Back to Journals » Therapeutics and Clinical Risk Management » Volume 13
Clinical patterns and outcomes of status epilepticus in patients with tuberous sclerosis complex
Authors Shehata HS ![]() , AbdelGhaffar HM, Nasreldin M, Elmazny A
, AbdelGhaffar HM, Nasreldin M, Elmazny A ![]() , Abdelalim A
, Abdelalim A ![]() , Sabbah A
, Sabbah A ![]() , Shalaby NM
, Shalaby NM
Received 1 April 2017
Accepted for publication 25 May 2017
Published 30 June 2017 Volume 2017:13 Pages 779—785
DOI https://doi.org/10.2147/TCRM.S138576
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Garry Walsh
Hatem S Shehata,1 Hadeer Mahmoud AbdelGhaffar,2 Mohammed Nasreldin,3 Alaa Elmazny,1 Ahmed Abdelalim,1 Asmaa Sabbah,1 Nevin M Shalaby1
1Department of Neurology, Cairo University, Giza, 2Department of Paediatrics, Fayoum University, Fayoum, 3Department of Psychiatrics, Cairo University, Giza, Egypt
Introduction: Refractory epilepsy is a common clinical manifestation in patients with tuberous sclerosis complex (TSC), which can be complicated by many life-threatening conditions, such as status epilepticus (SE). However, very few reports mention the patterns and semiology of SE in those patients.
Objective: To study the clinical characteristics and outcomes of SE in TSC patients.
Materials and methods: This observational, prospective study was carried out on 36 Egyptian children with definite TSC. Clinical history, general and neurological examination and psychometric evaluation by standard questionnaires were used to explore characteristics of epileptic manifestations and clinical patterns of SE. All included patients were required to have long-term video electroencephalograms (EEGs) and brain MRI performed.
Results: A total of 32 attacks of SE were recorded in 21 patients (58.3%) in our cohort during a follow-up period of 2.8±1.1 years; of those patients, 15 had convulsive status, 7 had non-convulsive SE, 6 had refractory/super-refractory SE and 14 patients had a history of infantile spasms (epileptic spasms). The duration of status ranged from 40 to 150 min (mean ± standard deviation: 90±15). Fourteen patients with SE had severe mental retardation, 9 had autistic spectrum disorder and 22 had severe epileptogenic EEG findings. Patients with SE had higher tuber numbers (mean: 9.6), 5 patients had subependymal giant cell astrocytomas and 2 patients had their SE after receiving everolimus.
Conclusions: The incidence of SE in our patient sample is high (>50%); severe mental retardation, autistic features, history of infantile spasm (epileptic spasms) and high tuber burden are risk factors for developing SE.
Keywords: Egyptian children, tuberous sclerosis, status epilepticus, infantile spasms, autistic spectrum
Introduction
Tuberous sclerosis complex (TSC) is an autosomal dominant genetic disorder with multiorgan involvement, primarily affecting the central nervous system, skin, kidney and heart.1 It has an estimated incidence of 1 in every 5,000 to 10,000 live births.2–4 The disease is caused by mutation in either TSC1 or TSC2 genes5 encoding for tuberin and hamartin proteins, respectively, which serve as functional protein complexes in the mammalian target of rapamycin complex 1 (mTOR-1). mTOR-1 controls cell growth and proliferation,6 accounting for the neuropathological findings of the disease, including the characteristic cortical tubers, subependymal nodules and subependymal giant cell atrocytomas (SEGAs).7 TSC is a neurocutaneous disorder that often causes disabling neurological disorders, including epilepsy, intellectual disability and autism. A significant association of having intractable seizures and disruptive behavior disorder, mood disorders, attention deficits and sleep alteration has been documented.8,9
Epilepsy is the most prevalent and challenging presentation, affecting more than 80%–90% of patients.10 All types of seizures have been reported, with infantile spasms (IS) (epileptic spasms) being the most common presentation (30%–60% of cases).11 Seizures are mostly refractory to pharmacological treatment and require polytherapy; this is believed to be a result of increased expression of multidrug resistance gene-1 P-glycoprotein (MDR1) and multidrug resistance–associated protein 1 (MRP1) in cortical tubers,12 which are believed to be directly related to epileptogenesis.10 Refractory epilepsy and status epilepticus (SE) are known to be associated with increased mortality13 and morbidity such as learning difficulties, behavioral problems and sleep alteration.14 Despite being a common life-threatening complication, there is insufficient literature discussing SE in TSC patients, and it is mostly confined to single case reports.15
The aim of this study is to assess the frequency, clinical patterns, risk factors and outcomes of SE in 36 Egyptian patients with TSC.
