Back to Archived Journals » Research and Reports in Biochemistry » Volume 5
Cell–matrix interactions governing skin repair: matricellular proteins as diverse modulators of cell function
Authors Walker J, Kim S, Michelsons S, Creber K, Elliott C, Leask A, Hamilton D
Received 18 December 2014
Accepted for publication 7 January 2015
Published 5 March 2015 Volume 2015:5 Pages 73—88
DOI https://doi.org/10.2147/RRBC.S57407
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Nikolay Dokholyan
John T Walker,1,* Shawna S Kim,1,* Sarah Michelsons,1,* Kendal Creber,2,* Christopher G Elliott,1,* Andrew Leask,3 Douglas W Hamilton1–3
1Department of Anatomy and Cell Biology, 2Graduate Program of Biomedical Engineering, 3Division of Oral Biology, Schulich School of Medicine and Dentistry, The University of Western Ontario, London, ON, Canada
*These authors contributed equally to this paper
Abstract: With the classification first proposed by the Bornstein group in 1995, matricellular proteins represent a diverse and expanding class of molecules that contribute to cell phenotype and regulate interactions with the extracellular matrix. Based on initial analysis, matricellular protein expression was thought to be limited to development, but in the intervening 20 years it has become apparent that it plays a pivotal role during healing in several different tissue types. Furthermore, while considered to modulate cell behavior, it is now apparent that matricellular proteins also function in the organization and crosslinking of the extracellular matrix during healing. The focus of this review is to discuss matricellular proteins in the context of skin healing, which in healthy individuals occurs through four overlapping temporal phases. We will also discuss matricellular proteins as potential therapeutics for the treatment of impaired skin healing.
Keywords: matricellular proteins, skin healing, inflammation, cell adhesion, microenvironment
Introduction
When skin is injured, a highly complex and coordinated set of processes is initiated to restore tissue structure and barrier function1 (for a review of skin healing, see Reinke and Sorg2). Involving multiple and diverse cell populations, four temporal phases occur, encompassing hemostasis (blood clotting), inflammation, proliferation, and remodeling, which result in re-establishment of skin integrity3 (Figure 1). As will be highlighted, concomitant with the inflammatory, proliferative, and remodeling phases of healing, keratinocytes proliferate and migrate across the wound bed to restore the epithelium and barrier function. In this review, we will consider re-epithelialization as a separate process concurrent with the four phases of healing shown in Figure 1. In its simplest form, skin healing can be considered an initial host immune response that triggers mesenchymal cell infiltration, re-epithelialization, and blood vessel ingrowth and is followed finally by temporal remodeling of the newly formed tissue.4–6 In most healthy individuals, this pattern of skin healing is evident, but nonhealing or “chronic” dermal wounds are a growing and significant clinical complication associated with diabetes/ischemia7,8 and immobility.9 Nonhealing skin wounds are a major burden for health care systems around the world, and the cost of treatment within North America is currently approaching $6 billion per annum.10 It is estimated that up to 20% of patients in long-term care facilities in Canada have chronic skin wounds,11,12 and the lifetime incidence of foot ulcers in diabetic patients is between 15% and 25%.13 The dysfunction leading to nonhealing wounds is extremely complex and multifactorial,14–19 but, fundamentally, the cells do not receive the necessary molecular signals to be able to progress to the proliferative and remodeling phases of wound healing.20 As a result of low clinical efficacy of current treatments, new therapeutics are needed, and this has driven intensive research to increase our understanding of the molecular signals that regulate skin healing.21
 | Figure 1 Schematic representation of the phases and timeline of the skin healing process in mice. Four overlapping phases shown schematically result in wound closure. The cellular processes at each stage are described in detail in “Normal skin healing process in mice”. |
With the discovery of growth factors, our understanding of wound healing increased significantly, particularly with respect to the regulation of inflammation22 and the proliferative phases of healing.23 However, clinical studies with local delivery of growth factors such as transforming growth factor beta (TGF-β) were not as effective at stimulating healing of stalled or chronic wounds as would have been anticipated,24 highlighting that growth factors alone may not be sufficient to regulate skin healing. First described in 1995 by the Bornstein group25 at the University of Washington, matricellular proteins (MPs) specifically modulate cell–matrix interactions and cell function (adhesion, spreading, migration, proliferation, and differentiation)26 by interacting with cell surface receptors (eg, integrins) and other bioeffector molecules, as well as with structural matrix proteins such as collagens, fibronectin, and other MPs.27 MPs are expressed and act in a temporal and spatial manner to control different aspects of skin repair.3,28–33 While fibrin, collagen, and fibronectin provide structural support to the cells and matrix during healing, MPs modulate the adhesion, migration, proliferation, and differentiation of inflammatory cells, perivascular cells, dermal fibroblasts, and keratinocytes.3,29 Initial description of MP regulation and function was based on analysis of in vivo expression patterns in mice and rats predominantly, with in vitro assays used to assess potential functions.34–37 While providing important information, this type of approach did not allow direct assessment of protein function in vivo.
The advent of genetic knockout (KO) mice was a significant step to allow analysis of MP function in physiologically relevant models. Of great significance, although somewhat confounding, was that as each MP genetic KO was derived, only two exhibited severe phenotypes; CCN1 deletion38 is embryonic lethal, and CCN2 deletion results in perinatal lethality.39 Indeed, many of the MP KO mice exhibited no obvious developmental phenotype. With the advent of these murine genetic KO models, it did, however, allow rigorous analysis of the roles of MPs in development, pathological insults, and, as is the focus of this review, skin healing.28,30,40–44
Normal skin healing process in mice
As skin healing in mice follows a very predictable and temporal pattern, deletion of individual MPs can alter the kinetics of skin excisional wound closure. As such, it is first important to define the normal temporal process of healing in mice (shown schematically in Figure 1). Experimentally, it is possible to assess skin healing using incisional or excisional wounds. In incisional injuries, re-epithelialization is the process responsible for healing, but in excisional wounds where the tissue is removed to the fascia planes, the healing process is much more involved.45 For the purpose of this review, we will describe the healing process following full-thickness excisional wounding.
Upon initial injury, hemostasis occurs within a few minutes, and after cessation of bleeding with platelets forming a hemostatic plug, the initial inflammatory phase of repair is triggered. As a result of the coagulation process, a provisional matrix of fibrin and fibronectin forms, which will mature into granulation tissue.46 During the initial phase of inflammation (>48 hours postinjury), neutrophils, monocytes, and macrophages enter the wound bed, facilitating debridement of damaged extracellular matrix (ECM).47 The role of macrophages is particularly pivotal in the healing response; deletion of macrophages significantly impedes the skin healing process.47,48 Macrophage phenotype during normal skin healing is highly variable49,50 and, as was eloquently highlighted by Charles Mills,50 macrophages are at the crossroads of immunity and stimulating tissue healing through a “fight (M1) or fix (M2)” polarization. Macrophages entering the wound bed at the early hours of inflammation have a classical proinflammatory cytokine signature also known as M1 polarization, secreting iNOS, high levels of interleukin (IL)-12, and low levels of IL-10.51 However, as the inflammatory phase progresses, macrophage phenotype switches to an alternatively activated state known as M2, characterized by secretion of TGF-β152 and platelet-derived growth factor (PDGF).22 Secretion of these cytokines is in part responsible for initiating the proliferative phase of repair and the initial migration of fibroblast and perivascular progenitor cells (also known as pericytes) into the developing granulation tissue matrix.3 PDGF destabilizes perivascular progenitor cells, which are associated with blood vessels, allowing them to migrate into the wound bed,53 where they are pivotal in new blood vessel formation, wound contraction, and ECM synthesis. As fibroblasts and pericytes migrate into the wound bed on this newly developed ECM, they undergo a phenotypic change triggered by TGF-β1, becoming α-smooth muscle actin (α-SMA)-expressing myofibroblasts.54 Myofibroblasts are contractile and highly migratory, facilitating contraction of the wound edge.54,55 Moreover, myofibroblasts secrete new ECM during the remodeling phase, primarily fibronectin and type I collagen,54 which, in combination with re-epithelialization, results in wound resolution.
As will be evident from this review, the first information on expression patterns of MPs during skin healing arose from temporal analysis of healing skin in wild type mice, as well as from studies in rats, pigs and humans. While the expression patterns implied potential involvement of certain MPs in skin healing, as will be illustrated, genetic deletion did not necessarily confirm these findings. Based on the temporal nature of skin healing, using genetic deletion of each MP allows assessment of changes in kinetics of closure, which points to which particular phase of the repair process may be affected (Figure 1). We will discuss the role of each MP within the context of each stage of healing, with certain MPs potentially showing different effects depending on cell types present in the wound. These findings are summarized in Table 1.
 | Table 1 Influence of genetic deletion of matricellular proteins on skin healing in mice |
Hemostasis and the inflammatory phase of healing
During the hemostatic phase, after cessation of bleeding with platelets forming a hemostatic plug, neutrophils, monocytes, and macrophages enter the wound bed, facilitating debridement of damaged ECM47 and triggering the inflammatory phase of repair. Only one MP, thrombospondin-2 (TSP-2), has been linked to changes in hemostasis;41,56 TSP-2 platelets show a defect in aggregation, although this appears to be an indirect effect.41 Instead, megakaryocytes, which give rise to platelets, contain high levels of TSP-2 in the milieu of the bone marrow, and deletion of TSP-2 results in defects in megakaryocyte structure and function. Platelets do not themselves express TSP-2, and although the KO animals have an increased susceptibility to bleeding and fewer platelets, it does not manifest in altered blood coagulation.
