Back to Journals » Clinical Interventions in Aging » Volume 12
A case of possibly pathogenic PSEN2 R62C mutation in a patient with probable early-onset Alzheimer’s dementia supported by structure prediction
Authors Park KW ![]() , An SS
, An SS ![]() , Bagyinszky E, Kim SY
, Bagyinszky E, Kim SY ![]()
Received 28 November 2016
Accepted for publication 13 January 2017
Published 13 February 2017 Volume 2017:12 Pages 367—375
DOI https://doi.org/10.2147/CIA.S128884
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Richard Walker
Kyung Won Park,1,* Seong Soo An,2,* Eva Bagyinszky,2 SangYun Kim3
1Department of Neurology, Busan Metropolitan Dementia Center, Dong-A University College of Medicine, Busan, 2Department of BioNano Technology and Gachon BioNano Research Institute, Gachon University, 3Department of Neurology, Seoul National University College of Medicine and Neurocognitive Behavior Center, Seoul National University Bundang Hospital, Seongnam, South Korea
*These authors contributed equally to this work
Abstract: A 49-year-old Korean male patient with dementia was diagnosed with probable early-onset Alzheimer’s disease (AD). He presented with memory problems, personality changes, and disorientation. His family history of dementia was probably negative, since no family member with dementia was found or mentioned. Mild cortical atrophy was observed upon magnetic resonance imaging analyses of his brain, and the single-photon emission computed tomography analysis revealed hypoperfusion in the frontal, temporal, and limbic lobes. The patient was tested for mutations in APP, PSEN1, PSEN2, PGRN, MAPT, and PRNP genes. Genetic analysis revealed R62C mutation in PSEN2 gene. PSEN2 R62C mutation was previously reported in European populations, including Dutch and Belgian families with AD. Herein, we present the first case report of PSEN2 R62C mutation in Asia. PolyPhen-2 and SIFT software analyses predicted this mutation as “possibly damaging”, suggesting its potential involvement with AD. In silico protein structural prediction analyses of PSEN2 R62 and C62 revealed two divergent structures, suggesting that large perturbations of R62C mutation might cause dysfunctions of PSEN2, which may alter the normal amyloid production.
Keywords: Alzheimer’s disease, PSEN2 mutation, dementia, PET, MRI, presenilin-2
Introduction
Alzheimer’s disease (AD) is a complex neurodegenerative disease, which can be divided into two major forms: early-onset AD (EOAD) and late-onset AD (LOAD) with dividing age of 65 years, respectively. Three dominant causative genes for EOAD are amyloid precursor protein (APP), presenilin-1 (PSEN1), and presenlin-2 (PSEN2). Mutations in these genes could increase the production of amyloid beta (Abeta, Aβ) peptide, resulting in their aggregation and deposition, leading to neuronal toxicity and loss. Five to ten percent of all AD cases under 65 years of age can be categorized as EOAD with an autosomal dominant inheritance pattern (familial EOAD). However, family history of the disease could also be absent in several EOAD patients, in which it could be described as occurring due to de novo mutations.1 Even though majority of pathogenic EOAD mutations (almost 200 mutations) were found in PSEN1, only limited numbers of pathogenic mutations were reported in APP (25 cases) and PSEN2 (14 cases; http://www.alzforum.org/mutations).
Mutations in PSEN2 (ch1 227,058,272–227,083,804) are rare, and majority of the reported cases were from patients of European ancestry.2 Only a few PSEN2 mutations were previously reported in Asia, but emerging recent studies found several novel mutations in Korean and Chinese patients.2–5 Phenotypes in AD patients with PSEN2 mutations were milder than those in with PSEN1 mutations. Additional interesting symptoms were also associated with PSEN2-mutation carriers, including atypical dementia, dementia with Lewy bodies, and frontotemporal dementia (http://www.alzforum.org/mutations). Age of disease onset could range between 45 and 88 years.6
Here, we present the detailed case report of a first Korean EOAD patient with a PSEN2 R62C mutation in Asia. In the two main databases, AD and frontotemporal dementia (FTD) mutation database (http://www.molgen.ua.ac.be/ADMutations/) and Alzgene database http://www.alzforum.org/mutations) database, this mutation was previously reported in patients from Europe with LOAD and “pathogenic nature unclear”.7
Materials and methods
Patient information
A 49-year-old, right-handed man visited our memory clinic in April 2013 with memory complaints, which were noticed in 2011 by the patient and his family members. His memory disturbances were gradually increasing, frequently making him lose his belongings without noticing and exhibiting severe memory loss. Other personality changes appeared, such as forgetting important promises to his friends, easily confusing the detailed “must do-lists” or difficulties in the calculations. Prominent naming impairment combined with hesitation to speak became apparent. Sometimes, he asked other people to repeat their conversations due to his failures in comprehending the talks. Geographic disorientations and visuospatial difficulties were also observed during driving with the family members.