Methodology
Study design and participant characteristics
This was an observational, prospective, multicenter study that was conducted on 36 Egyptian children with TSC recruited between September 2009 and October 2015. Patients were recruited from Neuropediatrics Clinic of Cairo and Fayoum University Hospitals, and from Neurology Outpatient Department of Cairo University Hospitals. A written informed consent was obtained from the parents of all children included in this study. The study protocol was approved by the Neurology Department Review Boards at Cairo University.
The diagnosis of TSC was established clinically according to the National Institutes of Health consensus conference revised diagnostic criteria for TSC,16 which were based on the presence of brain MRI finding of cortical tubers and/or subependymal nodules, associated with dermatological findings of TSC (two major criteria). The basic eligibility requirement is definite diagnosis of tuberous sclerosis.
Clinical assessment
Medical evaluation
All patients underwent 1) general and neurological examinations, 2) a detailed developmental history and 3) analysis of seizures characteristics (onset and duration of epilepsy, seizures semiology and evolution analyzed by eyewitness parents/reliable relatives and seizure clusters). For patients who developed SE (n=21), a standard questionnaire was applied to evaluate for the antecedent events, type, lateralizing features, duration, days of hospital stay, complications and management plans. SE was defined as ≥5 min of 1) continuous clinical and/or electrographic seizure activity, or 2) recurrent seizure activity without recovery (returning to baseline) between seizures.17 Refractory status was defined as seizures lasting longer than 60 min despite treatment with a benzodiazepine and an adequate loading dose of a standard intravenous anticonvulsant drug.18 Super-refractory SE was defined as SE that continued for ≥24 h after the onset of anesthesia, including those cases in which the SE recurs on the reduction or withdrawal of anesthesia.19 Echocardiography and abdominal ultrasound were requested for all included children.
Psychological assessment and psychometric tests
Estimates of intellectual capabilities were made using the following tests, appropriate for age: 1) The mean developmental index of Bayley Scales of Infant Development–Second Edition20 for children aged ≤3 years, 2) Wechsler Preschool and Primary Scale of Intelligence–Revised21 for children aged >3 to 7 years and 3) Wechsler Intelligence Scale for Children-III22 for children aged >7 years. Mental retardation was diagnosed if the intelligence quotient was <70.
A clinical interview with parents was performed by a psychiatrist (MN) to screen for autism spectrum disorders (ASDs) or attention deficit hyperactivity disorders (ADHD). Children with a possible ASD were assessed using Autism Diagnostic Observation Schedule-Generic,23 whereas those with possible ADHD were subjected to Vanderbilt Assessment Scale – Parent Informant.24 This interview was repeated 1 month after each SE.
Clinical response and outcomes
Responders were defined as those who had clinical and/or electroencephalogram (EEG) seizure cessation within 20 min of infusion without the need for rescue medication. Breakthrough seizures were defined as recurrence of SE during the treatment, despite initial control, resulting in the need for a change of therapy (and did not include those cases in which seizures recurred and control was gained by increasing the dose). Intolerable side effects necessitate institution of alternative therapy.25 Outcomes were categorized into 1) short-term (during status and within 30 days of its occurrence) and 2) long-term (after 30 days) outcomes.
