Back to Archived Journals » International Journal of Interferon, Cytokine and Mediator Research » Volume 7
Bruton’s tyrosine kinase in chronic inflammation: from pathophysiology to therapy
Authors Hartkamp L, Radstake T, Reedquist K
Received 10 April 2015
Accepted for publication 29 May 2015
Published 18 September 2015 Volume 2015:7 Pages 27—34
DOI https://doi.org/10.2147/IJICMR.S71779
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Randall Davis
Linda M Hartkamp, Timothy RDJ Radstake, Kris A Reedquist
Department of Rheumatology and Clinical Immunology, Laboratory of Translational Immunology, University Medical Center Utrecht, Utrecht, the Netherlands
Abstract: Bruton’s tyrosine kinase (Btk) not only plays a role in differentiation and activation of B-cells but is also involved in regulating signaling in myeloid cell populations, mast cells, platelets, and osteoclasts. Btk plays a critical role in B-cell receptor signaling and has been shown to be involved in CD40 ligation, Toll-like receptor triggering, and Fc-receptor signaling as well, suggesting that targeting Btk might be particularly useful in autoimmune diseases characterized by pathologic antibodies, macrophage activation, and myeloid-derived type I interferon responses. Btk knockout and X-linked immunodeficiency mouse material and X-linked agammaglobulinemia patient samples are powerful tools to examine the role of Btk in health and disease. In addition, the recent development of several covalent and noncovalent small-molecule inhibitors specifically targeting Btk can contribute to our understanding of the functional role of Btk in several cell types and disease mechanisms. Specific Btk inhibitors have been used in both in vitro and in vivo models for autoimmune diseases such as rheumatoid arthritis, systemic lupus erythematosus, multiple sclerosis, systemic sclerosis, and Sjögren's syndrome. The current small-molecule Btk inhibitors have demonstrated potent and selective Btk inhibition in preclinical studies, and some inhibitors have propelled the clinical development of such molecules toward clinical trials in patients with rheumatoid arthritis. Future studies on the effectiveness of Btk inhibition in other autoimmune diseases could further broaden the understanding of the disease mechanisms as well as lead to new targeted therapies.
Keywords: Bruton’s tyrosine kinase, rheumatoid arthritis, systemic lupus erythematosus, multiple sclerosis, systemic sclerosis, Sjögren’s syndrome
Introduction
Autoimmune disorders arise from the dysregulation of the immune system and the subsequent abnormal immune response to self-antigens. Genetic factors and environmental factors contribute to the pathogenesis of autoimmune disorders but the triggers that initiate disease remain to be elucidated. Bruton’s tyrosine kinase (Btk) is involved in the differentiation and activation of B-cells and myeloid cell populations, suggesting that targeting Btk might be particularly useful in autoimmune diseases characterized by pathologic antibodies, macrophage activation, and myeloid-derived type I interferon (IFN) responses. Here, we discuss the potential role of Btk and inhibition thereof in various disease conditions.
Tec family kinases
Btk, the best-known member of the Tec (tyrosine kinase expressed in hepatocellular carcinoma) family kinases, was identified as a new protein kinase, mutations in which caused X-linked agammaglobulinemia (XLA) in men and X-linked immunodeficiency (XID) in mice. The Tec family of kinases forms the second largest class of cytoplasmic protein tyrosine kinases after the Src family kinases (SFKs) and consists of five mammalian members: Btk, Bmx (bone marrow kinase on the X-chromosome, also known as Etk), Itk (IL-2 inducible T-cell kinase), Rlk (resting lymphocyte kinase, also known as Txk), and Tec.1 Most of the Tec family kinases are primarily expressed in the hematopoietic system, although both Tec and Bmx are also expressed in stromal tissues such as liver and endothelial cells, respectively.1–3 Two characteristic features that set the Tec family kinases apart from SFKs are the presence of a pleckstrin homology (PH) domain, located at the N-terminus of the protein, and a proline-rich region that is part of the Tec homology domain. The PH domain is able to bind phospholipids and signaling proteins such as the βγ-subunits of heterotrimeric G proteins and protein kinase C and is required for Tec family kinase membrane localization and molecular activation. The proline-rich region, which can interact with SFKs and other Src homology (SH) 3 domain-containing proteins, is implicated in autoregulation. In addition to the PH domain and the Tec homology domain, Tec family kinases contain a phospho-tyrosine-binding SH2 domain and SH3 and catalytic domains (SH1).
Activation of Tec family kinases upon cell-surface receptor triggering requires relocalization of the protein to the plasma membrane, which is mediated by the interaction of the PH domain with the lipid phosphatidylinositol (3,4,5)P3, formed by activated phosphatidylinositol-3 kinase. Subsequent phosphorylation by SFKs and autophosphorylation of tyrosine 223 result in the complete activation of Tec family kinases. Although translocation to the membrane seems to be a prerequisite for activation, Btk, Itk, and Rlk are also found in the nucleus upon cell activation.2–4 In addition to the well-studied role of Tec family kinases in PLCγ activation and Ca2+ mobilization, they can also act downstream of numerous cell-surface receptors that influence a wide range of signaling pathways involved in proliferation, differentiation, apoptosis, cell migration, transcriptional regulation, cellular transformation, and inflammation.1,3
Btk not only plays a critical role in B-cell receptor (BCR) signaling in B-cells but has also been shown to function downstream of CD40, affecting B-cell activation and function.5 Btk has also been demonstrated to directly interact with cytoplasmic Toll/IL-1 receptor domains of most Toll-like receptors (TLRs) and can interact with downstream adaptors MYD88, TRIF, and MYD88 adaptor-like protein (MAL; also known as TIRAP), and IL-1R-associated kinase 1. Btk regulates signaling by TLR2, 3, 4, 7, 8, and 9 in macrophages, dendritic cells (DCs), and B-cells, activating transcription factors such as NF-κB and IRF3 ultimately leading to cytokine and type I IFN production.6–15 Furthermore, it has been shown that Btk signals downstream of Fc receptors, leading to increased cytokine production in myeloid cells and mast cells and degranulation in mast cells.16–18
Btk regulates production of many cytokines in myeloid cells. In DCs, Btk has been reported to regulate type I IFN, IL12, IL18, and IL10.12,13,19,20 In monocytes/macrophages, pro-inflammatory cytokines such as TNF, IL1 and IL6, and MMPs are affected by Btk.8,16,21–23 As in monocytes/macrophages, IL6 and IL8 production are also regulated by Btk in platelets.24,25 In mast cells, degranulation and TNF production are Btk dependent.17,18 The involvement of Btk in the production of these cytokines and inflammatory mediators suggests that targeting Btk could be beneficial in various immune-mediated inflammatory disorders.
