")
Back to Journals » International Journal of Nanomedicine » Volume 19
BMP7-Loaded Human Umbilical Cord Mesenchymal Stem Cell-Derived Small Extracellular Vesicles Ameliorate Liver Fibrosis by Targeting Activated Hepatic Stellate Cells
Authors Zhu D, Sun Z, Wei J, Zhang Y, An W, Lin Y, Li X
Received 1 December 2023
Accepted for publication 23 March 2024
Published 11 April 2024 Volume 2024:19 Pages 3475—3495
DOI https://doi.org/10.2147/IJN.S450284
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor R.D.K. Misra
Dan Zhu,1 Zongbin Sun,1 Jiayun Wei,2 Yulin Zhang,2 Wenjing An,2 Yan Lin,1 Xun Li1– 3
1First Clinical Medical College, Lanzhou University, Lanzhou, People’s Republic of China; 2Gansu Province Key Laboratory of Biotherapy and Regenerative Medicine, First Hospital of Lanzhou University, Lanzhou University, Lanzhou, People’s Republic of China; 3General Surgery Department, First Hospital of Lanzhou University, Lanzhou University, Lanzhou, People’s Republic of China
Correspondence: Xun Li, First Hospital of Lanzhou University, 1st West Donggang Road, Chengguan District, Lanzhou, Gansu Province, 730000, People’s Republic of China, Tel +86-931-8356056, Fax +86-931-8629797, Email [email protected]
Purpose: Human umbilical cord mesenchymal stem cell (hucMSC)-derived small extracellular vesicles (sEVs) are natural nanocarriers with promising potential in treating liver fibrosis and have widespread applications in the fields of nanomedicine and regenerative medicine. However, the therapeutic efficacy of natural hucMSC-sEVs is currently limited owing to their non-specific distribution in vivo and partial removal by mononuclear macrophages following systemic delivery. Thus, the therapeutic efficacy can be improved through the development of engineered hucMSC-sEVs capable to overcome these limitations.
Patients and Methods: To improve the anti-liver fibrosis efficacy of hucMSC-sEVs, we genetically engineered hucMSC-sEVs to overexpress the anti-fibrotic gene bone morphogenic protein 7 (BMP7) in parental cells. This was achieved using lentiviral transfection, following which BMP7-loaded hucMSC-sEVs were isolated through ultracentrifugation. First, the liver fibrosis was induced in C57BL/6J mice by intraperitoneal injection of 50% carbon tetrachloride (CCL4) twice a week for 8 weeks. These mice were subsequently treated with BMP7+sEVs via tail vein injection, and the anti-liver fibrosis effect of BMP7+sEVs was validated using small animal in vivo imaging, immunohistochemistry (IHC), tissue immunofluorescence, and enzyme-linked immunosorbent assay (ELISA). Finally, cell function studies were performed to confirm the in vivo results.
Results: Liver imaging and liver histopathology confirmed that the engineered hucMSC-sEVs could reach the liver of mice and aggregate around activated hepatic stellate cells (aHSCs) with a significantly stronger anti-liver fibrosis effect of BMP7-loaded hucMSC-sEVs compared to those of blank or negative control-transfected hucMSC-sEVs. In vitro, BMP7-loaded hucMSC-sEVs promoted the phenotypic reversal of aHSCs and inhibited their proliferation to enhance the anti-fibrotic effects.
Conclusion: These engineered BMP7-loaded hucMSC-sEVs offer a novel and promising strategy for the clinical treatment of liver fibrosis.
Keywords: hucMSCs, sEVs, aHSCs, nanocarrier, BMP7
Graphical Abstract:
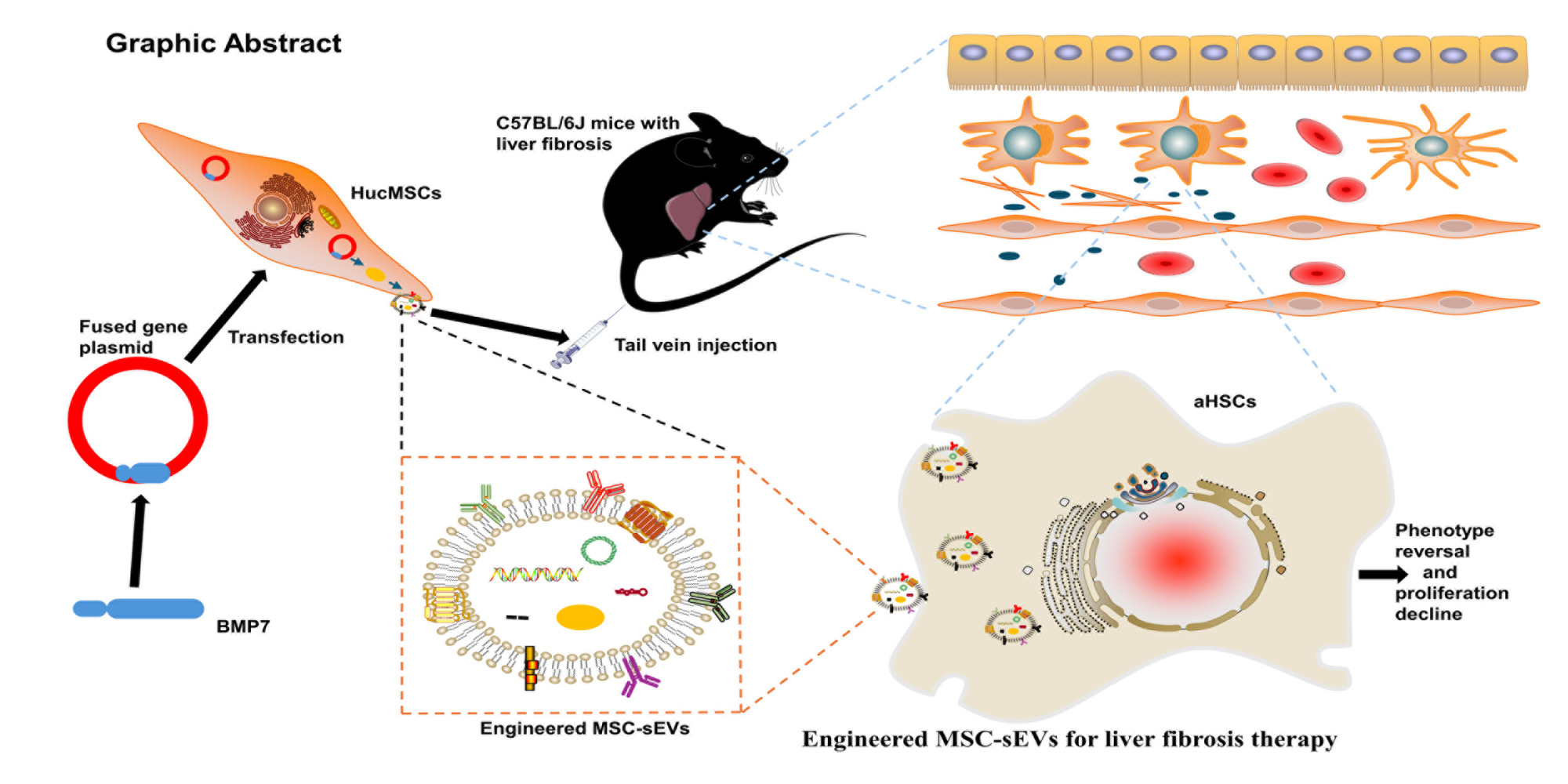
Introduction