Interictal digital EEG records
EEGs were recorded in at least two settings with a minimum 3 months apart for all included children. At least one of them was a long-term video recording using a 32-channel Neurofax, EEG–9000 Version 05–71 (EEG-9200k, CE; NIHON KOHDEN, Tokyo, Japan), based on the international 10–20 system of electrode placement. All EEGs were performed either under standard conditions or after sedative premedications (chloral hydrate) in noncooperative children. Hyperventilation for 3 min was used whenever possible, and intermittent photic stimulation was used for all patients. The EEG records were interpreted based on visual analysis by two of the authors (HA and HS) in the same setting, according to definitions of the International Federation of Clinical Neurophysiology.26 EEG traces were classified into 1) normal record, 2) focal/multifocal epileptiform abnormalities, 3) generalized paroxysms, 4) dysrhythmia and 5) encephalopathic patterns that included typical or modified hypsarrhythmia or suppression burst.
Brain imaging
All patients were submitted to brain MRI on a 1.5T Philips Intera scanner (Philips Medical Systems, Best, The Netherlands). The image protocol included axial T1, T2 and fluid-attenuated inversion recovery (at 5 mm slices thickness); sagittal T1 weighted image (at 5 mm slices thickness) and coronal T2 weighted image (at 3 mm slices thickness). For contrast enhancement, intravenous administration of gadolinium (0.1 mmol/kg) was performed. Images were interpreted by two neuroradiologists regarding the presence of cortical tubers and subependymal nodules. Both were classified according to their number and size; cortical tubers were further classified according to their location (follow-up period: range, 1.4–5.1 years; mean ± SD, 2.8±1.1).
Data analysis
Data management was performed using the Statistical Package for Social Sciences (version 15.0; SPSS Inc., Chicago, IL, USA). Normal distribution of data was assessed using Kolmogorov–Smirnov test. Descriptive data were expressed by numbers and percentages. Computing the standard descriptive statistics (eg, mean, SD) was used to summarize the data. Nominal data were analyzed using simple Chi-square test, whereas independent sample t-test procedure was used to compare means for the two groups of cases. A probability value (P-value) <0.05 was considered significant.
Results
Clinical data
The present series included 36 Egyptian patients with definite TSC and epilepsy, with an age range from 2.5 to 10.9 years (mean ± SD: 6.5±2.1) and a male-to-female ratio of 1.12:1. All enrolled children had the characteristic dermatological findings as inclusionary criteria. Negative consanguinity with absence of family history was detected in 86.1% of cases (31/36). The follow-up duration of the cohort was 2.8±1.1 years. Clinical features of included patients are shown in Table 1.
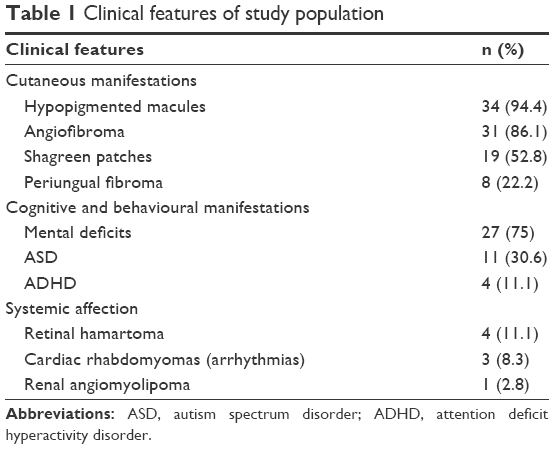 | Table 1 Clinical features of study population |
Seizure semiology and evolution
Seventeen patients of our series had a history of IS (47.2%); 6 of them (35.3%) developed definite or probable Lennox–Gastaut syndrome (LGS) and 9 patients (52.9%) developed other seizure types (generalized tonic clonic seizures [GTCs] [n=7], complex partial seizures [CPS]/(focal impaired awareness) [n=5], simple partial seizures [SPS] [n=2] and tonic seizures [n=1]). We had 2 patients with a history of IS (11.8%) who did not develop other seizure types. Twenty-five patients of the study population (69.4%) had more than 1 seizure type; 8 of them (22.2%) had definite or probable LGS. At least 1 seizure phenotype with clear focal onset (without or with secondary generalization) was documented in 27/36 patients (75%). Four patients (4/36) (11.1%) achieved seizure remission for at least 12 months at their last clinic visit. Seizure semiology of included patients is shown in Figure 1.