Consequences of Btk mutations
Btk is required for B-cell development and function and is the only known member of the family that causes disease in humans. XLA is a primary immunodeficiency resulting from mutations in Btk. Over 1,000 mutations have been described that render Btk nonfunctional or prevent protein expression. XLA is characterized by the complete absence of circulating B-cells due to a developmental block between pro- and pre-B-cell stages and a severe reduction in serum immunoglobulin levels.26,27 A similar, although considerably milder syndrome in the mouse, XID, is also caused by a mutation in Btk.27 Apart from its role in B-cells, it has also been shown to regulate monocyte, macrophage and DC function, mast cell and platelet activation, and osteoclast differentiation.18,28–32
The role of Btk in autoimmune diseases
Btk knockout and XID models combined with biological specimen from patient with XLA form a powerful tool to examine the role of Btk under steady-state and pathological conditions. In addition, the recent development of several covalent and noncovalent small-molecule inhibitors specifically targeting Btk can contribute to our understanding of the functional role of Btk in several cell types and disease mechanisms. Later, we will discuss the potential use of Btk inhibitors in several autoimmune diseases and their potential path toward clinical development.
Rheumatoid arthritis
Rheumatoid arthritis (RA) is a chronic and progressive inflammatory disease characterized by the recruitment and accumulation of activated immune cells along with hyperplastic growth of the intimal lining layer fibroblast-like synoviocytes in the synovial compartment.33 Whereas the initiation of RA likely involves antigen-presenting cell activation due to so-called dangers signals, the perpetuation of persistent inflammation involves the recognition and activation of B- and T-cells by autoantigens, which results in autoantibody production. Autoantibody formation precedes clinical signs and symptoms by years, and this does not coincide with subclinical inflammation in the joint, suggesting that multiple pathological events (hits) are needed to develop breakthrough of tolerance and chronic persistent synovitis.34 When synovitis does occur and pro-inflammatory cytokines are produced, in combination with the formation of immune complexes, this can lead to the activation of synovium-infiltrating and tissue-resident myeloid cells, such as macrophages, monocytes, DCs, neutrophils, and mast cells. Eventually, synovitis leads to erosion of the joint surface, causing deformity and loss of function. The prevalence rate is 1% population, with women affected three to five times as often as men. Commonly used animal models include the murine collagen-induced arthritis (CIA) and K/BxN serum transfer models.35
Initial evidence that Btk might play a role in RA arose from observations that XID mice are resistant to inflammation and joint destruction in CIA.36 This spurred further research and when small-molecule inhibitors specifically targeting Btk were developed, they were studied in both in vivo and in vitro models of arthritis. LFM-A13 (leflunomide metabolite analogue alpha-cyano-beta-hydroxy-beta-methyl-N-[2,5-dibromophenyl]-propenamide) was one of the first rationally designed Btk selective inhibitors. LFM-A13 binds to the catalytic domain and inhibits Btk in a reversible manner. Even though LFM-A13 has been described as a highly selective inhibitor targeting Btk, it has also been shown to target the activity of other kinases such as JAK2 and the erythropoietin receptor. Despite its antileukemia activity and the lack of any major toxicity in preclinical studies, rather high doses are needed for a pharmacological effect which prohibited LFM-A13 to enter clinical development.37–39
Following up on the success of LFM-A13 several covalent and noncovalent small-molecule inhibitors for Btk have been developed (Table 1). One of the first new chemotypes of small-molecule Btk inhibitor, CGI1746, uniquely binds preferentially to inactive Btk and stabilizes Btk in an inactive nonphosphorylated enzyme conformation and inhibits both auto- and transphosphorylation steps needed for Btk kinase activation. It was demonstrated that CGI1746 blocks BCR-mediated B-cell proliferation and decreases autoantibody levels in murine CIA.16 Pro-inflammatory cytokines such as TNFα, IL1β, and IL6 induced by Fc-receptor triggering were inhibited in mouse macrophages and human monocytes. CGI1746 decreased cytokine levels in the joints and ameliorated disease in myeloid and FcγR-dependent autoantibody-induced arthritis. This study provided direct evidence that Btk inhibition could suppress experimental arthritis via effects on both B lymphocyte and myeloid cell populations.16 Another reversible Btk inhibitor, RN486, potently and selectively inhibits Btk in human cell-based assays as well as animal models of arthritis. It was shown to efficiently block FcεR crosslinking-induced degranulation of mast cells, and BCR-induced B-cell activation.25,40 Furthermore RN486 inhibited inflammatory mediator production in response to multiple agonists relevant to inflammatory arthritis (ie, FcγR, TLR, and CD40 ligation) in human macrophages, as well as genes induced by the synovial fluid of patient with RA. Importantly, RN486 was able to suppress spontaneous cytokine and matrix metalloproteinase production in RA synovial tissue explants.21 Finally, RN486 was shown to reduce levels of platelet p-PLCγ2 upon GPVI stimulation with convulxin and could block IL6 and IL8 production in a human platelet-fibroblast-like synoviocyte coculture system.21 It displayed similar functional activity in animal models of arthritis like mouse CIA and rat adjuvant-induced arthritis. RN486 inhibited both joint and systemic inflammation, reducing paw swelling and inflammatory markers in peripheral blood.21,24,25,40
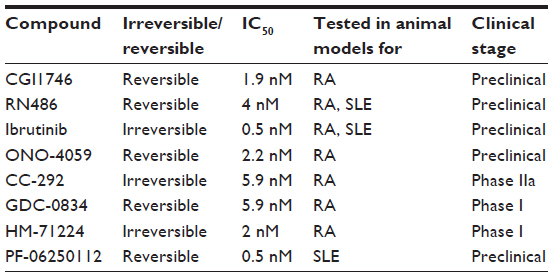 | Table 1 Btk inhibitors in development for immune-mediated inflammatory disorders |
