")
Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Optimization, ex vivo permeation, and stability study of lipid nanocarrier loaded gelatin capsules for treatment of intermittent claudication
Received 19 February 2015
Accepted for publication 16 April 2015
Published 13 July 2015 Volume 2015:10(1) Pages 4459—4478
DOI https://doi.org/10.2147/IJN.S83123
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Marwa Ahmed Sallam,1 María Teresa Marín Boscá2
1Department of Industrial Pharmacy, Faculty of Pharmacy, Alexandria University, Alexandria, Egypt; 2Department of Pharmacy and Pharmaceutical Technology, Faculty of Pharmacy, Granada University, Granada, Spain
Abstract: In this study, an optimized nanodispersible oral dosage form (containing a lactate ester) was developed for cilostazol (CZL). CZL is a phosphodiesterase inhibitor used for intermittent claudication. We aimed to improve the dissolution rate and absorption of CZL giving it a better chance of oral bioavailability, and to evaluate its stability on storage. Suitable compositions of nanoemulsion preconcentrate formulations were screened via solubility and compatibility tests. Response surface methodology and a desirability approach were applied to optimize preconcentrates containing minimum amount of surfactant mixture, maximum amount of lipid, and possessing the smallest globule size, with the highest emulsification and dissolution rates and minimum risk of drug precipitation. As part of the optimization process, the main effect, interaction effects and quadratic effects of amounts of lipid, and surfactant/co-surfactant ratio on % transmittance, globule size, emulsification time, drug precipitation, and drug release were investigated. The optimized formulation consisting of 28.9% butyl lactate, 28.9% Capryol®, 27.82% Solubilisant Gamma® 2429, and 14.18% Transcutol® possessing a globule size of 60 nm was mixed with Aerosil® 200. This gave uniform free flowing granules, which were characterized for surface and powder properties. The self-nanoemulsifying granules (SNEGs) filled into hard gelatin capsules showed two- and threefold increase in CZL released compared with conventional tablet and pure drug, respectively. The amount of drug permeated using non-everted sac technique from the SNEGs was twofold higher than that permeated from the tablet suspension. The shelf life was 526 days at 25°C. Our study illustrated that the developed SNEGs, with bioenhancing ingredients, held great potential as a superior alternative to traditional oral formulations of CZL.
Keywords: butyl lactate, preconcentrates, cilostazol, desirability, Solubilisant Gamma
Introduction
Oral route has been the major preferred route of drug delivery for the chronic treatment of many diseases, due to convenience and improved patient safety. However, nearly 40% of new drug candidates exhibit low solubility in water, which leads to poor oral bioavailability, high intra- and inter-subject variability, and lack of dose proportionality. In such compounds, the absorption rate from the gastrointestinal lumen is controlled by dissolution.1
In recent years, the most popular approach to increase the drug solubility and dissolution properties of the active poorly water soluble drug is the incorporation into inert lipid vehicles such as surfactant dispersions, liposomes, microemulsions, and nanoemulsions. Much attention has been focused on self-emulsifying formulation and self-nanoemulsifying formulation (SNEF). The latter systems are isotropic mixtures of natural or synthetic oils, solid or liquid surfactants, or alternatively, one or more hydrophilic solvents and co-solvents/surfactants.2
Such systems spread readily in the gastrointestinal tract (GIT) and under the agitation provided by the digestive motility in the stomach and intestine form fine oil in water emulsions or nanoemulsions with nanometric globule size in the range of 20–200 nm.3 Nanoemulsion preconcentrates (PCs) are characterized by their superior stability compared with solid lipid nanoparticles4 and liposomes.
In addition, they present drugs in small globule size and well-proportioned distribution and increase the dissolution and permeability.5 Furthermore, since the drugs can be loaded in the inner phase and delivered by lymphatic bypass share, such PCs protect drugs against hydrolysis by enzymes in the GIT and reduce the presystemic clearance in the GIT mucosa and hepatic first pass metabolism.6
Generally, nanoemulsion PCs are administered either as liquid dosage forms or incorporated into soft gelatin capsules. This method of administration some shortcomings especially in the manufacturing process, leading to high production costs.7 Moreover, these dosage forms may be inconvenient to use in addition to incompatibility problems with the soft gelatin shells.
Incorporation of a liquid self-emulsifying formulation into a solid dosage form may combine the advantages of self-emulsifying drug delivery systems with those of a solid dosage form and overcome the disadvantages of liquid formulations.8 Some trials were made to formulate liquid SNEF into solid dosage forms.9
Cilostazol (CZL), 6-[4-(1-cyclohexyl-1H-tetrazol-5-yl)butoxy]-3,4-dihydro-2(1H)-quinolinone, is an inhibitor of phosphodiesterase III. It also suppresses platelet aggregation and is a direct arterial vasodilator.10 CZL is a medication used in the alleviation of the symptom of intermittent claudication in individuals with peripheral vascular disease. The therapeutic efficacy of CZL is limited by its poor water solubility (3×10−3 mg/mL) resulting in limited oral bioavailability. Being a Class II drug, it often shows dissolution rate-limited oral absorption and high variability in pharmacological effects. CZL has a log P-value of 4.04 and its absorption is increased if taken with a high fat meal.11 This makes the use of lipid-based systems promising for improving its oral bioavailability. Various attempts to enhance the dissolution rate and bioavailability of CZL have been reported such as its preparation in a nanocrystalline form12 or as an inclusion complex.13 Recently, liquid self-nanoemulsifying drug delivery systems (SNEDDS) have been investigated for oral and parenteral administration of CZL.14 However, the literature lacks any data about the use of pre nanoemulsion solid dosage form systems for improving the dissolution and absorption of CZL. Also, lactate esters were not studied as ingredients in such formulations despite their use as food additives and their good emulsification properties. Most importantly is that the stability of CZL in such systems has not been previously studied.
Generally, the type of each composition in nanoemulsion PC formulations can be determined by solubility studies and phase behavior investigations. In addition, the oil percentage and the ratio of surfactant to co-surfactant (S/Co-s) seem to be closely related to the qualities of the formulation.15 In this regard, it is necessary to know exactly how the formulation characteristics are influenced by the formulation factors and potential interactions between them. Therefore, an appropriate method is needed to analyze this issue and to find the optimum PC formulation achieving a required property.
A computer optimization technique based on response surface methodology (RSM) has been reported to be efficient and satisfactory to acquire the necessary information to understand the relationship between controllable (independent) variables and performance or quality (dependent variables) in a formulation. Furthermore, desirability approach was commonly employed to optimize all the responses simultaneously and find the best compromise condition.16
The aim of this study was to optimize and characterize a solid nanoemulsion PC containing a lactate ester for improving the dissolution rate and absorption of CZL, which can be further processed to form tablets, or simply filled into hard gelatin capsules (HGCs) and to assess the ex vivo intestinal permeation and the stability of CZL in such a convenient final dosage form.
RSM and desirability approach were applied to optimize PC that contains a maximum amount of lipid, minimum amount of surfactant, and possesses the smallest globule size with highest solubilization capacity, emulsification, and dissolution rates.
As part of the optimization process, the main effect, interaction effects and quadratic effects of amounts of lipid, S/Co-s ratio on drug release, globule size, and emulsification time were investigated. The optimized formulation was then mixed with Aerosil® 200 to get uniform free flowing granules. These were characterized for surface and powder properties to establish the feasibility of formation of dosage form.
Optimized granulated PCs were filled into HGCs and their dissolution profile was compared with pure CZL powder and conventional tablet. The physical and chemical stabilities were also studied.
Materials and methods
Materials
CZL was kindly supplied by Pharco Pharmaceutical Inc (Alexandria, Egypt). Polyglycolized glycerides (Maisine® 35-1, Labrafil® M 1944 CS, Transcutol® P, Capryol® 90 [CP], Lauroglycol® 90 [LG], Labrafac®, and Solubilisant Gamma® 2429 [SG]) were kindly donated by Gattefossé (Nanterre, France). Butyl lactate (BL), Triacetin, Ethyl oleate, and Ethyl caprylate were purchased from Sigma-Aldrich Co. (St Louis, MO, USA) and Miglyol® 812 from Sasol (Hamburg, Germany). Aerosil® 200 and Aeroperl® 300 (amorphous and granular silicon dioxide) were from Degussa (Marl, Germany). Acetonitrile high performance liquid chromatography (HPLC) grade from Romil Ltd (Cambridge, England). Avicel PH 101 (microcrystalline cellulose) from JRS (Rosenberg, Germany). Absolute ethanol (propanol) from Merck KGaA (Darmstadt, Germany). Propylene glycol from Al Gomhuria Medicine Trade Co. (Cairo, Egypt). All other chemicals used were of analytical grade.
Methods
Solubility studies
Screening of excipients was done by determining the equilibrium solubility of CZL in different oils and co-surfactants. A known excess amount of CZL was added to a glass vial containing 2 mL of each of the tested vehicles. The vials were stoppered with rubber closures and vortexed for 1 minute (Vortex Mixer VM-300, Gemmy Indusrial Co., Taipei, Taiwan). The vials were then rotated in thermostatically controlled rotating bottle apparatus (Alexandra, Egypt) for 12 hours at room temperature (25°C) followed by equilibrium for 24 hours. The mixtures were centrifuged at 4,000 rpm for 10 minutes, followed by filtration of the supernatant through a Millipore membrane filter (0.45 μm HAWP 074700; EMD Millipore, Billerica, MA, USA). Samples from the filtrate were suitably diluted with ethanol and quantified spectrophotometrically for the dissolved CZL using a validated method at 257 nm against a blank plain of the vehicle in ethanol. The experiment was performed in triplicate. Results were expressed as mean value (mg/mL) ± standard deviation (SD).
Compatibility tests
Selection of surfactant
To assess the compatibility, a mixture of 1:1 CP and BL (BL:CP) was mixed with equal weight of each of the selected surfactants using a vortex mixer. The mixtures were gently heated at 50°C for homogenization of the components. The self micro-emulsifying properties of each blend were assessed by visual test. Accurately weighed 50 mg of each mixture was diluted with distilled water to 50 mL in a stoppered volumetric flask. The tendency of spontaneous microemulsification was observed. Emulsions were allowed to stand for 2 hours and their % transmittance was evaluated at 638.2 nm by UV-160A double beam spectrophotometer (PerkinElmer Inc., Waltham, MA, USA) using distilled water as a blank.
Selection of co-surfactant
Different co-surfactants (ethanol, propanol, propylene glycol, and Plurol Oleique® [PO] CC 497) were screened for emulsification ability with the selected surfactant and oily phases. Mixtures of oil:surfactant:co-surfactant in a ratio of 3:2:1 were prepared and evaluated in a similar manner as described in the previous section.17 The ratio of oil to surfactant and co-surfactant was decided on the basis of requirements stated by Porter et al3 for spontaneously emulsifying systems and represented Type III system.
Construction of ternary phase diagrams
The phase diagrams were constructed to identify the existence region of SNEF that could self-emulsify under dilution and gentle agitation. The concentration of the oil was varied from 20% to 90%, the surfactant from 10% to 80%, while the concentration of the co-surfactant was varied from 0% to 50%. For each diagram, 32 mixtures were prepared. For any mixture, the total of surfactant, co-surfactant, and oil concentrations always added to 100%.