The patient was working as a physical education teacher in a middle school, and showed significant impairments and carried out inappropriate decision making and social functions. He could not manage his work as a teacher, for example, creating tests and making decisions at school. Impairments appeared in his daily life activities, such as mistakes in transferring money through telebanking. He presented abnormal behavioral symptoms with an obsessive personality, for example, being stingy with money and also did not allow anyone to spend his money without prior discussions and permission. Patient did not have any medical disease prior to his cognitive decline, and family history was unremarkable since neither his parents nor his siblings showed any cognitive impairment or dementia so far. However, his 14-year-old daughter was mentally handicapped (Figure 1).
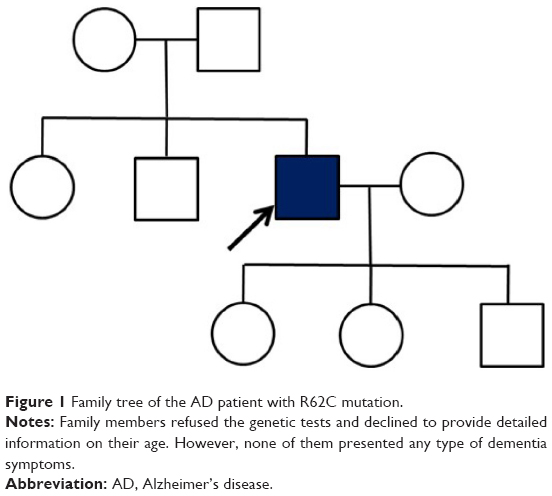 | Figure 1 Family tree of the AD patient with R62C mutation. |
Neurologic examination did not reveal any focal or lateralizing neurologic deficit. Routine laboratory tests, including blood chemistry, electrolyte, and urine analyses, were normal. Serum Venereal Disease Research Laboratory (VDRL) results, including those for Treponema pallidum particle agglutination assay (TPHA), were negative. Thyroid function tests were normal, as with vitamin B12 and folic acid levels. The patient’s apolipoprotein E (APOE) genotype was ε3/ε3. This patient was diagnosed according to the diagnostic criteria for “probable AD” by the National Institute of Neurological and Communicative Diseases and Stroke/Alzheimer’s Disease and Related Disorder Association, which were ascertained by clinical features, neuropsychological test, and brain magnetic resonance imaging (MRI) imaging with single-photon emission computed tomography (SPECT). Brain MRI revealed mild cortical atrophy without any ischemic change or other lesions (Figure 2A). Brain SPECT showed moderate hypoperfusion in the frontal, limbic, and temporal areas (Figure 2B). The reduced cerebral blood flow from the images of brain SPECT also supported the AD diagnosis.
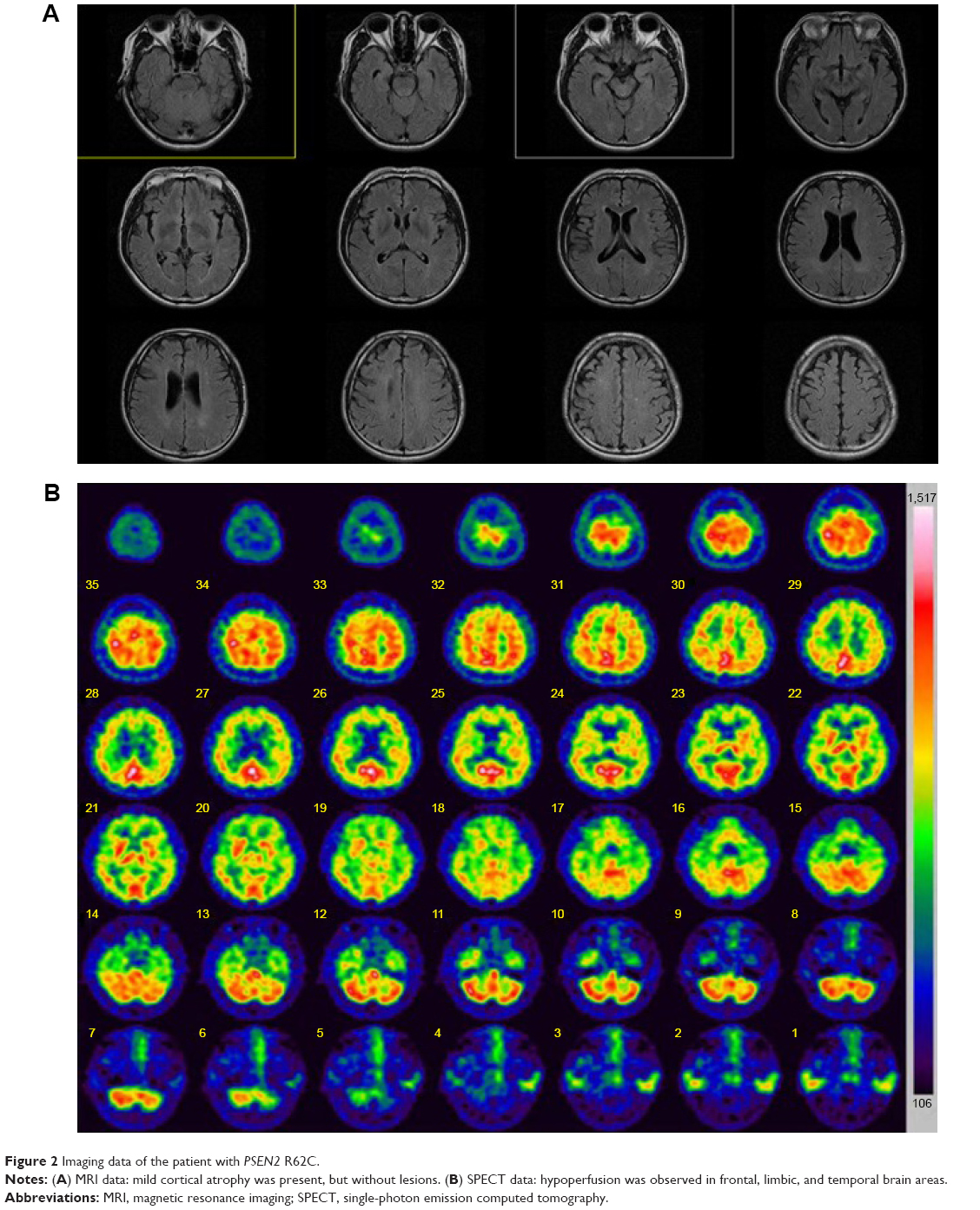 | Figure 2 Imaging data of the patient with PSEN2 R62C. |