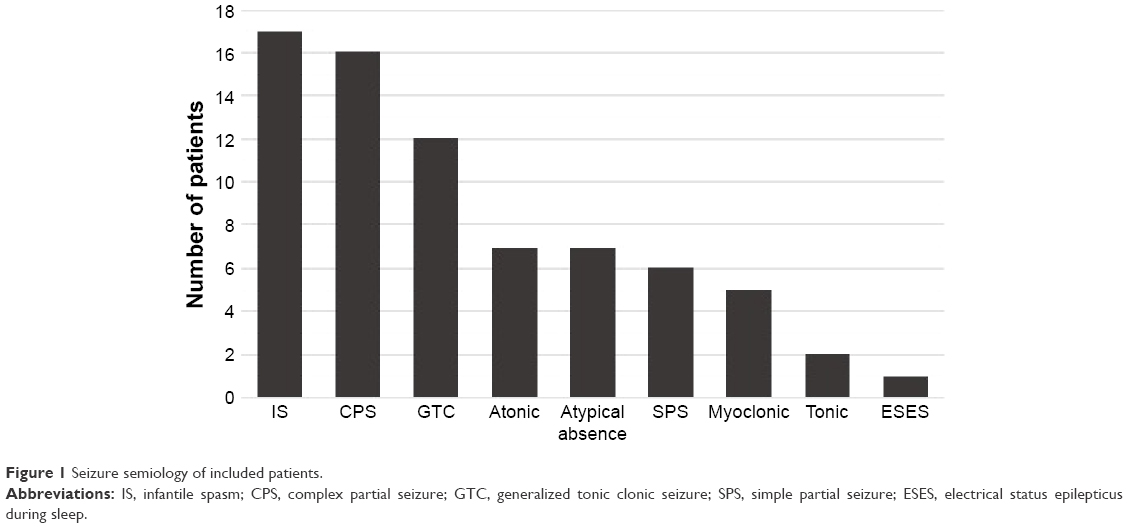 | Figure 1 Seizure semiology of included patients. |
Characteristics of SE episodes
A total of 32 episodes of SE were recorded in 21 patients (58.3%) of our cohort; 7/21 patients developed more than 1 episode of SE and 3/21 developed two different types of SE (convulsive status and status with CPS, non-convulsive SE). History of IS was detected in 14/21 patients, and all patients with definite or probable LGS (n=8) developed SE. Patients who achieved seizure remission for at least 12 months at last clinic visit (4/36) (11.1%) did not develop SE. The characteristics of reported SE are shown in Table 2.
 | Table 2 Characteristics of status epilepticus in study population |
Outcome of SE
Nineteen episodes of SE responded to first-line therapies, whereas 5 responded to second-line therapies and 2 responded on enteral Topamax through nasogastric tube. All patients received the maximum doses according to body weight.
Five patients presented with RSE, and 1 patient experienced super-refractory SE during the period of follow-up (range: 1.4–5.1 years; mean ± SD; 2.8±1.1). The short-term and long-term outcomes are shown in Table 3.
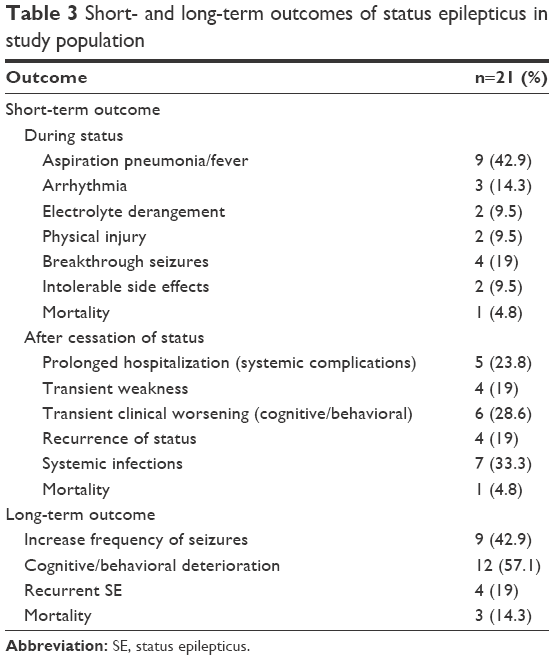 | Table 3 Short- and long-term outcomes of status epilepticus in study population |
Comparing patients with SE to those without SE
Clinical features, EEG findings and radiological data in patients with and without SE are shown in Table 4. In our cohort, 77 EEG records were analyzed; 45 were long-term EEGs (6–8 h) and 32 were routine interictal EEGs. None of the long-term EEGs were normal, whereas normal records were detected in 9 routine EEGs. MRI findings included tubers, nodules and SEGAs (Figures 2 and 3).
