")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 19
Genetic Insights into the Gut-Lung Axis: Mendelian Randomization Analysis on Gut Microbiota, Lung Function, and COPD
Authors Cheng ZX , Hua JL, Jie ZJ, Li XJ, Zhang J
Received 15 October 2023
Accepted for publication 21 February 2024
Published 4 March 2024 Volume 2024:19 Pages 643—653
DOI https://doi.org/10.2147/COPD.S441242
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Richard Russell
Zi-Xuan Cheng,1 Jian-Lan Hua,1 Zhi-Jun Jie,2 Xing-Jing Li,3 Jing Zhang1
1Department of Pulmonary and Critical Care Medicine, Zhongshan Hospital, Shanghai Medical College, Fudan University, Shanghai, People’s Republic of China; 2Department of Respiratory and Critical Care Medicine, the Fifth People’s Hospital of Shanghai, Fudan University, Shanghai, People’s Republic of China; 3Department of Respiratory Medicine, Zhongshan Hospital Wusong Branch, Fudan University, Shanghai, People’s Republic of China
Correspondence: Jing Zhang, Email [email protected]
Background: Chronic obstructive pulmonary disease (COPD) is a respiratory disorder with a complex etiology involving genetic and environmental factors. The dysbiosis of gut microbiota has been implicated in COPD. Mendelian Randomization (MR) provides a tool to investigate causal links using genetic variants as instrumental variables. This study aims to employ MR analysis to explore the causal relationship between gut microbiota, lung function, and COPD.
Methods: We utilized genome-wide association study (GWAS) data from MiBioGen, UK Biobank and FinnGen, which were related to gut microbial taxa, lung function parameters including forced vital capacity in one second (FEV1), forced vital capacity (FVC), and percentage of predicted FEV1 (FEV1%pred), as well as GWAS data for COPD. MR analysis was conducted to assess the causal effects of gut microbiota on lung function and the risk of COPD. Sensitivity analysis was utilized to examine the stability of the causal relationships. Multiple testing and reverse analysis were employed to evaluate the robustness of these relationships.
Results: Using the IVW method, 64 causal correlations were identified. Through conducting sensitivity analysis, multiple testing, and reverse analysis, we identified 14 robust and stable causal relationships. The bacterial taxa that showed a positive association with lung function included Desulfovibrionaceae, Erysipelotrichales, Desulfovibrionales, Clostridiales, Clostridia, Deltaproteobacteria and Erysipelotrichia, while Selenomonadales and Negativicutes showed a negative association with lung function. The abundance of Holdemanella were positively correlated with the risk of COPD, while FamilyXIII exhibited a negative correlation with the risk of COPD.
Conclusion: Several microbial taxa were discovered to have a positive causal correlation with lung function, offering potential insights into the development of probiotics. The presence of microbial taxa negatively correlated with lung function and positively correlated with COPD emphasized the potential impact of gut microbiota dysbiosis on respiratory health.
Keywords: COPD, lung function, gut-lung axis, gut microbiota, Mendelian randomization analysis
Introduction
Chronic obstructive pulmonary disease (COPD) is a prevalent respiratory disorder characterized by progressive airflow limitation and persistent respiratory symptoms. It presents a substantial global health burden and is a leading cause of morbidity and mortality.1 The development of COPD involves a complex interplay between genetic factors, such as predisposition, and environmental factors, including smoking, air pollution, and respiratory infections. Despite the diverse causes of COPD, current diagnostic and therapeutic approaches are limited, highlighting the need for a deeper understanding of its underlying mechanisms.2
In recent years, there has been growing interest in the role of the gut microbiota, which has been found to influence various aspects of human health, including immune regulation and systemic inflammation.3 Accumulating evidence suggests a potential link between the dysbiosis of the gut microbiota and the development of COPD.4,5 Observational studies have identified gut microbiome dysbiosis in COPD patients, and rodent models have demonstrated its contribution to COPD development.6,7 Furthermore, there have been reports on the associations between gut microbial dysbiosis and a decline in lung function in individuals with COPD.8–10
However, establishing a causal relationship between the gut microbiota and COPD remains challenging. The dynamic nature of the gut microbiome introduces challenges when attempting to characterize it through a single cross-sectional study design.11 This is due to the potential variability in microbial sequencing results, which can be influenced by various confounders and covariates. It is crucial to acknowledge that factors such as the use of inhalers, particularly inhaled corticosteroids (ICS), and antibiotics can act as confounding variables in these studies.12
Mendelian Randomization (MR) is an innovative and robust analytical approach that can provide insights into causal relationships between exposures and outcomes using genetic variants as instrumental variables (IV).13 By leveraging naturally occurring genetic variations unaffected by confounders, MR analysis circumvents some limitations of traditional observational studies, including confounding factors and reverse causality, and provides more reliable evidence for causal inference. The utilization of naturally occurring genetic variations in MR analysis offers a valuable approach to overcome limitations commonly encountered in traditional observational studies. MR analysis can effectively sidestep confounding factors and mitigate the issue of reverse causality.13
MR studies have been employed to establish genetic evidence supporting the causal link between gut microbiota and respiratory diseases.14–16 In a recent study conducted by Wei et al, they employed MR analysis to examine the causal connection between the gut microbiome and COPD. They identified 9 bacterial taxa associated with the risk of COPD. However, none of their MR results were found to be statistically significant after applying multiple testing corrections. This lack of significant findings may potentially be attributed to the relatively small sample size of the Genome-Wide Association Study (GWAS) utilized in their analysis.15 Additionally, while several observational studies have previously reported associations between the gut microbiota and lung function in COPD,8–10 the causal relationship involving the interplay between the gut microbiota, lung function, and COPD remains uncertain. Therefore, the objective of this study is to investigate the causal relationship between the gut microbiota, lung function, and COPD within the framework of MR analysis. To achieve this, we have incorporated the latest GWAS datasets, allowing for a more comprehensive analysis. By utilizing an extensive dataset and employing the MR approach, this research aims to shed light on the intricate causal relationships between these variables, providing insights into the pathogenesis and potential intervention strategies for COPD.