In all, 2 g of each mixture was prepared by the addition of variable proportions of the oil, surfactant, and co-surfactant into a capped glass vial. The components were mixed by vortex mixer (Vortex Mixer VM-300, GEMMY) for 1 minute and homogenized by gentle heating to 50°C. In all, 50 mg of each mixture was then diluted with distilled water to 50 mL in a volumetric flask. The tendency to emulsify spontaneously and the progress of emulsion globules spreading were visually assessed according to the number of flask inversions required to yield a homogeneous dispersion and were graded using the following grading criteria: A) Formulation spreads rapidly in water forming a clear and transparent nanoemulsion spontaneously without any flask inversions; B) Formulation forms transparent, gel-like intermediate structure prior to dispersing completely and requires several flask inversions to form clear transparent nanoemulsion; C) Formulation forming slightly less clear emulsion that has bluish-white appearance; and D) Formulation exhibits poor emulsification with large oil globules on the surface. Only clear dispersions having globule size less than 200 nm and % transmittance above 99% were considered in the nanoemulsion region of the phase diagram. All studies were performed in triplicate.
Preparation and characterization of CZL-loaded PCs
Some formulations were selected from self-nanoemulsifying region of the phase diagram (Figure 1). The compositions of the investigated formulations are presented in Table 1.
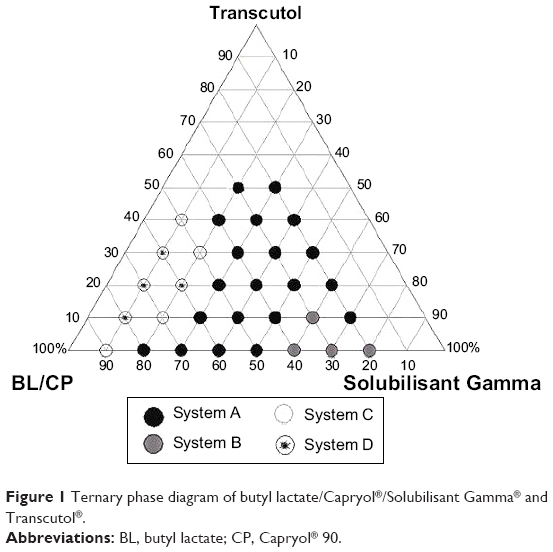 | Figure 1 Ternary phase diagram of butyl lactate/Capryol®/Solubilisant Gamma® and Transcutol®. |
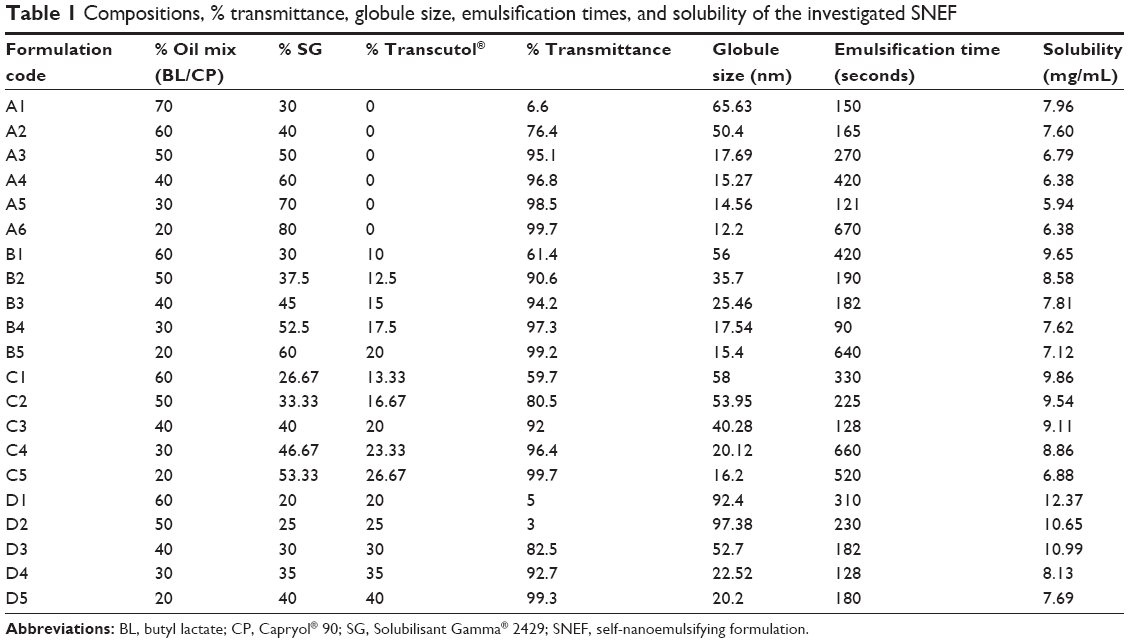 | Table 1 Compositions, % transmittance, globule size, emulsification times, and solubility of the investigated SNEF |
The solubilizing capacity of the formulations was tested. A known excess amount of CZL was added to 1 g of each of the tested formulations and the solubility study was performed as previously described in solubility studies.
Formulation dispersion – drug precipitation test
In all, 50 mg of each of the investigated CZL-loaded PCs was accurately weighed and diluted to 50 mL with 0.1 N HCl in a stoppered volumetric flask. The flasks were shaken to fully disperse the formulation and left to stand for 4 hours at room temperature. After 4 hours, 10 mL was taken from each of the obtained dispersions, transferred to a centrifuge tube, and centrifuged at 3,000 rpm for 20 minutes to separate any precipitated drug. The supernatant was filtered through 0.45 μm Millipore filter. The drug concentration was measured spectrophotometrically at 257 nm after appropriate dilution with 0.1 N HCl.
Spectroscopic characterization of optical clarity
One gram of each of the PC formulations was diluted to 100 mL with distilled water and the % transmittance of the resultant dispersion was measured as previously described in construction of ternary phase diagrams.
Globule size measurement
The globule size of each formulation was measured after 100-fold dilution with distilled water using a Zetasizer 300 HS (Malvern Instruments, Malvern, UK).
Determination of emulsification time
The emulsification time of the liquid PC was evaluated according to United State Pharmacopeia (USP) XXIII, dissolution apparatus II. In all, 250 mg of each formulation was loaded onto a glass support and placed at the bottom of dissolution vessel containing 500 mL distilled water at 37°C. Gentle agitation was provided by a standard dissolution paddle rotating at 50 rpm. The emulsification time was assessed visually by disappearance of the formulation from the surface of the glass support and formation of clear dispersion in the dissolution vessel.
In vitro dissolution study
The dissolution behavior of CZL from the PC and from crude CZL powder was compared in simulated intestinal fluid (phosphate buffer pH 6.8) according to the USP XXII paddle method.
In all, 500 mg of each of CZL-loaded PC was introduced into 900 mL phosphate buffer pH 6.8 at 37°C±0.5°C in type II USP dissolution apparatus. The paddle speed was kept at 100 rpm. Aliquots of 5 mL were withdrawn at 1, 5, 10, 20, 30, 45, and 60 minutes, filtered through 0.22 μm membrane filters and replaced with equal volume of fresh dissolution medium. The concentration of dissolved CZL was determined spectrophotometrically at 257 nm. The dissolution of pure CZL powder was similarly determined. Each experiment was performed in triplicate and the average percentage of CZL dissolved was plotted against time.
Formulation optimization of CZL-loaded PCs
Data analysis and statistical optimization
A computer optimization technique, based on a RSM utilizing polynomial equation was used to develop an optimum CZL-loaded PC. Based on the preliminary experiments, two formulation parameters; the oil percentage (X1) and S/Co-s (X2), were identified as key factors responsible for the properties of the PCs. This allows the evaluation of the three formulation components; % oil, % surfactant, and % co-surfactant by changing their concentrations simultaneously and keeping their total concentration constant. In the RSM analysis, the responses used for optimization, including highest drug loading capacity (Y1), the globule size (Y2), % transmittance (Y3), emulsification time (Y4), minimum percentage of drug precipitation (Y5), and highest percentage of drug dissolved after 1 minute (Y6) of the tested formulations, were treated by Design-Expert® software (version 9; Stat-Ease, Inc., Minneapolis, MN, USA).
The data obtained for the four responses in each trial were fitted to different mathematical models. The best fitting mathematical model was selected based on the comparisons of several statistical parameters including the SD, the multiple correlation coefficient (R2), adjusted multiple correlation coefficient (adjusted R2), and the predicted residual sum of square, proved by the Design-Expert software. Among them, predicted residual sum of square indicates how well the model fits the data, and for the chosen model it should be small relative to the other models under consideration.18 F-test was used to evaluate lack of fit within each model. Response surface delineation was performed according to the fitting model. Graphs of surface responses were plotted with the response variation against the two experimental variables.
Preparation and characterization of the optimized PCs
The optimized SNEF consisting of 28.9% BL, 28.9% Capryol®, 27.82% SG, and 14.18% Transcutol® was prepared by mixing the components in a closed glass vial. The solubility (Y1), the globule size (Y2), emulsification time (Y4), % drug precipitation (Y5), and in vitro (Y6) dissolution profile were determined as previously described. In addition, the optimized formulation was evaluated for the following criteria.
Robustness to dilution
The effects of the type and volume of the dilution medium on the appearance and the globule size of the optimized PC were investigated. The globule size was measured after 50- and 100-fold dilution with water and after 1,000-fold dilution with each of water, simulated intestinal fluid (phosphate buffer pH 6.8), and simulated gastric fluid (0.1 N HCl, pH 1.2). The resultant dispersions (nanoemulsions) were stored at room temperature for 24 hours and monitored for any physical changes such as precipitation or loss of clarity.
Cloud point measurement
The optimized CZL-loaded PC was diluted with water in the ratio of 1:250. The sample was placed in a water bath and the temperature was increased gradually, at 5°C intervals. Spectrophotometric analysis was carried out to measure the sample transmittance using distilled water as a blank.
Transmission electron microscopy
The morphology of the optimized PC was observed by transmission electron microscopy (JEM-100 CX electron microscope; JEOL, Tokyo, Japan). The sample was diluted with water (1:1,000) and a drop was placed on a copper grid. The excess was drawn off with a filter paper. Samples were subsequently stained with uranyl acetate solution for 30 seconds.
Preparation of self-nanoemulsifying granules
Selection of a suitable inert carrier
One gram of the liquid PC was placed in a small mortar, and then the inert carrier was added in increments of 10% w/w, mixed with the liquid PC using a small pestle till the formation of a granular mass. Different carriers (Aerosil® 200, Aeroperl® 300, and Avicel® PH 101) were tested for their ability to absorb PC, forming free flowing granules or powder. The amount of carrier material was calculated according to the following equation:
Lf = W/Q, |
where, Lf is the liquid loading factor; W and Q are the weights of liquid PC and the carrier material, respectively.19
Optimization and characterization of self-nanoemulsifying granules
Aerosil 200 was selected as a suitable inert carrier and mixed with the PC in a ratio of 0.6 Aerosil:1 SNEF. The mixture was then passed through 400 μm mesh to get uniform free flowing granules.20 The granules were then evaluated for their powder characteristics and ability to form nanoemulsion upon dilution. The bulk (δbulk) and tapped densities (δtap) were determined and the Hausner ratio (HR) was calculated according to the equation; HR = δtap/δbulk. Arithmetic mean particle size of powder was determined by sieve analysis (Retsch, Haan, Germany).