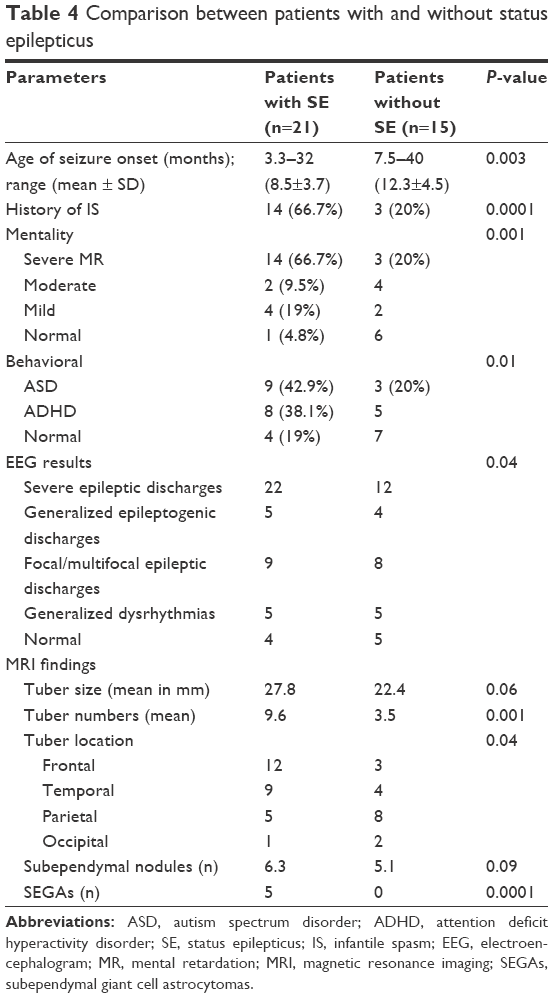 | Table 4 Comparison between patients with and without status epilepticus |
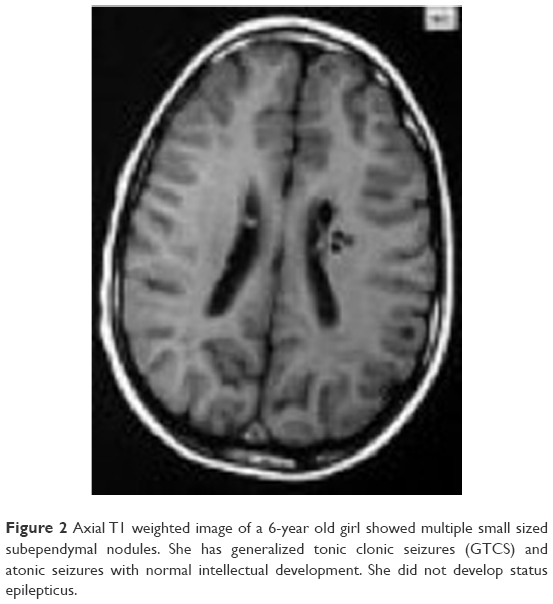 | Figure 2 Axial T1 weighted image of a 6-year old girl showed multiple small sized subependymal nodules. She has generalized tonic clonic seizures (GTCS) and atonic seizures with normal intellectual development. She did not develop status epilepticus. |
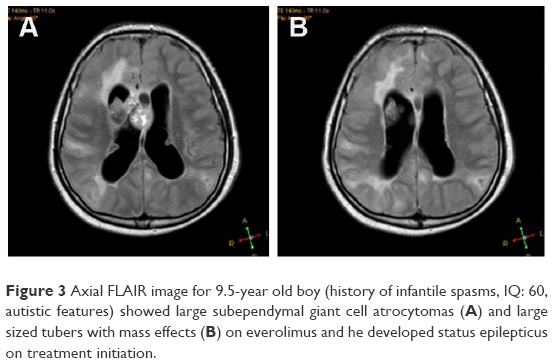 | Figure 3 Axial FLAIR image for 9.5-year old boy (history of infantile spasms, IQ: 60, autistic features) showed large subependymal giant cell atrocytomas (A) and large sized tubers with mass effects (B) on everolimus and he developed status epilepticus on treatment initiation. |
Discussion
This multicenter observational prospective study confirmed that SE is a common complication in patients with TSC (58.3%). It also provided data about well-characterized risk factors for developing different types of status in such cohort. To the best of our knowledge, this is the first study to address SE in TSC, which is a leading cause of genetic epilepsy that tends to be progressive with multiple semiologies and increased frequency.27 At least one-third of TSC patients have intractable seizures refractory to both medical and surgical treatment.28 These patients are at particular risk of developing SE, which is a chief cause of death among tuberous sclerosis patients.29
Early seizure onset is often associated with high risk of developing intractable epilepsy among TSC patients; this observation was documented by Cusmai et al,30 and this was also the case in the present study because patients who developed SE had earlier age of seizure onset.
Seizure intractability is an important factor in the developmental trajectory of children with TSC because it is closely intertwined with both cognitive and behavioral disorders; 57.1% (12/21) of patients in the present study experienced deterioration of their cognitive and behavioral symptoms after experiencing SE. Moreover, Samir et al31 demonstrated an association between intellectual disability and poor seizure outcome, which is in accordance with our results; 66.7% of the patients who experienced SE suffered from severe mental retardation. Furthermore, according to an earlier study, Asano et al32 documented that refractory epilepsy was significantly correlated with ASD. This finding was also emphasized by van Eeghen et al,33 and it agreed with the results of our study.