The patient underwent comprehensive neuropsychological tests, and he scored 23 out of 30 (2 percentile) on the mini-mental state examination. The subscores for time orientation and seven-serial calculations were 4 out of 5 and 1 out of 5, respectively. The delayed word recall score was 1 out of 3, and he scored 1 out of 3 in the task of clock drawing (Figure 3). His score fell in the abnormal range on the attention task of backward digit span (2.5 percentile). Significant impairment appeared in his confrontation naming ability (<0.01 percentile). Performances in copying the intersecting pentagon test and Rey–Osterrieth Complex Figure test were normal, but his performances on clock drawing were severely impaired (5 percentile). On the Seoul verbal learning test, he was able to recall 13 items (0.02 percentile) in three consecutive immediate recall trials, but only 3 items (0.3 percentile) in the 20-minute delayed recall. Poor visual memory score of 10 out of 36 (1 percentile) was obtained in the delayed recall of the Rey–Osterrieth Complex Figure test. He showed abnormal performances in several tasks of frontal and executive functions, especially in the controlled oral word association test, Stroop test, and trail making test.
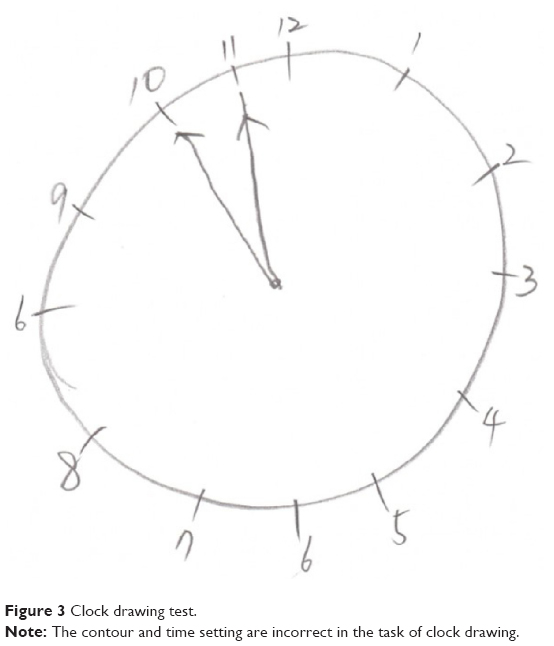 | Figure 3 Clock drawing test. |
Patient was diagnosed as probable EOAD, based on clinical assessment, detailed neuropsychological tests, and neuroimaging studies including brain MRI and SPECT.
Genetic screening
Genetic tests were performed in the patient to evaluate possible mutations. We were unable to make segregation analysis, since all family members and relatives refused the genetic test. The patient declined the genetic test for his children too. None of his family members presented any cognitive decline or dementia phenotypes, but they did not agree in releasing other family information. This patient was included in the Clinical Research Center for Dementia of South Korea study, which screened potential causative mutations in 100 patients with EOAD.8 Details of the clinical and scientific analyses of EOAD mutations in the Clinical Research Center for Dementia of South Korea study have not been included previously. A detailed clinical investigation of this patient with detailed personal interview, MRI, SPECT imaging data, and bioinformatic analyses are presented.