Ibrutinib, or PCI-32765, was the first Btk inhibitor to advance into clinical trials for the treatment of B-cell malignancies. This compound was unusual, as it is a covalent irreversible, Btk inhibitor, which binds the cysteine 481 residue in the ATP binding domain of the catalytic domain and prevents full activation of Btk. Irreversible inhibitors raised concerns of toxicity issues, especially in cases where the compound target protein has a long half-life – however, ibrutinib quickly saturates its target and has a short half-life.41,42 While Ibrutinib was chosen for clinical development because of its high selectivity (IC50: 0.5 nM), a small number of other kinases are known to be targeted as well (eg, EGFR, HER2, Itk, and Tec) albeit at higher IC50s. It still remains unknown as to what extent targeting these off-target kinases contributes to the efficacy and toxicity of ibrutinib. Upon approval by the US Food and Drug Administration, ibrutinib will be marketed under the name “Imbruvica”. Ibrutinib was shown to reverse arthritic inflammation in murine CIA and prevented clinical arthritis in the collagen antibody-induced arthritis murine model. It inhibited infiltration of immune cells to the joints, bone resorption, cartilage destruction, and inflammation. Ibrutinib was also demonstrated to be efficacious in cell-based assays, as it prevented BCR-activated B-cell proliferation, inhibited cytokine induction by FcγR triggering, and prevented degranulation of mast cells upon activation.43–45
A second compound entering clinical trials for B-cell malignancies is ONO-4059. A Phase I clinical trial for oral treatment of B-cell lymphoma was started in 2012. This compound binds covalently to Btk and reversibly blocks BCR signaling and B-cell proliferation. ONO-4059 was demonstrated to suppress production of inflammatory chemokines and cytokines by monocytes in a murine CIA model and prevented cartilage and bone destruction.45–47
While Ibrutinib and ONO-4059 advanced quickly in clinical trials for hematological malignancies, three different small-molecule inhibitors targeting Btk are now in first-stage clinical trials for RA. CC-292, also described as AVL-292, is similar to Ibrutinib in that it covalently binds cysteine 481 in the kinase domain. It showed promising results in animal models of arthritis with a 95% reduction in clinical scores and a decrease in inflammation and cartilage and bone destruction. CC-292 is the first Btk inhibitor reported appropriate for use in a human clinical setting of autoimmune disease. Healthy volunteers were treated with 2 mg/kg orally, and the compound was shown to consistently engage all circulating Btk protein. This first trial provided rapid insight into safety, pharmacokinetics (PK), and pharmacodynamics (PD), and CC-292 has currently progressed into a Phase IIa clinical trial. This study will test the clinical effectiveness and safety of an orally administered dose of CC-292 compared with placebo in patients on methotrexate with active RA.37,48,49
GDC-0834, another Btk specific inhibitor, has been shown to ameliorate disease in a rat CIA model. This study suggested that a high level of inhibition of phosphorylation of Btk was required for it to affect inflammatory arthritis in rats.50 A recent Phase I study in healthy volunteers showed that the inhibitor is metabolized to an inactive metabolite in the liver via amide hydrolysis. It is currently undergoing further clinical development for RA.47,51
Finally, HM-71224 is an orally active and irreversible Btk inhibitor showing a strong efficacy in mice and rat CIA models. The first clinical trial was started to evaluate the safety, tolerability, PK, PD, and food effect of HM-71224 following single ascending doses and multiple ascending doses in healthy volunteers. An interim report demonstrated a well-tolerated safety profile in healthy volunteers and desirable PK and PD properties supporting sustained target inhibition. A Phase II study in patients with active RA is reported to be initiated soon.47,52,53
Systemic lupus erythematosus
Systemic lupus erythematosus (SLE) is an autoimmune disease, which is characterized by autoantibodies recognizing nuclear self-antigens. Autoreactive T- and B-cells evade negative selection and, once activated, collaborate to produce autoantibodies, which culminate in immune complex deposition on tissues leading to organ damage. Deposition occurs most often in skin and kidneys (lupus nephritis), activating complement and promoting local inflammation by activating macrophages and DCs. Activation of plasmacytoid DCs (pDCs) leads to production of type I IFNs, major contributors to pathology in SLE. Various animal models of spontaneous lupus are commonly used and include the classical F1 hybrid of New Zealand Black and New Zealand White strains (NZB × NSW F1) and the MRL/lpr lupus-prone model.47,54,55
Various Btk murine knockout models have suggested that Btk plays a role in SLE. In SLE, murine models Btk has been demonstrated to be required for the initial loss of tolerance to DNA and the ensuing production of anti-DNA antibodies.56,57 Furthermore, it has been shown that specific inhibition of Btk by RN486 can regulate TLR9 signaling in pDCs and thus inhibit type I IFN production.7 In addition to in vitro studies, in vivo studies have also been conducted with RN486 in the NZB × NZW F1 model. The administration of RN486 completely blocked disease progression and this study suggested that Btk inhibition may simultaneously target autoantibody producing cells and impact the effector function of autoantibodies on monocyte activation in SLE.58 The results of this study were consistent with results of a study that used a different Btk inhibitor, PF-06250112. PF-06250112 binds residue cysteine 481 covalently but reversibly and was identified as a potent, orally bioavailable, small-molecule inhibitor of Btk. This study made use of the same mouse model (NZB × NZW F1) and showed that Btk inhibition prevented the development of proteinuria and reduced glomerular injury and inflammatory infiltrates. Furthermore, it showed a significant reduction in serum IgG anti-dsDNA antibodies and complement deposition.59 Ibrutinib has also been shown to be efficacious in animal models of SLE. Treatment of B6.Sle1 and B6.Sle1.Sle3 lupus-prone mice with Ibrutinib resulted in decreased renal damage and lymphocyte infiltration. Moreover, a significant reduction in autoantibodies was shown. In the MRL/lpr mouse model, Ibrutinib treatment resulted in reduced proteinuria and blood urea nitrogen and showed a trend toward improvement of glomerulonephritis.43,60 The clinical therapeutic efficacies have not been studied in human SLE thus far, and further evaluation of the potential of targeting Btk is of interest.