Drug content estimation
In all, 100 mg of self-nanoemulsifying granules (SNEGs) was dispersed in 10 mL ethanol, mixed thoroughly to dissolve the drug and then centrifuged at 3,000 rpm for 15 minutes to separate the colloidal silicon dioxide particles. The supernatant was filtered, suitably diluted, and analyzed spectrophotometrically at 257 nm.
Scanning electron microscopy of SNEGs
The morphological features of Aerosil® 200 particles and of SNEGs were investigated by Jeol JSM-840 scanning electron microscope. Gold sputter coating of all the samples was done to render the surface of particles electro-conductive.
In vitro drug release
SNEGs were filled into size “0” HGCs (Parke-Davis, Detroit, MI, USA) and gently agitated inside standard stainless steel rotating basket at 50 rpm in 900 mL phosphate buffer pH 7 at 37°C. The dissolution of commercial CZL tablets was similarly performed at the same time. Additionally, pure CZL powder was filled into HGCs and the dissolution was measured in buffer pH 7 containing 280 mg SG (equivalent amount to that in the SNEGs). Aliquots of 5 mL were withdrawn at 15, 20, 25, 30, 45, and 60 minutes, filtered through 0.22 μm membrane filters and replaced with fresh medium. The concentration of CZL was determined spectrophotometrically at 257 nm against the corresponding blank. All dissolution experiments were performed in triplicate.
Ex-vivo intestinal permeation
Twelve male Wistar rats (weighing 200–250 g) were housed in a temperature and humidity controlled room (23°C, 55% air humidity) with free access to water and standard diet. The rats were fasted overnight but supplied with water only before the experiment. Animals were sacrificed by spinal dislocation. The small intestine was immediately removed after sacrifice by cutting across the duodenal upper end and the lower end of the ileum and stripping the mesentery. The small intestine was washed out carefully with oxygenated saline solution (0.9%, weight/volume, NaCl) using a syringe equipped with a blunt end. The clean intestinal tract was cut into 8±0.2 cm long sacs. The optimized SNEGs were dispersed with 1 mL of phosphate buffer (pH 6.8). A suspension of marketed tablet formulation was prepared in 1 mL of phosphate buffer at an equivalent CZL concentration. Six sacs were filled via a blunt needle with SNEGs (equivalent to 10 mg of the drug) and the other six sacs were filled with equivalent amount of the tablet suspension. The two sides of the intestine were tied tightly with a thread. Each sac was placed in a glass conical flask containing 10 mL of Ringer’s solution. The entire system was maintained at 37°C in a shaking water bath operated at 100 rpm and aerated using a laboratory aerator. Samples were withdrawn from outside the sac and the medium was totally replaced by fresh medium every 15 minutes for 2 hours. Samples were analyzed by HPLC. The HPLC system (PerkinElmer) was equipped with reversed phase C18 analytical column (220×4.6 mm). A mobile phase of acetonitrile (60:40) was pumped at a flow rate of 0.8 mL/min. The injection volume was 20 μL and the effluent was monitored at 257 nm and the retention time was 6.2 minutes.
Stability studies
Stability studies were performed at three different temperatures for the determination of the shelf life. CZL-loaded SNEGs were filled into HGCs. Six capsules were packed in aluminum foil and stored at 40°C (stability chamber), 25°C (room temperature), and 5°C (refrigerator). Samples were withdrawn periodically at predetermined time intervals (0, 15, 30, 45, 60, and 90 days) and the globule size was measured after reconstitution. Zero time samples were used as controls. The remaining drug content was analyzed using a stability indicating HPLC method. The order of the reaction was determined. The reaction rate constant (K) for the degradation was measured from the slope of the lines at each temperature using the equation:
|

Arrhenius plot was constructed, K value at 25°C was determined and was used to calculate shelf life by substituting in the equation:
|
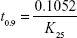
where t0.9 is the time required for 10% degradation of the drug and is referred to as shelf life.
Comparative bioavailability study
The in vivo absorption study was performed using New Zealand rabbits (weighing 1.5–2 kg). Before the experiment, the animals were fasted overnight, but had free access to water. The study was performed in three groups of six rabbits each. One group received SNEGs containing CZL, the other group received conventional tablet suspension, and the third group received drug-free SNEGs. The developed SNEGs and conventional tablet powder were dispersed in water and were administered orally to the rabbits at a dose equivalent to 4.6 mg/kg.13 The blood samples were collected from the marginal ear vein using heparinized needles at 1, 2, 3, 4, 6, 12, 18, and 24 hours after oral administration. The heparinized blood samples were immediately transferred to centrifuge tubes and centrifuged at 3,000 rpm for 15 minutes. The supernatant layer of plasma was separated into another centrifuge tube and stored at -20°C until analysis. CZL was extracted out of the plasma samples by adding 3 mL of methyl tertiary butyl ether to 0.5 mL of the thawed plasma, followed by vortexing for 2 minutes. The supernatant was collected by centrifugation for 20 minutes at 2,000 rpm at 4°C and then evaporated under a gentle stream of nitrogen. The residue was reconstituted with the mobile phase, and 20 μL of this mixture was injected into the HPLC system. CZL concentration in the plasma was determined by reverse phase HPLC described previously. The mean plasma drug concentration ± SD was plotted versus time. Experiments were performed in accordance with the European Community guidelines for the use of experimental animals and were approved by the institutional ethics committee.
Results and discussion
Excipient screening and formulation optimization of nanoemulsion PCs should be based on the following criteria: 1) the formulation composition should be simple and safe (ie, using the least amounts of surfactant and co-surfactant to produce the nanoemulsion); 2) it should afford a large nanoemulsion area in the phase diagram and no phase separation should be visible after storage; 3) it should be able to achieve a high drug loading; and 4) has a small and uniform globule size (ie, <200 nm) and rapidly disperses upon dilution with an aqueous medium.21
Therefore, the selection of oil, surfactant, and co-surfactant as well as the mixing ratio of oil to the surfactant/co-surfactant mixture plays an important role in the formulation.
Solubility study
The oil is the most important component of nanoemulsion PC formulations, as it can solubilize marked amounts of lipophilic drug, increasing its fraction transported via the intestinal lymphatic system, thereby increasing absorption from the GIT.2 Hence, to formulate robust PCs, oil with best solubilizing capacity should be selected as it determines the drug loading efficiency and the soluble portion of drug during storage and in vivo dilution. If surfactant or co-surfactant contributes to the drug solubilization, then there could be a risk of drug precipitation from PC during dilution in GIT due to lowering of the solvent capacity.22
The solubility of CZL was measured in 10 different oils with variable chemical compositions and hydrophilic–lipophilic balance (HLB) values. Figure 2 demonstrates the solubility of CZL in different oils. BL was found to possess the highest solubility for CZL followed by CP. However, BL could not be used alone as an oil phase due to its partial miscibility with water arising the risk of drug precipitation after dilution. So, it was used with each of CP and LG at a ratio of 1:1. Additionally, the use of mixed oils was reported to possess greater solubility in the applied surfactant system.23
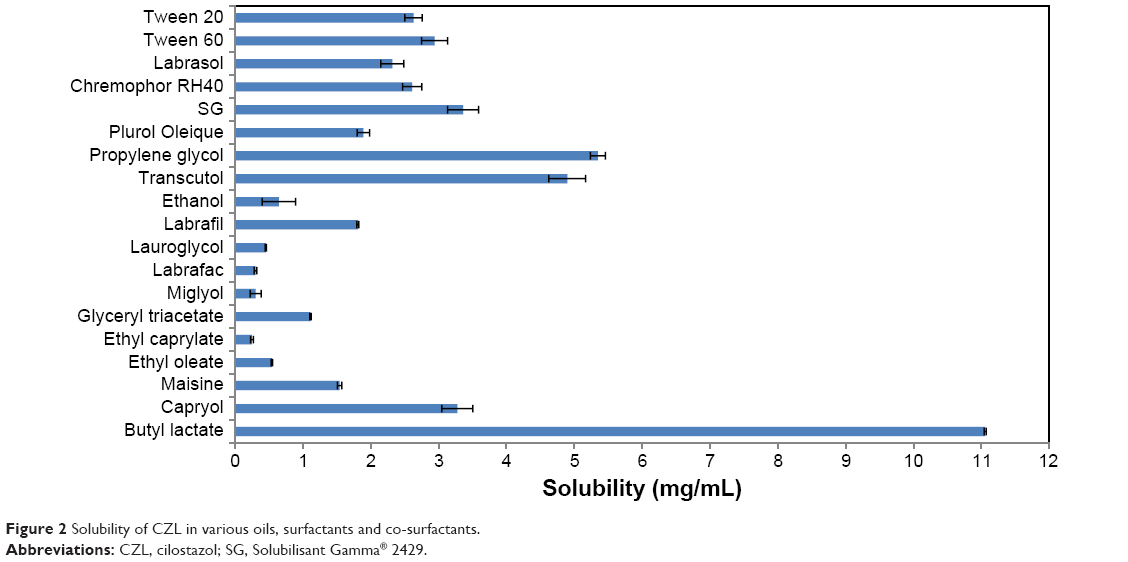 | Figure 2 Solubility of CZL in various oils, surfactants and co-surfactants. |
Final selection of the oily phase was then based on its emulsification properties with other ingredients. Regarding the surfactant and co-surfactant selection, all the surfactants tested showed good solubility for the drug with SG ranking the first. It is not necessary that the same surfactant that has a good solubilizing power for drugs would have equally a good affinity for the oil phase. Therefore, the selection was based not only on solubility but also on emulsification efficiency.17
Compatibility tests
Compatibility tests were critical steps in the whole formulation design by which it would be easy to know the characteristics and choose the optimal combination from the numerous oils and surfactants.
Screening of surfactants
Non-ionic surfactants are generally regarded as safe and biocompatible. They are less affected by pH and changes in ionic strength. In the present study, the five selected non-ionic surfactants (Labrasol®, Chremophor® RH40, Tween® 60, Tween® 20, and SG) were reported to possess bio-enhancing activities. This includes effects on tight junction allowing para cellular transport such as Labrasol® and inhibitory effects on P-gp and CYP450 enzymes such as Chremophor® and Tween® 80.24
The transmittance values of the different mixtures are demonstrated in Table 2. The results indicated that a mixture of CP:BL (1:1) followed by CP (HLB 6) exhibited the highest emulsification efficiency with all the surfactants employed particularly Chremophor® RH40 and SG (>99%). On the other hand, LG (HLB 5) exhibited the least emulsification properties with all the surfactants employed and when mixed with CP in a ratio of 1:1, it lowered the emulsification ability of the latter.
