")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 17
IL-36 is Closely Related to Neutrophilic Inflammation in Chronic Obstructive Pulmonary Disease
Authors Huang S, Feng T , Wang J, Dong L
Received 5 January 2022
Accepted for publication 1 May 2022
Published 7 June 2022 Volume 2022:17 Pages 1339—1347
DOI https://doi.org/10.2147/COPD.S357151
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Richard Russell
Siyuan Huang,1,* Tao Feng,2,* Jing Wang,1 Liang Dong3
1Department of Respiratory, Shandong Qianfoshan Hospital, Cheeloo College of Medicine, Shandong University, Jinan, People’s Republic of China; 2Department of Respiratory Medicine, Shengli Oilfield Central Hospital, Dongying, People’s Republic of China; 3Department of Respiratory, Shandong Provincial Qianfoshan Hospital, Shandong University, The First Affiliated Hospital of Shandong First Medical University, Shandong Institute of Respiratory Diseases, Jinan, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Liang Dong, Department of Respiratory, Shandong Provincial Qianfoshan Hospital, Shandong University, The First Affiliated Hospital of Shandong First Medical University, Shandong Institute of Respiratory Diseases, Jinan, 250014, People’s Republic of China, Tel +86-13505401207, Email [email protected]
Background: Interleukin (IL)-36α, IL-36β, and IL-36γ belong to the IL-36 family and play an important role in the pathogenesis of many diseases. Chronic obstructive pulmonary disease (COPD) may be correlated with IL-36; however, the specific role of IL-36 in COPD is unclear. In this study, we aimed to clarify whether IL-36 could be an indicator for determining COPD severity and the specific nature of the pro-inflammatory effects of IL-36 in COPD.
Methods: A total of 70 patients with COPD and 20 control subjects were included in this study. We collected peripheral blood samples from both the groups, analyzed the blood cell fractions by routine blood examination, and measured the serum levels of IL-36α, IL-36β, and IL-36γ by performing polymerase chain reaction and enzyme-linked immunosorbent assay. In addition, the correlation between the number of neutrophils and eosinophils and the level of IL-36 was also analyzed.
Results: We found that level of IL-36 in patients with COPD was positively correlated with the number of neutrophils but not with eosinophils, whereas the correlation was not found in the control group. Moreover, the level of IL-36 was negatively correlated with the level of lung function of patients with COPD, and the levels of IL-36α, IL-36β, and IL-36γ increased with advancing disease severity.
Conclusion: In COPD, the pro-inflammatory effect of IL-36 is closely related to neutrophils, and hence, IL-36 might be considered a novel biomarker for determining COPD severity.
Keywords: chronic obstructive pulmonary disease, IL-36, neutrophils, inflammation
Introduction
Chronic obstructive pulmonary disease (COPD) is a heterogeneous, inflammatory-airway disease. As the disease is characterized by high morbidity and mortality, approximately 200 million people worldwide are affected and more than 3 million deaths occur each year.1,2 Unfortunately, epidemiological data could not adequately reflect the widespread impact and disease burden caused by COPD because, in many lagging regions, the diagnosis rate of COPD is much lower than the true picture.3 The most critical feature of COPD is irreversible airflow limitation, and its typical pathogenesis includes airway remodeling, emphysematous lung parenchymal destruction, and airway inflammatory response.4 Airway inflammation in COPD involves various cells such as neutrophils, lymphocytes, monocytes, and dendritic cells.5,6 Among these cells, the association of neutrophils with COPD has been widely studied; neutrophil levels are significantly higher in sputum and bronchoalveolar lavage fluid of patients with COPD than those in healthy individuals.7–9 In addition, the number and distribution range of neutrophils were significantly increased in animal models of COPD.10,11 As neutrophils are closely associated with COPD, neutrophilic inflammation is undoubtedly an important part of the pathogenesis of COPD. Therefore, investigating the effect of related cytokines on neutrophilic inflammation will certainly enhance the understanding of COPD pathogenesis.
As a member of the interleukin (IL)-1 family, IL-36 has two classes of four isomers, namely IL-36α, IL-36β, IL-36γ, and IL-36Ra.12 IL-36α, IL-36β, and IL-36γ belong to the group of agonists that promote many diseases such as psoriasis, arthritis, and allergic rhinitis.13–15 On the other hand, IL-36Ra belongs to the group that mainly plays a role in counteracting the effects of IL-36.16 IL-36 is widely distributed in several vital organs such as the heart, brain, and kidney.17–19 Different IL-36 isoforms play different roles in diseases, either synergistically or counteracting each other.20 For example, IL-36α could trigger the activation of the IL-23/IL-17A signaling axis and thus induce an inflammatory response leading to psoriasis,21 whereas IL-36Ra could suppress skin inflammation and provide protection.22 IL-36β could increase the expression of IL-6 and CXCL8 in human lung fibroblasts and bronchial epithelial cells,23 and IL-36γ could promote the recruitment of Th17 cells and the activation of fibroblasts.24 As a typical chronic airway disease, the association of COPD with IL-36 has been demonstrated in several studies. For example, some studies reported that IL-36α and IL-36γ levels in sputum were significantly increased in patients with COPD,25 whereas the level of IL-36 decreased in patients with COPD with eosinophilic phenotype.26 Combined with the importance of neutrophils in COPD pathogenesis, we inferred that IL-36 is correlated with neutrophils in COPD pathogenesis. To further elucidate the mechanism underlying COPD pathogenesis, the correlation between IL-36 and neutrophils should be investigated.