Multiple sclerosis
Multiple sclerosis (MS) is a chronic inflammatory demyelinating disease of the central nervous system caused by an autoimmune response to self-antigens. Large, confluent plaques of demyelination are formed in the white and the gray matter. Even though MS is extensively studied, key aspects of MS etiology and pathophysiology still remain to be elucidated. Active tissue injury in all stages of MS is associated with inflammation, with the inflammatory infiltrates being composed of T lymphocytes, B lymphocytes, and plasma cells. This leads to active demyelination and axonal or neuronal damage at sites of microglia activation and macrophage infiltration of the tissue. Experimental autoimmune encephalomyelitis (EAE) is the main animal model for MS and is also characterized by auto-reactive T-cells.61–63 Although Btk inhibition has not been studied in EAE models, Btk-deficient XID mice develop less severe EAE.26 In line with this, Btk inhibition might target and inhibit B-cell and macrophage activation that are both implicated in the pathogenesis of the disease. However, the Btk targets TNF and type I IFN are hypothesized to play a dual role in MS. TNF has been described to be protective in lymphoid organs yet contribute to pathology in the central nervous system during EAE. And, while in multiple autoimmune disorders like SLE and Sjögren’s syndrome type I IFNs are increased and contribute to disease, treatment with or induction of type I IFN has proven beneficial.64–67 Both could have a protective effect, and this should be taken into account when considering targeting Btk in MS. Nonetheless, as current treatments are only partially effective, studying the role of Btk in MS pathology could provide an alternative approach to therapeutic intervention.
Type 1 diabetes
Type 1 diabetes (T1D) is characterized by immune-mediated destruction of insulin-producing pancreatic β cells by autoreactive T-cells. Autoantibodies are produced and pancreatic islets are infiltrated by inflammatory cells such as macrophages and CD4+ T-cells, leading to lifelong dependence on exogenous insulin and an increased risk for kidney failure, heart disease, blindness, limb loss, and episodes of hypo- and hyperglycemia. Both genetic and environmental factors play a role in the pathogenesis of T1D.68–71 The majority of our knowledge on the pathogenesis and etiology of T1D comes from the study of spontaneous disease in the nonobese diabetic (NOD) mouse model. The NOD mouse model showed that also B-cells have flaws in tolerance mechanisms.71 B-cells contribute to the disease by producing autoantibodies and activating autoreactive T-cells. Elimination of B-cells has proven successful at preventing disease, and Btk deficiency in NOD mice protects against T1D. Btk deficiency led to a failure to produce insulin autoantibodies by decreasing relative availability of mature autoreactive B-cells.72,73 This suggests that by targeting Btk, also T-cell-mediated autoimmunity could be alleviated in T1D. However, one case report described the development of T1D in a patient with XLA, implying that neither autoantibodies nor B-cell function is absolutely required for T1D onset.74 No studies examining pharmacological inhibition of Btk in T1D animal models have been reported thus far and would represent the next step in examining the role of Btk in this disease.
Systemic sclerosis
Systemic sclerosis (SSc), or scleroderma, is an autoimmune disease, which is characterized by vasculopathy, immune dysfunction, and fibrosis leading to multiorgan failure. Heterogeneity is caused by variable expression of these three pathological features that makes early diagnosis challenging. Historically, scleroderma research has been focused on the altered function of fibroblasts. Excessive extracellular matrix and collagen deposition by fibroblast activation and activation of Th2 cells were believed to play a prominent role. However, recent findings suggest a complex interplay among immune cells, endothelial cells, and fibroblasts indicating an important role for immune cells in the pathogenesis of SSc. Genetic association studies indeed revealed that among the most highly associated susceptibility markers, genes encoding immune signaling molecules such as T bet, IRF5, and STAT4 were included.75 Recently, pDCs have also been shown to play a prominent role in SSc pathology. These cells have the unique ability to trigger both the innate and the adaptive immune system, are key producers of type I IFNs, and in SSc, produce high levels of CXCL4, which might directly promote fibrosis.76 Autoantibodies are commonly observed in patients with SSc, although there is debate as to whether they contribute to disease or are an epiphenomenon. There are several animal models known for this disease, including the bleomycin-induced fibrosis and the tight skin-1 mouse models.77–79 As mentioned above, specific inhibition of Btk by RN486 can regulate TLR9 signaling in pDCs and thus inhibit type I IFN production.7 Therefore, one could speculate Btk inhibition would also prove beneficial in the treatment of SSc, but such studies have not yet been reported.