Liver fibrosis is a pathological change caused by excessive extracellular matrix (ECM) deposition in the liver due to various factors (eg, alcohol, drugs, viruses, and immunity), ultimately leading to abnormalities in the structure and function of the liver.1 Currently, 1.5 billion people worldwide are affected by liver fibrosis, which is a typical pathological phase of several chronic liver diseases, representing a considerable threat to global health.2 The main features of liver fibrosis include a large number of activated hepatic stellate cells (aHSCs) and the accumulation of excess ECM.3 Quiescent hepatic stellate cells (qHSCs) are characterized by abundant lipid droplets (LDs) in their cytoplasm that can store triglycerides and vitamin A, which are crucial for liver regeneration, lipid metabolism, and immunological regulation.4 However, when the liver sustains injury, HSCs undergo activation and migrate to the site of injury, where aHSCs acquire a myofibroblast-like phenotype, lose LDs, and express high levels of α-smooth muscle actin (α-SMA) and collagen type I (COLI).5 The reversibility of liver fibrosis was reported to involve the conversion of the active HSCs phenotype into the quiescent phenotype.6 Therefore, aHSCs are important targets for anti-fibrotic therapy.
Mesenchymal stem cells (MSCs) are adult stem cells with immunomodulatory and tissue repair properties.7 With the development of regenerative medicine, MSCs have been extensively exploited as superior seed cells in several fields of medical research. MSCs can alleviate liver fibrosis through various mechanisms.8–10 However, a few studies have shown that there is a risk of immune rejection, microvascular occlusion, and tumorigenicity when exogenous MSCs enter the body.11–13 Additionally, the aging, differentiation, and contamination issues caused by the massive expansion of MSCs in vitro do not guarantee the quality of MSCs injections.14 Additionally, their application in regenerative medicine remains limited owing to their short in vivo survival times.15 MSCs can repair damaged tissues and organs through various mechanisms,16,17 and the tissue repair function of MSCs is mainly mediated by self-secreted soluble cell factors and sEVs (30–200 nm in diameter).18 MSC-sEVs can effectively transfer functional cargo (such as microRNAs, mRNAs, proteins, and cytokines) from MSCs into target cells; thus, demonstrating the capability to act as natural anti-fibrotic agents.19 Moreover, MSC-sEVs are excellent natural nanocarriers that can be engineered to load small-molecule cargos and enhance their anti-fibrotic effects.20 Furthermore, MSC-sEVs exhibit robust abilities as signaling molecule transporters and regulatory factors owing to their small size, simple structure, and low immunogenicity.21 Therefore, MSC-sEVs offer clear advantages of greater stability than unmodified donor cells and can reduce the safety risk associated with live-cell administration, providing a novel strategy for achieving tissue and organ repair while avoiding the risks associated with cell therapy.22–24 Thus, MSC-sEVs are viewed as a promising therapeutic agent for liver fibrosis; however, their effects are restricted owing to their limited targeting ability.25,26 Therefore, several studies have used surface engineering techniques to express targeted peptides on the surface of sEVs, thereby enhancing the anti-fibrotic effect of MSC-sEVs. For example, strategies such as targeting bone marrow MSC-sEVs (IMTP-Exos), umbilical cord MSC-sEVs (HSTP1-Exos), and utilizing Vitamin A+sEVs have all demonstrated improved targeting capabilities for MSC-sEVs.27–29
Although engineered MSC-sEVs can carry target peptides, there are off-target effects, and the non-specific distribution of MSC-sEVs in target tissues in vivo as well as the poor production scalability in vitro limit their therapeutic effects.30 Therefore, enhancing the antifibrotic effect of MSC-sEVs is crucial to improving their anti-fibrotic effect. Further, sEVs not only carry most of the genetic information of the parental cell but also are natural nanocarriers with low immunogenicity and high biocompatibility that can be loaded with numerous types of small molecules (nucleic acids, proteins, and