Hence, we hypothesized that IL-36 could promote COPD pathogenesis by promoting a neutrophilic inflammatory response, and the level of IL-36 is closely related to the severity of COPD. To verify this hypothesis, we performed a polymerase chain reaction and enzyme-linked immunosorbent assay. In addition, the correlation between the number of neutrophils and eosinophils and the level of IL-36 was also analyzed. This study is expected to bring a new perspective to the diagnosis and treatment of COPD.
Materials and Methods
Subjects
Between March and September 2021, we enrolled patients with COPD (n = 70) and control subjects (n = 20) from the Shandong Provincial Qianfoshan Hospital. The characteristics of patients with COPD and control subjects are shown in Table 1. The diagnosis of COPD was in accordance with the Global Initiative for Chronic Obstructive Lung Disease (GOLD, 2021 edition). According to the GOLD criteria for the severity of disease, we divided the patients with COPD into 4 subgroups: GOLD 1 (n = 6), GOLD 2 (n = 21), GOLD 3 (n = 33), and GOLD 4 (n = 10). For the enrolled patients with COPD, all the following conditions were excluded: (i) Pulmonary disease other than COPD such as lung cancer, nodular disease, active tuberculosis, pulmonary fibrosis, and cystic fibrosis; (ii) Previous acute exacerbation of COPD within 4 weeks; (iii) Inflammatory diseases other than COPD such as rheumatoid arthritis, systemic lupus erythematosus, and inflammatory bowel disease; (iv) Lung surgery or recently diagnosed malignant tumor; (v) Unable to walk; (vi) Received blood transfusion within 4 weeks; (vii) Receiving systemic hormone therapy; (viii) Participating in any double-blind drug clinical trial. The subjects in the control group showed normal lung function and had no airflow limitation. For the enrolled control subjects, the following conditions were excluded: asthma, bronchiectasis, pulmonary abscess, interstitial lung disease, tuberculosis, central lung mass, systematic disease such as congestive heart failure, autoimmune disease, and infection. All patients voluntarily entered the study and signed the written informed consent. The study was conducted in accordance with the declaration of Helsinki. The study was approved by the Ethics Committee of the Shandong Provincial Qianfoshan Hospital.
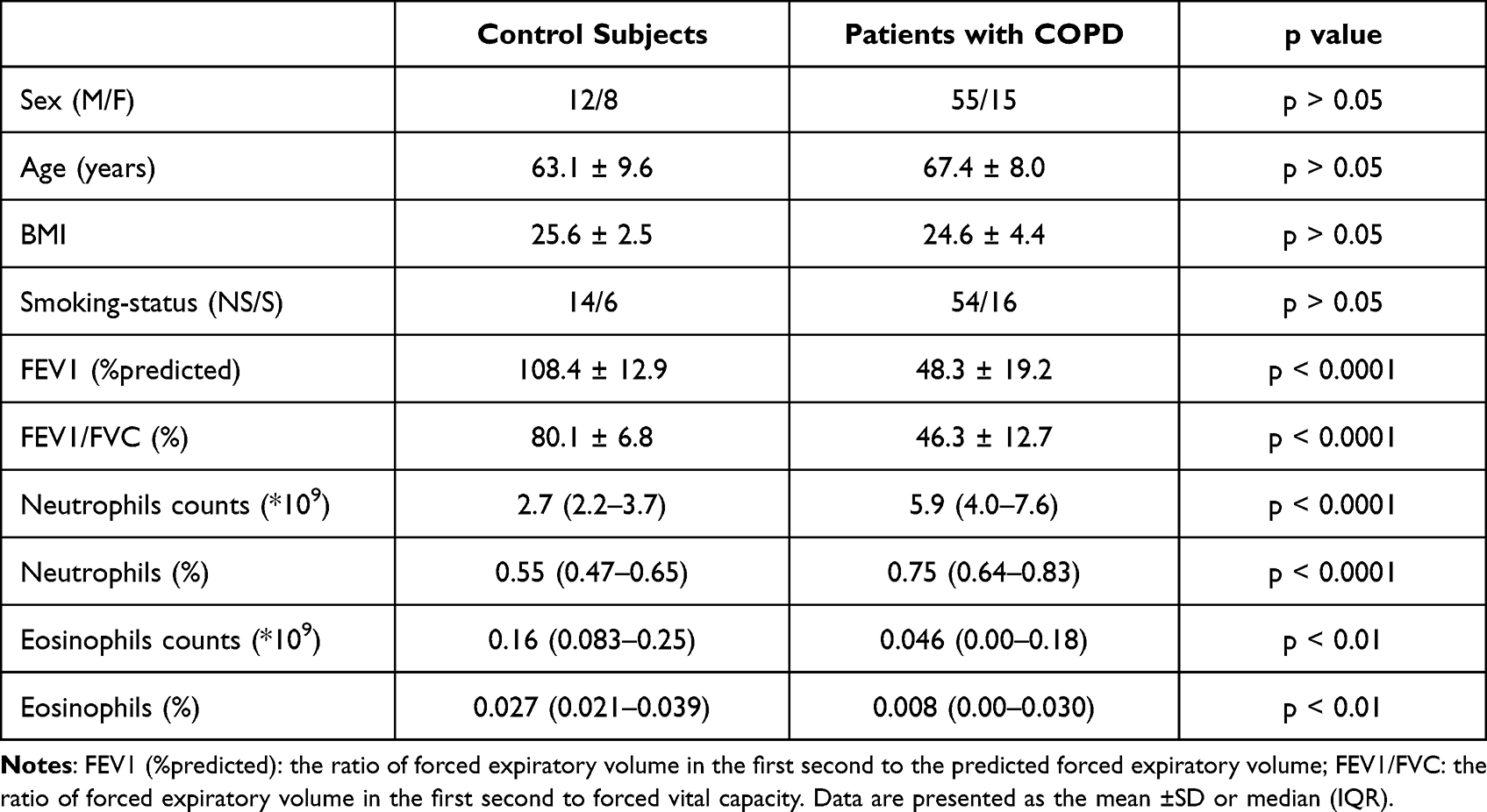 |
Table 1 Characteristics of COPD Patients and Control Subjects |
Peripheral Venous Blood Processing
We collected peripheral blood samples from all the enrolled individuals. The amount of blood samples was generally 5–10 mL due to the different cooperation of each person. One portion of the blood sample was centrifuged for 10 min at 1500 g at 4°C for basal experiments, and the remaining was used for routine blood examination. After separating the upper serum layer, we performed density-gradient centrifugation to collect the peripheral blood mononuclear cells (PBMCs).