Method
Study Exposures
The summary statistics for gut microbiota abundance were obtained from a comprehensive GWAS conducted by the MiBioGen consortium. This study involved the analysis of host genetic variations in 18,340 participants from 24 cohorts, primarily of European descent. The study combined 16S rRNA gene sequencing profiles and human genotyping data from the participants. The dataset included a total of 211 taxa, encompassing 9 phyla, 16 classes, 20 orders, 35 families, and 131 genera, identified through 16S rRNA gene sequencing techniques (Supplementary Table S1).
Study Outcomes
The GWAS summary statistics for the outcomes were extracted from the large-scale biomedical databases of UK biobank and FinnGen biobank, both of which contain comprehensive health and genetic information from a large population of participants. The summary statistics for lung function, including FEV1 (forced expiratory volume in 1 second), FVC (forced vital capacity), and percentage of predicted FEV1, were extracted from the second analytical round of the UK biobank database. This extraction was performed on July 2, 2023. The GWAS summary statistics for COPD were obtained from the eighth analytical round of the FinnGen biobank database, accessed on April 29, 2023. It is worth noting that there was minimal overlap between the individuals included in the exposure and outcome samples. Supplementary Table S1 provides details about the GWAS summary statistics for reference.
Selection of Instrumental Variables
Human SNPs that were associated with the abundance of gut microbiota, which demonstrated genome-wide significance with a p-value of less than 1×10−5 were included. To assess potential confounding factors associated with the identified SNPs, we employed the Phenoscanner. SNPs found to be linked to confounding factors or outcomes were excluded from our analysis. (Supplementary Table S2) Clumping of these variants was performed to ensure the independence of the IVs. A clumping window of 10,000 kb and a pairwise linkage disequilibrium (LD) threshold of r2 < 0.001 were used for this purpose. To avoid weak instrument bias and ensure the strength of the IVs, we calculated the F-statistic. SNPs with an F-statistic below 10 were excluded from the analysis. Harmonization of variants was undertaken by aligning the effect alleles of different studies to the same reference allele using the TwoSampleMR package in R. Given the variability in genotyping platforms utilized in GWAS, it is possible that certain SNPs associated with the gut microbiota may not be present in the outcome dataset. Consequently, these missing SNPs were excluded from this study’s analysis.
Mendelian Randomization Analyses
The primary method used for causal inference in this study was the inverse variance weighted (IVW) method. This method combines the ratio estimates obtained from each genetic instrument in a meta-analysis model.17 To ensure the robustness of our findings, we employed additional analysis methods, including MR-Egger, weighted median, simple mode, weighted mode, and MR-PRESSO. MR-Egger detects violations of MR assumptions, such as horizontal pleiotropy, and provides an effect estimate that is not affected by these violations.18 The weighted median method combines the ratio estimates from genetic instruments using a median-based approach, which can provide reliable estimates even if up to 50% of the instruments are invalid. The simple mode and weighted mode methods consider the majority or weighted majority of genetic instrument estimates, respectively, to determine the direction and strength of the causal relationship.17 MR-PRESSO, a general test for the presence of outliers, was used to identify and remove genetic variants that significantly contributed to heterogeneity through a simulation approach.19 By incorporating these supplementary analysis methods, we can evaluate the consistency of the results and gain a more comprehensive understanding of the causal associations while considering potential violations of MR assumptions. For associations with an IVW-MR p-value < 0.05, we applied the Benjamini-Hochberg (BH) correction for multiple testing to reduce the likelihood of false-positive findings. Additionally, we conducted reverse analysis to examine the reverse causal association. All statistical analyses were performed using R version 4.2.3.
Sensitivity Analyses
In order to ensure the robustness of our primary causal estimate for associations with an IVW-MR p-value < 0.05, we performed sensitivity analyses. Cochran’s Q statistic was utilized with both the IVW and MR-Egger methods to assess the heterogeneity of effects. Additionally, we employed the MR-Egger intercept and MR-PRESSO Global test to evaluate the presence of horizontal pleiotropy. If any of these tests indicated the presence of pleiotropy with a significance level of p < 0.05, the corresponding results were excluded.18,19 Furthermore, a leave-one-out analysis was conducted to identify outliers and assess the stability of the results. These sensitivity analyses were crucial in ensuring the reliability and robustness of our findings.
Result
The purpose of this MR study was to investigate the causal effects of specific gut microbial taxa on lung function and COPD. (Figure 1) There are three main assumptions of MR analysis. First, the IVs should be strongly associated with the exposure. To address this assumption, we selected only exposures that had at least three independent genetic instruments at minimum p-value < 1×10−5. Additionally, we have excluded SNPs with mean F statistics < 10. The SNPs were then clumped by LD to ensure the independence of the IVs. Second, the genetic variants used as IVs are independent of any confounding factors that may influence both the exposure and outcome. Thus, we utilized the Phenoscanner to examine potential confounding factors associated with the SNPs. Any SNPs found to be related to confounding factors or outcomes were excluded from our analysis. The third assumption requires that the IVs only affect the outcome through their effect on the exposure and not through any alternative pathways. In order words, no pleiotropy should be presented. Any causal relationships that were detected to have pleiotropy would be excluded. After selecting SNPs according to the above criteria, we proceeded to harmonize the effect sizes of these variants on the exposure and the outcome for consistency and comparability. Among the MR results we obtained (Supplementary Table S3), a total of 64 potential associations were identified. These findings are summarized in Supplementary Table S4.