Venous blood samples (5 mL) were collected from the patient with an ethylenediaminetetraacetic acid blood drawing tube. Blood sample was centrifuged at 1,500 × g for 5 min, followed by separation of white blood cells (buffy coat) for its storage at −80°C. DNA was purified using GeneAll Blood Protocol kit (GeneAll Biotechnology, Seoul, Korea), and purified DNA was kept at −20°C until the analysis.
A polymerase chain reaction (PCR)-based analysis was performed using primers, as previously described.2 Targeted potential EOAD genes were as following: APP exons 16 and 17, PSEN1, and PSEN2.9–12 Since the diagnosis of EOAD is complicated due to the pathologic overlaps within diverse neurodegenerative disorders, this patient was also screened for progranulin (PGRN), microtubule-associated protein tau (MAPT), and prion (PRNP) genes.13–15
The PCR products were purified using GeneAll Expin PCR kit (GeneAll Biotechnology). Single-strand conformation polymorphism was performed.16 Samples were mixed with formamide and incubated for 10 minutes at 95°C to generate single-stranded DNA bands. For separation, native polyacrylamide electrophoresis (BioRad, Seoul, Korea) was performed for 21 hours. SYBR Gold (Thermo Fisher Scientific, Waltham, MA, USA) staining was used to visualize the DNA bands.
To confirm the mutation, all PCR products were sequenced with the same primer sets in both directions by BioNeer Inc. (Dajeon, Korea). Before sequencing, we purified the PCR products by using GeneAll PCR kit (GeneAll Biotechnology). Big Dye Terminator Cyclic sequencing was performed with the ABI 3730XL DNA Analyzer (http://eng.bioneer.com/home.aspx; BioNeer Inc.). Sequencing data were screened with DNA BASER (http://www.dnabaser.com) software. Sequence variants were identified by NCBI (http://www.ncbi.nlm.nih.gov/gene) and UniProt (http://www.uniprot.org) databases.
Mutations were analyzed by PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) online in silico prediction program, which predicted the pathogenic nature of missense mutations. PolyPhen-2 used homology search, multiple sequence alignment, profiling, identity-based scores, and structure-based search parameters (such as accessible surface area and hydrophobicity features). These predictions could define the putative role of missense variants and could predict whether they were probably/possibly damaging or benign. PolyPhen-2 also provided the multiple sequence alignment, which compared the homologous sequences from different organisms. Two types of datasets were used for the prediction: HumDiv and HumVar scores. HumDiv was used for testing the pathogenicty of rare alleles, where mildly damaging variants must be treated as possibly damaging. HumVar was used in case of Mendelian disorders, where more damaging alleles should be distinguished from the less damaging ones.17
We also analyzed the mutation with SIFT algorithm (http://sift.jcvi.org/), which used several databases such as SWISS-PROT, SWISS-PROT/TrEMBL, and NCBI Protein Database. The software analyzed the possibility on the pathogenic properties of mutations by comparing the mutant and normal variants. SIFT scores of point mutation were calculated for weighting the mutations as possibly damaging allele or tolerated (nondamaging), with a dividing score of 0.05, respectively.18
Protein structure prediction analysis of PSEN2 was performed by Raptor X program at the University of Chicago (http://raptorx.uchicago.edu). Normal PSEN2 was composed of 488 amino acids, starting at 61 residues and continuing till residue 448. The prediction was performed at Raptor X web server for both normal PSEN2 and R62C mutation. Based on PSEN2 information from the UniProt database, R62C mutation belonged to the topological domain, positions 1–87, including the mutation site. Superimposed images were visualized by Discovery studio 3.5 software from Accelrys.19
Results
PCR-single-strand conformation polymorphism showed different mobility patterns for this patient’s sample, which suggested the presence of mutation exon 4 of PSEN2 (Figure 4A). Sanger sequencing revealed a heterozygosity of arginine (CGC, Arg, R) to cysteine (TGC, Cys, C) exchange at codon 62, which was previously reported in Europe as possibly a pathogenic variant (Figure 4B and C).7,20–24 We did not observe any pathogenic mutation in APP, PSEN1, PRNP, PGRN, or MAPT genes.