Owing to the fact that cortical tubers and the perituberal cortex are considered substrates for seizures, multiple epileptic foci coexist with the presence of multiple cortical tubers;10 in the present study, patients who experienced SE had significantly more cortical tubers compared with patients who did not. These results were previously emphasized by Samir et al,31 who documented that greater number of tubers was associated with poor seizure outcome. Moreover, tubers in areas of the brain that become functionally mature earlier can become epileptogenic before tubers located in brain regions that mature more slowly,34 which may explain why, in the present study, a statistically significant difference was found when comparing the location of tubers between patients who developed SE with patients who did not.
In contrast, SEGAs are not considered epileptogenic, but, as documented by Holmes and Stafstrom,10 it might heighten the excitability of the adjacent tissues, which might also explain the significant relation between the number of SEGAs and the development of SE in the present study.
In the present study, abnormal EEGs with severe epileptic discharges were significantly correlated with occurrence of SE. Husain et al35 also previously demonstrated that an abnormal EEG is an important predictor of eventual seizure control. Therefore, all TSC patients should undergo a baseline EEG evaluation. If it is abnormal, or in the case of patients exhibiting TSC-associated neuropsychiatric disorders, a 24 h video EEG should be considered to assess for unrecognized or subclinical seizure activity because early recognition and management of seizures is important in preventing subsequent cognitive and neuropsychiatric consequences.36
Several recent studies have documented promising results regarding seizure control in TSC after starting mTOR inhibitors;37–39 however, in the present study, two patients experienced seizure aggravation and SE after starting everolimus treatment. This finding was previously reported by Wiemer-Kruel et al40 in a 14-year-old patient after doubling her everolimus dose.
Limitations and strengths of the study
The current study points out a significant association between certain risk factors and the occurrence of SE during a mean follow-up period of 2.8 years, which can be considered a predominant strength of our study. There are, however, a number of limitations that included absence of genetic tests of included patients and the small sample size, which leads to restriction in using some statistical analysis.
In conclusion, early seizure onset, poor intellectual abilities and behavioral disorders and history of IS (epileptic spasms) are significant clinical determinants for SE, whereas increased tuber burden and presence of SEGAs are radiological predisposing factors for developing SE in patients with TSC. Thus, close follow-up with early recognition and control of seizures is recommended.