Sjögren’s syndrome
Primary Sjögren’s syndrome (pSS) is considered to be the second most common systemic autoimmune disease after RA. pSS is characterized by the infiltration of immune cells in the exocrine glands, leading to ocular and mouth dryness but also includes arthralgia and fatigue as frequent disabling symptoms. Approximately one-third of the patients also develop systemic complications such as arthritis, lung interstitial disease, neurological manifestations, or tubular nephropathy. This latter group is at a higher risk for lymphoma development, possibly driven by ongoing B-cell proliferation and/or stimulation. Although the underlying etiology has yet to be fully elucidated, in the past decade major advances have been made in understanding the pathogenesis of pSS. T-cells have been shown to play a major role in the pathogenesis, and the innate system seems to be involved in early stages of disease through the production of type I IFN. Besides their role in autoantibody production, B-cells are now recognized to have multiple roles in pSS pathophysiology and might play a central role in the development of the disease. There also appears to be a crucial interplay among T-cells, DCs, and exocrine gland epithelial cells. Various mouse models exist for pSS. The earliest models were strains that develop disease spontaneously (NZB × NZW F1 and MRL/lpr mice also possess SLE features), but transgenic (Tg) overexpression of many immune-related genes (as observed in BAFF Tg, TGFβ Tg or IL12 Tg mice) and genetic deficiencies can lead to the disease phenotype as well. Induced models include ro-peptide-induced or murine cytomegalovirus CMV-induced pSS.80–84 Btk knockout or inhibition has yet to be studied in the context of pSS. However, based on the ability of Btk inhibition to decrease autoantibody production and inhibit myeloid type I IFN responses in other disease models, a rationale to study the role of Btk in pSS is readily apparent.
Concluding remarks
Extensive research has established a critical role for Btk in several disease-relevant signaling pathways in B- and myeloid cells, including BCR, FcγR, and cytokine receptor signaling, making it an appealing drug discovery target. By targeting Btk, multiple signaling pathways in different cell populations can be regulated with one inhibitor. The current small-molecule Btk inhibitors have demonstrated potent and selective Btk inhibition in preclinical studies, and some inhibitors have propelled the clinical development of such molecules toward clinical trials in patients with RA. Future studies testing the therapeutic potential of Btk inhibition in chronic inflammatory diseases will need to address several key issues. First, Btk inhibitors have only been studied in animal models of RA and SLE, and experimental data are lacking in other disease models. Second, with the exception of RA, Btk activation status or contribution of Btk to gene expression in patient-derived cells has yet to be examined. Third, Btk can undergo multiple post-translational modifications other than tyrosine phosphorylation, such as serine phosphorylation, which regulate its activity.27 As cell or disease-specific modifications could influence efficacy of Btk inhibition, proteomic analysis of patient B lymphocytes and myeloid cells could be useful in understanding the potential effects of Btk inhibitors in the clinic. Finally, as observed in myeloid-derived cells, the ability of Btk to couple to multiple agonistic signaling pathways in a cell-specific manner and also differentially regulate target genes commonly induced by multiple agonists,16,21,44 will likely necessitate a nonhypothesis-based approach to examining the effects of Btk inhibition on highly purified patient cell subsets. Such studies on the effectiveness of Btk inhibition in other autoimmune diseases could further broaden the understanding of disease mechanisms and lead to the broadening of our therapeutic armamentarium.
Disclosure
The authors report no conflicts of interest in this work.