As a result of blood coagulation, a provisional matrix of fibrin and fibronectin is formed that will subsequently mature into granulation tissue and act as a scaffold for cell infiltration.46 With respect to MPs, in inflammatory events (up to 5 days postwounding), galectin-3,57 osteopontin,58–60 hevin,61 and TSP-162 are expressed in this phase of inflammation and, in particular, regulate neutrophil and monocyte recruitment as well as macrophage polarization and function.
Galectin-3 is initially expressed highly in the developing granulation tissue and at the wound edge during the inflammatory phase,36 with messenger ribonucleic acid (mRNA) levels peaking at day 1 postwounding (Hamilton lab, unpublished data, 2015). Although the KO mice have been created, a full assessment of genetic deletion of galectin-3 on inflammatory processes in excisional wounds has yet to be performed. However, galectin-3 has been implicated in having a number of roles that would be expected to support or regulate the innate immune response, which plays a critical role during initial wound healing.57,63,64 In skin healing in rat models, the number of galectin-3-positive cells peaks at day 1, but these cells persist through to day 20.65 The majority of these cells represented ED1+ (CD68) macrophages, which are associated with high lysosomal activity, wound debridement, and a classic proinflammatory phenotype. In the initial “fight” stage of inflammation, some studies suggest that galectin-3 may also enhance neutrophil infiltration in response to the presence of bacteria in vivo, although this was dependent on the specific pathogen used,66 and this hypothesis was not tested specifically in a skin wound healing model. Galectin-3 does, however, stimulate neutrophil migration, although it does not act specifically as a chemoattractant for neutrophils in vitro.66 Thus, at initial stages of inflammation, galectin-3 is linked to infiltration of neutrophils and macrophages and regulation of the essential proinflammatory response. However, even though its expression peaks at day 1, galectin-3 expression does persist, with additional influence evident in later inflammatory processes. Once the acute phase of inflammation has peaked, neutrophils undergo apoptosis and are phagocytosed to prevent tissue damage from intracellular matrix-cleaving enzymes. The binding of galectin-3 to apoptotic neutrophils greatly enhances macrophage engulfment and clearance of these neutrophils from the wound bed.50 This clearance is significant, as neutrophils are also a potent source of tumor necrosis factor alpha (TNF-α), a cytokine that prolongs inflammation, inhibits the proliferative phase of healing,67,68 and is abundant in a nonhealing skin wound and wound fluid.69
During late inflammation, macrophage phenotype changes from M1 to the “fix” M2 polarization, and galectin-3 is also implicated in this process. Although bone marrow-derived macrophages from galectin-3 KO and WT mice show similar release of the proinflammatory cytokines TNF-α and IL-6 in response to stimulation with lipopolysaccharide or interferon-γ,51 macrophages isolated from galectin-3 KO mice exhibit reduced IL-4/IL-13-induced alternative macrophage activation in vitro compared with those from WT mice.64 Following treatment with IL-4, the KO macrophages showed significantly lower mRNA levels of the mouse M2 macrophage markers, mannose receptor, arginase I, FIZZ-1, and Ym-1. Confirmation of this has, however, not been established in vivo during skin healing and remains a significant point yet to be addressed. However, galectin-3 still stands as a potentially important matricellular regulator of neutrophil and macrophage behavior in skin healing.
Osteopontin has been linked to macrophage infiltration in injury models,70–72 and addition of osteopontin-neutralizing antibodies reduces macrophage infiltration by up to 60% in response to an intradermal injection of N-formyl-met-leu-phe (macrophage chemotactic peptide).58 With respect to skin healing, osteopontin is upregulated 6 hours postincisional wounding in WT mice, and localizes primarily to the wound margin, as assessed through in situ hybridization.73 This corresponds to areas containing high levels of leukocytes and neutrophils and, to a lesser extent, macrophages. Interestingly, analysis of osteopontin KO mice demonstrated no significant difference in the number of macrophages infiltrating the wounds; instead, it appears that the macrophages show a reduced ability to debride the wound bed, one of their primary functions at this stage of healing,73 and the macrophages from KO animals expressed lower levels of the mannose receptor. This receptor has been shown to be involved in collagen degradation74 but is more typically associated with M2 alternatively activated macrophages rather than the typical proinflammatory phagocytosis phenotype.75 It is becoming clear that macrophage polarization results in a heterogeneous population, such that delineating M1 and M2 cells in a wound is complex based on markers identified thus far.75 Knockdown of osteopontin using antisense oligodeoxynucleotides in excisional wound healing results in faster healing,60 which is concomitant with a reduction in macrophages and neutrophils at 3 and 7 days postwounding. Expression of osteopontin in inflammatory cells has also been demonstrated in horses during skin healing,59 but functional significance was not determined. In summary, osteopontin appears to regulate macrophage function.
TSP-1 is a known chemoattractant for macrophages, and its expression peaks at day 3 postwounding.62 Deletion of TSP-1 significantly reduces the number of macrophages present in the wound bed at day 7 postwounding, suggesting a significant impairment in monocyte or macrophage recruitment.76 In support of this, TSP-1 mice exhibit increased inflammation in acute cutaneous hypersensitivity.77 Interestingly, hevin, a member of the secreted protein acidic and rich in cysteine (SPARC) family,61 has also been implicated in the regulation of macrophage recruitment. Hevin KO mice show increased macrophage infiltration during skin healing, although this does not manifest in wound area changes until day 10.
Overall, MPs in the context of the inflammatory phase appear to predominantly influence neutrophil and macrophage infiltration and/or phenotype. When many of these initial studies were performed, our knowledge of inflammation was considerably less, and the importance of M1/M2 macrophage polarization was not understood. Based on our current, increased understanding of inflammation and macrophage polarization and their role in the healing process, re-evaluation of the role of MPs in these processes may be warranted.
Proliferative phase: crossroads of myofibroblasts and ECM formation
The proliferative phase of skin repair is characterized by mesenchymal cell infiltration (eg, resident dermal fibroblasts, pericytes, and progenitor cells) beginning at day 3 followed by a proliferative response, angiogenesis, and matrix deposition (Figure 1). TGF-β1 secreted by macrophages52 is in part responsible for this initial mesenchymal cell migration into what represents relatively loose and compliant granulation tissue.3 At this stage, MPs modulate the adhesion, migration, proliferation, and differentiation of dermal fibroblasts, pericytes, and progenitor cells up to 15 days postwounding in mice.3,29 Tenascin (TN)-C, SPARC, TSP-2, hevin, periostin, CCN2, and CCN3 are expressed at this stage of healing.
As a class of molecules, TNs were first shown to be upregulated in skin healing in 1988, before their classification as MPs.34,35 TNs represent a family of ECM glycoproteins, with five members.78 Using a rat model, Mackie et al34 were the first to assess TN (in this case TN-C) expression postwounding, observing upregulation in the dermis and particularly in the basement membrane under the unwounded epithelium. The latter will be discussed in the section on re-epithelialization. TN appeared in the granulation tissue at day 3 postwounding and persisted at day 6, although TN did not strongly colocalize with myofibroblast populations. By day 10, it was downregulated significantly in the regenerating dermis and was not associated with scar tissue formation. Interestingly, TN-C was one of the first MPs to be studied in skin healing using whole body genetic deletion in mice.79 TN-C KO mice exhibited no defects in development, adult tissue function, or lifespan. During skin healing, no major structural abnormalities or differences in healing kinetics were observed in KO animals, except a reduction in fibronectin content in the granulation tissue. In summary, neither study pointed to TN-C as being important or required for the wound healing process as its initial expression patterns had.