 | Figure 4 Genetic analysis of a patient with PSEN2 R62C. |
PSEN2 R62C mutation was checked against the Korean Center for Diseases and Controls database (http://www.cdc.go.kr/CDC/contents/), a database of whole genome sequences from 622 healthy control individuals in Korea, but this mutation was not found. Mutation was also checked in the Exome Aggregation Consortium (ExAC) (http://exac.broadinstitute.org/) Browser for reporting all variants from 60,706 unrelated individuals. ExAC was useful for the disease-associated genetic analyses. Frequency of PSEN2 R62C was 0.0001653 (1/10,000–0.001) with heterozygosity only. The previous association studies revealed that this mutation was found in both AD patients and in a few healthy controls.7,21–24
PolyPhen-2 prediction supported the data of Sala Frigerio et al, suggesting PSEN2 R62C as a possibly damaging variant with HumDiv and HumVar scores of 0.877 and 0.513, respectively.22 PolyPhen-2 multiple sequence alignment revealed that R62 might not be conserved among vertebrates. Multiple sequence alignment suggested that PSEN2 sequences of African elephants and giant pandas were similar to human PSEN2, with Arg at the same position. However, in PSEN-like genes of some vertebrate, different amino acids took place of Arg: glycine in Tasmanian devil and short-tailed opossum, threonine in while duckbill platypus, and asparagine in Turkey, respectively. SIFT scores were 0.05, categorizing PSEN2 R62C as a “damaging” variant.
Three-dimensional structure modeling data revealed that PSEN2 1–87 topological domain had an N-terminal loop structure up to residue 71 (Figure 5), suggesting divergent structures between normal PSEN2 and its mutant R62C. The mutation may not have strong defined structure, but reveals dynamic motion in the loop structure. The highly distinct properties of Arg and Cys could have dramatic impact on the structure and functions of PSEN2. Arg is positively charged with high hydrophilicity and bulk volume, while Cys presents a smaller volume and has a sulfhydryl (−SH) group with negative charge, which can form disulfide bond with other Cys residues inside the PS2 protein or with other proteins. This new SH-bonding potential could result in altered protein functions, which may influence the altered A production.
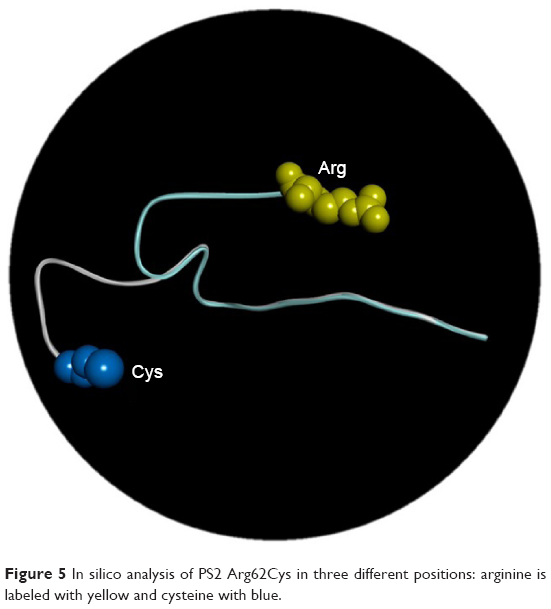 | Figure 5 In silico analysis of PS2 Arg62Cys in three different positions: arginine is labeled with yellow and cysteine with blue. |
Discussion
Previously, PSEN2 R62C mutation was reported in LOAD patients and one healthy individual from the Netherlands in Europe.21–24 PSEN2 R62C was also reported in a Caucasian Belgian AD patient with age of onset at 70 years without a detailed clinical report.20,21,23 Sala Frigerio et al detected PSEN2 R62C in one AD patient, whose brother was also diagnosed with AD. Again, no detailed report on the clinical symptoms or disease progression was available, including their age of onset, phenotypes, or genetic testing of their relatives. Hence, the previous reports on PSEN2 R62C did not segregate the relatives of the patients with the disease status.7,21–24
Here, we discovered the same mutation in a Korean male patient with probable EOAD for the first time in Asia. Even though his family did not present with any previous neurologic diseases, this mutation could not be categorized as a probable de novo case, because his family did not agree in revealing any information about their ages nor allowed further genetic testing of siblings and descendants. Hence, we could not completely rule out the possible hereditary traits.
PSEN R62C is located in the N-terminal loop region of PSEN2 protein. Exchange from Arg to Cys could allow PSEN2 to form a disulfide bond with another Cys inside the PSEN2 protein or with other proteins near PSEN2 at cytoplasmic pH (pH 7.2) level. These new SH bonds could have the potential to create novel intermolecular or intramolecular structures that could be involved in pathogenic mechanisms.