drugs) and translocated to target cells.31 Therefore, a simple, efficient, and clinically available nanoengineering technique is required to enhance the anti-liver fibrotic effects of MSC-sEVs. Bone morphogenetic protein 7 (BMP7) is a negative regulator of TGF-β/Smad signaling,32,33 which can protect against organ fibrosis by acting on membrane receptors and activating intracellular signaling pathways to interfere with TGF-β1-mediated fibrogenesis.34 Importantly, studies have shown that BMP7 can inhibit HSCs activation through various mechanisms, thereby alleviating liver fibrosis.35–37 As such, we hypothesized that MSC-sEVs loaded with the anti-fibrotic protein BMP7 would exhibit enhanced anti-liver fibrosis effects.
To test this possibility, we used a genetic engineering strategy to overexpress BMP7 in hucMSCs and obtained BMP7-loaded MSC-sEVs (BMP7+sEVs) via ultracentrifugation. We further evaluated the physical and chemical properties of BMP7+sEVs and tested their anti-liver fibrosis efficacy after systemic administration in mice with liver fibrosis as well as their ability to target aHSCs in vitro. We anticipate that these engineered BMP7+sEVs can represent a new breakthrough in anti-liver fibrosis therapy and the expansion of stem cell therapy in regenerative medicine.
Materials and Methods
Cultivation of hucMSCs
One fresh umbilical cord was obtained from a healthy donor, and hucMSCs were isolated from umbilical cord tissue via tissue block culture as previously described.38 All procedures were approved by our institutional ethics committee (Ethics Committee of LZU No. l Hospital, no. LDYYLL-2023-459). After the umbilical cord was cut, it was immediately clamped and loaded into the tissue collection bottle and stored at 4 °C. The umbilical cord with the clip was removed from the bottle in a biosafety cabinet. It was then placed for 15 s in a wide-mouthed recipient containing 300 mL of 75% alcohol. The umbilical cord was subsequently rinsed three times with normal saline for 3 min each time. The cleaned umbilical cord was placed in a Petri dish with normal saline before being cut into 1 cm pieces. The vessels were avulsed in a Petri dish, and the cord was severed lengthwise. The tissue blocks were consecutively placed in 3 beakers filled with normal saline solution for three rounds of washing. Each round lasted for 3 min, with the aim of removing blood cells. The tissue blocks were subsequently cut into approximately 1 mm3 sections, evenly distributed in T75 cell culture flasks and culture dishes, and placed upside down in a cell culture incubator (37 °C, 5% CO2) for 3 h until dry and adherent to the wall. After turning over the cell culture bottles and culture dishes, 15 mL of Dulbecco’s modified Eagle medium (DMEM)-F12 (Gibco, Grand Island, NY, USA) with 15% fetal bovine serum (FBS) (Gibco) was added to the cell culture chamber to allow the cells to climb out of the tissue. Cells began to creep on the seventh day. The crawling cells were digested with trypsin and reseeded on the 14th day to cultivate primary hucMSCs. Primary hucMSCs passaging was used to culture passage-1 hucMSCs after 2 days. A portion of passage-1 hucMSCs was cryopreserved, while another portion was further cultured for bacterial detection and flow cytometry-based phenotyping.
Identification of hucMSCs
After culturing to the passage-3, hucMSCs were dissociated and incubated for 40 min at 25 °C with fluorescein isothiocyanate (FITC)-labeled antibodies against CD19 and CD73; phycoerythrin (PE)-labeled antibodies against CD34, CD105, and HLA-DR; and allophycocyanin (APC)-labeled antibodies against CD45, CD11b, and CD90 (BioLegend, San Diego, CA, USA). Samples were evaluated using a flow cytometer (Agilent, Santa Clara, CA, USA) after cleaning and resuspension in phosphate-buffered saline (PBS).