Reverse Transcription and Quantitative-Polymerase Chain Reaction (qPCR)
All steps for the qPCR were performed according to the instructions provided with the kit. Total RNA from PBMCs was extracted using the RNAfast200 kit (Fastagen, Shanghai, China). The Evo M-MLV RT kit was used to synthesize cDNA (AG, Hunan, China). The SYBR® Green Premix Pro Taq HS qPCR kit (AG, Hunan, China) was used to quantify mRNA. GAPDH served as the internal reference for this experiment. The 2−ΔΔCT method was used to calculate the experimental results. The primer sequences used in this study are listed in Table 2.
 |
Table 2 Primers for qRT-PCR |
Enzyme-Linked Immunosorbent Assay (ELISA)
Blood samples were obtained from every participant, and serum was obtained and stored at −80°C until use. The serum levels of IL-36α (Solarbio, Beijing, China), IL-36β (Solarbio, Beijing, China), and IL-36γ (Abcam, Cambridge, UK) were measured using the corresponding ELISA kits following the manufacturers’ protocols.
Statistical Analyses
The patient characteristics were expressed as the mean ± standard deviation (SD) or the median (IQR). Count data were analyzed using the Chi-square test, and the measurement data were based on the distribution using the unpaired t-test for normal distribution and the Mann–Whitney test for skewed distribution. Correlations were calculated using Spearman’s rank correlation analyses. All statistical analyses were performed using the SPSS 25 (Abbott Laboratories, USA). The differences were considered to be statistically significant at the two-sided p-value of <0.05.
Results
Patient Characteristics
No significant differences were found between the COPD and control groups in terms of gender, age, BMI, and smoking status. Lung function was significantly worse in the COPD group than in the control group, which is not surprising (Table 1).
Peripheral Venous Blood Cell Counts
To determine the association of neutrophils and eosinophils with COPD, we compared the differences between the COPD and control groups in terms of neutrophil count and eosinophil count obtained by routine blood examination. The results showed that the COPD group had a higher number and proportion of neutrophils but lower eosinophil content compared with the control group. This result indicated the possible association of neutrophils and eosinophils with COPD (Table 1).
IL-36 Could Promote COPD Development and is an Indicator for Determining COPD Severity
To clarify the association between IL-36 and COPD, we measured the levels of IL-36α, IL-36β, and IL-36γ in the serum of patients and control subjects by performing ELISA. The results showed that the levels of IL-36α, IL-36β, and IL-36γ were higher in patients than in control subjects, and the levels of IL-36α, IL-36β, and IL-36γ increased with the progression of the disease (Figure 1).