At the same locus of PSEN2 R62, another mutation to histidine (His, H) was previously reported with a frequency of 0.009897 (0.001–0.01) in ExAC database. PSEN2 R62H appeared as both homozygous and heterozygous stages, suggesting for the potential of higher number of unaffected individuals and less or no pathogenicity. For H62R, the exchange of Arg to His might not have a dramatic effect on PSEN2, since His and Arg are both positively charged and large amino acids. On the other hand, exchange with Cys could be very different as mentioned in the result. Both PSEN2 R62C and R62H mutations have unclear pathogenicity. Nevertheless, PolyPhen-2 and SIFT analyses suggested that PSEN2 R62C could be more pathogenic than R62H. Both algorithms revealed PSEN2 R62C as possibly damaging, but the R62H was predicted as benign.25 In addition, the ExAC database showed much lower frequency for R62C in unaffected individuals than R62H. Sleegers et al revealed that since both R62C and R62H mutations were rare variants, it would be difficult to perform association study with them; however, they may occur more frequently in patients than in unaffected individuals.21
Guerreiro et al designed an algorithm for classifying PSEN1 and PSEN2 mutations as definitely pathogenic, probably pathogenic, possibly pathogenic, or nonpathogenic groups. If the mutation was found in larger number of patients than controls, it still could be categorized as a “risk factor” or “possibly pathogenic” mutations.26 PSEN2 R62C and R62H are found in higher frequency of patients with dementia than in controls. The measured Aβ levels in the cellular in vitro or in vivo studies of many of the mutations may help in verifying the effect of mutations on increasing Aβ production. PSEN2 R62H mutation did not affect the A production or A 42/40 ratio.27,28 Ertekin-Taner et al examined the correlation of plasma Aβ42 levels between R62C and R62H.7 In this study, a significant association was found between PSEN2 polymorphisms and plasma Aβ42 levels in first-degree relatives with LOAD at a level of 0.03.4 This study was repeated in a large community-based LOAD setting, but no significant association was detected. These findings suggested that even though these PSEN2 mutations might increase the levels of Aβ42 in AD patients, they might not be prominent genetic factors in disease onset. Since the analyses for PSEN2 R62C were not performed thoroughly, the mutational effects on Aβ levels, its metabolism, or altered functions of PSEN2 function cannot be conclusive. In addition, PSEN2 R62C and R62H could be also affected potentially from the gene–environment interactions.7
Besides R62C and R62H, seven missense mutations in the N-terminal region of PSEN2 protein are described in Table 1. Majority of them in the N-terminus loop region are categorized as mutations with unclear pathogenicity or nonpathogenic variants. PSEN2 T18M was discovered in a German patient with Parkinson’s disease at 62 years of age, and AD symptoms did not appear until the age of 70 years. No family member was affected with Parkinson’s disease.29 PSEN2 R29H was discovered in one African individual. The functional effect R29H variant remained unknown, but PolyPhen-2 suggested that A29H might have some pathogenic effect.26 PSEN2 G34S was discovered in a Caucasian LOAD patient from the Netherlands, but this mutation did not segregate the family members completely with the disease. PSEN2 G34S did not show clear association with increased Aβ42/Aβ40 ratio.21 PSEN2 P69A was found in a LOAD patient where segregation also could not be assessed, and Dobricic et al suggested that P69A might be a nonpathogenic variant.30 PSEN2 R71W was detected in a LOAD patient without segregation of relatives with the disorder. Previously, PolyPhen-2 predictions suggested that this mutation might be involved in EOAD, but we could not confirm it.21,24,26,31 PSEN2 K82R was found in a Chinese EOAD patient, who presented additional symptoms such as depression and language impairment. She developed memory loss at the age of 53, and family history remained unclear.26 PSEN2 A85V from a Sardinian family was the only pathogenic mutation in the N-terminal region of PSEN2, which was verified from affected family members with AD or dementia with Lewy bodies. The disease usually occurred in 1960s or early 1970s.32