References
Schmidt U, Boucheron N, Unger B, Ellmeier W. The role of Tec family kinases in myeloid cells. Int Arch Allergy Immunol. 2004;134(1):65–78. | |
Smith CI, Islam TC, Mattsson PT, Mohamed AJ, Nore BF, Vihinen M. The Tec family of cytoplasmic tyrosine kinases: mammalian Btk, Bmx, Itk, Tec, Txk and homologs in other species. Bioessays. 2001;23(5):436–446. | |
Takesono A, Finkelstein LD, Schwartzberg PL. Beyond calcium: new signaling pathways for Tec family kinases. J Cell Sci. 2002;115(Pt 15):3039–3048. | |
Mohamed AJ, Vargas L, Nore BF, Backesjo CM, Christensson B, Smith CI. Nucleocytoplasmic shuttling of Bruton’s tyrosine kinase. J Biol Chem. 2000;275(51):40614–40619. | |
Brunner C, Avots A, Kreth HW, Serfling E, Schuster V. Bruton’s tyrosine kinase is activated upon CD40 stimulation in human B lymphocytes. Immunobiology. 2002;206(4):432–440. | |
Jefferies CA, Doyle S, Brunner C, et al. Bruton’s tyrosine kinase is a Toll/interleukin-1 receptor domain-binding protein that participates in nuclear factor kappaB activation by Toll-like receptor 4. J Biol Chem. 2003;278(28):26258–26264. | |
Wang J, Lau KY, Jung J, Ravindran P, Barrat FJ. Bruton’s tyrosine kinase regulates TLR9 but not TLR7 signaling in human plasmacytoid dendritic cells. Eur J Immunol. 2014;44(4):1130–1136. | |
Jefferies CA, O’Neill LA. Bruton’s tyrosine kinase (Btk)-the critical tyrosine kinase in LPS signalling? Immunol Lett. 2004;92(1–2):15–22. | |
Herbst S, Shah A, Mazon MM, et al. Phagocytosis-dependent activation of a TLR9-BTK-calcineurin-NFAT pathway co-ordinates innate immunity to Aspergillus fumigatus. EMBO Mol Med. 2015;7(3):240–258. | |
Liljeroos M, Vuolteenaho R, Morath S, Hartung T, Hallman M, Ojaniemi M. Bruton’s tyrosine kinase together with PI 3-kinase are part of Toll-like receptor 2 multiprotein complex and mediate LTA induced Toll-like receptor 2 responses in macrophages. Cell Signal. 2007;19(3):625–633. | |
Lee KG, Xu S, Kang ZH, et al. Bruton’s tyrosine kinase phosphorylates Toll-like receptor 3 to initiate antiviral response. Proc Natl Acad Sci U S A. 2012;109(15):5791–5796. | |
Li YF, Lee KG, Ou X, Lam KP. Bruton’s tyrosine kinase and protein kinase C micro are required for TLR7/9-induced IKKalpha and IRF-1 activation and interferon-beta production in conventional dendritic cells. PLoS One. 2014;9(8):e105420. | |
Lougaris V, Baronio M, Vitali M, et al. Bruton tyrosine kinase mediates TLR9-dependent human dendritic cell activation. J Allergy Clin Immunol. 2014;133(6):1644–1650. | |
Kubo T, Uchida Y, Watanabe Y, et al. Augmented TLR9-induced Btk activation in PIR-B-deficient B-1 cells provokes excessive autoantibody production and autoimmunity. J Exp Med. 2009;206(9):1971–1982. | |
Kenny EF, Quinn SR, Doyle SL, Vink PM, van EH, O’Neill LA. Bruton’s tyrosine kinase mediates the synergistic signalling between TLR9 and the B cell receptor by regulating calcium and calmodulin. PLoS One. 2013;8(8):e74103. | |
Di Paolo JA, Huang T, Balazs M, et al. Specific Btk inhibition suppresses B cell- and myeloid cell-mediated arthritis. Nat Chem Biol. 2011;7(1):41–50. | |
Ellmeier W, Abramova A, Schebesta A. Tec family kinases: regulation of FcepsilonRI-mediated mast-cell activation. FEBS J. 2011;278(12):1990–2000. | |
Hata D, Kawakami Y, Inagaki N, et al. Involvement of Bruton’s tyrosine kinase in FcepsilonRI-dependent mast cell degranulation and cytokine production. J Exp Med. 1998;187(8):1235–1247. | |
Ni Gabhann J, Spence S, Wynne C, et al. Defects in acute responses to TLR4 in Btk-deficient mice result in impaired dendritic cell-induced IFN-gamma production by natural killer cells. Clin Immunol. 2012;142(3):373–382. | |
Horwood NJ, Urbaniak AM, Danks L. Tec family kinases in inflammation and disease. Int Rev Immunol. 2012;31(2):87–103. | |
Hartkamp LM, Fine JS, van Es IE, et al. Btk inhibition suppresses agonist-induced human macrophage activation and inflammatory gene expression in RA synovial tissue explants. Ann Rheum Dis. Epub April 24, 2014. | |
Horwood NJ, Mahon T, McDaid JP, et al. Bruton’s tyrosine kinase is required for lipopolysaccharide-induced tumor necrosis factor alpha production. J Exp Med. 2003;197(12):1603–1611. | |
Horwood NJ, Page TH, McDaid JP, et al. Bruton’s tyrosine kinase is required for TLR2 and TLR4-induced TNF, but not IL-6, production. J Immunol. 2006;176(6):3635–3641. | |
Hsu J, Gu Y, Tan SL, Narula S, DeMartino JA, Liao C. Bruton’s Tyrosine Kinase mediates platelet receptor-induced generation of microparticles: a potential mechanism for amplification of inflammatory responses in rheumatoid arthritis synovial joints. Immunol Lett. 2013;150(1–2):97–104. | |
Xu D, Kim Y, Postelnek J, et al. RN486, a selective Bruton’s tyrosine kinase inhibitor, abrogates immune hypersensitivity responses and arthritis in rodents. J Pharmacol Exp Ther. 2012;341(1):90–103. | |
Mangla A, Khare A, Vineeth V, et al. Pleiotropic consequences of Bruton tyrosine kinase deficiency in myeloid lineages lead to poor inflammatory responses. Blood. 2004;104(4):1191–1197. | |