Lentivirus Packaging and Infection of Target Cells
Plasmid GV492 (20 µg; element sequence Ubi-MCS-3FLAG-CBh-gcGFP-IRES-puromycin), Helper 1.0 (15 µg), and Helper 2.0 (10 µg) (all from Jikai Gene Medical Technology Co. Ltd., Shanghai, China) were mixed with 1 mL viral transfection reagent and incubated at 25 °C for 20 min. HEK-293T cells (Procell Life Science & Technology Co., Ltd, Wuhan, China) were seeded in 6-well plates at a density of 1.0×104 cells/well, and the medium (DMEM without FBS) was replaced when the cell density reached 70%. The plasmid mixture was slowly added to the medium of HEK-293T cells and cultured in a cell incubator (37 °C, 5% CO2). Six hours after transfection, the cells was cultured with the new medium (DMEM with 10% FBS). After 48 h of culturing, the cell supernatant containing lentiviral particles was collected and centrifuged at 3000 × g and 4 °C for 15 min to remove cell debris. Samples were further centrifuged using an ultracentrifuge (Beckman, Pasadena, CA, USA) at 30,000 × g and 4 °C for 10 min. The viral precipitate was retained and the viral solution was filtered using a 0.25 µm filter to eliminate bacteria after resuspension with the addition of viral storage solution. Next, 2×105 passage-3 hucMSCs were evenly distributed into two wells of 6-well plates to achieve 30% confluence on the day of transfection. Cells were fully adherent to the wall and stretched after 8 h for LV-BMP7 and LV-negative vector transfection; 960 µL of complete medium and 40 µL of infection solution A were added for transfection, and the virus was inserted vertically in a dot dispersion. After 48 h of transfection, hucMSCs were screened with 2 µg/mL puromycin. After 48 h, 100% of the cells in the normal group and 10% of the cells in the negative control and intervention groups had died. On the third day after transfection, green fluorescent proteins in the cytoplasm of hucMSCs were observed using a fluorescence microscope (Olympus, Tokyo, Japan).
Isolation of hucMSC-sEVs
The purification of sEVs was optimized based on previously reported ultracentrifugation protocols.39 Three types of hucMSCs (Blank control, Negative control, and BMP7+) were cultured using DMEM-F12 for 48 h in a cell incubator (37 °C, 5% CO2) before the supernatants were collected separately. The conditioned media (CM) was centrifuged for 5 min at 1000 × g and 4 °C to eliminate floating cells before being centrifuged for 10 min at 3000 × g and 4 °C to remove dead cells. To eliminate any remaining cellular debris and impurities from the CM, it was centrifuged at 10,000 × g for 30 min at 4 °C before being filtered through a 0.22 µm filter. The CM was then centrifuged at 100,000 × g, 4 °C for 70 min in an ultracentrifuge (Beckman), and the milky white precipitate was retained after another centrifugation step. The milky white precipitate was subsequently resuspended in 10 mL of PBS and centrifuged for 90 min at 110,000 × g and 4 °C to wash and purify sEVs. Finally, these purified sEVs were resuspended in pre-cooled PBS before storing at –80 °C for next use.
Nanoparticle Tracking Analysis (NTA), Transmission Electron Microscopy (TEM) and Flow Cytometry
The sEVs precipitate was resuspended in a trace amount of fresh PBS (200 µL). This suspension was used for NTA, TEM, and flow cytometry. The 50 µL of sEVs stock solution was diluted to 1 mL and the particle size was determined using NTA (Malvern, Malvern City, UK). For TEM,10 µL of the sample was fixed overnight at 4 °C with 2.5% glutaraldehyde solution before being placed onto a copper grid and incubated for 5 min at 25 °C. Next, the copper grid was stained with three drops of saturated uranium dioxide acetate solution for 1 min and then washed with three drops of ddH2O for 5 min at 25 °C. This washing process was repeated once. After drying at 25 °C, the samples were imaged using a TEM (FEI, Hillsboro, Oregon, USA). For flow cytometry, the sEVs stock solution was diluted to 60 µL and by adding 20 µL of FITC-CD9 and FITC-CD63 (BD, Franklin Lakes, NJ, USA) antibodies before being incubated at 25 °C for 40 min. After the addition of 1 mL of pre-cooled PBS, the excess antibody was washed by centrifugation at 100,000 × g for 70 min and the washing step was repeated once more. Samples were resuspended with 100 µL of pre-cooled 1× PBS and detected using flow cytometry (BD).
RT-qPCR and Western Blot Analyses
Total RNA of hucMSCs was extracted using the TRIzol reagent (TaKaRa, Tokyo, Japan). Reverse transcription was performed using a reagent kit (TaKaRa) following the manufacturer’s guidelines for synthesis of cDNA. Real-time PCR was performed using a Mx3000 real-time PCR detection system (Agilent). The relative expression levels of the sample mRNA were calculated using the 2−ΔΔCt method. Total protein was extracted using cellular protein lysates (Solarbio, Peking, China) and the concentration of cell proteins was determined using the bicinchoninic acid protein assay kit (Beyotime, Shanghai, China). Proteins were denatured by boiling the samples for 15 min at 100 °C with sodium dodecyl sulfate. The hucMSCs proteins were denatured, separated using 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and transferred onto a polyvinylidene fluoride (PVDF) membrane. After blocking with 3% bovine serum albumin, the PVDF membrane (0.22 µm) was washed with 1× Tris buffered saline with 1% Tween (TBST). The expression of BMP7 was determined in hucMSCs and hucMSC-sEVs with antibodies against BMP7 (dilution ratio = 1:200) and GAPDH (dilution ratio = 1:2000) (SAB, Sioux Falls, South Dakota, USA) using Western blotting.