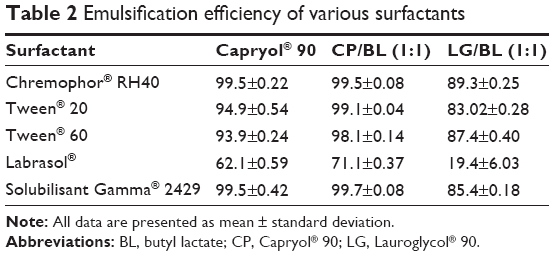 | Table 2 Emulsification efficiency of various surfactants |
The superior emulsification ability of CP to LG can be attributed to the smaller molecular volume of the former and its higher HLB value. These results are in line with those reported by Rao and Shao,25 in that: oils of higher HLB values are better to form SNEDDS than those with lower HLB values. Among the surfactant tested, Labrasol® exhibited the lowest emulsification efficiency as indicated by the lowest transmittance values with all the tested oily phases. This contends the results obtained by Basalious et al24 who reported the inferior emulsification efficiency of Labrasol® compared with Chremophor® RH40 and Tween® 80 when used with Miglyol® and Labrafil® in the formulation of SNEDDS for lacidipine.
Although the HLB values of the screened surfactants are close in the range of 14–16, the difference observed in their emulsifying ability could be attributed to the difference in their structure and chain length and hence, the lipid surfactant affinity.17
Screening of co-surfactants
Co-surfactants are added to surfactant containing formulations to improve dispersibility and drug absorption.3 Ethanol, propylene glycol, PO, and Transcutol® were screened as co-surfactants. Short- to medium-chain-length alcohols are reported to reduce the interfacial tension, fluidize the hydrocarbon region of the interfacial film, and decrease the bending stress of the interface, resulting in the improvement in spontaneity of emulsification.26,27
Both PO and Transcutol® are reported to increase permeation when incorporated in formulations. Except for PO, all the tested co-surfactants showed good emulsification properties (>99%) (Table 3). These results are inconsistent with those obtained by Borhade et al28 who reported that PO being a lipophilic long chain co-surfactant was less effective than short chain amphiphiles (Transcutol® and propylene glycol) and could not improve the emulsification of different surfactants.
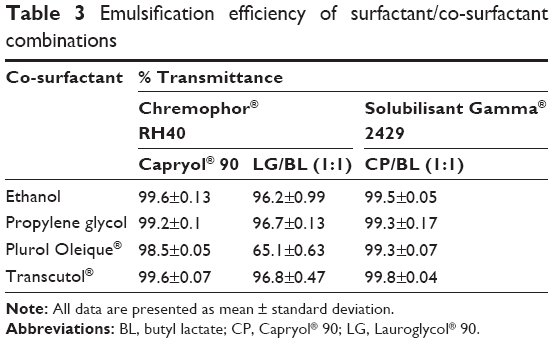 | Table 3 Emulsification efficiency of surfactant/co-surfactant combinations |
Alcohols have higher hydrophilicity, increasing the risk of destroying the nanoemulsion upon dilution compared with Transcutol® (HLB 4.3). In addition, Transcutol® provided the highest drug solubility among the co-surfactants tested in our study (Figure 2). Therefore, based on the aforementioned results, a mixture of CP:BL (1:1) was selected as oily phase. SG and Transcutol® were selected as surfactant and co-surfactant, respectively.
Construction of ternary phase diagrams
The phase diagram was constructed using SG, Transcutol®, and BL:CP (1:1) (Figure 3). Pseudo ternary phase diagrams are normally constructed with the oil phase, surfactant or mixture of surfactant and co-surfactant, and the aqueous phase, which will reveal the regions of liquid crystal, microemulsion (w/o or o/w) and coarse emulsion.
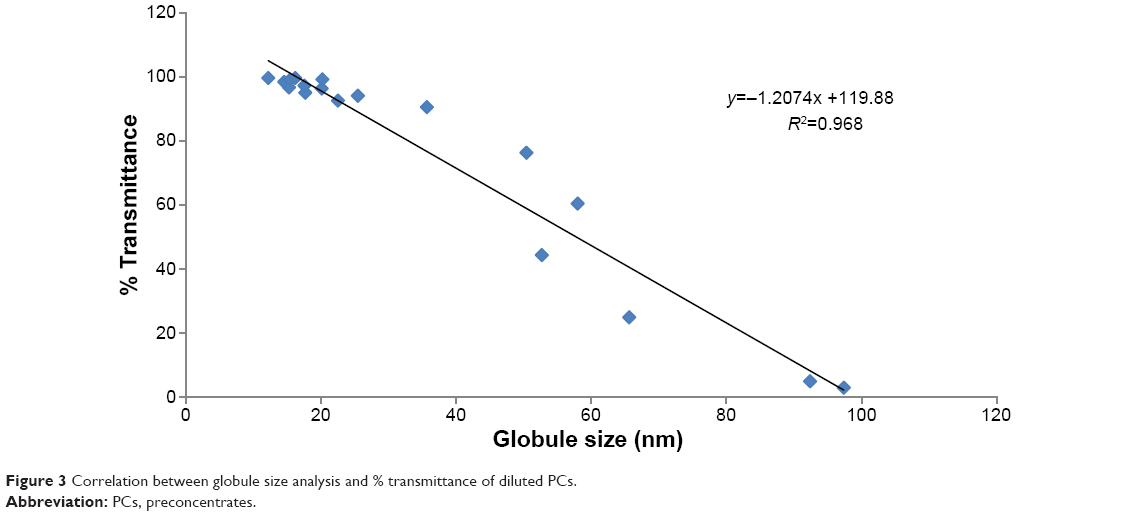 | Figure 3 Correlation between globule size analysis and % transmittance of diluted PCs. |
For simplicity, the present study has ignored the effect of the aqueous phase (1,000 times dilution by water), and used only the oil, surfactant, and co-surfactant components to identify the self-nanoemulsifying region. In addition, the concentration range of each component (oil, surfactant, and co-surfactant) was chosen to cover the various compositions of different types of lipid formulations.3
For systems of oil concentration 40% or more, the efficiency of emulsification was good when the surfactant concentration was more than 30%. Decreasing the amount of oil (30% or less of the formulation) decreases the amount of surfactant required for emulsification. Systems containing 60% or more surfactant required much longer time and more flask inversions to yield clear homogeneous dispersions (grade B). This may be due to the formation of lamellar liquid crystal on dilution.29
For such systems increasing the concentration of the co-surfactant (Transcutol®) within the self-nanoemulsifying region caused an increase in the spontaneity of the self-emulsification process due to lowering the interfacial tension and also influencing the interfacial film curvature and stability. On the other hand, factors of safety should be considered with the increasing concentration of surfactant and co-surfactant.
Characterization of PCs and data analysis
The experimental results are reported in Table 1. The selected responses were individually fitted to linear, two-factor interaction, quadratic, cubic, and quartic models. Each obtained model was validated by analysis of variance. For each response, the model, which generated a higher F value, was identified as the fitting model. The model statistical summary of the measured responses is given in Table 4. The values of the coefficients X1 and X2 are related to the effect of these variables on the response. A positive sign of coefficient indicates a synergistic effect, while a negative term indicates an antagonistic effect upon the response.18 The larger coefficient means the independent variable has a more potent influence on the response.
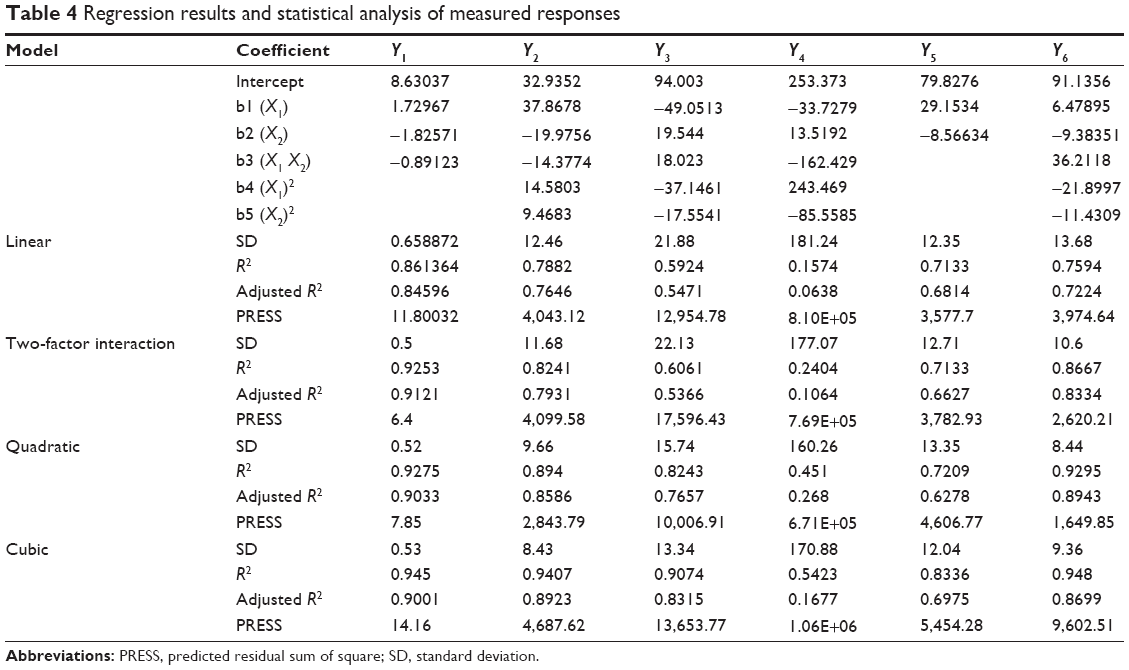 | Table 4 Regression results and statistical analysis of measured responses |
Determination of loading capacity
The solubilizing ability of the investigated PCs for CZL is given in Table 1. The solubility ranged from 5.94 to 12.37 mg/mL. Statistical analysis revealed that the solubilizing capacity of the PCs increased significantly with increasing the oil content as demonstrated by the positive coefficient of variable X1 (Table 4). Surprisingly, the surfactant mixture (Smix) had a negative influence on the solubilizing capacity of the PCs. Our result is in contrast with those reported by Pund et al30 who investigated a nanoemulsifying system for CZL, where the solubility of the drug in the formulation was mainly dependent on the Smix composed of Tween® 80 and Transcutol® while Campul MCM as an oil phase was not of significant effect on the solubilizing capacity. This might be an additional advantage for our system reducing the risk of drug precipitation after dilution which occurs if the drug is solubilized mainly by the Smix.
Formulation dispersion drug precipitation test
The most important parameter to be assessed is the possibility of drug precipitation as the formulation is dispersed in an aqueous medium. This can be achieved by dispersing the drug loaded PC in a simulated gastric fluid (0.1 N HCl) and quantification of the amount of drug that is highly dispersed in the aqueous phase and which does not precipitate after 4 hours. This provides an indication of the relative proclivity of the formulation with respect to in vivo precipitation.
The amount of the unprecipitated drug was high in high oil content PCs and it decreased by increasing the Smix content as indicated by the positive coefficient of the X1 and the negative coefficient of X2. The highest percentage of drug precipitation was observed in systems containing 20% oil and 80% Smix. Drug precipitation upon dilution of high Smix amount could be attributed to the relationship between drug solubility and co-solvent concentration, which is commonly approximated to a logarithmic relationship.