Mohamed AJ, Yu L, Backesjo CM, et al. Bruton’s tyrosine kinase (Btk): function, regulation, and transformation with special emphasis on the PH domain. Immunol Rev. 2009;228(1):58–73. | |
Melcher M, Unger B, Schmidt U, Rajantie IA, Alitalo K, Ellmeier W. Essential roles for the Tec family kinases Tec and Btk in M-CSF receptor signaling pathways that regulate macrophage survival. J Immunol. 2008;180(12):8048–8056. | |
Quek LS, Bolen J, Watson SP. A role for Bruton’s tyrosine kinase (Btk) in platelet activation by collagen. Curr Biol. 1998;8(20):1137–1140. | |
Atkinson BT, Ellmeier W, Watson SP. Tec regulates platelet activation by GPVI in the absence of Btk. Blood. 2003;102(10):3592–3599. | |
Shinohara M, Koga T, Okamoto K, et al. Tyrosine kinases Btk and Tec regulate osteoclast differentiation by linking RANK and ITAM signals. Cell. 2008;132(5):794–806. | |
Setoguchi R, Kinashi T, Sagara H, Hirosawa K, Takatsu K. Defective degranulation and calcium mobilization of bone-marrow derived mast cells from Xid and Btk-deficient mice. Immunol Lett. 1998;64(2–3):109–118. | |
Buckley CD, Filer A, Haworth O, Parsonage G, Salmon M. Defining a role for fibroblasts in the persistence of chronic inflammatory joint disease. Ann Rheum Dis. 2004;63(Suppl 2):ii92–ii95. | |
van de Sande MG, de Hair MJ, van der Leij C, et al. Different stages of rheumatoid arthritis: features of the synovium in the preclinical phase. Ann Rheum Dis. 2011;70(3):772–777. | |
McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med. 2011;365(23):2205–2219. | |
Jansson L, Holmdahl R. Genes on the X chromosome affect development of collagen-induced arthritis in mice. Clin Exp Immunol. 1993; 94(3):459–465. | |
Vargas L, Hamasy A, Nore BF, Smith CI. Inhibitors of BTK and ITK: state of the new drugs for cancer, autoimmunity and inflammatory diseases. Scand J Immunol. 2013;78(2):130–139. | |
Burger JA. Bruton’s tyrosine kinase (BTK) inhibitors in clinical trials. Curr Hematol Malig Rep. 2014;9(1):44–49. | |
Uckun FM. Clinical potential of targeting Bruton’s tyrosine kinase. Int Rev Immunol. 2008;27(1–2):43–69. | |
Lou Y, Owens TD, Kuglstatter A, Kondru RK, Goldstein DM. Bruton’s tyrosine kinase inhibitors: approaches to potent and selective inhibition, preclinical and clinical evaluation for inflammatory diseases and B cell malignancies. J Med Chem. 2012;55:4539–4550. | |
Hutchinson CV, Dyer MJ. Breaking good: the inexorable rise of BTK inhibitors in the treatment of chronic lymphocytic leukaemia. Br J Haematol. 2014;166(1):12–22. | |
Spaargaren M, de Rooij MF, Kater AP, Eldering E. BTK inhibitors in chronic lymphocytic leukemia: a glimpse to the future. Oncogene. 2015;34(19):2426–2436. | |
Honigberg LA, Smith AM, Sirisawad M, et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc Natl Acad Sci U S A. 2010;107(29):13075–13080. | |
Chang BY, Huang MM, Francesco M, et al. The Bruton tyrosine kinase inhibitor PCI-32765 ameliorates autoimmune arthritis by inhibition of multiple effector cells. Arthritis Res Ther. 2011;13(4):R115. | |
Shinohara M, Chang BY, Buggy JJ, et al. The orally available Btk inhibitor ibrutinib (PCI-32765) protects against osteoclast-mediated bone loss. Bone. 2014;60:8–15. | |
Yoshizawa T, Ariza Y, Ueda Y, Hotta S, Narita M, Kawabata K. Development of a Bruton’s tyrosine kinase (Btk) inhibitor, ONO-4059: efficacy in a collagen induced arthritis (CIA) model indicates potential treatment for rheumatoid arthritis (RA). Arthritis Rheum. 2012;64:S709. | |
Akinleye A, Chen Y, Mukhi N, Song Y, Liu D. Ibrutinib and novel BTK inhibitors in clinical development. J Hematol Oncol. 2013;6:59. | |
Evans EK, Tester R, Aslanian S, et al. Inhibition of Btk with CC-292 provides early pharmacodynamic assessment of activity in mice and humans. J Pharmacol Exp Ther. 2013;346(2):219–228. | |
Whang JA, Chang BY. Bruton’s tyrosine kinase inhibitors for the treatment of rheumatoid arthritis. Drug Discov Today. 2014;19(8):1200–1204. | |
Liu L, Di Paulo J, Barbosa J, Rong H, Reif K, Wong H. Antiarthritis effect of a novel Bruton’s tyrosine kinase (BTK) inhibitor in rat collagen-induced arthritis and mechanism-based pharmacokinetic/pharmacodynamic modeling: relationships between inhibition of BTK phosphorylation and efficacy. J Pharmacol Exp Ther. 2011;338(1):154–163. | |
Liu L, Halladay JS, Shin Y, et al. Significant species difference in amide hydrolysis of GDC-0834, a novel potent and selective Bruton’s tyrosine kinase inhibitor. Drug Metab Dispos. 2011;39(10):1840–1849. | |
Yoon YK, Hadi S, Iersel TV, et al. Safety, pharmacokinetics, pharmacodynamics and food effect of an oral Bruton’s tyrosine kinase inhibitor HM71224 in healthy subjects. Ann Rheum Dis. 2014;73(Suppl 2):231. | |
Park JK, Park JA, Lee YJ, et al. HM71224, a novel oral Btk inhibitor, inhibits human immune cell activation: new drug candidate to treat B-cell associated autoimmune diseases. Ann Rheum Dis. 2014;73(Suppl 2):355–356. doi: 10.1136/annrheumdis-2014-eular.2783. | |