In subsequent years, the role of another family member, TN-X, has been investigated in skin healing using genetic deletion.80 The deletion of TN-X did not alter the kinetics of wound closure compared with WT mice, and expression of the protein in WT mice was relatively low at 7 and 14 days postwounding. However, deletion of TN-X significantly reduced the breaking strength of healing skin at 7 days postwounding onwards, suggesting that TN-X is involved in matrix maturation. However, as it is downregulated in wound bed tissue compared with noninjured skin, the exact role of TN-X is yet to be determined. In healthy skin, the patterns of TN-C and TN-X expression are distinct and appear to be regulated independently of each other,81 but, in wound healing, TN-X appears to have more functional importance than TN-C.80
Genetic deletion of SPARC (also termed osteonectin) highlighted a defect in healing in the mice of 25 mm oblong excisional wounds; wounds resolved by day 24 in WTs, but in KOs this occurred at day 31.28 The study identified that loss of SPARC impaired ECM secretion and cell migration. However, one potential issue with this study is that histological analysis was actually performed using 6 mm dermal punch wounds, not 25 mm. The kinetics of wound closure could be significantly altered in wounds with such a size discrepancy, and the cellular processes involved could be very different between the two types of wounds. The role of SPARC in skin healing became contentious when a second manuscript showed the exact opposite finding, that SPARC deletion increased the speed of wound closure in KO mice in comparison with WT.82 Although this study also identified a significant decrease in collagen content in healing tissue in SPARC KOs compared with WTs, they suggested that this reduction enhanced contractibility of the fibroblast populations, with significant decreases in wound size evident by day 4 (beginning of the proliferative phase of healing) in SPARC KOs. No difference in cell proliferation or cell number in the granulation tissue was evident between SPARC KOs and WTs. Interestingly, they measured a faster migration in KO cells, contradicting the findings of Basu et al,28 who reported impaired migration in SPARC-null fibroblasts. As we have previously highlighted, in analyzing MPs in skin healing, the devil appears to be in the detail.83 When considering these two papers on SPARC alone, different wound sizes were used and methods of analysis were not the same. If we focus on analysis of SPARC-null and WT fibroblast migration, standardized methods were not followed in both papers. In scratch wound assays, to assess the influence of migration alone, it is necessary to inhibit proliferation, commonly achieved through addition of mitomycin-C to the culture medium. While the Basu et al28 study, which reported an inhibition of migration, used mitomycin-C, Bradshaw et al,82 who reported increased scratch wound closure, did not add mitomycin-C to their medium and, as a result, likely measured both proliferation and migration in their assays. As MP function is very much context dependent, we suggest that to eliminate these types of contradictory results, standardization of methods is pivotal as we further analyze the role of these proteins in skin healing.
Largely overlooked in the MP field but defined as an MP due to its described effects on cellular deadhesion, which is a hallmark of MPs,84 hevin has also been implicated in the proliferative phase of skin healing, and null mice were observed to close excisional wounds significantly faster than WT mice.61 Analysis of healing tissue in hevin KO mice demonstrated that loss of the protein affected the structure of ECM in the regenerating dermis indirectly through regulation of decorin levels and collagen fibril assembly. Addition of recombinant hevin to null fibroblasts recovered decorin production.85 Of significance is that this study highlights that the effects of MPs are often indirect.
The potential involvement of TSP-2 in aspects of the proliferative phase of healing was first published by Kyriakides et al86 in 1999, assessed through a full-thickness 6 mm excisional wound model. Although wound closure kinetics were not measured in this study, deletion of TSP-2 resulted in altered collagen orientation in the regenerating dermis at day 14 in KOs, as well as an increase in blood vessel number.
Osteopontin, as as described in Hemostasis and the inflammatory phase of healing, is upregulated at 6 hours postwounding and appears to have direct influence over inflammatory events postwounding.73 At the proliferative phase of healing, loss of osteopontin manifests in a greater disorganization of the matrix and an alteration of collagen fibrillogenesis, leading to smaller diameter collagen fibrils.73 As osteopontin is downregulated between 4 and 6 days postwounding, which corresponds to the first 2 days of the proliferative phase of healing, any effects of the protein on the proliferative and remodeling phases of healing appear to be indirect. Indeed, it appears that these alterations in ECM structure specifically arise as a result of the deficits in the inflammatory phase in the absence of osteopontin.
Of the MPs studied in the context of the proliferative phase of repair, periostin,32,33,42,83 CCN2 (formerly known as connective tissue growth factor),87 and CCN3 (formerly known as NOV) appear to be pivotal. These proteins first appear in the developing granulation tissue within the wound bed at days 3–5 postwounding, showing an expression profile that is clearly different from galectin-3, hevin, and TSPs, which all peak in the inflammatory phase. At day 7 postwounding, unlike several other MPs that are downregulated at this phase,3 the expression of CCN2,87,88 CCN3, and periostin33,42,83 peaks.
As shown by our group, periostin becomes detectable at the mRNA and protein level in the granulation tissue at day 3 postwounding, where it functions as a key modulator of myofibroblast differentiation of mesenchymal cells during wound healing.32,42,83 As fibroblasts, pericytes, and progenitor cells migrate into the wound bed on the newly developed granulation tissue, they undergo a phenotypic change triggered by TGF-β1, becoming α-SMA-expressing myofibroblasts.54 Myofibroblasts are contractile and highly migratory, facilitating contraction of the wound edge.54,55 Our analysis of the periostin KO mouse demonstrated that at day 7, when α-SMA peaks in WT mice, there is no corresponding peak in the KOs at the mRNA or protein level.42 Furthermore, temporal analysis of the protein demonstrated that this peak never occurs. In vitro, periostin KO dermal fibroblasts showed reduced ability to generate force and were unable to contract collagen gels, although contraction could be recovered by the addition of recombinant periostin.42 Interestingly, α-SMA was expressed at the edge of the wounds in the KO animals, an area that corresponds to the highest level of stiffness, but not in the granulation tissue (Figure 2). This suggested that periostin may be required to induce myofibroblast differentiation in the more compliant granulation tissue. Using polyacrylamide gels of different stiffness, we demonstrated that as substrate stiffness increased, periostin KO dermal fibroblasts were able to differentiate into myofibroblasts, which correlated with the in vivo findings. Of potential significance, addition of periostin into wounds in KO animals was sufficient to recover α-SMA expression in the granulation tissue at day 7.42 A further study, by Ontsuka et al89 in 2012, demonstrated that exogenous periostin stimulated fibroblast migration, although our analysis of the KO mouse showed no difference in cell recruitment in KO animals compared with WT.42 However, the contradictory results represented an interesting finding, as they highlighted that addition of recombinant protein may have different effects on cells if the protein is endogenously expressed.
 | Figure 2 α-Smooth muscle actin expression is significantly reduced in periostin knockout mice compared with wild-types at 7 days postwounding. Red arrows mark the wound edge. |
CCN2, a member of the CCN family of MPs,90 was first shown to be upregulated in cutaneous tissue repair in 1993.88 As will be highlighted, the role of CCN2 represents a cautionary tale with respect to exactly what cellular processes MPs may be involved with postwounding. In healing, CCN2 was thought to be involved in pericyte recruitment and secretion of proangiogenic factors,91–93 but it has also been shown to enhance myofibroblast differentiation and ECM synthesis in response to TGF-β1.92,94 In the dermal punch mouse model, CCN2 promoter activity is induced by day 3 and in essentially all fibroblasts in the healing wound by day 7; conversely, CCN2 promoter activity was absent in epithelia.87 In day 7 wounds, CCN2-expressing fibroblasts are α-SMA positive, and approximately one-third of these cells are also positive for the progenitor/pericyte marker NG2.87 Expression of CCN2 by fibroblasts, although not required for the overall kinetics of cutaneous tissue repair,43 is required for the recruitment of progenitor cells such as pericytes to the wound.44 Thus, it appears that the mode of action of CCN2 is on pericyte or skin progenitor cells, not dermal fibroblasts.
CCN3 is significantly upregulated in 6 mm excisional wounds at day 5, peaks at day 7, and is downregulated by day 14.95 CCN3 was detectable in migrating keratinocytes but was predominantly expressed by fibroblasts and endothelial cells in the granulation tissue. Based on in vitro assays with CCN3, it altered fibroblast adhesion and induced chemotaxis and gene expression related to matrix turnover.95
MP modulation of the proliferative phase of healing appears to fall into two categories: 1) effects arising indirectly from MP functions during inflammation or 2) direct and indirect effects due to upregulation of MPs specifically within this phase of healing. As will be discussed next, the role of MPs in re-epithelialization, a process that spans both the inflammatory and proliferative phases of repair, complicates yet further the function of MPs in skin healing.
Re-epithelialization
For the purpose of this review, we will discuss re-establishment of the epidermis separately, as this is a temporal process that occurs during both the inflammatory and proliferative phases of healing. In healthy skin, the epithelial layer is separated from dermis through a basement membrane. Upon injury, keratinocytes respond rapidly to re-establish the barrier function of the skin, and it is necessary to understand the cellular processes that occur in the epithelium to put the expression of MP in context (for an in-depth appraisal of re-epithelialization, see Pastar et al96). In homeostasis, basal keratinocytes are tightly bound to their basement membrane through hemidesmosomes, and the lateral migration of keratinocytes across the developing granulation tissue requires these adhesion sites to be severed. A hallmark of MPs is stimulating intermediate states of cell adhesion that facilitate migration,84 such that expression of MPs would be anticipated. To this point in the review, we have been discussing MPs in the context of granulation tissue, but for re-epithelialization to be initiated, upregulation of MPs under the keratinocytes at the wound edge would be hypothesized. Some MPs, such as osteopontin, have been shown to have no effects on re-epithelialization rates.73 However, TN-C, TSP-2, galectin-3, and periostin have been directly or indirectly implicated in the regulation of keratinocyte migration postwounding.