Optical clarity and globule size measurement
Globule size is a critical value for assessing nanoemulsion PCs. The smaller globule size provides a larger interfacial surface area for drug absorption and is suggested to permit a faster release rate. In view of the feasibility of formation at the extreme values, formulations were selected from the self-nanoemulsifying region of the phase diagram. The compositions of the selected formulations are presented in Table 1. Each formula is given a code. The alphabetical letter denotes the S/Co-s such that codes A, B, C, and D denote S/Co-s of 1:0, 3:1, 2:1, and 1:1, respectively, while the % oil was 70%, 60%, 50%, 40%, 30%, and 20%. The % transmittance of the formulations varied between 3% and 99.7%. The % transmittance was significantly reduced by increasing the % oil and reducing the S/Co-s (ie, increasing the co-surfactant amount). This is clear from the values of the coefficients shown in Table 4. As expected, compositions with the highest % transmittance showed the smallest globule size and were optically clear where the oil globules are thought to be in a state of finer dispersion.
As shown in Figure 3, there is a good correlation between the % transmittance of diluted PCs and the globule size analysis (R2=0.968). Thus, the spectrophotometric characterization of optical clarity of the diluted nanoemulsion PCs could be used as a simple technique for predicting the globule size, rather than the expensive and sophisticated equipment used for measurement of globule size.
Based on the calculated models for the % transmittance and globule size, the 3D response surface plots are shown in Figure 4. At a fixed S/Co-s ratio, the decrease in the % oil and subsequent increase in the surfactant mixture (% Smix) resulted in decrease in the globule size. This effect could be explained as being the result of an increased availability of surfactant for adsorption around the oil–water interface of a droplet, and decreased interfacial tension in the system. Both effects favor the formation of nanoemulsions with smaller droplets.31 Similarly, the increase in the globule size by increasing the oil content in the SNEDDS has been reported by other authors.16
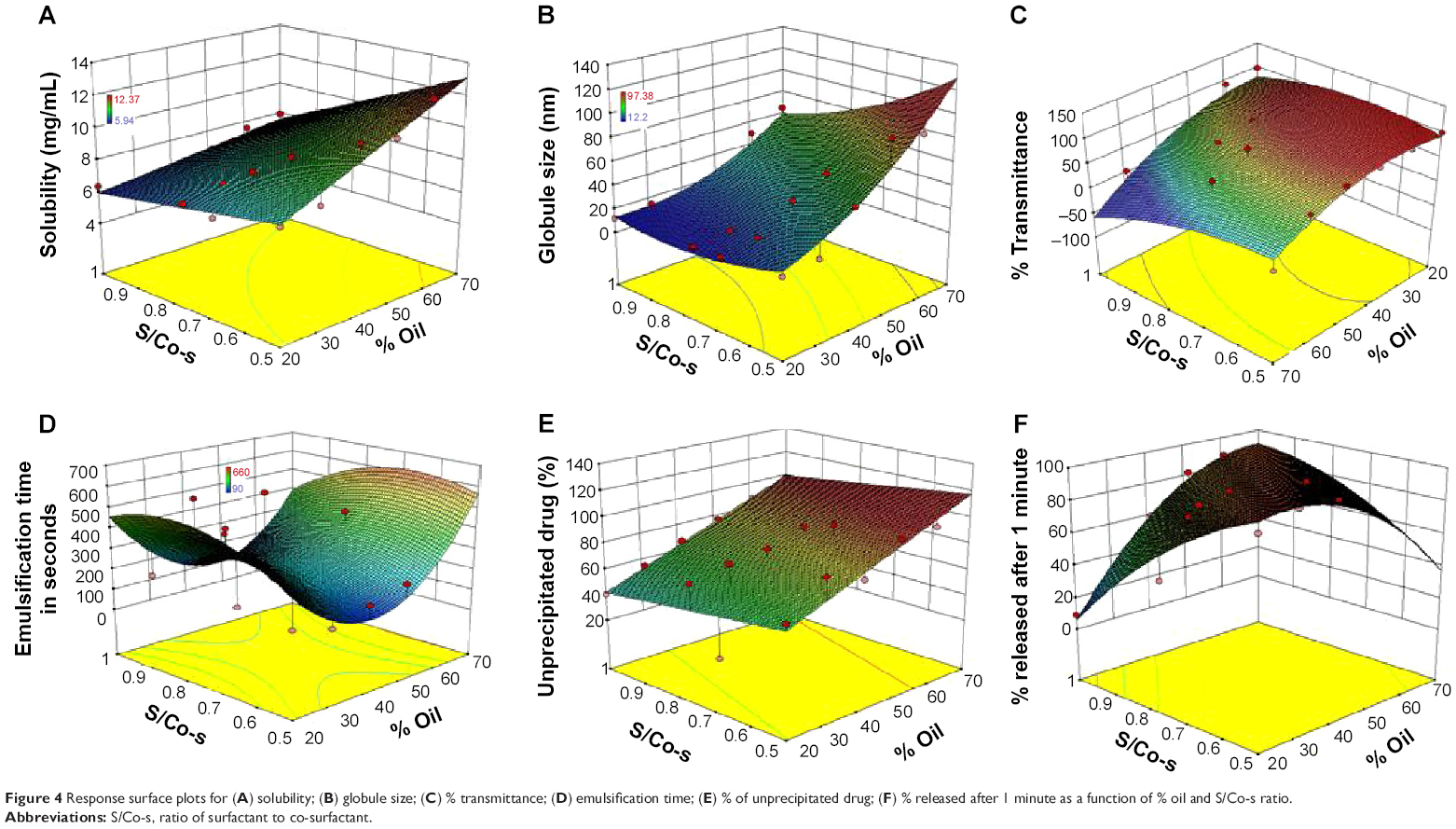 | Figure 4 Response surface plots for (A) solubility; (B) globule size; (C) % transmittance; (D) emulsification time; (E) % of unprecipitated drug; (F) % released after 1 minute as a function of % oil and S/Co-s ratio. |
At fixed oil concentration, the increase in the % co-surfactant (decrease in the S/Co-s ratio) resulted in an increase in the globule size. Figure 5 is the 2D contour plot showing the effect of each of the three components on the globule size. The red region representing the highest globule size values is in the Transcutol® rich area. This effect is more pronounced in high oil content systems. For instance, decreasing the S/Co-s ratio from 1:0 to 3:1 (or the addition of Transcutol® to co-surfactant free system) at oil concentration of 50% resulted in increase in globule size from 17.69 nm (A3) to 35.7 nm (B2). Further increase in the amount of Transcutol® at the same % oil resulted in an increase in globule size to 53.95 nm ([C2] S/Co-s 2:1) and finally to 97.38 nm ([D2] S/Co-s 21:1).
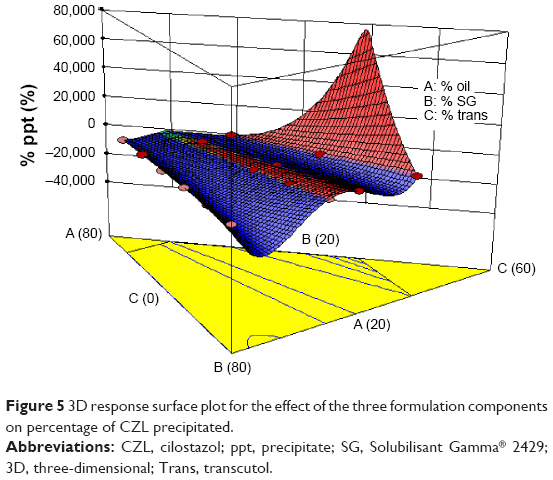 | Figure 5 3D response surface plot for the effect of the three formulation components on percentage of CZL precipitated. |
The increase in the globule size by increasing the concentration of co-surfactant has been reported in previous studies.21 Such an increase in the globule size could be attributed to the expansion of the interfacial film by the co-surfactant present. On the other hand, these results are in contrast to those reported by Pund et al30 who reported a decrease in the globule size by increasing the amount of Transcutol® in a system composed of Capmul® MCM, Tween® 80, and Transcutol® HP. The conflicting results may be due to the different behavior of the oily phases employed on the interfacial tension and fluidity by penetrating as well as the bending stress at the globule interface.
Determination of emulsification time
The rate of emulsification is taken as an important index for the assessment of the efficiency of self-nanoemulsification. This means that the PC should disperse completely and quickly when subjected to dilution under mild agitation. The emulsification time was significantly increased by increasing the percentage of oil (P<0.0001) and by increasing the S/Co-s ratio, ie, increasing the surfactant concentration (Table 4). This may be due to the formation of highly viscous hexagonal liquid-crystalline phase at high concentration of surfactant in the aqueous phase, which decreased the rate of transport or adsorption of surfactant at the interface.26 The addition of Transcutol® reduces the emulsification time due to the decrease in the viscosity of the PCs.
In vitro dissolution study
In this study, the ultra-filtration method was used to determine the dissolution of CZL from different PCs.32 The dissolution profiles of CZL from the different investigated PCs as well as pure CZL powder are shown in Figure 6. Pure drug powder showed only 26.8% released after 60 minutes.
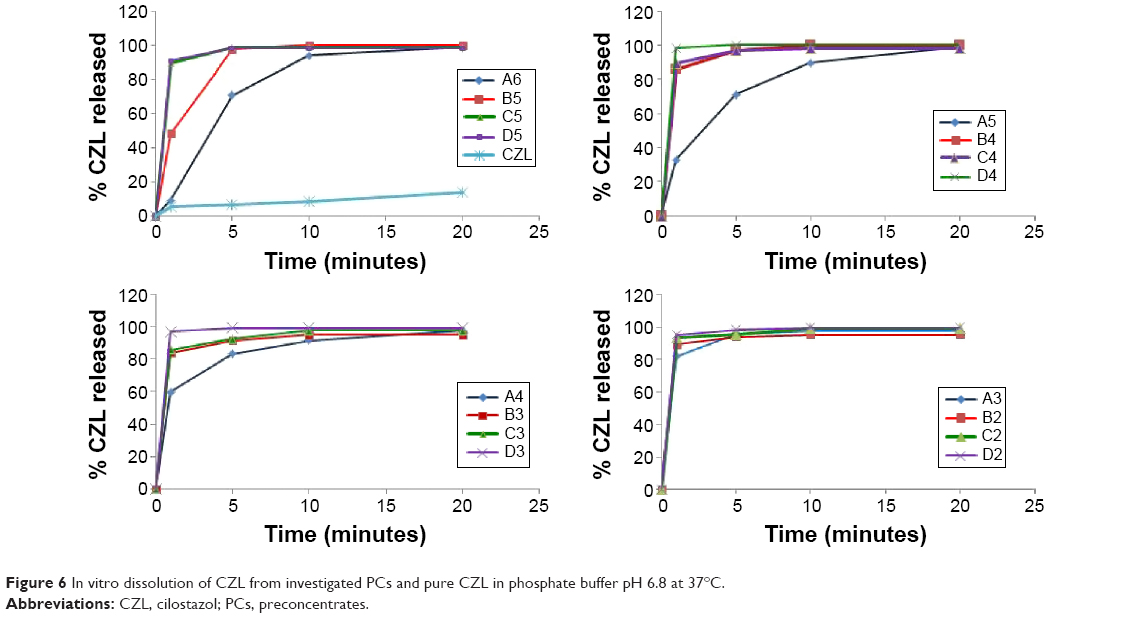 | Figure 6 In vitro dissolution of CZL from investigated PCs and pure CZL in phosphate buffer pH 6.8 at 37°C. |
The dissolution results represent the total amount of free drug molecules and drug in nanoemulsion with diameter below 220 nm (diameter of the filter membrane). This could represent to some extent the release of CZL-PCs in the in vivo circumstances. The dissolution of CZL was markedly different from one formulation to another and there was a good inverse correlation between the drug release rate and the emulsification time.