Mok CC, Lau CS. Pathogenesis of systemic lupus erythematosus. J Clin Pathol. 2003;56(7):481–490. | |
Pathak S, Mohan C. Cellular and molecular pathogenesis of systemic lupus erythematosus: lessons from animal models. Arthritis Res Ther. 2011(5);13:241. | |
Halcomb KE, Musuka S, Gutierrez T, Wright HL, Satterthwaite AB. Btk regulates localization, in vivo activation, and class switching of anti-DNA B cells. Mol Immunol. 2008;46(2):233–241. | |
Whyburn LR, Halcomb KE, Contreras CM, Lowell CA, Witte ON, Satterthwaite AB. Reduced dosage of Bruton’s tyrosine kinase uncouples B cell hyperresponsiveness from autoimmunity in lyn-/- mice. J Immunol. 2003;171(4):1850–1858. | |
Mina-Osorio P, LaStant J, Keirstead N, et al. Suppression of glomerulonephritis in lupus-prone NZB x NZW mice by RN486, a selective inhibitor of Bruton’s tyrosine kinase. Arthritis Rheum. 2013;65(9):2380–2391. | |
Rankin AL, Seth N, Keegan S, et al. Selective inhibition of BTK prevents murine lupus and antibody-mediated glomerulonephritis. J Immunol. 2013;191(9):4540–4550. | |
Hutcheson J, Vanarsa K, Bashmakov A, et al. Modulating proximal cell signaling by targeting Btk ameliorates humoral autoimmunity and end-organ disease in murine lupus. Arthritis Res Ther. 2012;14(6):R243. | |
Ciccarelli O, Barkhof F, Bodini B, et al. Pathogenesis of multiple sclerosis: insights from molecular and metabolic imaging. Lancet Neurol. 2014;13(8):807–822. | |
Nylander A, Hafler DA. Multiple sclerosis. J Clin Invest. 2012;122(4):1180–1188. | |
Lassmann H. Multiple sclerosis: lessons from molecular neuropathology. Exp Neurol. 2014;262 Pt A:2–7. | |
Kruglov AA, Lampropoulou V, Fillatreau S, Nedospasov SA. Pathogenic and protective functions of TNF in neuroinflammation are defined by its expression in T lymphocytes and myeloid cells. J Immunol. 2011; 187(11):5660–5670. | |
Dendrou CA, Bell JI, Fugger L. A clinical conundrum: the detrimental effect of TNF antagonists in multiple sclerosis. Pharmacogenomics. 2013;14(12):1397–1404. | |
Trinchieri G. Type I interferon: friend or foe? J Exp Med. 2010;207(10):2053–2063. | |
Verweij CL, Vosslamber S. Relevance of the type I interferon signature in multiple sclerosis towards a personalized medicine approach for interferon-beta therapy. Discov Med. 2013;15(80):51–60. | |
Atkinson MA. The pathogenesis and natural history of type 1 diabetes. Cold Spring Harb Perspect Med. 2012;2(11):pii:a007641. | |
Gillespie KM. Type 1 diabetes: pathogenesis and prevention. CMAJ. 2006;175(2):165–170. | |
Li M, Song LJ, Qin XY. Advances in the cellular immunological pathogenesis of type 1 diabetes. J Cell Mol Med. 2014;18(5):749–758. | |
Luo X, Herold KC, Miller SD. Immunotherapy of type 1 diabetes: where are we and where should we be going? Immunity. 2010;32(4):488–499. | |
Bonami RH, Sullivan AM, Case JB, et al. Bruton’s tyrosine kinase promotes persistence of mature anti-insulin B cells. J Immunol. 2014; 192(4):1459–1470. | |
Kendall PL, Moore DJ, Hulbert C, Hoek KL, Khan WN, Thomas JW. Reduced diabetes in btk-deficient nonobese diabetic mice and restoration of diabetes with provision of an anti-insulin IgH chain transgene. J Immunol. 2009;183(10):6403–6412. | |
Martin S, Wolf-Eichbaum D, Duinkerken G, et al. Development of type 1 diabetes despite severe hereditary B-lymphocyte deficiency. N Engl J Med. 2001;345(14):1036–1040. | |
van Bon L, Cossu M, Radstake TR. An update on an immune system that goes awry in systemic sclerosis. Curr Opin Rheumatol. 2011;23(6):505–510. | |
van Bon L, Affandi AJ, Broen J, et al. Proteome-wide analysis and CXCL4 as a biomarker in systemic sclerosis. N Engl J Med. 2014; 370(5):433–443. | |
Avouac J, Elhai M, Allanore Y. Experimental models of dermal fibrosis and systemic sclerosis. Joint Bone Spine. 2013;80(1):23–28. | |
Katsumoto TR, Whitfield ML, Connolly MK. The pathogenesis of systemic sclerosis. Annu Rev Pathol. 2011;6:509–537. | |
O’Reilly S, Hugle T, van Laar JM. T cells in systemic sclerosis: a reappraisal. Rheumatology (Oxford) 2012;51(9):1540–1549. | |
Cornec D, Devauchelle-Pensec V, Tobon GJ, Pers JO, Jousse-Joulin S, Saraux A. B cells in Sjogren’s syndrome: from pathophysiology to diagnosis and treatment. J Autoimmun. 2012;39(3):161–167. | |
Delaleu N, Nguyen CQ, Peck AB, Jonsson R. Sjogren’s syndrome: studying the disease in mice. Arthritis Res Ther. 2011;13(3):217. | |
Hillen MR, Ververs FA, Kruize AA, Van Roon JA. Dendritic cells, T-cells and epithelial cells: a crucial interplay in immunopathology of primary Sjogren’s syndrome. Expert Rev Clin Immunol. 2014;10(4):521–531. | |
Huang YF, Cheng Q, Jiang CM, et al. The immune factors involved in the pathogenesis, diagnosis, and treatment of Sjogren’s syndrome. Clin Dev Immunol. 2013;2013:160491. | |
Nocturne G, Mariette X. Advances in understanding the pathogenesis of primary Sjogren’s syndrome. Nat Rev Rheumatol. 2013;9(9):544–556. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.