for chiral anticancer drugs. J Mater Chem B Mater Biol Med. 2014;2(32):5238–5248. | ||
Dong LF, Freeman R, Liu J, et al. Suppression of tumor growth in vivo by the mitocan alpha-tocopheryl succinate requires respiratory complex II. Clin Cancer Res. 2009;15(5):1593–1600. |
Supplementary materials
Synthesis and characterization of encapsulation materials and bovine serum albumin (BSA)-folate acid (FA)
Synthesis procedure of cationic amphiphilic starch was published in Biomaterials.1 Synthesis procedure of BSA-FA followed literature of Meng et al.2 Briefly, 80 mg of BSA, 40 mg of N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC), 35 mg of N-hydroxysuccinimide (NHS) and 50 mg of FA were added to dimethyl sulfoxide (DMSO), and the mixture was stirred for 12 h in the dark at 25°C. Then, the sample was centrifuged at 10,000 rpm for 30 min to remove the excess FA and EDC/NHS. In this reaction, EDC and NHS were used to form FA-NHS, which could conjugate with amino group of BSA. The product was assayed by 1H-nuclear magnetic resonance (1H-NMR).
The tumor cell lines and their folate receptor (FR) expression
RNA extraction, complementary DNA (cDNA) reverse transcription and real-time polymerase chain reaction (PCR) were performed according to our previous work.3
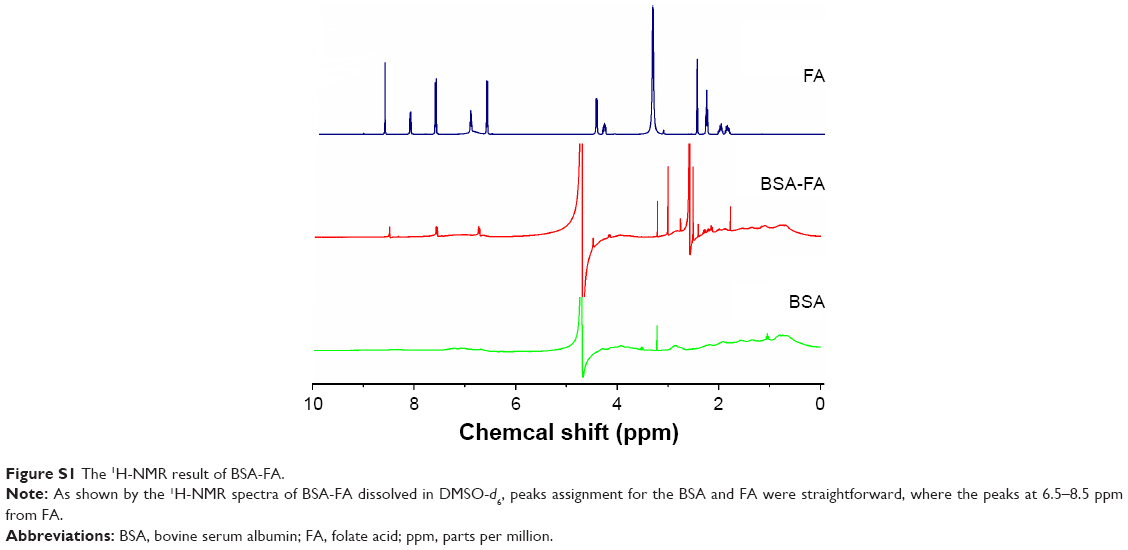 | Figure S1 The 1H-NMR result of BSA-FA. |
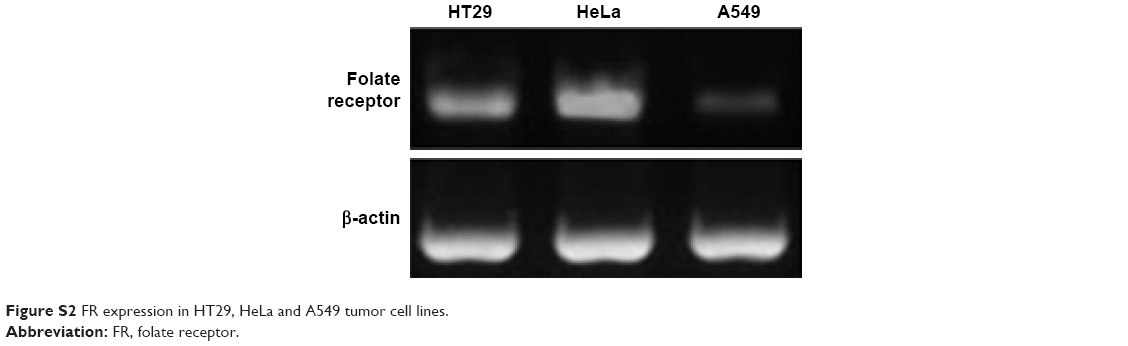 | Figure S2 FR expression in HT29, HeLa and A549 tumor cell lines. |
 | Table S1 LC50 values of α-TOS for apoptosis in tumor cell lines |
References
Li K, Liu H, Gao W, et al. Mulberry-like dual-drug complicated nanocarriers assembled with apogossypolone amphiphilic starch micelles and doxorubicin hyaluronic acid nanoparticles for tumor combination and targeted therapy. Biomaterials. 2015;39:131–144. | ||
Meng H, Chen JY, Mi L, et al. Conjugates of folic acids with BSA-coated quantum dots for cancer cell targeting and imaging by single-photon and two-photon excitation. J Biol Inorg Chem. 2011;16(1):117–123. | ||
Feng F, Jiang Y, Lu H, et al. Rab27A mediated by NF-κB promotes the stemness of colon cancer cells via up-regulation of cytokine secretion. Oncotarget. 2016;7(39):63342–63351. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.