Acknowledgment
The abstract of this paper was presented in a poster session at The 5th London-Innsbruck Colloquium on Status Epilepticus and Acute Seizures, held in London, UK, on April 9–11, 2015.
Disclosure
The authors report no conflicts of interest in this work.
References
Northrup H, Krueger DA. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol. 2013;49(4):243–254. | ||
Osborne JP, Fryer A, Webb D. Epidemiology of tuberous sclerosis. Ann N Y Acad Sci. 1991;615:125–127. | ||
O’Callaghan FJ. Tuberous sclerosis. BMJ (Clin Res ed). 1999;318(7190):1019–1020. | ||
Curatolo P, Bombardieri R, Jozwiak S. Tuberous sclerosis. Lancet. 2008;372(9639):657–668. | ||
Rosset C, Netto CB, Ashton-Prolla P. TSC1 and TSC2 gene mutations and their implications for treatment in Tuberous Sclerosis Complex: a review. Genet Mol Biol. 2017;40(1):69–79. | ||
Child ND, Benarroch EE. mTOR: its role in the nervous system and involvement in neurologic disease. Neurology. 2014;83(17):1562–1572. | ||
Mizuguchi M, Takashima S. Neuropathology of tuberous sclerosis. Brain Devel. 2001;23(7):508–515. | ||
Parisi L, Filippo T, La Grutta S, et al. Sturge-weber syndrome: a report of 14 cases. Ment Illn. 2013;5(1):e7. | ||
Staley BA, Vail EA, Thiele EA. Tuberous sclerosis complex: diagnostic challenges, presenting symptoms, and commonly missed signs. Pediatrics. 2011;127(1):e117–e125. | ||
Holmes GL, Stafstrom CE. Tuberous sclerosis complex and epilepsy: recent developments and future challenges. Epilepsia. 2007;48(4):617–630. | ||
Curatolo P, Seri S, Verdecchia M, Bombardieri R. Infantile spasms in tuberous sclerosis complex. Brain Devel. 2001;23(7):502–507. | ||
Lazarowski A, Lubieniecki F, Camarero S, et al. Multidrug resistance proteins in tuberous sclerosis and refractory epilepsy. Pediatr Neurol. 2004;30(2):102–106. | ||
Boggs JG. Mortality associated with status epilepticus. Epilepsy Curr. 2004;4(1):25–27. | ||
Carotenuto M, Parisi P, Esposito M, Cortese S, Elia M. Sleep alterations in children with refractory epileptic encephalopathies: a polysomnographic study. Epilepsy Behav. 2014;35:50–53. | ||
Diaz-Corpas T, Mateu-Puchades A, Rojo-Espana R, Marquina-Vila A. Poliosis and status epilepticus as the presentation of tuberous sclerosis in an infant. Actas Dermosifiliogr. 2010;101(9):812–814. | ||
Roach ES, Gomez MR, Northrup H. Tuberous sclerosis complex consensus conference: revised clinical diagnostic criteria. J Child Neurol. 1998;13(12):624–628. | ||
Lowenstein DH, Bleck T, Macdonald RL. It’s time to revise the definition of status epilepticus. Epilepsia. 1999;40(1):120–122. | ||
Tripathi M, Vibha D, Choudhary N, et al. Management of refractory status epilepticus at a tertiary care center in a developing country. Seizure. 2010;19(2):109–111. | ||
Shorvon S, Ferlisi M. The outcome of therapies in refractory and super-refractory convulsive status epilepticus and recommendations for therapy. Brain. 2012;135(Pt 8):2314–2328. | ||
Bayley N. Bayley Scales of Infant Development. 2nd ed. San Antonio, TX: The Psychological Corporation; 1993. | ||
Wechsler D. Manual for the Wechsler Preschool and Primary Scale of Intelligence-Revised. New York, NY: Psychological Corporation; 1989. | ||
Wechsler D. Wechsler Intelligence Scale for Children (III). San Antonio, TX: Psychological Corporation; 1991. | ||
Lord C, Risi S, Lambrecht L, et al. The autism diagnostic observation schedule-generic. J Autism Dev Disord. 2000;30(3):205–223. | ||