Historically, the first MPs assessed in the context of re-epithelialization were TNs. Sparsely expressed in healthy basement membrane between the epidermis and dermis, Mackie et al34 in 1988 were the first to describe TN upregulation in the wound edge following incisional wounding in rats, and using explant cultures demonstrated that TN was upregulated at the migrating edge. This expression pattern was confirmed in human healing,36 and further studies demonstrated that it is basal keratinocytes that express TN-C based on in situ hybridization.97 While both these studies strongly implicated TNs in keratinocyte migration, analysis of the TN-C KO mouse found no defect in re-epithelialization,79 with the caveat that wound closure kinetics were not actually measured.
TSP-2 was implicated in re-epithelialization by Kyriakides et al86 in 1999. Deletion of TSP-2 resulted in altered epithelial structure. In both WT and KO animals, the epithelium had completely covered the wound bed at day 7, re-establishing the barrier function of the epidermis, but, in KO mice, the epidermis had formed rete pegs. Rete pegs are invaginations of the epidermis into the underlying dermis, increasing attachment and strength of the skin. However, if re-epithelialization is considered to be 100% coverage of the wound bed by the migrating epithelium, then no apparent difference was evident between TSP-2 KOs and WTs. Wound closure kinetics were not measured in either WTs or KOs in this study, such that although the epithelial structure was more mature in KOs by day 7, barrier function was also present in WT animals, suggesting that no changes in actual wound closure rates were present.
Galectin-3 has been postulated to play an important role in re-epithelialization of skin wounds in mice.98,99 Using an excisional partial-thickness wound in miniature pigs, Klima et al100 in 2009 observed galectin-3 in activated keratinocytes, suggesting a potential role in migration. Previously shown to be an important mediator of epithelial migration in corneal healing,98 galectin-3 KO epidermal keratinocytes showed impaired migration in vitro, assessed by a reduced capacity to close scratch wounds compared with WT cells.99 In this study, epithelial migration was also measured in vivo in KO animals but was only measured at day 2, to avoid the confounding factor of wound contraction. Although the epithelial tongue was bigger in area in WTs compared with galectin-3 KOs, no measure of closure kinetics was performed and, as such, no direct evidence was presented that showed that the in vitro impairment in migration actually translated to an in vivo reduction in closure of the epithelium. Nevertheless, in this system, intracellular galectin-3 was found to regulate trafficking of epidermal growth factor receptor, enabling endocytosed receptor to be recycled back to the plasma membrane after stimulation.
In 2010, we first implicated periostin in keratinocyte response after wounding.33 Using an incisional wound model, we showed that periostin protein expression increases in the basal lamina under the undamaged skin at the wound edge, which correlated spatially with increasing proliferation in the basal keratinocyte layer. As keratinocyte migration and proliferation are required for re-epithelialization of the wound, we concluded that periostin could trigger these processes.33 Alternatively, as MPs are known to stimulate an intermediate state of cell adhesion associated with increased migration, periostin may act in this capacity to stimulate keratinocyte migration into the wound area. The first direct evidence presented that periostin was functionally required for re-epithelialization was reported by Nishiyama et al101 in 2011 from their analysis of healing in periostin-null mice. By measuring percentage re-epithelialization in periostin KO and WT mice at day 3 and day 5, they found significantly lower rates of epithelial closure in the KOs. Using HaCAT cells transfected with periostin–HA vector in vitro, they demonstrated that cells overexpressing periostin closed scratch wounds faster than those with the control vector, but that this was likely due to proliferation, not migration.101 While this work pointed to a role for periostin in re-epithelialization, independently, using a different derivative of the KO mouse, we were not able to identify a defect in epithelial migration. Although the overall closure kinetics we measured were the same as those quantified by Nishiyama et al,101 we measured the distance migrated by the epithelium and found no difference between periostin KO and WT animals.42 We concluded that due to a defect in wound contraction, to fully re-epithelialize the wound, periostin KO keratinocytes had to migrate further, as the edges of the wounds were not contracting together (for a detailed explanation, see Elliott et al83).
Context of MP function: does redundancy exist?
As is evident from this review, deletion of different MPs can affect similar processes; for example, galectin-3,57,65 osteopontin,73 and TSP-176 all influence macrophage behavior and recruitment. However, analysis of the phenotypes reveals very subtle variations in the way the genetic deletions each manifest (see “Hemostasis and the inflammatory phase of healing”). This suggests that each MP has a specific function and that redundancy may be very low; the specificity of each MP is extremely context dependent and depends specifically on how it is measured. However, it should also be pointed out that a comprehensive analysis of all MP expression profiles in mice with specific MP deletions has never been performed; that is, in an osteopontin-null mouse, do the expression patterns of galectin-3 or TSP-1 change? This could provide direct evidence for redundancy of function if expression patterns of these molecules change. A more rigorous approach is the use of double KOs, although this has been sparsely applied to skin healing. To our knowledge, wound healing has been assessed in two double KOs, TSP-1/TSP-262 and SPARC-TSP-2.30 In the TSP-1/TSP-2 double KO, TSP-1 was the limiting factor. Interestingly, they did not measure significant differences in the double KO compared with TSP-1 KOs, which may not be overly surprising based on the expression patterns of the proteins; TSP-1 is upregulated at 6 hours and TSP-2 is upregulated at 7 days postwounding. Therefore, it would be expected that the loss of TSP-1 early in healing would be the rate-limiting step, but TSP-2 clearly could not compensate for the loss of TSP-1, suggesting no functional overlap.62 Individually, both SPARC82 and TSP-286 mice have been reported to show enhanced wound closure, which is also evident in the SPARC-TSP-2 double KOs.30 Exactly whether these effects were additive was not definitively tested, as the double KOs were not compared with the single KOs, as was performed in the TSP-1/TSP-2 case.30 Both of these studies, however, show that the effects of MPs may not necessarily overlap, and compensatory effects could be minimal. Generation of further double KOs will be required as we dissect further the roles of MPs in skin repair.
In vitro versus in vivo: can we really dissect MP function in monolayer cultures?
The effects of MPs are clearly highly context dependent. In our analysis of the periostin KO mouse, we observed a clear defect in myofibroblast differentiation in vivo, but initially we could not recreate this defect in KO cells in vitro.42 All KO dermal fibroblasts in culture differentiated into myofibroblasts, and only when we changed the compliance of the culture substrate did the defect become apparent in the KO cells.42,83 At this stage, we were then able to address how periostin influenced the cells and myofibroblast differentiation. In the case of CCN2, initial analysis suggested that the protein regulated myofibroblast differentiation in vitro based on cell culture assays,102 but analysis of the phenotype in vivo eventually demonstrated that it is not required for dermal fibroblasts to assume a myofibroblast phenotype in vivo43 but is required for recruitment of Sox2 progenitor cells.44 These examples from our previous work highlight that in vitro assays must be carefully chosen and certainly not over-interpreted without a clear analysis of the KO animal.
MP expression in human skin healing: what do we know?