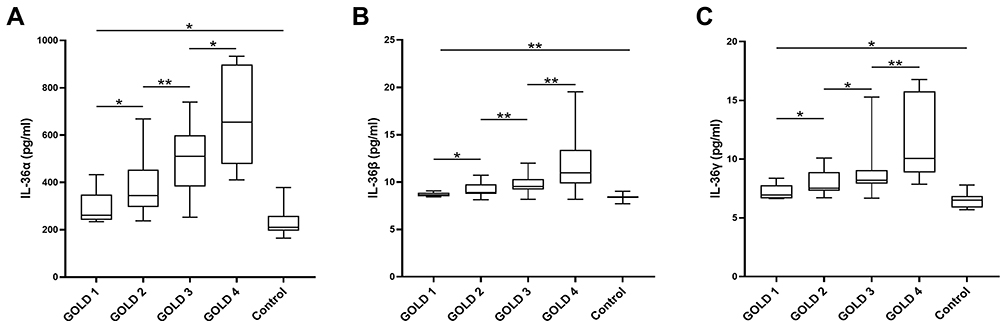 |
Figure 1 IL-36 was highly expressed in patients with COPD and was related to the severity of COPD. The levels of the IL-36α (A), IL-36β (B), and IL-36γ (C) in the serum of patients with COPD were measured using the ELISA kit. The number of samples in each group was as follows, GOLD 1 (n = 6), GOLD 2 (n = 20), GOLD 3 (n = 20), GOLD 4 (n=9), Control (n = 15). Data were pooled from at least 3 independent experiments and are presented as the mean ±SD. *p < 0.05, **p < 0.01. |
To further confirm the association between IL-36 and COPD, PBMCs from both groups were analyzed by performing PCR. The PCR results are consistent with the ELISA results. The results of PCR showed that the mRNA level of IL-36 increased with the GOLD grading. The mRNA levels of IL-36 were statistically different in COPD patients with different GOLD grading. Thus, IL-36 could promote the development of COPD and the level of IL-36 might be considered an indicator for determining COPD severity (Figure 2).
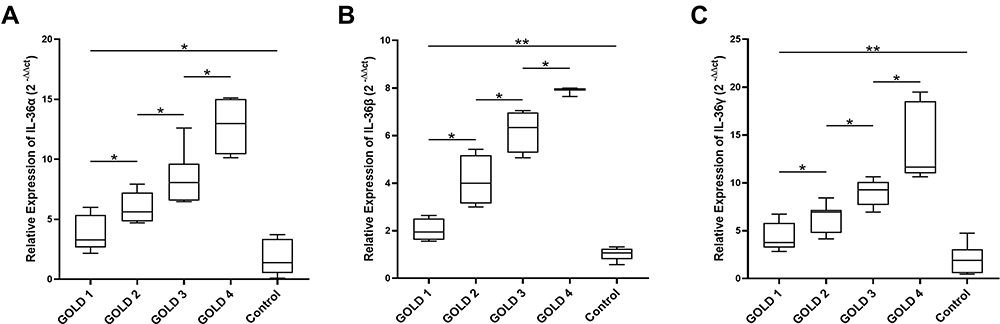 |
Figure 2 IL-36 was highly expressed in patients with COPD and was related to the severity of COPD. The mRNA levels and significant differences in IL-36α (A), IL-36β (B), and IL-36γ (C) in PBMCs of patients with COPD were determined by PCR. The number of samples in each group was as follows, GOLD 1 (n = 6), GOLD 2 (n = 20), GOLD 3 (n = 20), GOLD 4 (n=9), Control (n = 15). Data were pooled from at least 3 independent experiments and are presented as the mean ±SD. *p < 0.05, **p < 0.01. |
COPD, IL-36 is Closely Associated with Neutrophils
As IL-36 is a pro-inflammatory factor, the association between IL-36 and COPD is most likely based on neutrophils or eosinophils. To elucidate the mechanism underlying the pro-inflammatory effect of IL-36 in COPD, we analyzed the correlation between the levels of IL-36α, IL-36β, and IL-36γ and the number of neutrophils and eosinophils. The results showed that the levels of IL-36α (r = 0.5578, p < 0.0001), IL-36β (r = 0.4511, p < 0.01), and IL-36γ (r = 0.6908, p < 0.0001) in serum were positively correlated with the number of neutrophils, but not with the number of eosinophils in patients with COPD (Figure 3). In addition, IL-36 levels in patients with COPD were negatively correlated with their lung function levels. For control subjects, no significant association was found between IL-36 level and the number of neutrophils or eosinophils (Figure 4). To conclude, the pro-inflammatory factors IL-36α, IL-36β, and IL-36γ could promote neutrophilic inflammatory responses in COPD and hence might be considered novel, potential targets for determining COPD severity.