The values of the coefficients of X1 and X2 as well as the analysis of variance indicated that the effect of S/Co-s ratio on the release was highly significant (P=0.01), while that of the oil was not significant (P=0.512).
It is obvious from Figure 6 that increasing the surfactant concentration reduced the release rate. The slowest rate was observed from formula “A6” containing 80% surfactant. It reached 98.7% drug release after 20 minutes. On the other hand, increasing the amount of co-surfactant increased the release rate. The highest rate was obtained by formula “D4” showing 98.4% released after 1 minute. The highest release rate observed with such formula was due to its higher amount of co-surfactant and lower amount of oil as well as the quicker self-emulsification nature of this composition. Regardless of the differences between the formulations, the release of CZL from such formulations was significantly higher than that from pure drug powder.
Optimization of CZL-loaded PCs
The aim of the optimization of pharmaceutical formulations is generally to determine the levels of the variables from which a robust product with high quality characteristics may be produced.
Our goal is to obtain nanoemulsion PCs with highest drug solubility, short emulsification time, highest amount of drug released within 1 minute, least amount of drug precipitated, and possessing the smallest globule size after dilution. Also, the amount of oil has to be maximized to facilitate absorption and lymphatic transport, while that of the surfactant and co-surfactant has to be minimized to avoid GIT irritation problems. However, it is almost impossible to optimize all the objectives simultaneously because they do not coincide with each other and conflict may occur between them. The optimum condition reached in one response may have an opposite influence on another response.
After obtaining the individual desirability for each response, the five responses were then combined by overlaying the plots to determine the overall optimum region or the maximum overall desirability through which the multi-criteria problem can be treated as single criterion problem (Figure 7). The predicted best compromising PC satisfying the desired criteria for all responses was prepared and evaluated.
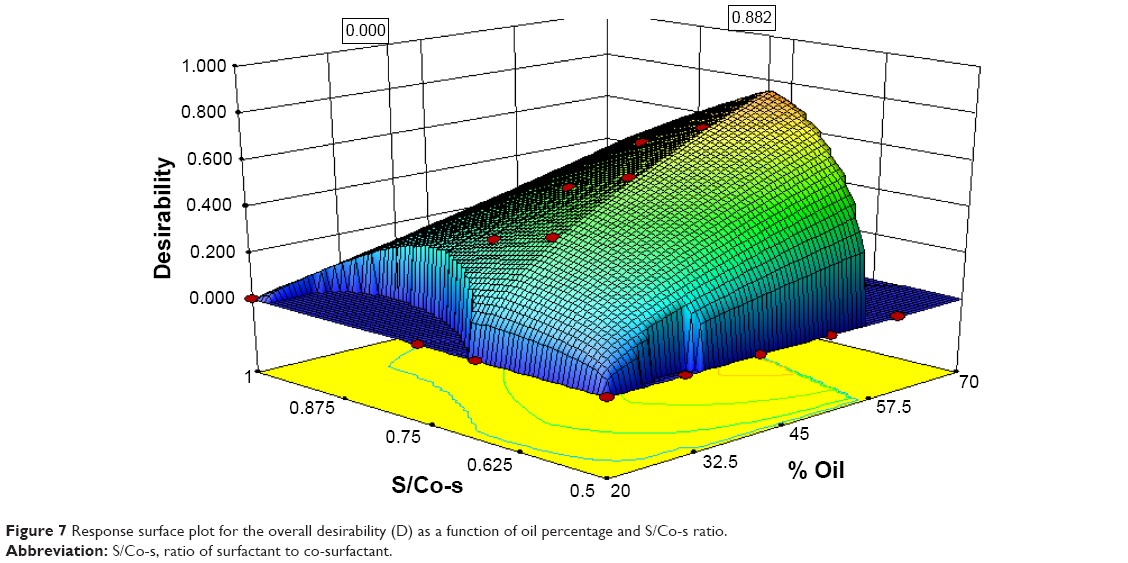 | Figure 7 Response surface plot for the overall desirability (D) as a function of oil percentage and S/Co-s ratio. |
Characterization of the optimized PC
The predicted and observed responses are reported in Table 5. The predicted and observed values of Y1, Y2, Y4, Y5, and Y6 of the optimum formulation showed small percentage errors, 2.22, 3.56, 2.53, 0.27, and 2.04, respectively. A good agreement is obtained between the model prediction and experimental observation.
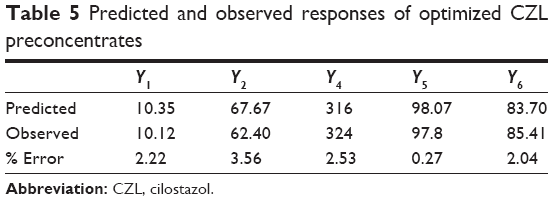 | Table 5 Predicted and observed responses of optimized CZL preconcentrates |
Robustness to dilution
PCs possess the risk of in vivo drug precipitation upon dilution in stomach and intestine, which can lead to failure in the bioavailability enhancement. Additionally, the alteration in environmental pH of the system may trigger the tendency for separation of the two phases of a nanoemulsion to reduce the interfacial area and, hence, the free energy of the system.27
Therefore, the stability of the optimized PCs was evaluated by measuring globule size after different folds dilution with water as well as after dilution with different media (water, 0.1 N HCl pH 1.2 and phosphate buffer pH 6.8) to the same volume (1,000-fold dilution).
The globule size was 60, 60, and 62 nm when measured in water, pH 1.2, and pH 6.8, respectively. This demonstrated that the optimized PCs were not affected by pH and were capable of maintaining a globule size of less than 200 nm regardless the pH of the medium. The dilution volume was found to have no effect on the resulting globule size, which remained 60 nm after 50-, 100- and 1,000-fold dilution. All the dispersions remained clear showing no precipitation, cloudiness, or separation for 4 hours. This indicated that the surfactant/co-surfactant blend used in the optimized PCs brought sufficient reduction in free energy of the system to resist thermodynamic instability under the employed alternations in environment of the nanoemulsion.
It provided sufficient mechanical barrier to coalescence of oil droplets.
Cloud point measurement
The cloud point is the temperature above which the dispersed PC turns cloudy due to irreversible phase separation. This occurs due to dehydration of the polyoxyethylene oxide moiety of the non-ionic surfactant and results in a decline in drug solubilization and absorption enhancement effect. Hence, the cloud point for PC should be above 37°C, which will avoid phase separation occurring in the GIT. The optimized CZL-loaded PC exhibited cloudiness at 63°C with a drop in % transmittance from 99.2 to 75.5. It was reversible upon returning to room temperature. This suggested the stability of CZL PC toward separation at physiological temperature in vivo.
Transmission electron microscopy
The morphology of the PCs as well as SNEGs was observed after 1,000-fold dilution with water. The photographs depicted in Figure 8 reveal that all droplets after dilution possessed nearly a spherical shape with the same size and that globules obtained from SNEGs were of larger size. This is consistent with the globule size analysis.
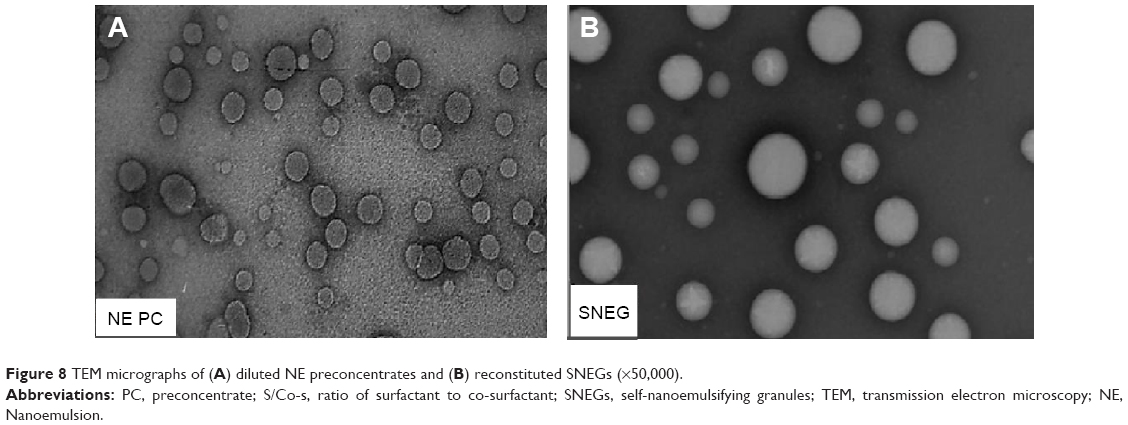 | Figure 8 TEM micrographs of (A) diluted NE preconcentrates and (B) reconstituted SNEGs (×50,000). |
Preparation and evaluation of CZL-loaded SNEGs
Selection of an inert carrier
Aerosil® 200 was selected as a suitable carrier because it provided the highest liquid loading factor Lf =1.667 (0.6 Aerosil®:1 SNEDDS) and the best free flowing granules after mixing with liquid PC. At lower amount of Aerosil® 200 gritty particles with poor flow ability were produced.
Characterization of SNEGs
The resultant granules had a bulk and tapped density of 0.472 and 0.575, respectively. The angle of repose was 15.6° and the HR was 1.18 indicating good flow properties. The mean particle size was found to be 148 μm.
Upon mixing SNEGs with water, stable nanoemulsion of globule size 85 nm was produced. This was slightly larger than the globule size produced from the liquid PCs. The slight increase in globule size may be due to partial adsorption of surfactant and co-surfactant onto Aerosil® 200.
Morphological analysis of SNEGs
The scanning electron micrographs of Aerosil® 200 and CZL-loaded SNEGs are shown in Figure 9. Aerosil® 200 appeared as aggregates of rough surfaced amorphous particles. The SNEGs appeared as smooth surfaced nearly spherical particles, indicating that the liquid PCs are absorbed inside the pores of Aerosil® 200. No crystals were evident on the surface of the granules.
 | Figure 9 Scanning electron micrographs of (A) Aerosil® 200 and (B) SNEGs. |
In vitro dissolution study
The in vitro dissolution profiles of SNEGs, pure CZL powder, and conventional tablets are shown in Figure 10. In case of pure drug powder, 278 mg SG was added to the dissolution medium. The dissolution profile of SNEGs filled in capsules showed a decrease in the rate of dissolution compared with the liquid PCs. A maximum of 82.42% of the drug was released after 45 minutes. This could be attributed to the additional disintegration step (time for the capsule to open) required for the drug dissolution, besides the time required for surfactant and co-surfactant to be released from the surface of silica. Some amount of surfactant and co-surfactant may also remain entrapped inside the granule. This may be the same reason for the observed increase in globule size of PCs after dispersing from SNEGs.