As described in this article, dissection of the roles of MPs in skin healing has involved the use of genetic deletion of the proteins in mice. It should be noted that several major differences exist between mice and human skin,103 but of most relevance is that skin healing in mice predominantly occurs as a result of wound contraction but in humans occurs by both re-epithelialization and contraction. While MP expression has been relatively well described in mice, the expression pattern of MPs in human skin healing is relatively uncharacterized. Here we will document what is known about MP expression patterns in human skin healing, information summarized in Table 2.104
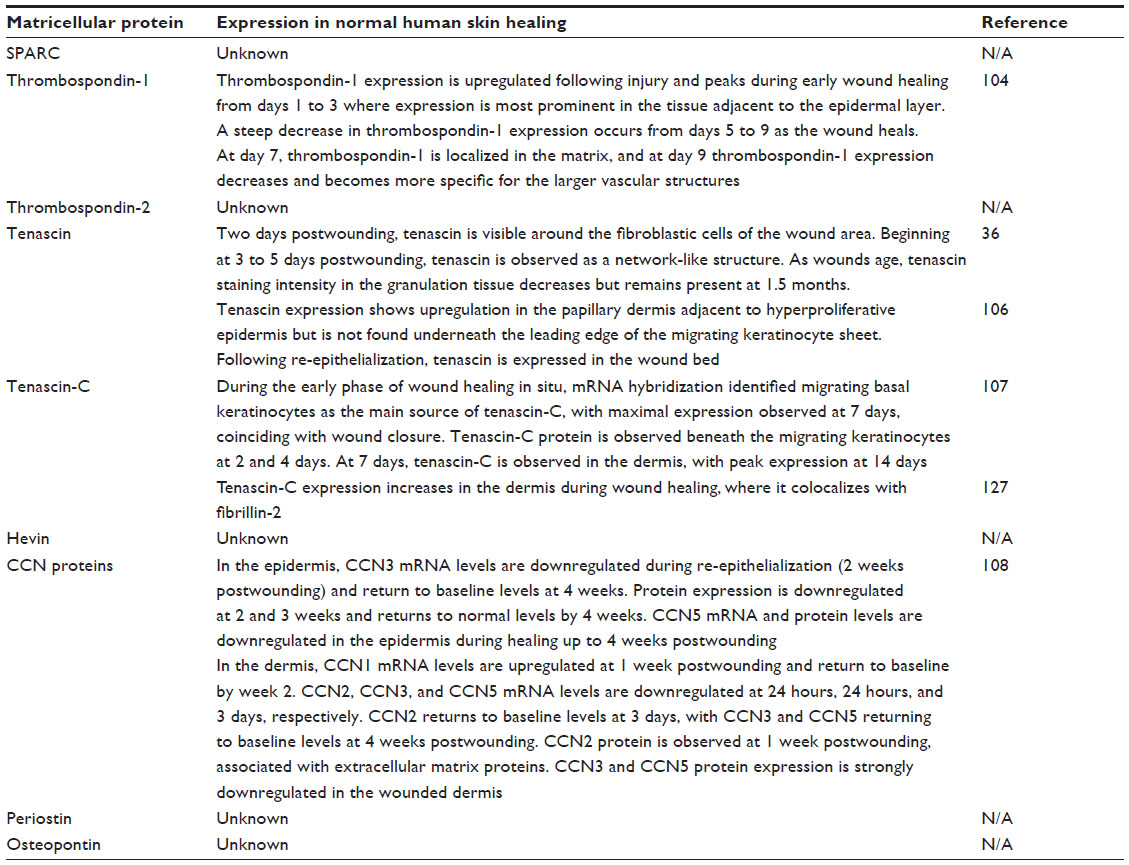 | Table 2 Known expression profiles of matricellular proteins in human wounds |
TSP-1 expression was assessed in wounds retrieved from burn patients.104,105 The pattern of expression was consistent with that evident in murine wounds; TSP-1 was upregulated in injured skin immediately after the injury, particularly in stromal cells.105 It was observed to be rapidly downregulated as the tissue healed and was not present in scar tissue that formed. This is similar to mice, where TSP-1 peaks at day 3 postwounding. TN was also studied in 56 surgically treated human skin wounds ranging in age from 8 hours to 7 months postwounding.36 TN first appeared in lesions 2 days postwounding, but in all wounds of greater than 5 days, it was identified localizing to the dermal–epidermal junction and wound edge.36 A further study showed that TN is not expressed under the leading edge of migrating keratinocytes in human excisional wounds but is upregulated in the granulation tissue at later time points.106 As highlighted previously, basal epidermal keratinocytes are the main source of TN-C during wound healing,107 but it does not appear to be a substrata promoting migration.106 Finally, the only other MPs to be assessed in human skin healing are the members of the CCN family, namely CCN1, 2, 3, 4, and 5.108 Interestingly, Rittie et al108 demonstrated that each CCN family member is temporally regulated during the different phases of healing in human partial-thickness wounds created using a CO2 laser. CCN2, which we highlighted earlier in the review, is downregulated in the inflammatory phase but peaks at day 7 in human wounds, mirroring almost exactly the expression pattern seen during excisional skin healing in mice. In contrast to CCN2, CCN3 is significantly downregulated in human healing and does not return to baseline levels until 4 weeks postwounding. In mice, CCN3 peaks at day 7, suggesting that the protein may not be as important in human skin healing, although it must be noted that the murine wounds were full-thickness wounds and the human wounds partial thickness, the latter requiring little formation of granulation tissue where CCN3 was predominantly expressed in mice.
In summary, our knowledge of the expression pattern of MPs in human skin healing is limited, and observations to date highlight some similarities but some contrasting findings in expression patterns between mice and humans. Performing full-thickness wound healing experiments on healthy volunteers could provide important information on MP expression during normal healing, but depending on the country, human ethics approval represents a significant roadblock to this type of work. For this reason, we have focused part of our program on assessment of MP expression in situations of abnormal healing, with a focus specifically on nonhealing skin wounds.
Nonhealing skin wounds and MPs as potential therapeutics
Nonhealing or “chronic” dermal wounds are a significant and growing clinical complication associated with vascular disease, diabetes, and immobility. The field of regenerative medicine has made considerable strides toward the development of alternative strategies for the regeneration of chronic wounds,109 but significant problems persist and no reproducible strategies exist yet. At the cellular level, chronic wounds remain in a proinflammatory state, which subsequently manifests in an inhibition of keratinocyte migration, fibroblasts failing to migrate into the wound bed and differentiate into myofibroblasts, impaired pericyte recruitment, and a correlative reduction in ECM synthesis and secretion, including collagen type I. Impaired formation of granulation tissue is a well-characterized feature of chronic wounds.110 As we have highlighted, the provisional matrix formed upon hemostasis would normally provide the essential signals regulating inflammation, with many processes associated with neutrophils and macrophages in mice regulated or modulated by MPs (see “Hemostasis and the inflammatory phase of healing”). Persistence of neutrophils and proinflammatory macrophages within the wound prevent wound transition, although the exact reasons why the wounds remain trapped in the inflammatory phase are not fully elucidated. When considering chronic wound repair, it is important to consider the microenvironmental factors present that result from a prolonged inflammatory response within the damaged tissue; this is not a normal skin repair situation. Hypoxia (low oxygen),111 infection,112 wound fluid,113 and high levels of TNF-α secreted by neutrophils114 all impact negatively on the quality of healing, ultimately with the wounds unable to transition from a proinflammatory state.
To assess the potential involvement of MPs in healing, we have assessed the expression patterns of galectin-3115 and periostin (Figure 3) in noninvolved skin as well as within the wound bed. Galectin-3 expression is significantly decreased in the wound bed, and interestingly there is an increase in the presence of advanced glycation end products;115 galectin-3 has been shown to be a receptor for the clearance of advanced glycation end products.116 Moreover, as galectin-3 has been implicated in regulation of macrophage phenotype and neutrophil clearance,63–65 it is interesting to note that galectin-3 mRNA and protein expression is absent in the wound bed, an area associated with a high inflammatory cell infiltrate.117–119 Therefore, although we have yet to confirm it, it is possible that this downregulation of galectin-3 in nonhealing wounds results in a failure of macrophages to polarize to an M2 phenotype, hindering TGF-β release and subsequent transition to the proliferative and remodeling phases of healing. Similarly to galectin-3, periostin, which is important in myofibroblast differentiation,42 is also downregulated in the wound bed compared with noninvolved skin (Figure 3). This correlates with an absence of myofibroblast differentiation in and surrounding the wound bed, which is detrimental for skin healing. Of direct relevance is that we33 and others120,121 have shown that periostin is overexpressed in hypertrophic and keloid scarring in skin, where it promotes myofibroblast persistence.120 While the downregulation of periostin in nonhealing wounds is a correlative finding, based on our studies in mice showing that periostin modulates myofibroblast differentiation during the proliferative phase of healing, it is certainly intriguing.
 | Figure 3 Periostin expression is reduced in human nonhealing skin wounds and correlates with a reduction in α-SMA expression and collagen content. Tissue was isolated at elective amputation from an 83-year-old female with type 2 diabetes. |
It is clear that mechanisms to stimulate mesenchymal cell recruitment, proliferation, differentiation, and ECM synthesis within the wound are still not being adequately addressed by current wound care products. Design of scaffolds that mimic the structure of the ECM while possessing the necessary biological signals is one of the major hurdles in regenerative medicine. Whether MPs represent new therapeutics has yet to be elucidated, but they certainly represent potential targets for nonhealing wounds.
Conclusion
Over the last two decades, MPs have emerged as a significant factor governing cellular and matrix interactions during skin healing. While much progress has been made, we consider this field to be very much still in its infancy, and considerable efforts need to be made to increase our understanding of these molecules. We have revised Midwood et al’s3 2004 expression profile based on studies performed in the intervening years (Figure 4), but, as can be seen, much is still to be learnt. We suggest that standardization of wound models would significantly aid in our interpretation of the roles of each MP, especially considering their spatiotemporal expression, which is often extremely context dependent and also varies depending on which cell type the MPs are acting on. As our understanding increases, we suggest that local delivery of MPs may represent a new therapeutic for nonhealing skin lesions.