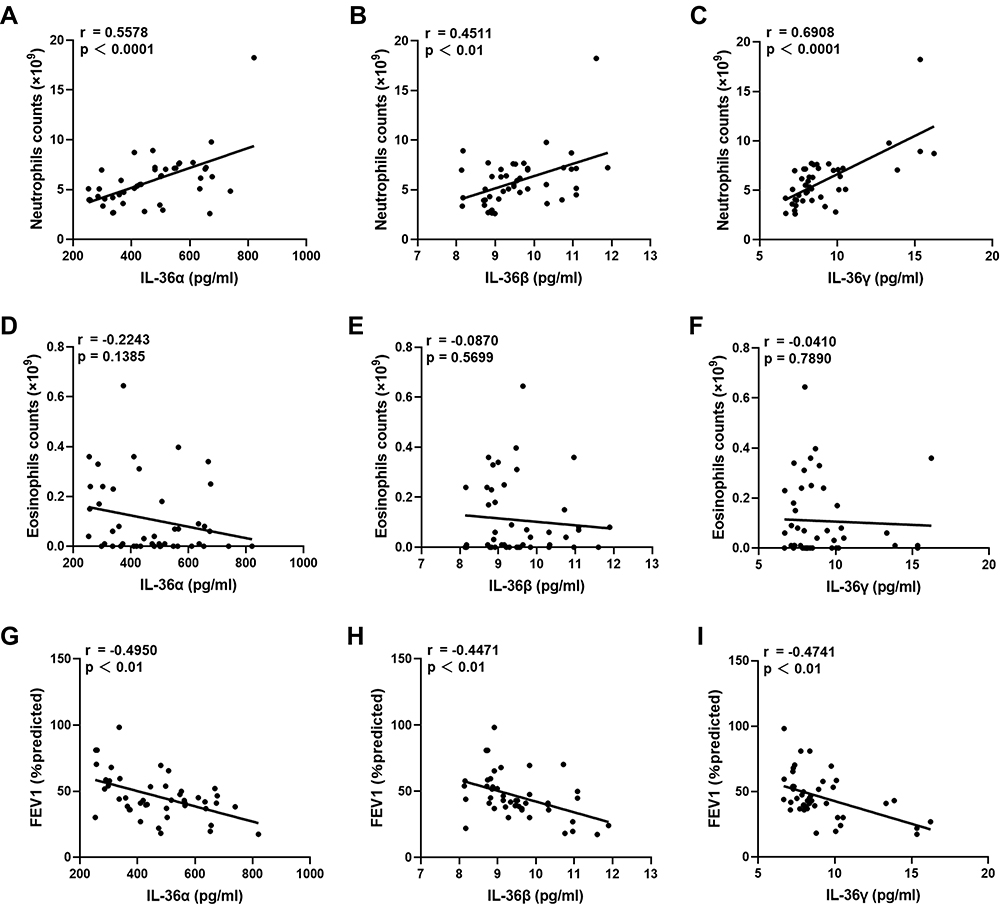 |
Figure 3 In COPD, IL-36 was closely related to neutrophils. (A–C) Correlation analysis of IL-36 and neutrophils in the COPD group. (D–F) Correlation analysis of IL-36 and eosinophils in the COPD group. (G–I) Correlation analysis of IL-36 and the lung functions in the COPD group. |
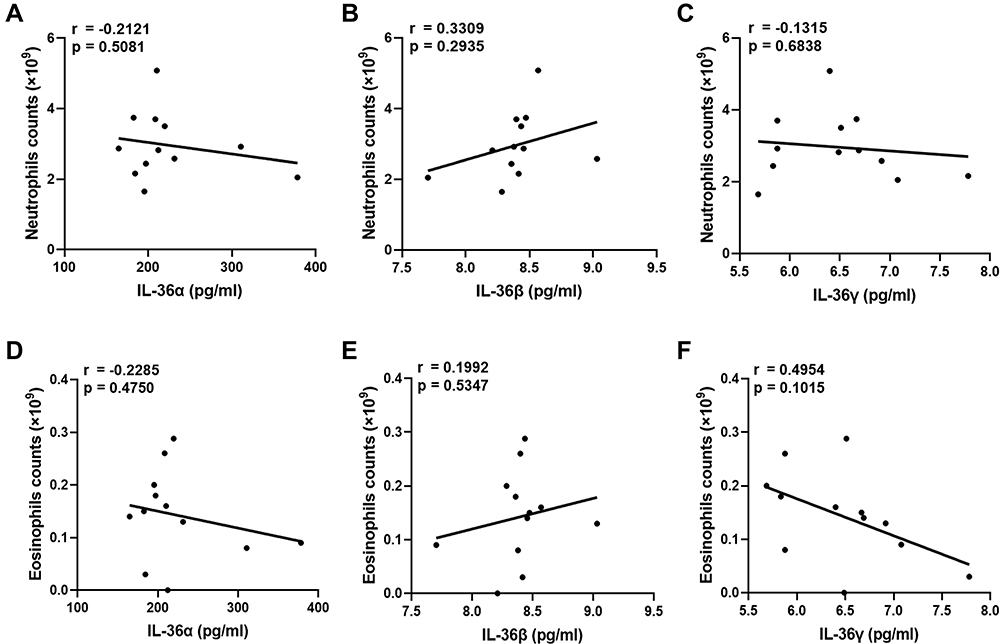 |
Figure 4 For control subjects, the IL-36 levels were not related to the number of inflammatory cells. (A–C) Correlation analysis of IL-36 and neutrophils in the control group. (D–F) Correlation analysis of IL-36 and eosinophils in the control group. |
Discussion
COPD is a chronic airway disease with high rates of morbidity, disability, and mortality. Despite its low diagnosis rate in economically disadvantaged regions, an epidemiological survey conducted in 2015 showed that COPD ranked third in age-standardized mortality rates for men and women worldwide, after ischemic heart disease and cerebrovascular disease.27 To reduce the heavy disease burden associated with COPD, studies on the pathogenesis of COPD are required. Several important factors have been identified that trigger COPD, such as smoking, air pollution, and the presence of susceptibility genes.28,29 However, the identification of these factors has not facilitated substantial progress in COPD treatment because each factor has its limitations, such as the presence of several nonsmokers among patients with COPD and the difficulty of implementing effective interventions at the genetic level. Hence, COPD is difficult to treat, and even though some studies have shown that inhaling glucocorticoids combined with dual bronchodilators can reduce the incidence of acute exacerbations in COPD, the effect is less pronounced than that in the treatment of other diseases such as asthma.30,31 To improve the efficiency of COPD diagnosis and treatment, exploring its pathogenesis and identifying new targets are crucial.