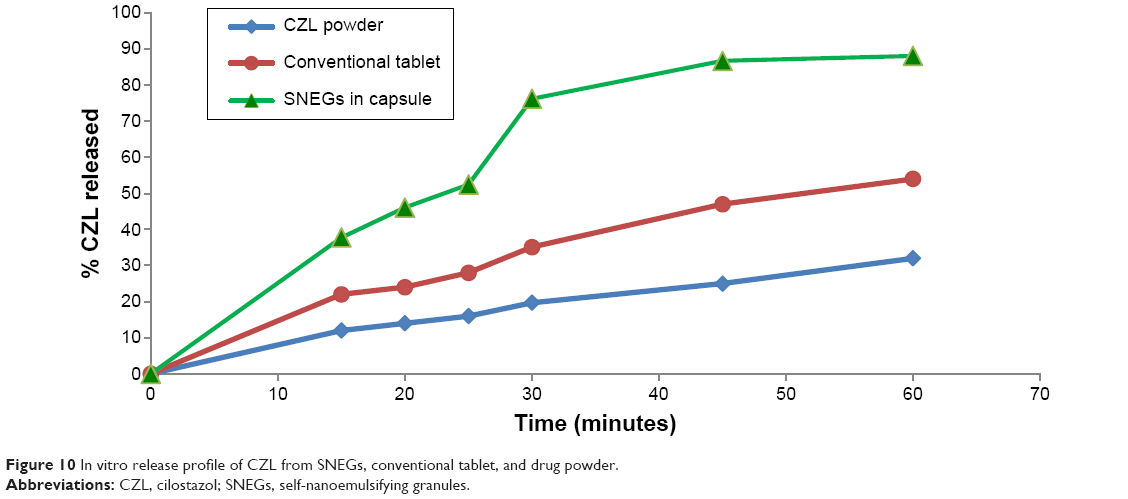 | Figure 10 In vitro release profile of CZL from SNEGs, conventional tablet, and drug powder. |
The release of CZL from the SNEGs was significantly higher than that from the conventional tablet and pure drug powder ensuring that the SNEGs presented an improvement of the in vitro dissolution. After 25 minutes, the SNEGs showed two- and threefold increase in CZL released compared with tablet and pure drug, respectively.
After 30 minutes, 76.22% of the drug was released from the SNEGs compared with only 35% from the conventional tablet and 19.7% from CZL powder in the presence of equivalent amount of surfactant as that in the SNEF. This proves that the enhanced dissolution of CZL is not due to the presence of surfactant but rather due to both the presence of the drug in solubilized form and the formation of nanosized oil globules that rapidly dissolve in the dissolution medium. This illustrates the advantage of SNEGs as convenient dosage form for the delivery of poorly water soluble drugs.
Ex vivo intestinal permeation
The everted and non-everted rat intestinal sacs are commonly used ex vivo absorption models to assess transport mechanisms, and to predict in vivo drug absorption. They offer the advantage of reduced labor and experimental costs compared with in vivo animal studies.33 In this work, the non-everted sac technique was adopted to assess ex vivo intestinal permeation of the elaborated SNEGs compared with the conventional tablets. Figure 11 shows the cumulative amount of CZL permeated over a period of 2 hours. The amount of drug permeated from the SNEGs was twofold higher than that permeated from the tablet suspension.
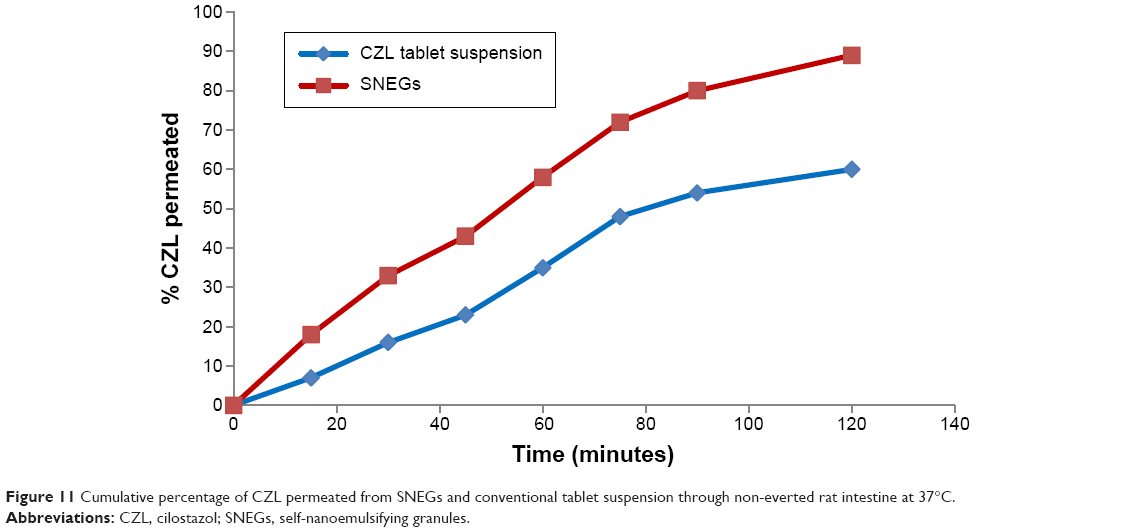 | Figure 11 Cumulative percentage of CZL permeated from SNEGs and conventional tablet suspension through non-everted rat intestine at 37°C. |
Such an enhancement in the permeation could be ascribed to the rapid dissolution of CZL in the intestinal sac and a subsequent rapid diffusion. This is attributed to the nanometric globule size and the bioenhancing activities of the ingredients of the SNEGs.
Stability studies
Formulations kept at 40°C, 25°C, and 5°C were analyzed individually. The percentage of undecomposed CZL remaining in SNEGs after 90 days of storage was 95.06%, 96%, and 98.97% at 40°C, 25°C, and 5°C, respectively. The order of degradation of CZL was found to be first order as indicated by a graph between the logarithms of percentage drug remaining against time which gave a straight line (Figure 12). The shelf lives of the formulation were calculated from the degradation constant (K) values obtained from Figure 12. The reaction rate constant “K” for the degradation was measured from the slope of the lines at each temperature. Plot of the logarithm of K values at each temperature against the reciprocal of absolute temperature was drawn (Arrhenius plot) as shown in Figure 13. The shelf lives of CZL–SNEGs at 40°C, 25°C, and 5°C were found to be 350, 526, and 761 days, respectively. It was found that the shelf life of optimized formulation was significantly higher when stored at refrigerator temperature (P<0.05).
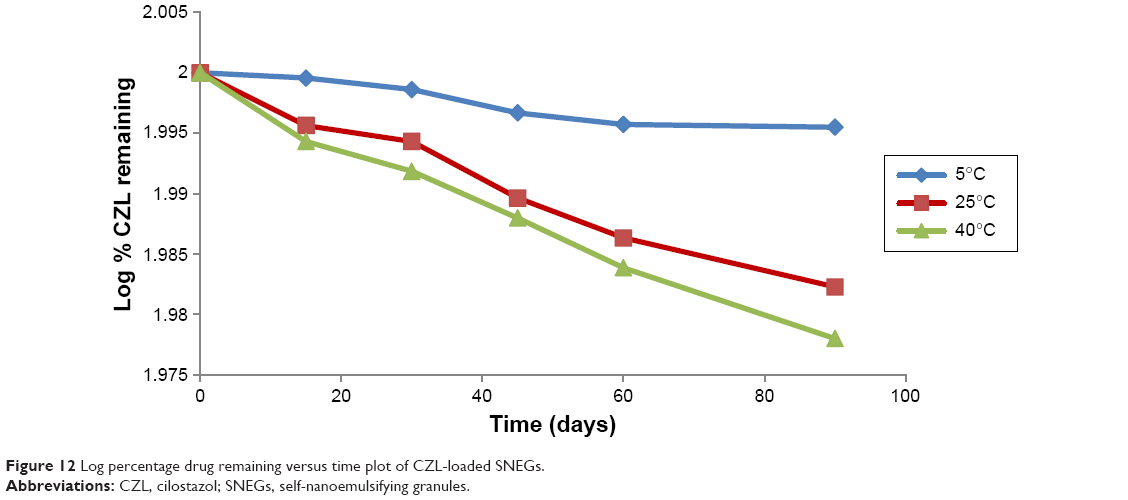 | Figure 12 Log percentage drug remaining versus time plot of CZL-loaded SNEGs. |
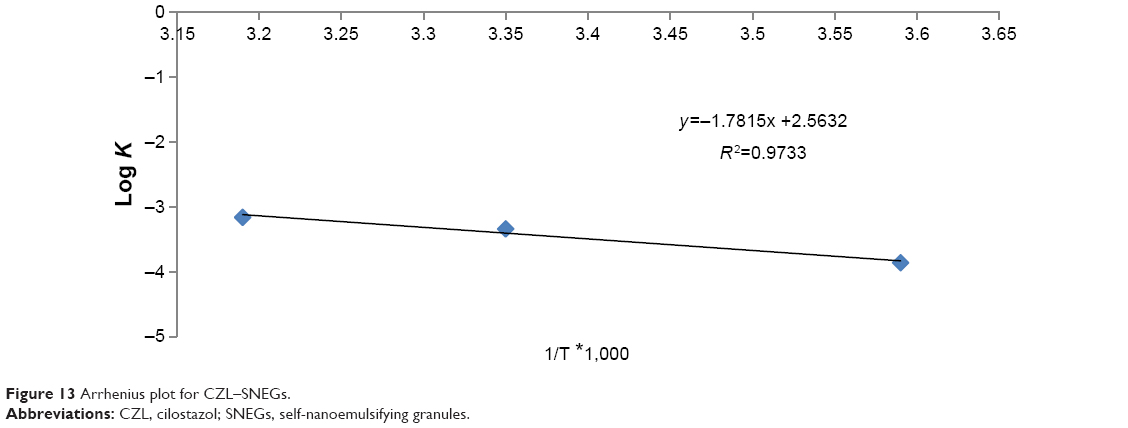 | Figure 13 Arrhenius plot for CZL–SNEGs. |
Comparative bioavailability study
The mean plasma concentration of CZL after oral administration in rabbits was plotted as a function of time and illustrated in Figure 14. The SNEGs showed higher Cmax and AUC0–24 values than those of the commercially available tablet. The mean values of the Cmax of CZL after 2 hours were 3,550.83±636.39 ng/mL and 1,675.21±671.75 ng/mL for the SNEGs and the commercial tablet, respectively. The AUC0–24 values were 26,900.12±10,394.47 ng·h/mL and 15,450.23±1,258.5 ng·h/mL for the SNEGs and the commercial tablet, respectively.