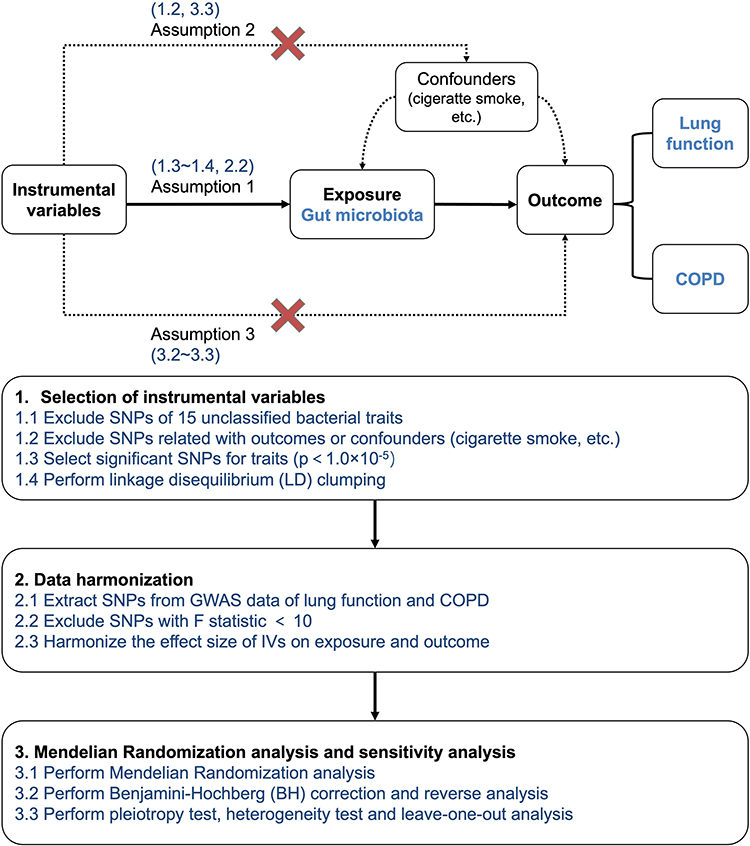 |
Figure 1 Overall MR framework and workflow of this study. |
Effect of Gut Microbial Abundance on Lung Function
Fifteen potential causal relationships were identified between the genetically predicted abundance of gut microbial taxa and FEV1 in the IVW analysis. Six relationships remained significant after conducting multiple correction. (Figure 2) (Table 1) Order Erysipelotrichales (β = 0.031, CI = 0.009–0.053, p = 0.032), order Desulfovibrionales (β = 0.029, CI = 0.010–0.049, p = 0.031), order Clostridiales (β = 0.037, CI = 0.015–0.060, p = 0.020), class Clostridia (β = 0.044, CI = 0.020–0.067, p = 0.003), class Deltaproteobacteria (β = 0.034, CI = 0.015–0.053, p = 0.003) and class Erysipelotrichia (β = 0.031, CI = 0.009–0.053, p = 0.026) were positively associated with FEV1. No significant horizontal pleiotropy was detected in the MR-Egger intercept and MR-PRESSO analysis (p>0.05). (Supplementary Tables S5 and S6) Results from Cochrane’s. Q test showed no significant heterogeneity in the relationships (p>0.05). (Supplementary Table S7) All the results remained robust after excluding the SNP one by one in leave-one-out analysis. (Supplementary Table S8) The reverse analysis did not reveal any reverse correlations. (Supplementary Table S9)
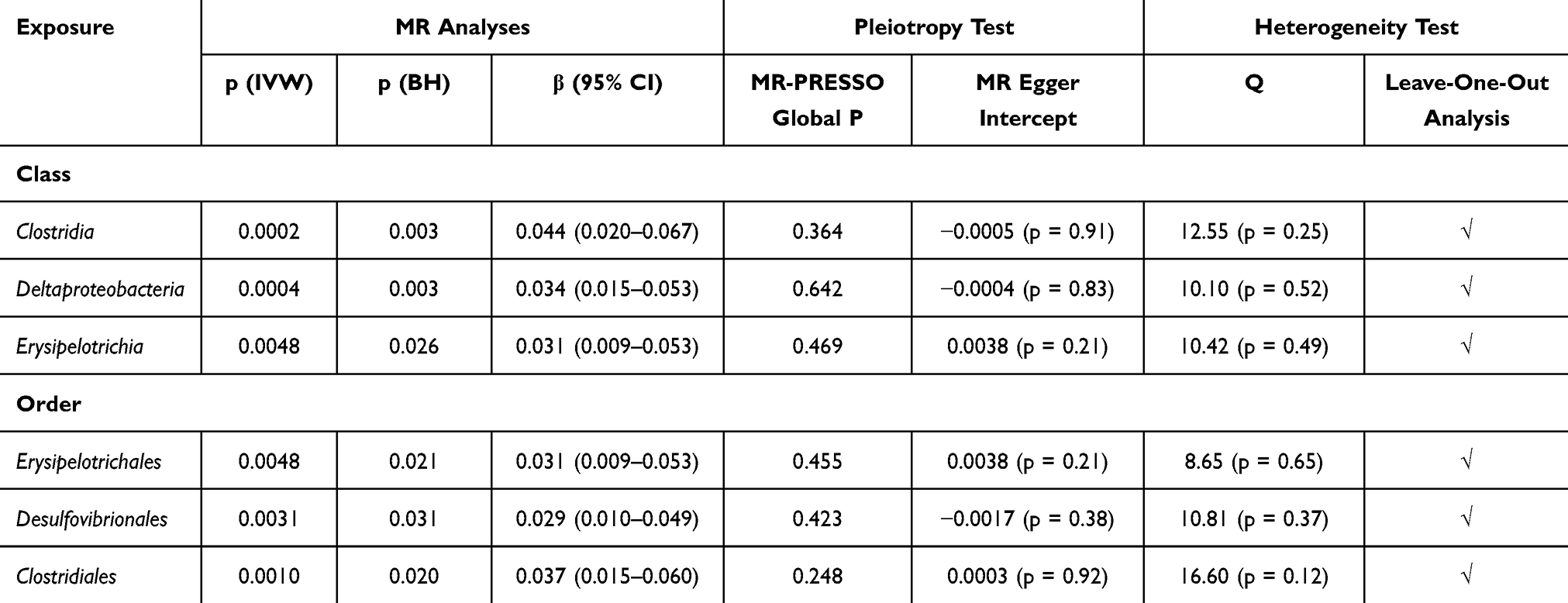 |
Table 1 Causal Relationships Between Gut Microbiota and FEV1 |
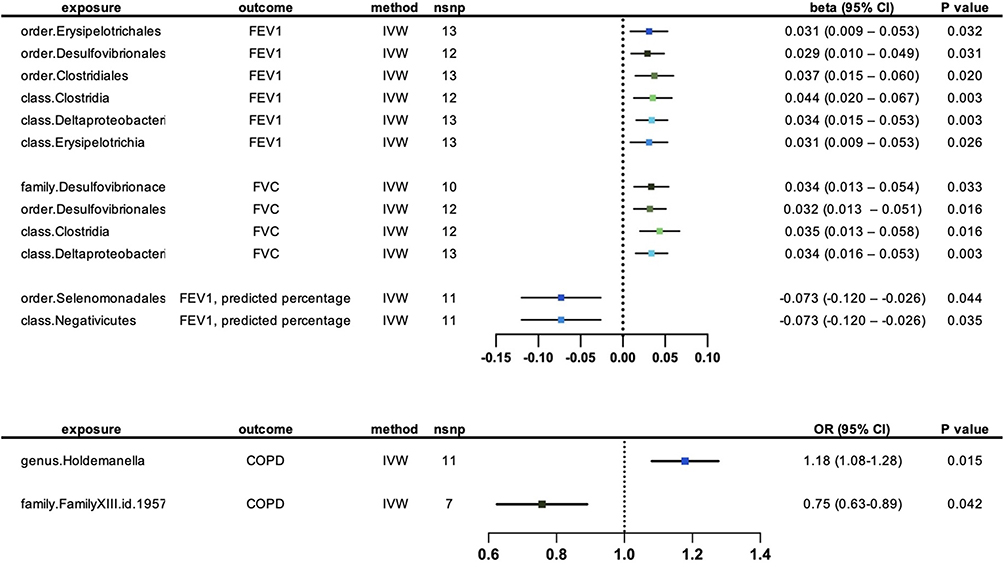 |
Figure 2 Forest plot for the causal effects of genetically predicted abundance of gut microbial taxa on lung function and COPD. |