As a member of the IL-1 superfamily, IL-36 contains two classes of mutually antagonistic factors.12 IL-36Ra belongs to one class, which plays an anti-inflammatory protective role in many tissues such as epithelial tissues.32,33 IL-36α, IL-36β, and IL-36γ belong to another class, and many studies have demonstrated the pro-inflammatory and pro-fibrotic functions of this class of molecules. For example, IL-36α activates the IL-23/IL-17A signaling pathway in psoriasis;21 IL-36γ promotes eosinophil activation and migration in allergic rhinitis;34 IL-36β plays a pro-inflammatory role in arthritis.35 A study has shown that IL-36 agonist molecules (IL-36α, IL-36β, and IL-36γ) exert their effects mainly via myeloid differentiation factor 88, mitogen-activated protein kinase, and nuclear factor kappa-B signaling pathways.36 The association of COPD with IL-36 agonist molecules has been demonstrated in a few studies;37,38 however, it is unclear. In this study, we attempted to explore the pro-inflammatory role of IL-36 agonist molecules in COPD and to determine whether an association exists between IL-36 levels and COPD severity.
To verify the association between IL-36 and COPD, the COPD and control groups with no difference in the baseline status (age, sex, BMI, and smoking status) were included in the study. We then collected serum samples from both the groups and measured IL-36α, IL-36β, and IL-36γ levels by performing ELISA. The results showed that the levels of IL-36 agonist molecules were significantly higher in the COPD group than in the control group, and the levels of IL-36 increased with the aggravation of the disease. To further confirm this association, PBMCs were extracted from peripheral blood obtained from both the groups, and PCR was performed. The PCR results and the ELISA results are consistent. These results suggested that the levels of IL-36 agonist molecules are correlated with COPD pathogenesis and hence might be considered as indicators for determining COPD severity.
Neutrophils and eosinophils are two critical types of cells in the airway inflammatory response.39 As IL-36 agonist molecules can exert their pro-inflammatory effects in COPD, we determined which cells among the two types are responsible for these pro-inflammatory effects. We analyzed the correlation between IL-36 levels and peripheral blood neutrophil and eosinophil counts in the two groups. The results demonstrated that levels of IL-36α, IL-36β, and IL-36γ correlated with the number of neutrophils but not with eosinophils in patients with COPD. The levels of IL-36 in control subjects were not correlated with the number of either cell type. Thus, IL-36 could induce COPD mainly by promoting neutrophilic inflammation.
Conclusion
Although the study is not complex and has some limitations such as the relatively small number of participants, the use of only serological experiments, and the absence of a wider variety of samples, the originality and value of this study are pronounced. This study is the first to report the possible association between il-36 and COPD at the circulating level. This study not only demonstrated that IL-36 induces COPD by promoting neutrophilic inflammation but also indicated the possibility of IL-36 as a novel predictor of COPD severity. Although this study focuses more on clinical findings than on the molecular mechanisms underlying this phenomenon, further research is expected to bring new breakthroughs in the treatment of COPD.
Data Sharing Statement
Experimental data related to this study can be obtained from the corresponding author upon reasonable request.
Ethical Approval
All studies involving human participants were conducted in accordance with the standards specified by the Ethics Committee of Shandong Provincial Qianfoshan Hospital. This study is in line with the Declaration of Helsinki.
Funding
This work was funded by the National Natural Science Foundation of China (Grant No. 81770029).