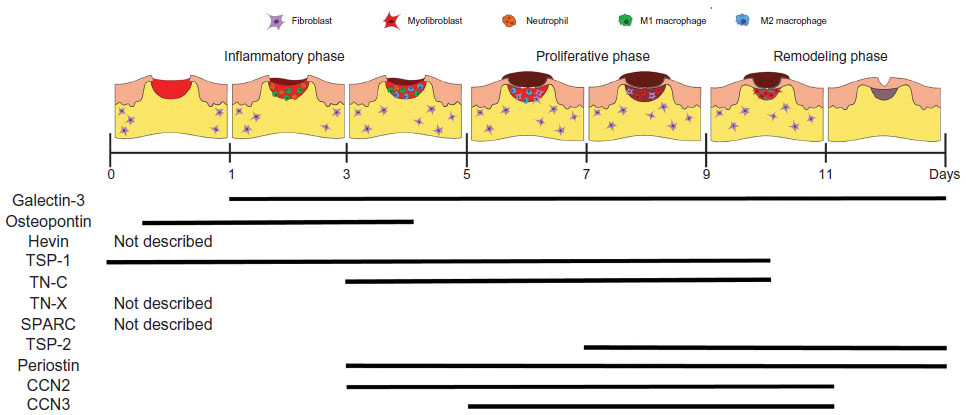 | Figure 4 Temporal expression profiles of matricellular proteins during skin healing. |
Disclosure
The authors report no conflicts of interest in this work.
References
Elliott CG, Hamilton DW. Deconstructing fibrosis research: do pro-fibrotic signals point the way for chronic dermal wound regeneration? J Cell Commun Signal. 2011;5(4):301–315. | |
Reinke JM, Sorg H. Wound repair and regeneration. Eur Surg Res. 2012; 49(1):35–43. | |
Midwood KS, Williams LV, Schwarzbauer JE. Tissue repair and the dynamics of the extracellular matrix. Int J Biochem Cell Biol. 2004;36:1031–1037. | |
Martin P. Wound healing – aiming for perfect skin regeneration. Science. 1997;276(5309):75–81. | |
Singer AJ, Clark RA. Cutaneous wound healing. N Engl J Med. 1999;341:738–746. | |
Hunt TK, Hopf H, Hussain Z. Physiology of wound healing. Adv Skin Wound Care. 2000;13(2 Suppl):6–11. | |
Jeffcoate WJ, Price PE, Phillips CJ, et al. Randomised controlled trial of the use of three dressing preparations in the management of chronic ulceration of the foot in diabetes. Health Technol Assess. 2009;13(54):1–86, iii–iv. | |
Driver VR, Fabbi M, Lavery LA, Gibbons G. The costs of diabetic foot: the economic case for the limb salvage team. J Vasc Surg. 2010;52:17S–22S. | |
Mustoe TA, O’Shaughnessy K, Kloeters O. Chronic wound pathogenesis and current treatment strategies: a unifying hypothesis. Plast Reconstr Surg. 2006;117(7 Suppl):35S–41S. | |
Markova A, Mostow EN. US skin disease assessment: ulcer and wound care. Dermatol Clin. 2012;30(1):107–111, ix. | |
Woodbury M, Houghton P. Prevalence of pressure ulcers in Canadian healthcare settings. Ostomy Wound Manage. 2004;50(10):22–24, 26, 28, 30, 32, 34, 36–38. | |
Woodbury M, Houghton P. The extent of chronic wounds in Canada. Wound Care Canada. 2005;3(1):18–52. | |
Reiber GE. The epidemiology of diabetic foot problems. Diabet Med. 1996;13 Suppl 1:S6–S11. | |
Gardner SE, Frantz RA. Wound bioburden and infection-related complications in diabetic foot ulcers. Biol Res Nurs. 2008;10(1):44–53. | |
Gary Sibbald R, Woo KY. The biology of chronic foot ulcers in persons with diabetes. Diabetes Metab Res Rev. 2008;24 Suppl 1:S25–S30. | |
Kravitz SR, McGuire J, Zinszer K. Management of skin ulcers: understanding the mechanism and selection of enzymatic debriding agents. Adv Skin Wound Care. 2008;21:72–74. | |
Wall IB, Moseley R, Baird DM, et al. Fibroblast dysfunction is a key factor in the non-healing of chronic venous leg ulcers. J Invest Dermatol. 2008;128(10):2526–2540. | |
Zgonis T, Stapleton JJ, Girard-Powell VA, Hagino RT. Surgical management of diabetic foot infections and amputations. Aorn J. 2008;87(5):935–946; quiz 947–950. | |
Sen CK, Gordillo GM, Roy S, et al. Human skin wounds: a major and snowballing threat to public health and the economy. Wound Repair Regen. 2009;17(6):763–771. | |
Acosta JB, del Barco DG, Vera DC, et al. The pro-inflammatory environment in recalcitrant diabetic foot wounds. Int Wound J. 2008;5(4):530–539. | |
Montfrans CV, Stok M, Geerkens M. Biology of chronic wounds and new treatment strategies. Phlebology. 2014;29(1 Suppl):165–167. | |
Barrientos S, Stojadinovic O, Golinko MS, Brem H, Tomic-Canic M. Growth factors and cytokines in wound healing. Wound Repair Regen. 2008;16(5):585–601. | |
Finnson KW, McLean S, Di Guglielmo GM, Philip A. Dynamics of transforming growth factor beta signaling in wound healing and scarring. Adv Wound Care. 2013;2(5):195–214. | |
Robson MC, Mustoe TA, Hunt TK. The future of recombinant growth factors in wound healing. Am J Surg. 1998;176(2A Suppl):80S–82S. | |
Bornstein P. Diversity of function is inherent in matricellular proteins: an appraisal of thrombospondin 1. J Cell Biol. 1995;130(3):503–506. | |
Bornstein P, Sage EH. Matricellular proteins: extracellular modulators of cell function. Curr Opin Cell Biol. 2002;14(5):608–616. | |
Murphy-Ullrich JE, Sage EH. Revisiting the matricellular concept. Matrix Biol. 2014;37:1–14. | |
Basu A, Kligman LH, Samulewicz SJ, Howe CC. Impaired wound healing in mice deficient in a matricellular protein SPARC (osteonectin, BM-40). BMC Cell Biol. 2001;2:15. | |
Kyriakides TR, Bornstein P. Matricellular proteins as modulators of wound healing and the foreign body response. Thromb Haemost. 2003;90(6):986–992. | |
Puolakkainen PA, Bradshaw AD, Brekken RA, et al. SPARC-thrombospondin-2-double-null mice exhibit enhanced cutaneous wound healing and increased fibrovascular invasion of subcutaneous polyvinyl alcohol sponges. J Histochem Cytochem. 2005;53(5):571–581. | |
Hamilton DW. Functional role of periostin in development and wound repair: implications for connective tissue disease. J Cell Commun Signal. 2008;2(1–2):9–17. | |
Jackson-Boeters L, Wen W, Hamilton DW. Periostin localizes to cells in normal skin, but is associated with the extracellular matrix during wound repair. J Cell Commun Signal. 2009;3(2):125–133. | |
Zhou HM, Wang J, Elliott C, Wen W, Hamilton DW, Conway SJ. Spatiotemporal expression of periostin during skin development and incisional wound healing: lessons for human fibrotic scar formation. J Cell Commun Signal. 2010;4(2):99–107. | |
Mackie EJ, Halfter W, Liverani D. Induction of tenascin in healing wounds. J Cell Biol. 1988;107(6 Pt 2):2757–2767. | |
Murakami R, Yamaoka I, Sakakura T. Appearance of tenascin in healing skin of the mouse: possible involvement in seaming of wounded tissues. Int J Dev Biol. Dec 1989;33(4):439–444. | |
Betz P, Nerlich A, Tubel J, Penning R, Eisenmenger W. Localization of tenascin in human skin wounds – an immunohistochemical study. Int J Legal Med. 1993;105(6):325–328. | |
Luomanen M, Virtanen I. Distribution of tenascin in healing incision, excision and laser wounds. J Oral Pathol Med. 1993;22(1):41–45. | |
Mo FE, Muntean AG, Chen CC, Stolz DB, Watkins SC, Lau LF. CYR61 (CCN1) is essential for placental development and vascular integrity. Mol Cell Biol. 2002;22(24):8709–8720. | |
Ivkovic S, Yoon BS, Popoff SN, et al. Connective tissue growth factor coordinates chondrogenesis and angiogenesis during skeletal development. Development. 2003;130(12):2779–2791. | |
Yang Z, Kyriakides TR, Bornstein P. Matricellular proteins as modulators of cell-matrix interactions: adhesive defect in thrombospondin 2-null fibroblasts is a consequence of increased levels of matrix metalloproteinase-2. Mol Biol Cell. 2000;11(10):3353–3364. | |
Kyriakides TR, Rojnuckarin P, Reidy MA, et al. Megakaryocytes require thrombospondin-2 for normal platelet formation and function. Blood. 2003;101(10):3915–3923. | |
Elliott CG, Wang J, Guo X, et al. Periostin modulates myofibroblast differentiation during full-thickness cutaneous wound repair. J Cell Sci. 2012;125(Pt 1):121–132. | |
Liu S, Thompson K, Leask A. CCN2 expression by fibroblasts is not required for cutaneous tissue repair. Wound Repair Regen. 2014;22(1):119–124. | |
Tsang M, Leask A. CCN2 is required for recruitment of Sox2-expressing cells during cutaneous tissue repair. J Cell Commun Signal. Epub October 18, 2014. | |
Dorsett-Martin WA, Wysocki A. Rat models of skin wound healing. In: Conn PM, editor. Sourcebook of Models for Biomedical Research. Totowa, NJ: Springer; 2008:631–638. | |