In the IVW analysis, we identified sixteen potential causal relationships between the abundance of distinct gut microbiota and FVC. Following multiple correction, a significant association remained for four out of sixteen relationships. (Figure 2) (Table 2) Family Desulfovibrionaceae (β = 0.034, CI = 0.013–0.054, p = 0.033), order Desulfovibrionales (β = 0.032, CI = 0.013–0.051, p = 0.016), class Clostridia (β = 0.035, CI = 0.013–0.058, p = 0.016) and class Deltaproteobacteria (β = 0.034, CI = 0.016–0.053, p = 0.003) were all positively correlated with FVC. No significant heterogeneity and horizontal pleiotropy were detected in MR-Egger intercept, MR-PRESSO and Cochrane’s Q tests. (Supplementary Tables S5–S7) Leave-one-out analysis revealed that some single SNPs might dominate the positive effects of class Clostridia. (Supplementary Table S8) No reverse correlations were found through reverse analysis. (Supplementary Table S9)
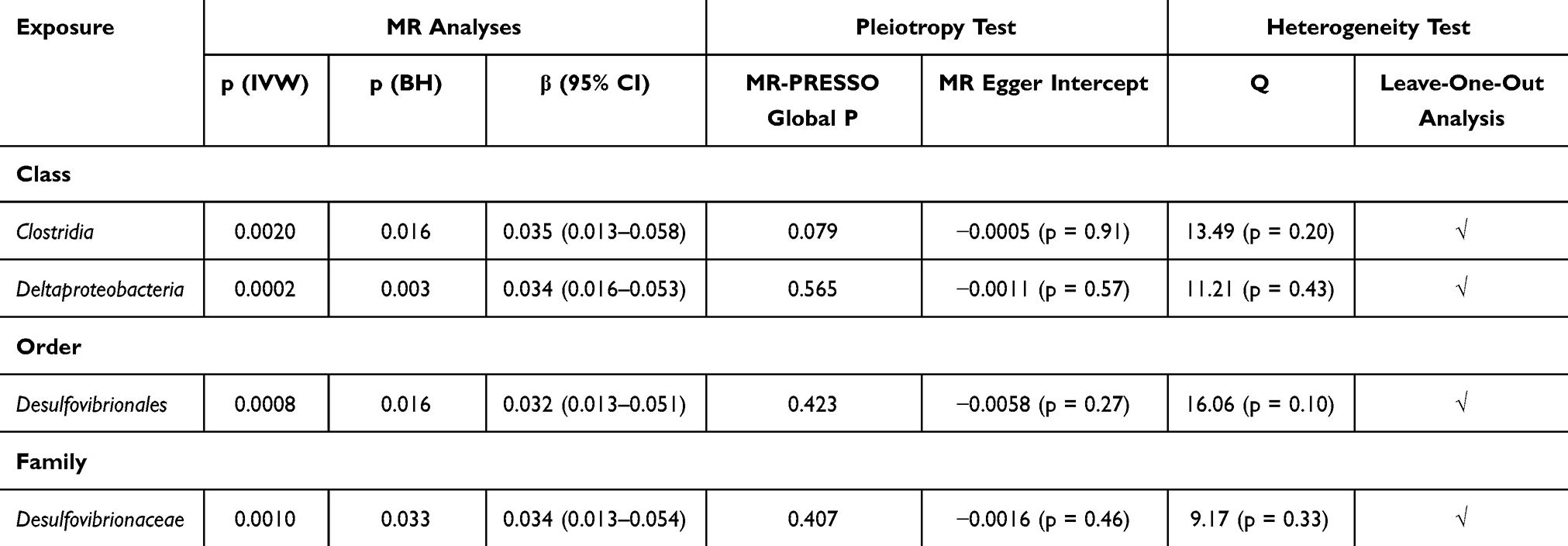 |
Table 2 Causal Relationships Between Gut Microbiota and FVC |
IVW analysis unveiled eighteen potential causal relationships linking the abundance of gut microbiota and percentage of predicted FEV1. After conducting multiple corrections, two of these relationships remained statistically significant. (Figure 2) (Table 3) Order Selenomonadales (β = −0.073, CI = −0.120 – −0.026, p = 0.044) and class Negativicutes (β = −0.073, CI = −0.120–0.0026, p = 0.035) were negatively correlated with percentage of predicted FEV1. MR-Egger intercept, MR-PRESSO and Cochrane’s Q tests indicated no evidence of horizontal pleiotropy and heterogeneity. (Supplementary Tables S5–S7) Furthermore, the stability and robustness of the results were confirmed through the leave-one-out analysis. (Supplementary Table S8) The reverse analysis indicated no reverse correlations. (Supplementary Table S9).