CCl4-Induced Liver Fibrosis Model and Animal Treatment
Five-week-old male C57BL/6J mice (Lanzhou Veterinary Research Institute, Chinese Academy of Agricultural Sciences, China) weighing 16–20 g were divided into control and experimental groups at random after acclimating to the specific pathogen-free laboratory for 1 week. The experimental group was intraperitoneally injected with 1 mL/kg CCl4/olive oil (1:1 v/v) twice weekly for 8 weeks to induce liver fibrosis. After establishing the model, six mice were randomly selected for model verification. The remaining mice from the experimental group were randomly and evenly divided into four groups: PBS, blank-sEVs, negative-sEVs, and BMP7+sEVs groups. Mice in the PBS group received 200 µL PBS via the tail vein every 2 days, while mice in the blank-sEVs, negative-sEVs, and BMP7+sEVs groups received 10 µg/g of the corresponding sEVs via the tail vein every 2 days for 2 weeks. After 1 week of treatment, two mice from each treatment group were injected with DiR-labeled blank-sEVs, negative-sEVs, or BMP7-sEVs via the tail vein, and the distribution of sEVs in tissues and organs in vivo was assessed using the VISQUE small animal in vivo imaging system (Vieworks, Pyeongtaek, South Korea) at 2, 12, 24, 48, and 72 h after injection. One mouse from each treatment group was injected with CM-Dil-labeled blank-sEVs, negative-sEVs, or BMP7+sEVs via the tail vein, and the distribution of sEVs in the liver was determined by paraffin sectioning. Tissue immunofluorescence was used to stain liver tissue sections with the α-SMA antibody and FITC-labeled secondary antibody (SAB). Tissue sections were imaged using a fluorescence microscope (Olympus). At the end of the experiment, the liver tissues were collected, treated with a tissue fixative, and embedded in paraffin. Hematoxylin and eosin (HE) and Masson staining were used to analyze the tissue structure and fibrosis area, respectively. The collagen-positive area was calculated using ImageJ software. Paraffin sections of the liver tissues were subjected to immunohistochemical analysis using mouse α-SMA (SAB) and COLIA1 (SAB) as primary targets. Blood was collected from mice in each group. After standing for 2 h, the serum was extracted by centrifugation at 2000 × g for 20 min. Liver function markers, alanine transaminase (ALT) and aspartate aminotransferase (AST), and liver fibrosis markers, hyaluronic acid (HA), collagen type IV (IV-C), and procollagen type III (PC-III) were tested using ELISA kits (Elabscience, Wuhan, China) according to the manufacturer protocols.
HSCs Culture
Rat HSCs (HSC-T6) and human HSCs (LX-2) (Procell Life Science & Technology Co., Ltd) were cultured with DMEM (BioInd, Kibbutz Beit Haemek, Israel) containing 10% FBS in a cell incubator (37 °C, 5% CO2). All HSCs were stimulated with 10 ng/mL TGF-β1 (Proteintech, Wuhan, China) for 24 h prior to the in vitro experiments to obtain aHSCs.
HucMSC-sEVs Uptake Analysis
To observe the uptake of BMP7+sEVs by aHSCs, the aHSCs (HSC-T6 and LX-2 cells) were co-cultured with blank-sEVs, negative-sEVs, and BMP7+sEVs labeled with CM-Dil (Bestbio, Shanghai, China) for 12 h. Next, the samples were fixed with 4% paraformaldehyde for 30min, washed three times with PBS, and the nuclei stained with DAPI (Beyotime) for 10 min. Samples were then rinsed thrice with PBS and examined under a fluorescence microscope (Olympus).
LDs Staining and Observation
The aHSCs (HSC-T6 and LX-2 cells) were divided into a control group (0 µg/mL sEVs), blank-sEVs group (100 µg/mL blank-sEVs), negative-sEVs group (100 µg/mL negative-sEVs), and BMP7+sEVs group (100 µg/mL BMP7+sEVs). After 48 h of the corresponding treatments, the cells in each group were scraped with a cell scraper before the samples were collected by centrifugation at 2000 × g for 3 min at 25 °C. After discarding the fixative, a new electron microscopy fixative was added. After cleaning, dehydration, resin infiltration, and embedding the sections in pure resin, the cells in each group were imaged using TEM after immunolabeling with colloidal gold.
Similarly, the aHSCs (HSC-T6 and LX-2 cells) were treated with 0 µg/mL sEVs, 100 µg/mL blank-sEVs, 100 µg/mL negative-sEVs, or 100 µg/mL BMP7+sEVs for 48 h before being stained with an Oil Red O kit (Solarbio). Specimens were observed and photographed under a microscope (Leica, Wetzlar, Germany) to detect red staining indicating the formation of lipid droplets.