Greiling D, Clark RA. Fibronectin provides a conduit for fibroblast transmigration from collagenous stroma into fibrin clot provisional matrix. J Cell Sci. 1997;110(Pt 7):861–870. | |
DiPietro LA. Wound healing: the role of the macrophage and other immune cells. Shock. 1995;4(4):233–240. | |
Leibovich SJ, Ross R. The role of the macrophage in wound repair. A study with hydrocortisone and antimacrophage serum. Am J Pathol. 1975;78(1):71–100. | |
Martinez FO, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Front Biosci. 2008;13:453–461. | |
Mills CD. M1 and M2 macrophages: oracles of health and disease. Crit Rev Immunol. 2012;32(6):463–488. | |
Jaguin M, Houlbert N, Fardel O, Lecureur V. Polarization profiles of human M-CSF-generated macrophages and comparison of M1-markers in classically activated macrophages from GM-CSF and M-CSF origin. Cell Immunol. 2013;281(1):51–61. | |
Assoian RK, Fleurdelys BE, Stevenson HC, et al. Expression and secretion of type beta transforming growth factor by activated human macrophages. Proc Natl Acad Sci U S A. 1987;84(17):6020–6024. | |
Morikawa S, Ezaki T. Phenotypic changes and possible angiogenic roles of pericytes during wound healing in the mouse skin. Histol Histopathol. 2011;26(8):979–995. | |
Gabbiani G. The myofibroblast in wound healing and fibrocontractive diseases. J Pathol. 2003;200(4):500–503. | |
Desmouliere A, Chaponnier C, Gabbiani G. Tissue repair, contraction, and the myofibroblast. Wound Repair Regen. 2005;13(1):7–12. | |
Kyriakides TR, Zhu YH, Smith LT, et al. Mice that lack thrombospondin 2 display connective tissue abnormalities that are associated with disordered collagen fibrillogenesis, an increased vascular density, and a bleeding diathesis. J Cell Biol. 1998;140(2):419–430. | |
Liu FT, Hsu DK. The role of galectin-3 in promotion of the inflammatory response. Drug News Perspect. 2007;20(7):455–460. | |
Giachelli CM, Lombardi D, Johnson RJ, Murry CE, Almeida M. Evidence for a role of osteopontin in macrophage infiltration in response to pathological stimuli in vivo. Am J Pathol. 1998;152(2):353–358. | |
Miragliotta V, Pirone A, Donadio E, Abramo F, Ricciardi MP, Theoret CL. Osteopontin expression in healing wounds of horses and in human keloids. Equine Vet J. Epub October 7, 2014. | |
Mori R, Shaw TJ, Martin P. Molecular mechanisms linking wound inflammation and fibrosis: knockdown of osteopontin leads to rapid repair and reduced scarring. J Exp Med. 2008;205(1):43–51. | |
Sullivan MM, Puolakkainen PA, Barker TH, Funk SE, Sage EH. Altered tissue repair in hevin-null mice: inhibition of fibroblast migration by a matricellular SPARC homolog. Wound Repair Regen. 2008;16(2):310–319. | |
Agah A, Kyriakides TR, Lawler J, Bornstein P. The lack of thrombospondin-1 (TSP1) dictates the course of wound healing in double-TSP1/TSP2-null mice. Am J Pathol. 2002;161(3):831–839. | |
Rabinovich GA, Baum LG, Tinari N, et al. Galectins and their ligands: amplifiers, silencers or tuners of the inflammatory response? Trends Immunol. 2002;23(6):313–320. | |
MacKinnon AC, Farnworth SL, Hodkinson PS, et al. Regulation of alternative macrophage activation by galectin-3. J Immunol. 2008;180(4):2650–2658. | |
Juniantito V, Izawa T, Yamamoto E, Murai F, Kuwamura M, Yamate J. Heterogeneity of macrophage populations and expression of galectin-3 in cutaneous wound healing in rats. J Comp Pathol. 2011;145(4):378–389. | |
Bhaumik P, St-Pierre G, Milot V, St-Pierre C, Sato S. Galectin-3 facilitates neutrophil recruitment as an innate immune response to a parasitic protozoa cutaneous infection. J Immunol. 2013;190(2):630–640. | |
Verrecchia F, Pessah M, Atfi A, Mauviel A. Tumor necrosis factor-α inhibits transforming growth factor-β/Smad signaling in human dermal fibroblasts via AP-1 activation. J Biol Chem. 2000;275(39):30226–30231. | |
Goldberg M, Han Y, Yan C, Shaw M. TNF-α suppresses α-smooth muscle actin expression in human dermal fibroblasts: an implication for abnormal wound healing. J Invest Dermatol. 2007;127(11):2645–2655. | |
Tarnuzzer RW, Schultz GS. Biochemical analysis of acute and chronic wound environments. Wound Repair Regen. 1996;4(3):321–325. | |
Nomiyama T, Perez-Tilve D, Ogawa D, et al. Osteopontin mediates obesity-induced adipose tissue macrophage infiltration and insulin resistance in mice. J Clin Invest. 2007;117(10):2877–2888. | |
Uaesoontrachoon K, Wasgewatte Wijesinghe DK, Mackie EJ, Pagel CN. Osteopontin deficiency delays inflammatory infiltration and the onset of muscle regeneration in a mouse model of muscle injury. Dis Model Mech. 2013;6(1):197–205. | |
Li XD, Chen J, Ruan CC, Zhu DL, Gao PJ. Vascular endothelial growth factor-induced osteopontin expression mediates vascular inflammation and neointima formation via Flt-1 in adventitial fibroblasts. Arterioscler Thromb Vasc Biol. 2012;32(9):2250–2258. | |
Liaw L, Birk DE, Ballas CB, Whitsitt JS, Davidson JM, Hogan BL. Altered wound healing in mice lacking a functional osteopontin gene (spp1). J Clin Invest. 1998;101(7):1468–1478. | |
Madsen DH, Leonard D, Masedunskas A, et al. M2-like macrophages are responsible for collagen degradation through a mannose receptor-mediated pathway. J Cell Biol. 2013;202(6):951–966. | |
Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. 2014;6:13. | |
DiPietro LA, Nissen NN, Gamelli RL, Koch AE, Pyle JM, Polverini PJ. Thrombospondin 1 synthesis and function in wound repair. Am J Pathol. 1996;148(6):1851–1860. | |
Velasco P, Huegel R, Brasch J, et al. The angiogenesis inhibitor thrombospondin-1 inhibits acute cutaneous hypersensitivity reactions. J Invest Dermatol. 2009;129(8):2022–2030. | |
Halper J, Kjaer M. Basic components of connective tissues and extracellular matrix: elastin, fibrillin, fibulins, fibrinogen, fibronectin, laminin, tenascins and thrombospondins. Adv Exp Med Biol. 2014;802:31–47. | |
Forsberg E, Hirsch E, Frohlich L, et al. Skin wounds and severed nerves heal normally in mice lacking tenascin-C. Proc Natl Acad Sci U S A. 1996;93(13):6594–6599. | |
Egging D, van Vlijmen-Willems I, van Tongeren T, Schalkwijk J, Peeters A. Wound healing in tenascin-X deficient mice suggests that tenascin-X is involved in matrix maturation rather than matrix deposition. Connect Tissue Res. 2007;48(2):93–98. | |
Matsumoto K, Saga Y, Ikemura T, Sakakura T, Chiquet-Ehrismann R. The distribution of tenascin-X is distinct and often reciprocal to that of tenascin-C. J Cell Biol. 1994;125(2):483–493. | |
Bradshaw AD, Reed MJ, Sage EH. SPARC-null mice exhibit accelerated cutaneous wound closure. J Histochem Cytochem. 2002;50(1):1–10. | |
Elliott CG, Kim SS, Hamilton DW. Functional significance of periostin in excisional skin repair: is the devil in the detail? Cell Adh Migr. 2012;6(4):319–326. | |
Murphy-Ullrich JE. The de-adhesive activity of matricellular proteins: is intermediate cell adhesion an adaptive state? J Clin Invest. 2001; 107(7):785–790. | |
Sullivan MM, Barker TH, Funk SE, et al. Matricellular hevin regulates decorin production and collagen assembly. J Biol Chem. 2006;281(37):27621–27632. | |
Kyriakides TR, Tam JW, Bornstein P. Accelerated wound healing in mice with a disruption of the thrombospondin 2 gene. J Invest Dermatol. 1999;113(5):782–787. | |