 |
Table 3 Causal Relationships Between Gut Microbiota And percentage of Predicted FEV1 |
Effect of Gut Microbial Abundance on COPD
Our Mendelian randomization analysis revealed that genetically predicted abundance of fifteen microbial taxa exhibited potential causal effects on COPD. After implementing multiple corrections, two of these relationships remained statistically significant. (Figure 2) (Table 4) The abundance of genus Holdemanella (OR = 1.176, CI = 1.082–1.278, p = 0.015) were positively correlated with the risk of COPD, while FamilyXIII. (OR = 0.750, CI = 0.629–0.894, p = 0.042) exhibited a negative correlation with the risk of COPD. MR-Egger intercept, MR-PRESSO, and Cochrane’s Q tests did not detect any significant heterogeneity or horizontal pleiotropy in the analysis. (Supplementary Tables S5–S7) The robustness and stability of the results were confirmed by the leave-one-out analysis. (Supplementary Table S8) The reverse analysis provided confirmation of the causal direction of the results. (Supplementary Table S9)
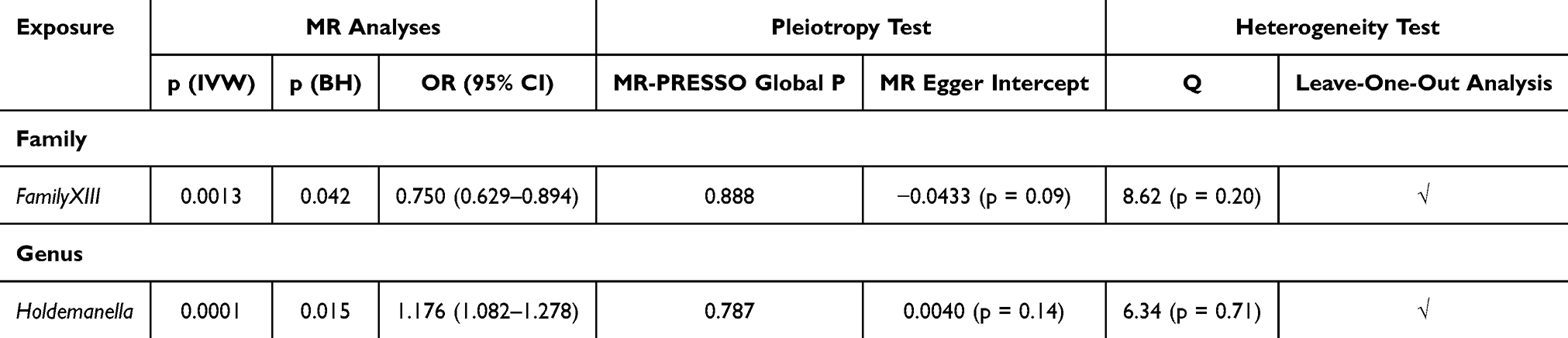 |
Table 4 Causal Relationships Between Gut Microbiota and the Risk of COPD |
Discussion
To our knowledge, this study is the first to explore the causal relationship between gut microbiota, lung function, and COPD. Following a rigorous analysis that included sensitivity analysis, reverse analysis, and multiple corrections, a total of fourteen robust and stable causal relationships were identified. There was no overlap found between the microbial taxa influencing lung function and those impacting COPD. However, several microbial taxa were discovered to have a positive causal correlation with lung function, offering potential insights into the development of probiotics. Furthermore, the presence of microbial taxa negatively correlated with lung function and positively correlated with COPD emphasized the potential impact of gut microbiota dysbiosis on lung function and COPD development. This contributes to the expanding understanding of the gut-lung axis.