Transwell Assay
The aHSCs in the control (0 µg/mL sEVs), blank-sEVs (100 µg/mL), negative-sEVs (100 µg/mL), and BMP7+sEVs (100 µg/mL) groups were seeded (3 × 104 cells per group for HSC-T6 cells and 2×104 cells per group for LX-2 cells) in the upper chamber of a Transwell with 300 µL (HSC-T6) or 200 µL (LX-2) DMEM without FBS; adding 700 µL DMEM containing 30% (HSC-T6) or 20% (LX-2) FBS to the lower chamber. Staining the migrating cells in the bottom of the filter membrane with 1% crystal violet for 15 min after 48 h of culture in a cell incubator (37 °C, 5% CO2). The phase contrast microscope (Olympus) was used to capture images of randomly selected fields.
Cellular Immunofluorescence Staining
The aHSCs (HSC-T6 and LX-2) of the control, blank-sEVs, negative-sEVs, and BMP7+sEVs groups were seeded in 6-well plates 48 h after treatment. After fixing with 4% paraformaldehyde, the cells were permeabilized for 40 min at 4 °C with 0.5% Triton X-100, then blocked with 3% bovine serum albumin for 40 min at 25 °C. Cells were further incubated with α-SMA (SAB) and proliferating cell nuclear antigen (PCNA) (SAB) antibodies at 25 °C for 2 h, and then washed with PBS trice before incubating with Texas Red-conjugated and FITC-conjugated secondary antibodies for 2 h. The nuclei were stained with DAPI for 10 min after washing with PBS, and the cells in the slides were using a fluorescence microscope (Olympus).
Real-Time Live-Cell Imaging
The aHSCs (HSC-T6 and LX-2) of the control, blank-sEVs, negative-sEVs, and BMP7+sEVs groups were seeded in 96-well plates (HSC-T6, 4×103 cells/well; LX-2, 2×103 cells/well). Each group of aHSCs was prepared in three replicate wells and treated with the corresponding hucMSC-sEVs. Cells were subsequently placed in a Cytation C5 system (BioTek, Winooski, Vermont, USA) for live-cell dynamic detection, and images as well as cell counts were obtained every 2 h for 61 cycles.
Statistical Analysis
All data are displayed as mean ± SD. To compare data between two groups, the independent t-test was used, and one-way analysis of variance (ANOVA) was used to analyze differences among multiple groups. Statistical analyses were carried out using Prism (GraphPad, Boston, MA, USA).
Results
Isolation and Identification of hucMSCs
Primary hucMSCs were isolated from umbilical cord tissues using tissue-block culture. One week later, the cells crawled out of the tissue blocks, which exhibited a pike or polygonal shape (Figure 1A). The hucMSCs tended to homogenize and stabilize after the third passage. Flow cytometry confirmed the characteristics of MSCs according to positive surface marker expression of CD105, CD90, and CD73, whereas CD11b, CD34, CD19, CD45, and HLA-DR were negative (Figure 1B). Furthermore, fluorescence microscopy at 24 and 48 h after staining the cytoplasm and nuclei of cells with CM-Dil and Hoechst 33342, respectively, demonstrated that the hucMSCs had a long pike shape with parallel or swirling growth at low density; however, they were arranged in a fishbone-like structure at high density with a morphology similar to that of fibroblasts (Figure 1C).