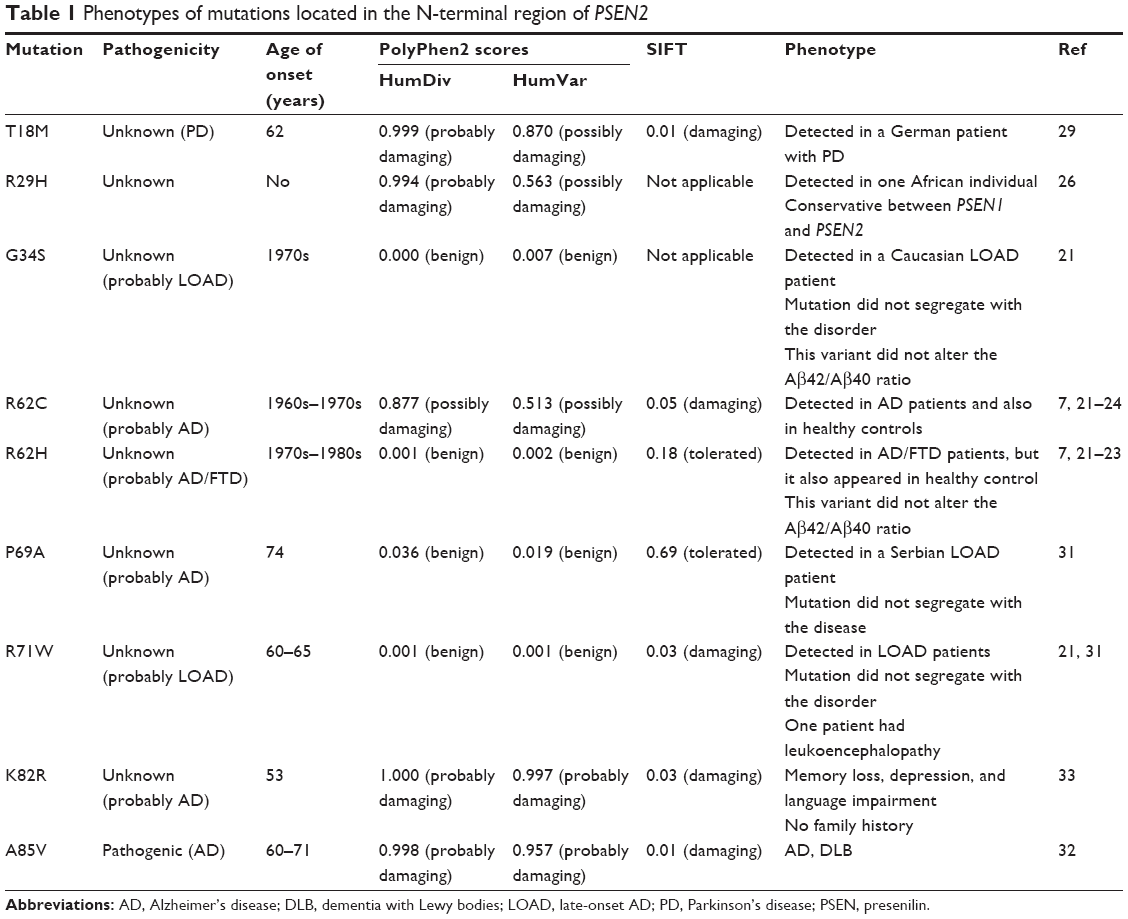 | Table 1 Phenotypes of mutations located in the N-terminal region of PSEN2 |
Conclusion
We discovered a PSEN2 R62C in a male Korean patient with EOAD for the first time in Asia. From the analyses against two major AD databases, AD and FTD mutation databases and the Alzgene databases, PSEN2 R62C was verified as a rare mutation with unclear pathogenic nature, which was similar to the His variant at residue 62, R62H. However, R62C might be involved in pathogenicity, since in silico analyses from PolyPhen-2, SIFT, and three-dimensional modeling suggested pathogenic effects without clear mechanisms. The free SH group in Cys could associate by forming novel disulfide bridge in the loop with nearby proteins or with other sulfurs or metals, leading to altered protein function. Limitation of our study was that we could not make segregation analysis from other family members, since all relatives and family members declined the genetic test, nor revealing other information. Since both PSEN2 R62C and R62H are relatively rare variants in Asia, the association studies may be difficult. Lastly, the functional/cellular studies of these mutations in future could be helpful in verifying their possible roles in AD pathogenicity.
Acknowledgments
Written informed consent for publication of their clinical details and/or clinical images was obtained from the patient.
This work was supported by grants from the Korea Health Technology R&D Project (HI14C3331) through the Korea Health Industry Development Institute (KHIDI), Korea Ministry of Health and Welfare.
Disclosure
The authors report no conflicts of interest in this work.
References
Bagyinszky E, Youn YC, An SS, Kim S. The genetics of Alzheimer’s disease. Clin Interv Aging. 2014;9:535–551. | ||
Youn YC, Bagyinszky E, Kim H, Choi BO, An SS, Kim S. Probable novel PSEN2 Val214Leu mutation in Alzheimer’s disease supported by structural prediction. BMC Neurol. 2014;14:105. | ||
Niu F, Yu S, Zhang Z, Yi X, et al. A novel mutation in the PSEN2 gene (N141Y) associated with early-onset autosomal dominant Alzheimer’s disease in a Chinese Han family. Neurobiol Aging. 2014;35(10):2420.e1–e5. | ||
Shi Z, Wang Y, Liu S, et al. Clinical and neuroimaging characterization of Chinese dementia patients with PSEN1 and PSEN2 mutations. Dement Geriatr Cogn Disord. 2015;39(1–2):32–40. | ||
Xia M, Chen S, Shi Y, et al. Probable novel PSEN2 Pro123Leu mutation in a Chinese Han family of Alzheimer’s disease. Neurobiol Aging. 2015;36(12):3334.e13–e18. | ||
Binetti G, Signorini S, Squitti R, et al. Atypical dementia associated with a novel presenilin-2 mutation. Ann Neurol. 2003;54(6):832–836. | ||
Ertekin-Taner N, Younkin LH, Yager DM, et al. Plasma amyloid beta protein is elevated in late-onset Alzheimer disease families. Neurology. 2008;70(8):596–606. | ||
An SS, Park SA, Bagyinszky E, et al. A genetic screen of the mutations in the Korean patients with early-onset Alzheimer’s disease. Clin Interv Aging. 2016;11:1817–1822. | ||
Tanzi RE, Vaula G, Romano DM, et al. Assessment of amyloid β-protein precursor gene mutations in a large set of familial and sporadic Alzheimer disease cases. Am J Hum Genet. 1992;51(2):273–282. | ||
Schellenberg GD, Pericak-Vance MA, Wijsman EM, et al. Linkage analysis of familial Alzheimer disease, using chromosome 21 markers. Am J Hum Genet. 1991;48(3):563–853. | ||