Based on our research, we found that our findings reinforced the existing body of observational evidence, lending further support to the established knowledge in this domain. In our results, the bacterial taxa that were positively associated with lung function were family Desulfovibrionaceae, order Erysipelotrichales, Desulfovibrionales, Clostridiales, class Clostridia, Deltaproteobacteria and Erysipelotrichia. On the other hand, order Selenomonadales and class Negativicutes were negatively correlated with lung function. Interestingly, various pulmonary disease states have been associated with the elimination of these taxa. Upon treating influenza-infected mice with either the Chinese herb formula GeGen QinLian or fecal microbiota transplantation, the intestinal flora was restored. This restoration led to an increase in the abundance of Desulfovibrio_C21_c20 and a subsequent decrease in both mortality and lung inflammation.20 Long-term consumption of allium tuberosum reduced inflammatory cell count, interleukin (IL)-5 and IL-13 in bronchoalveolar lavage fluid in asthmatic mice. This consumption also improved pulmonary histopathology. Additionally, Desulfovibrionaceae was revealed as a biomarker indicating the effectiveness of the treatment.21 Clinical observational studies have revealed a decrease in Clostridia within the gut microbial composition of adult asthma patients.22 Interestingly, among asthmatic patients, those with lower specific IgE levels to mites and Ascaris exhibited an enrichment of various members from the order Clostridiales.23 In a MR study, it was found that class Negativicutes and order Selenomonadales exhibited a notable association with COVID-19 hospitalization, susceptibility, and severity.14
Previous studies have provided evidence of the impact of gut microbiota on immune regulation, suggesting a potential mechanism by which dysbiosis of gut microbiota could affect lung function. Among the various mechanisms investigated, the role of short-chain fatty acids (SCFAs) has received significant attention. SCFAs are produced through the fermentation of dietary fibers and are released into the lumen and peripheral circulation. The binding of SCFAs to free fatty acid receptors on immune cells such as neutrophils and macrophages allows them to exert anti-inflammatory effects.24,25 This protective role of SCFAs has been observed in both animal models and clinical studies. In a mouse model of emphysema, SCFAs demonstrated notable preventive potential by reducing the progression and severity of emphysema.26–28 Furthermore, a comprehensive study revealed that individuals who consumed dietary fibers over a long term had a 30% reduced risk of developing COPD.29 Previous observational studies have also reported associations between changes in the gut microbiome and a decline in lung function in individuals with COPD. These associations may be linked to the loss of protective microbial taxa that are involved in SCFA pathways.8–10 In addition to the immune regulatory mechanisms associated with the secretion of SCFAs, the beneficial microbiota may be associated with the homeostasis of the gut microbiome, which attribute to the colonization of a more diverse microbiome, preventing the dominance of one potentially pathogenic microbiota. Furthermore, the maintenance of gut microbial homeostasis allows beneficial intestinal microorganisms to play a protective role. Therefore, the gut microbiota that exhibited positive associations with lung function in our study may potentially act as protective bacteria through similar mechanisms.
It is noteworthy that the bacterial taxa correlated with lung function in our study have not only been associated with pulmonary diseases in previous observational studies, but they have also been linked to inflammatory bowel diseases (IBD). In dogs with IBD, the abundance of Erysipelotrichia and Clostridia were notably diminished.30 Similarly, individuals with Crohn’s disease displayed a significant decrease in the abundance of Erysipelotrichales and Clostridiales within their gut microbial profile, which strongly correlated with their disease status.31 Interestingly, it has been reported that patients with COPD exhibit reduced integrity and function of the intestinal barrier, as well as a higher prevalence of IBD.32–35 Therefore, our results have offered insights into the intricate relationship between the gut and lungs, known as the gut-lung axis. The bacterial taxa that demonstrated positive causal correlations with lung function may potentially confer protective effects through this gut-lung axis.
In our MR results, the abundance of genus Holdemanella were positively correlated with the risk of COPD, while FamilyXIII exhibited a negative correlation with the risk of COPD. It is worth noting that previous findings have shown a intriguing negative correlation between the abundance of Holdemanella and propionate levels, one of the SCFAs, in individuals with diabetes and cognitive impairment.36 Furthermore, a recent MR analysis has observed a causal correlation between Holdemanella and the risk of developing asthma.16 These findings indicate that the genus Holdemanella could potentially play a role in the development of pulmonary diseases, possibly through the SCFA pathways. However, Holdemanella biformis, a specific strain belonging to the genus Holdemanella, has exhibited protective effect in mouse colitis.37 Therefore, it is important to note that different species within the same genus can have distinct impacts on health, though microbial taxa can only be classified up to the genus level when using 16S rRNA sequencing. As far as our knowledge goes, there have been no previous associations reported between FamilyXIII and either pulmonary or bowel diseases in animal or clinical studies. Therefore, the causal correlation we observed between FamilyXIII and COPD necessitates additional investigation. All in all, the causal relationships uncovered in our study likely involve intricate interactions between specific microbial taxa and host factors. To fully understand the mechanisms through which dysbiosis of the gut microbiota impacts lung function and COPD, future studies employing metagenomic and metabolic sequencing techniques are warranted. Further validation of the potentially beneficial bacteria we identified can be conducted in subsequent experiments. For instance, Lai et al conducted a study where they constructed a murine model of COPD, analyzed the intestinal bacterial profiles of COPD rats and normal rats, isolated a strain called Parabacteroides goldsteinii from the differing bacteria, and demonstrated that preparations of this strain had a beneficial effect in mitigating COPD. Thus, investigations into the functional capabilities of specific microbial taxa are necessary to gain further insights.
The inclusion of a MR design presents an advantage in our study. Through selection of SNPs significantly associated with the exposure, while excluding SNPs correlated with the outcome or potential confounders, we establish the validity of the IVs, thereby enhancing the reliability of our results. By conducting sensitivity analysis, multiple testing, and reverse analysis, we confirm the stability, robustness, and direction of the identified causal relationships in our MR results. Consequently, we overcome limitations commonly encountered in traditional observational studies, including confounding factors and reverse causality, and provide genetic evidence for causal inference. Furthermore, our study benefits from utilizing the largest publicly available GWAS datasets, ensuring a comprehensive and robust analysis. Incorporating these extensive datasets significantly enhances the statistical power of our MR analysis, enabling accurate estimation of causal effects.
It is important to acknowledge certain limitations in our study. Firstly, our analysis utilized GWAS data for microbiota that did not specifically target the complete 16S rRNA gene. This limitation poses a challenge in differentiating between microbial taxa with the desired taxonomic resolution. It is worth noting that within the same genus, microbial taxa can have opposing effects on the host, thus the absence of complete 16S rRNA gene sequencing data restricts our ability to identify potential therapeutic targets accurately. Furthermore, since 16S rRNA sequencing is not designed to target viruses and fungi, the impact of such microbiota components was not investigated in our study. Secondly, given the dynamic and complex nature of the gut microbiota, it is an exposure phenotype that influenced by numerous variants with relatively small effect size. To address this complexity and increase statistical power, we have used a less stringent p-value threshold of 1 × 10−5, thus incorporating a larger number of IVs into our analysis, consequently facilitating the use of sensitivity analysis and bolstering statistical power. However, this approach carries the risk of including false-positive variants. By only including SNPs with a F statistic above 10 and performing multiple correction, we have reduced the possibility of false-positive results. Thirdly, our study predominantly focused on European populations due to the availability of suitable genetic data. This limits the generalizability of our findings to other ethnic groups, and further investigations involving diverse populations are warranted. Lastly, our results only establish a limited causal relationship between one specific flora and the outcome. The intricate biological mechanisms, including the impact of short-term or long-term changes in this flora on the overall gut microbiome and the influence on microbial metabolites and host immunity remain unclear. These aspects require more comprehensive mechanistic studies to be conducted in the future.