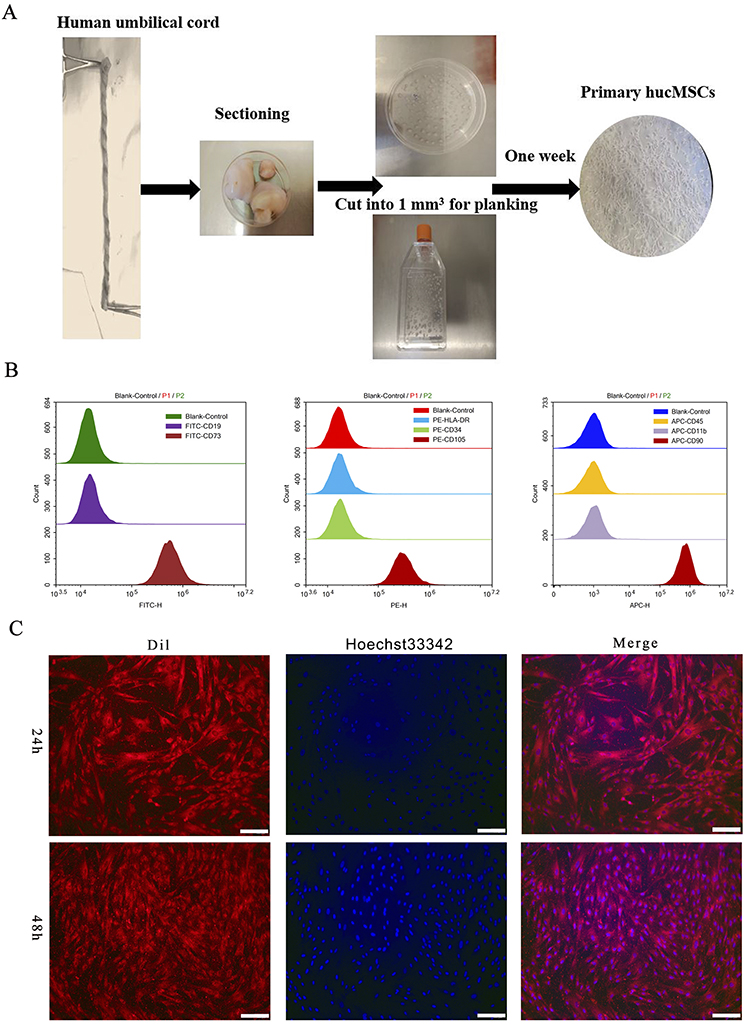 |
Figure 1 Isolation and identification of hucMSCs. (A) Brief procedure for isolating and cultivating primary hucMSCs. (B) Flow cytometry identification of primary hucMSCs. (C) Cell morphology of primary hucMSCs. After passaging, cell membranes and nuclei were stained with CM-Dil (Dil) and Hoechst 33342, and observed under a fluorescence microscope after 24 and 48 h. Scale bar: 400 µm. |
Modification of hucMSCs
The isolated hucMSCs were genetically modified to overexpress BMP7 using lentivirus transfection (multiplicity of infection = 100). Three days after transfection, fluorescence microscopy revealed a strong green fluorescent signal in the cells, and the infection rate of both the BMP7 overexpression lentivirus and negative-control lentivirus was 90% (Figure 2A). DAPI nuclear staining demonstrated that BMP7 was amplified in the cytoplasm of hucMSCs (Figure 2B). Reverse transcription-quantitative polymerase chain reaction (RT-qPCR) and Western blotting confirmed that BMP7 was successfully amplified in hucMSCs (Figure 2C) and efficiently translated into proteins (Figure 2D), respectively, with increased expression compared with that of the negative control-transfected cells (Figure 2E).
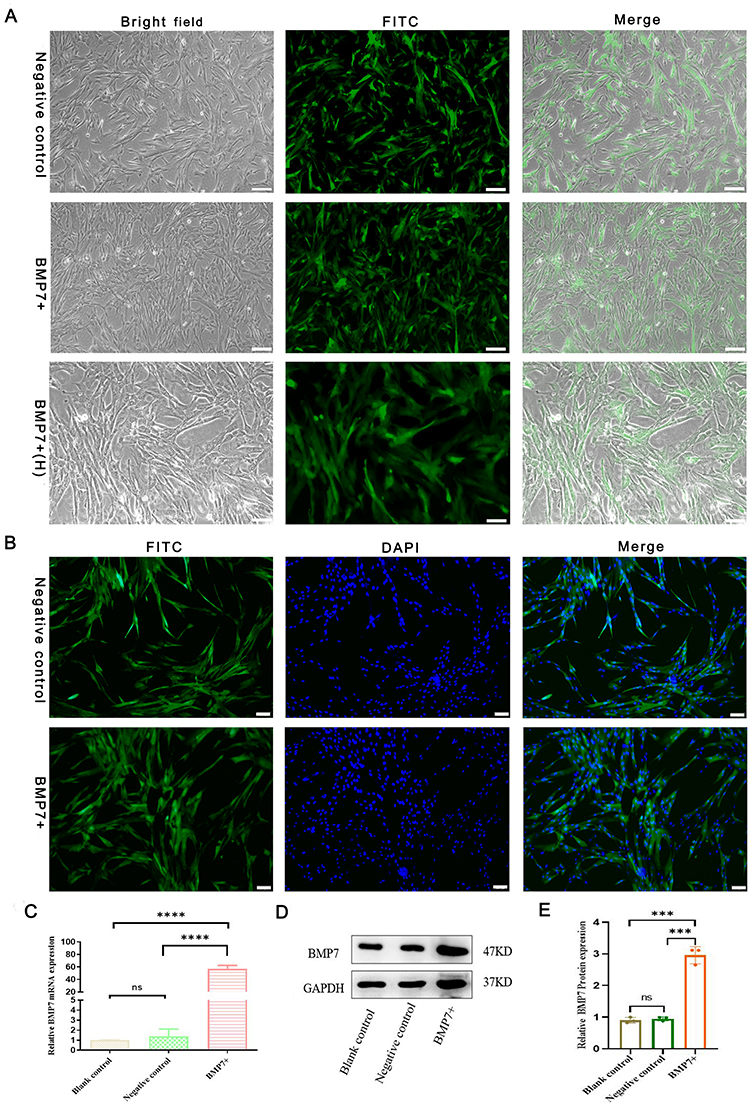 |
Figure 2 Modification of hucMSCs. (A) Infection efficiency of negative lentivirus (negative control) and BMP7 gene overexpression lentivirus (BMP7+) in hucMSCs. (B) Expression efficiency of negative lentivirus (negative control) and BMP7 gene overexpression lentivirus (BMP7+) in hucMSCs. BMP7+(H) scale bar: 100 µm. Other scale bars: 200 µm. (C) RT-qPCR results of BMP7 mRNA expression in blank control, negative control, and BMP7+ in hucMSCs. The following primers were used: forward: 5ʹ-TTCGTCAACCTCGTGGAACAT-3ʹ, reverse: 5ʹ-GAAGCGTTCCCGGATGTAGT-3ʹ. (D) Western blotting result of relative BMP7 protein expression qualification in hucMSCs. (E) Western blotting quantification. ns P > 0.05, ***P < 0.001, ****P < 0.0001. |
Characterization of hucMSC-sEVs
Ultracentrifugation-purified hucMSC-sEVs (Figure 3A) were characterized using TEM, NTA, and flow cytometry. TEM revealed that the sEVs were ring-shaped disks with lipid bilayers (Figure 3B), whereas the TEM and NTA results showed that the diameter of the sEVs was mostly in the 100–200 nm range (Figure 3C). Flow cytometry revealed that the sEVs were positive for the surface markers CD61 and CD9 (Figure 3D). In addition, Western blotting confirmed that the hucMSC-sEVs were successfully loaded with BMP7 (Figure 3E and F). These results confirmed that sEVs could be successfully engineered by modifying parental cells using genetic engineering techniques.