Cruts M, van Duijn CM, Backhovens H, et al. Estimation of the genetic contribution of presenilin-1 and −2 mutations in a population-based study of presenile Alzheimer disease. Hum Mol Genet. 1998;7(1):43–51. | ||
Kamimura K, Tanahashi H, Yamanaka H, Takahashi K, Asada T, Tabira T. Familial Alzheimer’s disease genes in Japanese. J Neurol Sci. 1998;160(1):76–81. | ||
Cruts M, Gijselinck I, van der Zee J, et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. 2006;442(7105):920–924. | ||
Rizzu P, Van Swieten JC, Joosse M, et al. High prevalence of mutations in the microtubule-associated protein tau in a population study of frontotemporal dementia in the Netherlands. Am J Hum Genet. 1999;64(2):414–421. | ||
Jeong BH, Ju WK, Huh K, et al. Molecular analysis of prion protein gene (PRNP) in Korean patients with Creutzfeldt-Jakob disease. J Korean Med Sci. 1998;13(3):234–240. | ||
Hayashi K. PCR-SSCP: a simple and sensitive method for detection of mutations in the genomic DNA. PCR Methods Appl. 1991;1(1):34–38. | ||
Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–249. | ||
Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31(13):3812–3814. | ||
Källberg M, Wang H, Wang S, et al. Template-based protein structure modeling using the RaptorX web server. Nat Protoc. 2012;7(8):1511–1522. | ||
Levy-Lahad E, Wasco W, Poorkaj P, et al. Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science. 1995;269(5226):973–977. | ||
Sleegers K, Roks G, Theuns J, et al. Familial clustering and genetic risk for dementia in a genetically isolated Dutch population. Brain. 2004;127(Pt 7):1641–1649. | ||
Sala Frigerio C, Lau P, Troakes C, et al. On the identification of low allele frequency mosaic mutations in the brains of Alzheimer’s disease patients. Alzheimers Dement. 2015;11(11):1265–1276. | ||
Levy-Lahad E, Wasco W, Poorkaj P, et al. Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science. 1995;269(5226):973–977. | ||
Brouwers N, Sleegers K, Van Broeckhoven C. Molecular genetics of Alzheimer’s disease: an update. Ann Med. 2008;40(8):562–583. | ||
Cai Y, Bagyinszky E, An SS, Kim SY. In silico modeling of pathogenic or possibly pathogenic point mutations in PSEN2. Mol Cell Toxicol. 2016;12(4):453–464. | ||
Guerreiro RJ, Baquero M, Blesa R, et al. Genetic screening of Alzheimer’s disease genes in Iberian and African samples yields novel mutations in presenilins and APP. Neurobiol Aging. 2010;31(5):725–731. | ||
Walker ES, Martinez M, Brunkan AL, Goate A. Presenilin 2 familial Alzheimer’s disease mutations result in partial loss of function and dramatic changes in Abeta 42/40 ratios. J Neurochem. 2005;92(2):294–301. | ||
To MD, Gokgoz N, Doyle TG, et al. Functional characterization of novel presenilin-2 variants identified in human breast cancers. Oncogene. 2006;25(25):3557–3564. | ||
Blauwendraat C, Wilke C, Jansen IE, et al. Pilot whole-exome sequencing of a German early-onset Alzheimer’s disease cohort reveals a substantial frequency of PSEN2 variants. Neurobiol Aging. 2016;37:208.e11–e17. | ||
Dobricic V, Stefanova E, Jankovic M, et al. Genetic testing in familial and young-onset Alzheimer’s disease: mutation spectrum in a Serbian cohort. Neurobiol Aging. 2012;33(7):1481.e7–e12. | ||
Cruchaga C, Haller G, Chakraverty S, et al. Rare variants in APP, PSEN1 and PSEN2 increase risk for AD in late-onset Alzheimer’s disease families. PLoS One. 2012;7(2):e31039. | ||
Piscopo P, Marcon G, Piras MR, et al. A novel PSEN2 mutation associated with a peculiar phenotype. Neurology. 2008;70(17):1549–1554. | ||
Shi Z, Wang Y, Liu S, et al. Clinical and neuroimaging characterization of Chinese dementia patients with PSEN1 and PSEN2 mutations. Dement Geriatr Cogn Disord. 2015;39(1–2):32–40. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.