In summary, our MR analysis presents genetic evidence supporting a causal link between alterations in gut microbiota, lung function, and COPD. Our findings highlight the potential involvement of the gut microbiota in the development and advancement of COPD. The findings of this study provide potential insights for future research, including the investigation of therapeutic approaches such as probiotics to modulate the gut microbiota and alleviate COPD progression. Further investigations, particularly utilizing metagenomic and metabolomic sequencing approaches, are warranted to elucidate the underlying mechanisms and enhance our understanding of the complex interplay within the gut-lung axis.
Abbreviations
COPD, Chronic obstructive pulmonary disease; ICS, inhaled corticosteroids; MR, Mendelian Randomization; IV, instrumental variables; GWAS, Genome-Wide Association Study; FEV1, forced expiratory volume in 1 second; FVC, forced vital capacity; LD, linkage disequilibrium; IVW, inverse variance weighted; BH, Benjamini-Hochberg; SCFA, short-chain fatty acid; IL, interleukin.
Data Sharing Statement
The datasets supporting the conclusions of this article are available in the MiBioGen [https://mibiogen.gcc.rug.nl/], FinnGen [https://www.finngen.fi/en/access_results] and UK biobank [https://www.nealelab.is/uk-biobank] repository.
Ethical Approval
Summary statistics for the studies used for analysis were composed and obtained from published studies. All studies have received prior approval from their respective institutional review boards (IRBs). The institutional Review Board of Zhongshan Hospital approved the protocol for this study, and as per their guidelines, this study exclusively utilized publicly available data without using any individual-level data. Therefore, no additional IRB approval was necessary.
Acknowledgments
We would like to extend our appreciation to the participants and investigators involved in the MiBioGen Consortium, UK biobank, and FinnGen study. Their invaluable contributions to the large-scale GWAS studies have significantly advanced our knowledge of the gut microbiome and its connection to lung function and COPD. We would also like to extend appreciation to Dr. Xicheng Gu from Huashan Hospital, Fudan University, for providing invaluable advices on code writing.
Funding
This work is supported by the Shanghai Science and Technology Committee (Project number 19DZ1920104).
Disclosure
The authors declare that they have no competing interests in this work.
References
1. Christenson SA, Smith BM, Bafadhel M, Putcha N. Chronic obstructive pulmonary disease. Lancet. 2022;399(10342):2227–2242. doi:10.1016/S0140-6736(22)00470-6
2. Stolz D, Mkorombindo T, Schumann DM, et al. Towards the elimination of chronic obstructive pulmonary disease: a Lancet Commission. Lancet. 2022;400:921–972.
3. Ruan W, Engevik MA, Spinler JK, Versalovic J. Healthy human gastrointestinal microbiome: composition and function after a decade of exploration. Dig Dis Sci. 2020;65(3):695–705. doi:10.1007/s10620-020-06118-4
4. Budden KF, Gellatly SL, Wood DLA, et al. Emerging pathogenic links between microbiota and the gut–lung axis. Nat Rev Microbiol. 2017;15(1):55–63. doi:10.1038/nrmicro.2016.142
5. Wypych TP, Wickramasinghe LC, Marsland BJ. The influence of the microbiome on respiratory health. Nat Immunol. 2019;20(10):1279–1290. doi:10.1038/s41590-019-0451-9
6. Lai H-C, Lin T-L, Chen T-W, et al. Gut microbiota modulates COPD pathogenesis: role of anti-inflammatory Parabacteroides goldsteinii lipopolysaccharide. Gut. 2022;71(2):309–321. doi:10.1136/gutjnl-2020-322599
7. Li N, Dai Z, Wang Z, et al. Gut microbiota dysbiosis contributes to the development of chronic obstructive pulmonary disease. Respir Res. 2021;22(1):274. doi:10.1186/s12931-021-01872-z
8. Chiu Y-C, Lee S-W, Liu C-W, Lan T-Y, Ls-h W. Relationship between gut microbiota and lung function decline in patients with chronic obstructive pulmonary disease: a 1-year follow-up study. Respir Res. 2022;23(1):10. doi:10.1186/s12931-022-01928-8
9. Chiu Y-C, Lee S-W, Liu C-W, et al. Comprehensive profiling of the gut microbiota in patients with chronic obstructive pulmonary disease of varying severity. PLoS One. 2021;16(4):e0249944. doi:10.1371/journal.pone.0249944
10. Wu Y, Luo Z, Liu C. Variations in fecal microbial profiles of acute exacerbations and stable chronic obstructive pulmonary disease. Life Sci. 2021;265:118738. doi:10.1016/j.lfs.2020.118738
11. Combrink L, Humphreys IR, Washburn Q, et al. Best practice for wildlife gut microbiome research: a comprehensive review of methodology for 16S rRNA gene investigations. Front Microbiol. 2023;14:1092216. doi:10.3389/fmicb.2023.1092216
12. Pragman AA, Kim HB, Reilly CS, Wendt C, Isaacson RE, Doern GV. Chronic obstructive pulmonary disease lung microbiota diversity may be mediated by age or inhaled corticosteroid use. J Clin Microbiol. 2015;53(3):1050. doi:10.1128/JCM.03320-14
13. Davies NM, Holmes MV, Davey Smith G. Reading Mendelian randomisation studies: a guide, glossary, and checklist for clinicians. BMJ. 2018;362:k601. doi:10.1136/bmj.k601