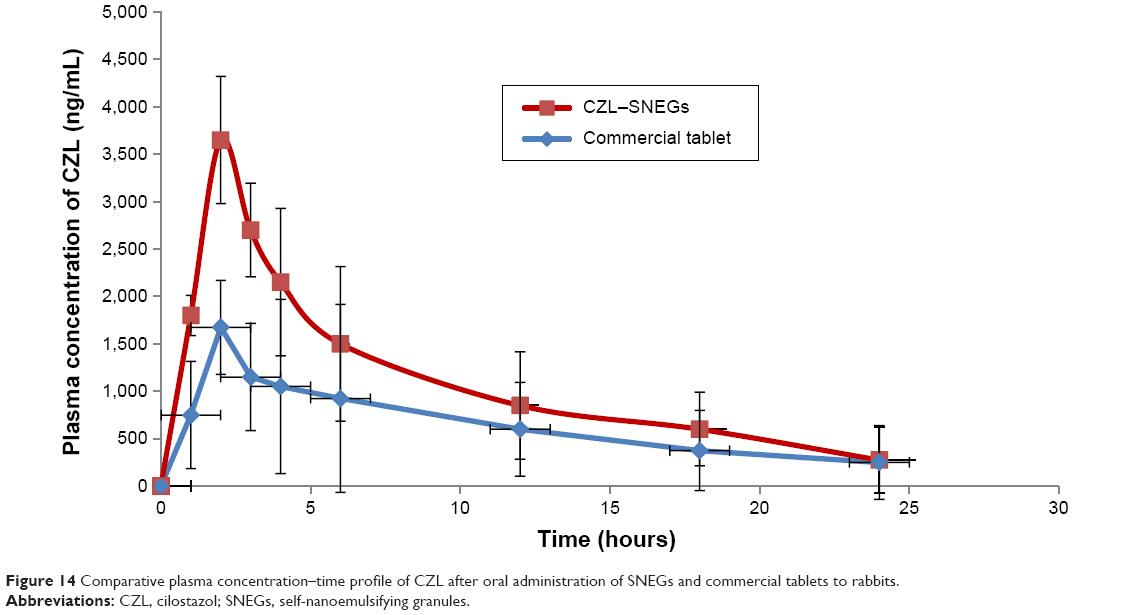 | Figure 14 Comparative plasma concentration–time profile of CZL after oral administration of SNEGs and commercial tablets to rabbits. |
Accordingly, the relative bioavailability of CZL-loaded SNEGs was 1.74 times higher than that of the commercial formulation. Such an enhancement of the oral bioavailability may be attributed to the enhanced dissolution of CZL resulting from the large specific surface area of the spontaneously formed nanoemulsion droplets and their stability in the GIT. This is in addition to the improved permeation because of the lymphatic transport in the presence of the surfactant and oil mixture.
Conclusion
In this study, SNEGs for CZL were developed and evaluated. All the formulations showed superior release profiles compared with pure drug. RSM and desirability approach were applied to obtain an optimal formulation with the highest oil and least surfactant content possessing the highest drug loading, the smallest globule size, and the highest emulsification and dissolution rates. An optimal formulation consisting of 28.9% Capryol®, 28.9% BL, 27.82% SG, and 14.18% Transcutol® was mixed with Aerosil® 200 to get free flowing SNEGs, which after dilution with water possessed a globule size of 85 nm. Capsules filled with SNEGs showed three- and fourfold enhancement in dissolution rate compared with conventional tablet and pure drug, respectively. The shelf life of CZL–SNEGs was 526 days at 25°C. Our results proposed that the developed SNEGs with bioenhancing agents could be promising to improve oral absorption of CZL. The in vivo evaluation of the developed SNEGs showed an enhancement in the oral bioavailability.
Acknowledgment
The authors are grateful for Gattefosse France for providing the polyglycolized glycerides essential for this study.
Disclosure
The authors report no conflicts of interest in this work.
References
Amidon GL, Lennernas H, Shah VP, Crison JR. A theoretical basis for a biopharmaceutical drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res. 1995;12:413–420. | ||
Gursoy RN, Benita S. Self-emulsifying drug delivery systems (SEDDS) for improved oral delivery of lipophilic drugs. Biomed Pharmacother. 2004;58:173–182. | ||
Porter CJH, Pouton CW, Cuine JF, Charman WN. Enhancing intestinal drug solubilisation using lipid-based delivery systems. Adv Drug Deliv Rev. 2008;60:673–691. | ||
Mueller RH, Maeder K, Gohla S. Solid lipid nanoparticles (SLN) for controlled drug delivery: a review of the state of the art. Eur J Pharm Biopharm. 2000;50:161–177. | ||
Kawakami K, Yoshikawa T, Morto Y, et al. Microemulsion formulation for enhanced absorption of poorly soluble drugs I. Prescription design. J Control Rel. 2002;81:65–74. | ||
Kossena GA, Charman WN, Boyd BJ, Dunstan DE, Porter CJH. Probing drug solubilization patterns in the gastrointestinal tract after administration of lipid based delivery systems: a phase diagram approach. Pharm Sci. 2004;93:332–348. | ||
Franceschinis E, Voinovich D, Grassi M, et al. Self-emulsifying pellets prepared by wet granulation in high-shear mixer: influence of formulation variables and preliminary study on the in vitro absorption. Int J Pharm. 2005;291:87–97. | ||
Yi T, Wan J, Xu H, Yang X. A new solid self-microemulsifying formulation prepared by spray-drying to improve the oral bioavailability of poorly water soluble drugs. Eur J Pharm Biopharm. 2008;70:439–444. | ||
Nazzal S, Smalyukh I, Lavrentovich OD, Khan MA. Preparation and in vitro characterization of a eutectic based semisolid self-nanoemulsified drug delivery system (SNEDDS) of ubiquinone: mechanism and progress of emulsion formation. Int J Pharm. 2002;235:247–265. | ||
Maruyama H, Fukuoka T, Deguchi I, et al. Dual antiplatelet therapy clopidogrel with low-dose cilostazol intensifies platelet inhibition in patients with ischemic stroke. Intern Med. 2013;52(10):1043–1047. | ||
Bramer SL, Forbes WP. Relative bioavailability and effects of a high fat meal on single dose cilostazol pharmacokinetics. Clin Pharmacokinet. 1999;37:13–23. | ||
Xiaoqing M, Changshan S, Tongying J, Li Z, Tianyi W, Siling W. Investigation of nanosized crystalline form to improve the oral bioavailability of poorly water soluble cilostazol. J Pharm Pharm Sci. 2011;14:196–214. | ||
Samir GP, Sadhana JR. Enhancement of oral bioavailability of cilostazol by forming its inclusion complexes. AAPS Pharm Sci Tech. 2009;10:660–669. | ||
Mahmoud DB, Shukr MH, Bendas ER. In vitro and in vivo evaluation of self-nanoemulsifying drug delivery systems of cilostazol for oral and parenteral administration. Int J Pharm. 2014;476:60–69. | ||
Zidan AS, Sammour OA, Hammad MA, Megrab NA, Habib MJ, Khan MA. Quality by design: understanding the product variability of a self nanoemulsifying drug delivery system of cyclosporine A. J Pharm Sci. 2007;96:2409–2423. | ||
Liu Y, Zhang P, Feng N, Zhang X, Wu S, Zhao J. Optimization and in situ intestinal absorption of self-microemulsifying drug delivery system of oridonin. Int J Pharm. 2009;365:136–142. | ||
Date AA, Nagarsenker MS. Design and evaluation of self-nanoemulsifying drug delivery systems (SNEDDS) for cefpodoxime proxetil. Int J Pharm. 2007;329:166–172. | ||
Huang YB, Tsai Y, Lee SH, Chang JS, Wu PC. Optimization of pH independent release of nicardipine hydrochloride extended-release matrix tablets using response surface methodology. Int J Pharm. 2005;289:87–95. | ||
Mahmoud EA, Bendas ER, Mohamed MI. Preparation and evaluation of self nanoemulsifying tablets of carvedilol. AAPS Pharm Sci Tech. 2009;10:183–192. | ||
Dixit RP, Nagarsenker MS. Self-nanoemulsifying granules of ezetimibe: design, optimization and evaluation. Eur J Pharm Sci. 2008;35:183–192. | ||
Zhao Y, Wang C, Chow AHL, et al. Self nanoemulsifying drug delivery system (SNEDDS) for oral delivery of Zedoary essential oil: Formulation and bioavailability studies. Int J Pharm. 2010;383:170–177. | ||
Shafiq S, Shakeel F, Talegaonka, S, Ahmad FJ, Khar RK, Ali M. Development and bioavailability assessment of ramipril nanoemulsion formulation. Eur J Pharm Biopharm. 2007;66:227–243. | ||
Rane SS, Anderson BD. What determines drug solubility in lipid vehicles: is it predictable. Adv Drug Deliv Rev. 2008;60:638–656. | ||
Basalious EB, Shawky N, Badr-Eldin SM. SNEDDS containing bioenhancers for improvement of dissolution and oral absorption of lacidipine. I:Development and optimization. Int J Pharm. 2010;391:203–211. | ||
Rao SV, Shao J. Self-nanoemulsifying drug delivery systems (SNEDDS) for oral delivery of protein drugs: I. Formulation development. Int J Pharm. 2008;362:2–9. | ||
Nepal PR, Han H, Choi H. Preparation and in vitro–in vivo evaluation of Witepsol® H35 based self-nanoemulsifying drug delivery systems (SNEDDS) of coenzyme Q10. Eur J Pharm Sci. 2010;39:224–232. | ||
Kommuru TR, Gurley B, Khan MA, Reddy IK. Self-emulsifying drug delivery systems (SEDDS) of Coenzyme Q10: formulation development and bioavailability assessment. Int J Pharm. 2001;212:233–246. | ||
Borhade V, Nair H, Hegde D. Design and evaluation of self-microemulsifying drug delivery system (SMEDDS) of tacrolimus. AAPS Pharm Sci Tech. 2008;9:13–21. | ||
Li P, Ghosh A, Wagner RF, Krill S, Joshi YM, Serajuddin ATM. Effect of combined use of nonionic surfactant on formation of oil-in-water microemulsions. Int J Pharm. 2005;288:27–34. | ||
Pund S, Shete Y, Jagadale S. Multivariate analysis of physicochemical characteristics of lipid based nanoemulsifying cilostazol-quality by design. Colloids Surf B Biointerfaces. 2014;115:29–36. | ||
Wang L, Dong J, Chen J, Eastoe J, Li X. Design and optimization of new self nanoemulsifying drug delivery system. J Colloid Interface Sci. 2009;330:443–448. | ||
Chen Y, Li G, Wu X, et al. Self microemulsifying drug delivery system (SMEDDS) of vinpocetine: formulation development and in vivo assessment. Biol Pharm Bull. 2008;31:118–125. | ||
Le Ferrec E, Chesne C, Artusson P, et al. In vitro models of the intestinal barrier. The report and recommendations of ECVAM Workshop 46. European Centre for the Validation of Alternative methods. Altern Lab Anim. 2001;29:649–668. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.