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Rabe KF, Watz H. Chronic obstructive pulmonary disease. Lancet. 2017;389(10082):1931–1940. doi:10.1016/S0140-6736(17)31222-9
2. GBD 2015 Disease and Injury Incidence and Prevalence Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990–2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet. 2016;388(10053):1545–1602. doi:10.1016/S0140-6736(16)31678-6
3. Lamprecht B, Soriano JB, Studnicka M, et al. Determinants of underdiagnosis of COPD in national and international surveys. Chest. 2015;148(4):971–985. doi:10.1378/chest.14-2535
4. O’Donnell DE. Hyperinflation, dyspnea, and exercise intolerance in chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2006;3(2):180–184. doi:10.1513/pats.200508-093DO
5. Barnes PJ, Shapiro SD, Pauwels RA. Chronic obstructive pulmonary disease: molecular and cellular mechanisms. Eur Respir J. 2003;22(4):672–688. doi:10.1183/09031936.03.00040703
6. Brusselle GG, Joos GF, Bracke KR. New insights into the immunology of chronic obstructive pulmonary disease. Lancet. 2011;378(9795):1015–1026. doi:10.1016/S0140-6736(11)60988-4
7. Ravi AK, Khurana S, Lemon J, et al. Increased levels of soluble interleukin-6 receptor and CCL3 in COPD sputum. Respir Res. 2014;15(1):103. doi:10.1186/s12931-014-0103-4
8. Martin TR, Raghu G, Maunder RJ, Springmeyer SC. The effects of chronic bronchitis and chronic air-flow obstruction on lung cell populations recovered by bronchoalveolar lavage. Am Rev Respir Dis. 1985;132(2):254–260. doi:10.1164/arrd.1985.132.2.254
9. Hunninghake GW, Gadek JE, Kawanami O, Ferrans VJ, Crystal RG. Inflammatory and immune processes in the human lung in health and disease: evaluation by bronchoalveolar lavage. Am J Pathol. 1979;97(1):149–206.
10. Dhami R, Gilks B, Xie C, Zay K, Wright JL, Churg A. Acute cigarette smoke-induced connective tissue breakdown is mediated by neutrophils and prevented by alpha1-antitrypsin. Am J Respir Cell Mol Biol. 2000;22(2):244–252. doi:10.1165/ajrcmb.22.2.3809
11. Lams BE, Sousa AR, Rees PJ, Lee TH. Immunopathology of the small-airway submucosa in smokers with and without chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1998;158(5 Pt 1):1518–1523. doi:10.1164/ajrccm.158.5.9802121
12. Yuan ZC, Xu WD, Liu XY, Liu XY, Huang AF, Su LC. Biology of IL-36 signaling and its role in systemic inflammatory diseases. Front Immunol. 2019;10:2532. doi:10.3389/fimmu.2019.02532
13. Ainscough JS, Macleod T, McGonagle D, et al. Cathepsin S is the major activator of the psoriasis-associated proinflammatory cytokine IL-36γ. Proc Natl Acad Sci U S A. 2017;114(13):E2748–E2757. doi:10.1073/pnas.1620954114
14. Frey S, Derer A, Messbacher ME, et al. The novel cytokine interleukin-36α is expressed in psoriatic and rheumatoid arthritis synovium. Ann Rheum Dis. 2013;72(9):1569–1574. doi:10.1136/annrheumdis-2012-202264
15. Qin X, Zhang T, Wang C, Li H, Liu M, Sun Y. IL-36α contributes to enhanced T helper 17 type responses in allergic rhinitis. Cytokine. 2020;128:154992. doi:10.1016/j.cyto.2020.154992
16. Yi G, Ybe JA, Saha SS, et al. Structural and functional attributes of the Interleukin-36 receptor. J Biol Chem. 2016;291(32):16597–16609. doi:10.1074/jbc.M116.723064
17. Queen D, Ediriweera C, Liu L. Function and regulation of IL-36 signaling in inflammatory diseases and cancer development. Front Cell Dev Biol. 2019;7:317. doi:10.3389/fcell.2019.00317
18. Berglöf E, Andre R, Renshaw BR, et al. IL-1Rrp2 expression and IL-1F9 (IL-1H1) actions in brain cells. J Neuroimmunol. 2003;139(1–2):36–43. doi:10.1016/S0165-5728(03)00130-9
19. Nishikawa H, Taniguchi Y, Matsumoto T, et al. Knockout of the interleukin-36 receptor protects against renal ischemia-reperfusion injury by reduction of proinflammatory cytokines. Kidney Int. 2018;93(3):599–614. doi:10.1016/j.kint.2017.09.017
20. Murrieta-Coxca JM, Rodríguez-Martínez S, Cancino-Diaz ME, Markert UR, Favaro RR, Morales-Prieto DM. IL-36 cytokines: regulators of inflammatory responses and their emerging role in immunology of reproduction. Int J Mol Sci. 2019;20(7):1649. doi:10.3390/ijms20071649