Kapoor M, Liu S, Huh K, Parapuram S, Kennedy L, Leask A. Connective tissue growth factor promoter activity in normal and wounded skin. Fibrogenesis Tissue Repair. 2008;1(1):3. | |
Igarashi A, Okochi H, Bradham DM, Grotendorst GR. Regulation of connective tissue growth factor gene expression in human skin fibroblasts and during wound repair. Mol Biol Cell. 1993;4(6):637–645. | |
Ontsuka K, Kotobuki Y, Shiraishi H, et al. Periostin, a matricellular protein, accelerates cutaneous wound repair by activating dermal fibroblasts. Exp Dermatol. 2012;21(5):331–336. | |
Perbal B. CCN proteins: a centralized communication network. J Cell Commun Signal. 2013;7(3):169–177. | |
Liu S, Shi-wen X, Abraham DJ, Leask A. CCN2 is required for bleomycin-induced skin fibrosis in mice. Arthritis Rheum. 2011;63(1):239–246. | |
Liu S, Shi-Wen X, Abraham DJ, Leask A. CCN2 is required for bleomycin-induced skin fibrosis. Arthritis Rheum. Epub October 8, 2010. | |
Kennedy L, Liu S, Shi-Wen X, et al. CCN2 is necessary for the function of mouse embryonic fibroblasts. Exp Cell Res. 2007;313(5):952–964. | |
Shi-Wen X, Leask A, Abraham D. Regulation and function of connective tissue growth factor/CCN2 in tissue repair, scarring and fibrosis. Cytokine Growth Factor Rev. 2008;19(2):133–144. | |
Lin CG, Chen CC, Leu SJ, Grzeszkiewicz TM, Lau LF. Integrin-dependent functions of the angiogenic inducer NOV (CCN3):implication in wound healing. J Biol Chem. 2005;280(9):8229–8237. | |
Pastar I, Stojadinovic O, Yin NC, et al. Epithelialization in wound healing: a comprehensive review. Adv Wound Care. 2014;3(7):445–464. | |
Aukhil I, Sahlberg C, Thesleff I. Basal layer of epithelium expresses tenascin mRNA during healing of incisional skin wounds. J Periodontal Res. 1996;31(2):105–112. | |
Cao Z, Said N, Amin S, et al. Galectins-3 and -7, but not galectin-1, play a role in re-epithelialization of wounds. J Biol Chem. 2002;277(44):42299–42305. | |
Liu W, Hsu DK, Chen HY, et al. Galectin-3 regulates intracellular trafficking of EGFR through Alix and promotes keratinocyte migration. J Invest Dermatol. 2012;132(12):2828–2837. | |
Klima J, Lacina L, Dvorankova B, et al. Differential regulation of galectin expression/reactivity during wound healing in porcine skin and in cultures of epidermal cells with functional impact on migration. Physiol Res. 2009;58(6):873–884. | |
Nishiyama T, Kii I, Kashima TG, et al. Delayed re-epithelialization in periostin-deficient mice during cutaneous wound healing. PLoS One. 2011;6(4):e18410. | |
Leask A, Abraham DJ. The role of connective tissue growth factor, a multifunctional matricellular protein, in fibroblast biology. Biochem Cell Biol. 2003;81(6):355–363. | |
Wong VW, Sorkin M, Glotzbach JP, Longaker MT, Gurtner GC. Surgical approaches to create murine models of human wound healing. J Biomed Biotechnol. 2011;2011:969618. | |
Roth JJ, Hughes WB, DeClement FA, Hensell DO, Granick MS, Tuszynski GP. The 1998 Moyer Award. Characteristics of thrombospondin-1 and its cysteine-serine-valine-threonine-cysteine-glycine receptor in burn wounds. J Burn Care Rehabil. 1998;19(6):487–493. | |
Roth JJ, Albo D, Rothman VL, et al. Thrombospondin-1 and its CSVTCG-specific receptor in wound healing and cancer. Ann Plast Surg. 1998;40(5):494–501. | |
Latijnhouwers MA, Bergers M, Van Bergen BH, Spruijt KI, Andriessen MP, Schalkwijk J. Tenascin expression during wound healing in human skin. J Pathol. 1996;178(1):30–35. | |
Latijnhouwers M, Bergers M, Ponec M, Dijkman H, Andriessen M, Schalkwijk J. Human epidermal keratinocytes are a source of tenascin-C during wound healing. J Invest Dermatol. 1997;108(5):776–783. | |
Rittie L, Perbal B, Castellot JJ Jr, Orringer JS, Voorhees JJ, Fisher GJ. Spatial-temporal modulation of CCN proteins during wound healing in human skin in vivo. J Cell Commun Signal. 2011;5(1):69–80. | |
Metcalfe AD, Ferguson MW. Bioengineering skin using mechanisms of regeneration and repair. Biomaterials. 2007;28(34):5100–5113. | |
Black E, Vibe-Petersen J, Jorgensen LN, et al. Decrease of collagen deposition in wound repair in type 1 diabetes independent of glycemic control. Arch Surg. 2003;138(1):34–40. | |
Thackham JA, McElwain DL, Long RJ. The use of hyperbaric oxygen therapy to treat chronic wounds: a review. Wound Repair Regen. 2008; 16(3):321–330. | |
Tsourdi E, Barthel A, Rietzsch H, Reichel A, Bornstein SR. Current aspects in the pathophysiology and treatment of chronic wounds in diabetes mellitus. Biomed Res Int. 2013;2013:385641. | |
Bucalo B, Eaglstein WH, Falanga V. Inhibition of cell proliferation by chronic wound fluid. Wound Repair Regen. 1993;1(3):181–186. | |
Wallace HJ, Stacey MC. Levels of tumor necrosis factor-alpha (TNF-alpha) and soluble TNF receptors in chronic venous leg ulcers – correlations to healing status. J Invest Dermatol. 1998;110(3):292–296. | |
Pepe D, Elliott CG, Forbes T, Hamilton DW. Detection of galectin-3 and localization of advanced glycation end products (AGE) in human chronic skin wounds. Histol Histopathol. 2014;29(2):251–258. | |
Zhu W, Sano H, Nagai R, Fukuhara K, Miyazaki A, Horiuchi S. The role of galectin-3 in endocytosis of advanced glycation end products and modified low density lipoproteins. Biochem Biophys Res Commun. 2001;280(4):1183–1188. | |
Raffetto JD. Inflammation in chronic venous ulcers. Phlebology. 2013;28 Suppl 1:61–67. | |
Oberyszyn TM. Inflammation and wound healing. Front Biosci. 2007;12:2993–2999. | |
Diegelmann RF, Evans MC. Wound healing: an overview of acute, fibrotic and delayed healing. Front Biosci. 2004;9:283–289. | |
Crawford J, Nygard K, Gan BS, O’Gorman DB. Periostin induces fibroblast proliferation and myofibroblast persistence in hypertrophic scarring. Exp Dermatol. Epub November 24, 2014. | |
Sidgwick GP, Bayat A. Extracellular matrix molecules implicated in hypertrophic and keloid scarring. J Eur Acad Dermatol Venereol. 2012;26(2):141–152. | |
Streit M, Velasco P, Riccardi L, et al. Thrombospondin-1 suppresses wound healing and granulation tissue formation in the skin of transgenic mice. EMBO J. 2000;19(13):3272–3282. | |
Bornstein P, Agah A, Kyriakides TR. The role of thrombospondins 1 and 2 in the regulation of cell-matrix interactions, collagen fibril formation, and the response to injury. Int J Biochem Cell Biol. 2004;36(6):1115–1125. | |
Agah A, Kyriakides TR, Letrondo N, Björkblom B, Bornstein P. Thrombospondin 2 levels are increased in aged mice: consequences for cutaneous wound healing and angiogenesis. Matrix Biol. 2004;22(7):539–547. | |
Maclauchlan S, Skokos EA, Agah A, et al. Enhanced Angiogenesis and Reduced Contraction in Thrombospondin-2–null Wounds Is Associated With Increased Levels of Matrix Metalloproteinases-2 and -9, and Soluble VEGF. J Histochem Cytochem. 2009;57(4):301–313. | |
Gendronneau G, Sidhu SS, Delacour D, et al. Galectin-7 in the Control of Epidermal Homeostasis after Injury. Mol Biol Cell. 2008;19(12):5541–5549. | |
Brinckmann J, Hunzelmann N, Kahle B, et al. Enhanced fibrillin-2 expression is a general feature of wound healing and sclerosis: potential alteration of cell attachment and storage of TGF-beta. Lab Invest. 2010;90(5):739–752. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.