Wolraich ML, Lambert W, Doffing MA, Bickman L, Simmons T, Worley K. Psychometric properties of the Vanderbilt ADHD diagnostic parent rating scale in a referred population. J Pediatr Psychol. 2003;28(8):559–567. | ||
Agarwal P, Kumar N, Chandra R, Gupta G, Antony AR, Garg N. Randomized study of intravenous valproate and phenytoin in status epilepticus. Seizure. 2007;16(6):527–532. | ||
Noachtar S, Binnie C, Ebersole J, Mauguiere F, Sakamoto A, Westmoreland B. A glossary of terms most commonly used by clinical electroencephalographers and proposals for the report for the EEG findings. In: Deuschl G, Eisen A, editors. Recommendation for the practice of Clinical Neurophysiology: Guidelines of the International Federation of Clinical Neurophysiology (EEG Suppl. 52). 1st ed. Amsterdam, the Netherlands: Elsevier Science B.V. Section 1. (chapters 1–5), 1999:21–44. | ||
Curatolo P, Jozwiak S, Nabbout R. Management of epilepsy associated with tuberous sclerosis complex (TSC): clinical recommendations. Eur J Paediatr Neurol. 2012;16(6):582–586. | ||
Krueger DA, Franz DN. Current management of tuberous sclerosis complex. Paediatr Drugs. 2008;10(5):299–313. | ||
Moavero R, Cerminara C, Curatolo P. Epilepsy secondary to tuberous sclerosis: lessons learned and current challenges. Childs Nerv Syst. 2010;26(11):1495–1504. | ||
Cusmai R, Moavero R, Bombardieri R, Vigevano F, Curatolo P. Long-term neurological outcome in children with early-onset epilepsy associated with tuberous sclerosis. Epilepsy Behav. 2011;22(4):735–739. | ||
Samir H, Ghaffar HA, Nasr M. Seizures and intellectual outcome: clinico-radiological study of 30 Egyptian cases of tuberous sclerosis complex. Eur J Paediatr Neurol. 2011;15(2):131–137. | ||
Asano E, Chugani DC, Muzik O, et al. Autism in tuberous sclerosis complex is related to both cortical and subcortical dysfunction. Neurology. 2001;57(7):1269–1277. | ||
van Eeghen AM, Pulsifer MB, Merker VL, et al. Understanding relationships between autism, intelligence, and epilepsy: a cross-disorder approach. Dev Med Child Neurol. 2013;55(2):146–153. | ||
Cusmai R, Chiron C, Curatolo P, Dulac O, Tran-Dinh S. Topographic comparative study of magnetic resonance imaging and electroencephalography in 34 children with tuberous sclerosis. Epilepsia. 1990;31(6):747–755. | ||
Husain AM, Foley CM, Legido A, Chandler DA, Miles DK, Grover WD. Tuberous sclerosis complex and epilepsy: prognostic significance of electroencephalography and magnetic resonance imaging. J Child Neurol. 2000;15(2):81–83. | ||
Bombardieri R, Pinci M, Moavero R, Cerminara C, Curatolo P. Early control of seizures improves long-term outcome in children with tuberous sclerosis complex. Eur J Paediatr Neurol. 2010;14(2):146–149. | ||
Krueger DA, Wilfong AA, Holland-Bouley K, et al. Everolimus treatment of refractory epilepsy in tuberous sclerosis complex. Ann Neurol. 2013;74(5):679–687. | ||
Miller JW. Treating epilepsy in tuberous sclerosis with everolimus: getting closer. Epilepsy Curr. 2014;14(3):143–144. | ||
Curatolo P. Mechanistic target of rapamycin (mTOR) in tuberous sclerosis complex-associated epilepsy. Pediatr Neurol. 2015;52(3):281–289. | ||
Wiemer-Kruel A, Woerle H, Strobl K, Bast T. Everolimus for the treatment of subependymal giant cell astrocytoma probably causing seizure aggravation in a child with tuberous sclerosis complex: a case report. Neuropediatrics. 2014;45(2):129–131. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.