14. Song J, Wu Y, Yin X, Ma H, Zhang J. The causal links between gut microbiota and COVID-19: a Mendelian randomization study. J Med Virol. 2023;95(5):e28784. doi:10.1002/jmv.28784
15. Wei Y, Lu X, Liu C. Gut microbiota and chronic obstructive pulmonary disease: a Mendelian randomization study. Front Microbiol. 2023;14:1196751. doi:10.3389/fmicb.2023.1196751
16. Jin Q, Ren F, Dai D, Sun N, Qian Y, Song P. The causality between intestinal flora and allergic diseases: insights from a bi-directional two-sample Mendelian randomization analysis. Front Immunol. 2023;14:1121273. doi:10.3389/fimmu.2023.1121273
17. Bowden J, Holmes MV. Meta-analysis and Mendelian randomization: a review. Res Synth Methods. 2019;10(4):486–496. doi:10.1002/jrsm.1346
18. Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol. 2015;44(2):512–525. doi:10.1093/ije/dyv080
19. Verbanck M, Chen C-Y, Neale B, Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat Genet. 2018;50(5):693–698. doi:10.1038/s41588-018-0099-7
20. Deng L, Shi Y, Liu P, et al. GeGen QinLian decoction alleviate influenza virus infectious pneumonia through intestinal flora. Biomed Pharmacother. 2021;141:111896. doi:10.1016/j.biopha.2021.111896
21. Zheng H-C, Liu Z-R, Li Y-L, et al. Allium tuberosum alleviates pulmonary inflammation by inhibiting activation of innate lymphoid cells and modulating intestinal microbiota in asthmatic mice. J Integr Med. 2021;19(2):158–166. doi:10.1016/j.joim.2020.11.003
22. B-h G, Choi J-P, Park T, et al. Adult asthma with symptomatic eosinophilic inflammation is accompanied by alteration in gut microbiome. Allergy. 2023;78(7):1909–1921. doi:10.1111/all.15691
23. Buendía E, Zakzuk J, San-Juan-Vergara H, Zurek E, Ajami NJ, Caraballo L. Gut microbiota components are associated with fixed airway obstruction in asthmatic patients living in the tropics. Sci Rep. 2018;8(1):9582. doi:10.1038/s41598-018-27964-3
24. He J, Zhang P, Shen L, et al. Short-chain fatty acids and their association with signalling pathways in inflammation, glucose and lipid metabolism. Int J Mol Sci. 2020;21(17):6356. doi:10.3390/ijms21176356
25. Ney L-M, Wipplinger M, Grossmann M, Engert N, Wegner VD, Mosig AS. Short chain fatty acids: key regulators of the local and systemic immune response in inflammatory diseases and infections. Open Biol. 2023;13(3):230014. doi:10.1098/rsob.230014
26. Jang YO, Kim O-H, Kim SJ, et al. High-fiber diets attenuate emphysema development via modulation of gut microbiota and metabolism. Sci Rep. 2021;11(1):7008. doi:10.1038/s41598-021-86404-x
27. Jang YO, Lee SH, Choi JJ, et al. Fecal microbial transplantation and a high fiber diet attenuates emphysema development by suppressing inflammation and apoptosis. Exp Mol Med. 2020;52(7):1128–1139. doi:10.1038/s12276-020-0469-y
28. Lee SH, Kim J, Kim NH, et al. Gut microbiota composition and metabolite profiling in smokers: a comparative study between emphysema and asymptomatic individuals with therapeutic implications. Thorax. 2023;78(11):1080–1089. doi:10.1136/thorax-2021-217923
29. Szmidt MK, Kaluza J, Harris HR, Linden A, Wolk A. Long-term dietary fiber intake and risk of chronic obstructive pulmonary disease: a prospective cohort study of women. Eur J Nutr. 2020;59(5):1869–1879. doi:10.1007/s00394-019-02038-w
30. Minamoto Y, Otoni CC, Steelman SM, et al. Alteration of the fecal microbiota and serum metabolite profiles in dogs with idiopathic inflammatory bowel disease. Gut Microbes. 2015;6(1):33–47. doi:10.1080/19490976.2014.997612
31. Gevers D, Kugathasan S, Denson LA, et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe. 2014;15(3):382–392. doi:10.1016/j.chom.2014.02.005
32. Rutten EPA, Lenaerts K, Buurman WA, Wouters EFM. Disturbed Intestinal Integrity in Patients With COPD. Chest. 2014;145(2):245–252. doi:10.1378/chest.13-0584
33. Kirschner SK, Deutz NEP, Jonker R, et al. Intestinal function is impaired in patients with Chronic Obstructive Pulmonary Disease. Clin Nutr. 2021;40(4):2270–2277. doi:10.1016/j.clnu.2020.10.010
34. Ekbom A, Brandt L, Granath F, Löfdahl C-G, Egesten A. Increased risk of both ulcerative colitis and Crohn’s disease in a population suffering from COPD. Lung. 2008;186(3):167–172. doi:10.1007/s00408-008-9080-z
35. Lee J, Im JP, Han K, et al. Risk of inflammatory bowel disease in patients with chronic obstructive pulmonary disease: a nationwide, population-based study. World J Gastroenterol. 2019;25(42):6354–6364. doi:10.3748/wjg.v25.i42.6354
36. Du Y, Li X, An Y, Song Y, Lu Y. Association of gut microbiota with sort-chain fatty acids and inflammatory cytokines in diabetic patients with cognitive impairment: a cross-sectional, non-controlled study. Front Nutr. 2022;9:930626. doi:10.3389/fnut.2022.930626
37. Pujo J, Petitfils C, Le Faouder P, et al. Bacteria-derived long chain fatty acid exhibits anti-inflammatory properties in colitis. Gut. 2021;70(6):1088–1097. doi:10.1136/gutjnl-2020-321173
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.