21. Milora KA, Fu H, Dubaz O, Jensen LE. Unprocessed Interleukin-36α regulates psoriasis-like skin inflammation in cooperation with Interleukin-1. J Invest Dermatol. 2015;135(12):2992–3000. doi:10.1038/jid.2015.289
22. Blumberg H, Dinh H, Trueblood ES, et al. Opposing activities of two novel members of the IL-1 ligand family regulate skin inflammation. J Exp Med. 2007;204(11):2603–2614. doi:10.1084/jem.20070157
23. Zhang J, Yin Y, Lin X, et al. IL-36 induces cytokine IL-6 and chemokine CXCL8 expression in human lung tissue cells: implications for pulmonary inflammatory responses. Cytokine. 2017;99:114–123. doi:10.1016/j.cyto.2017.08.022
24. Ramadas RA, Ewart SL, Medoff BD, LeVine AM. Interleukin-1 family member 9 stimulates chemokine production and neutrophil influx in mouse lungs. Am J Respir Cell Mol Biol. 2011;44(2):134–145. doi:10.1165/rcmb.2009-0315OC
25. Li W, Meng X, Hao Y, Chen M, Jia Y, Gao P. Elevated sputum IL-36 levels are associated with neutrophil-related inflammation in COPD patients. Clin Respir J. 2021;15(6):648–656. doi:10.1111/crj.13338
26. Moermans C, Damas K, Guiot J, et al. Sputum IL-25, IL-33 and TSLP, IL-23 and IL-36 in airway obstructive diseases. Reduced levels of IL-36 in eosinophilic phenotype. Cytokine. 2021;140:155421. doi:10.1016/j.cyto.2021.155421
27. GBD 2015 Mortality and Causes of Death Collaborators. Global, regional, and national life expectancy, all-cause mortality, and cause-specific mortality for 249 causes of death, 1980–2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet. 2016;388(10053):1459–1544. doi:10.1016/S0140-6736(16)31012-1
28. Eisner MD, Iribarren C, Yelin EH, et al. The impact of SHS exposure on health status and exacerbations among patients with COPD. Int J Chron Obstruct Pulmon Dis. 2009;4:169–176. doi:10.2147/COPD.S4681
29. Peacock JL, Anderson HR, Bremner SA, et al. Outdoor air pollution and respiratory health in patients with COPD. Thorax. 2011;66(7):591–596. doi:10.1136/thx.2010.155358
30. Wedzicha JA, Banerji D, Chapman KR, et al. Indacaterol-Glycopyrronium versus Salmeterol-Fluticasone for COPD. N Engl J Med. 2016;374(23):2222–2234. doi:10.1056/NEJMoa1516385
31. Pascoe SJ, Lipson DA, Locantore N, et al. A Phase III randomised controlled trial of single-dose triple therapy in COPD: the IMPACT protocol. Eur Respir J. 2016;48(2):320–330. doi:10.1183/13993003.02165-2015
32. Towne JE, Renshaw BR, Douangpanya J, et al. Interleukin-36 (IL-36) ligands require processing for full agonist (IL-36α, IL-36β, and IL-36γ) or antagonist (IL-36Ra) activity. J Biol Chem. 2011;286(49):42594–42602. doi:10.1074/jbc.M111.267922
33. Günther S, Sundberg EJ. Molecular determinants of agonist and antagonist signaling through the IL-36 receptor. J Immunol. 2014;193(2):921–930. doi:10.4049/jimmunol.1400538
34. Qin X, Liu M, Zhang S, Wang C, Zhang T. The role of IL-36γ and its regulation in eosinophilic inflammation in allergic rhinitis. Cytokine. 2019;117:84–90. doi:10.1016/j.cyto.2019.02.008
35. Magne D, Palmer G, Barton JL, et al. The new IL-1 family member IL-1F8 stimulates production of inflammatory mediators by synovial fibroblasts and articular chondrocytes. Arthritis Res Ther. 2006;8(3):R80. doi:10.1186/ar1946
36. Towne JE, Garka KE, Renshaw BR, Virca GD, Sims JE. Interleukin (IL)-1F6, IL-1F8, and IL-1F9 signal through IL-1Rrp2 and IL-1RAcP to activate the pathway leading to NF-kappaB and MAPKs. J Biol Chem. 2004;279(14):13677–13688. doi:10.1074/jbc.M400117200
37. Kovach MA, Che K, Brundin B, et al. IL-36 cytokines promote inflammation in the lungs of long-term smokers. Am J Respir Cell Mol Biol. 2021;64(2):173–182. doi:10.1165/rcmb.2020-0035OC
38. McCombs JE, Kolls JK. Walking down the “IL”: the newfound marriage between IL-36 and chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2021;64(2):153–154. doi:10.1165/rcmb.2020-0461ED
39. Bagdonas E, Raudoniute J, Bruzauskaite I, Aldonyte R. Novel aspects of pathogenesis and regeneration mechanisms in COPD. Int J Chron Obstruct Pulmon Dis. 2015;10:995–1013. doi:10.2147/COPD.S82518
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.