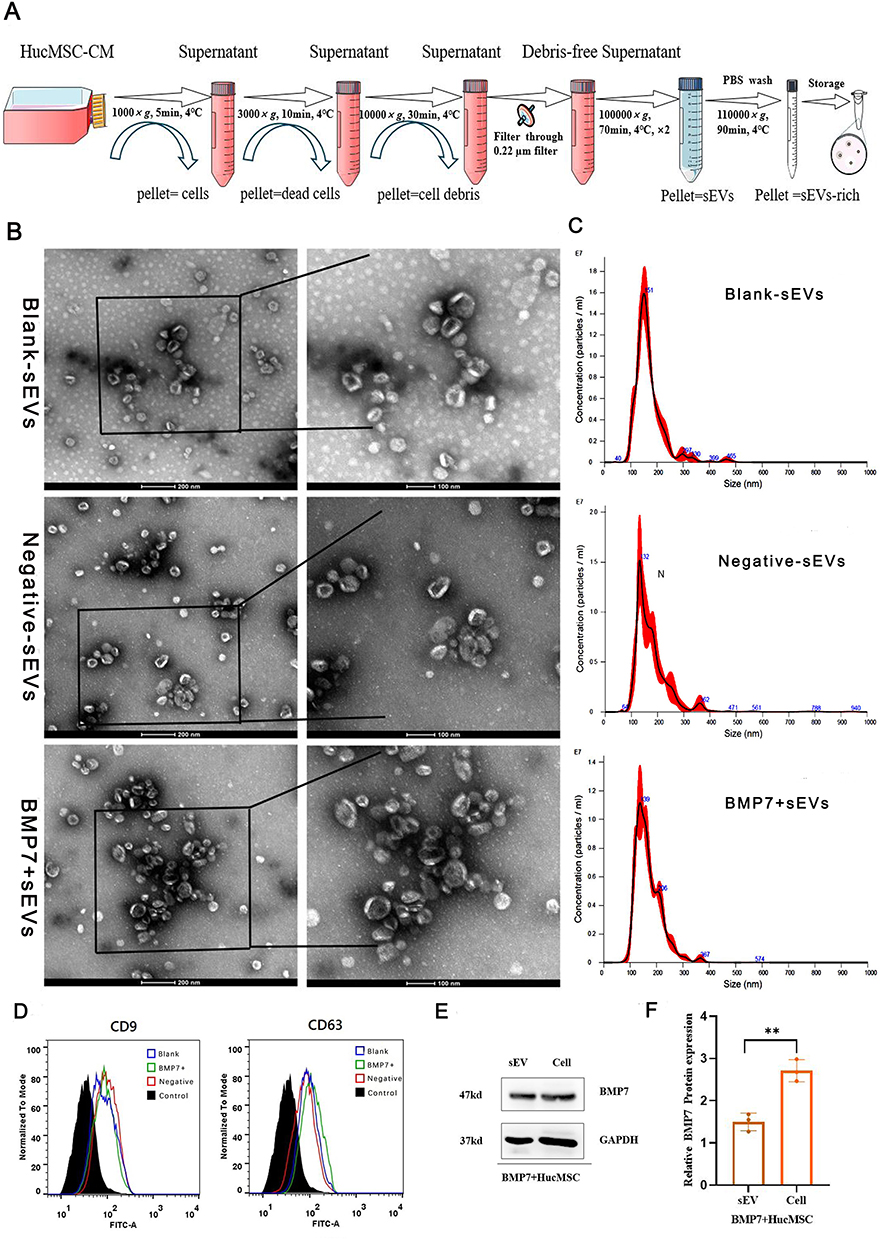 |
Figure 3 Isolation and identification of BMP7+sEVs. (A) Process of ultracentrifugation for hucMSC-sEVs purification. (B) TEM images of blank-sEVs, negative-sEVs, and BMP7+sEVs. (C) Size distribution of blank-sEVs, negative-sEVs, and BMP7+sEVs based on NTA. (D) Flow cytometry analysis of blank-sEVs, negative-sEVs, and BMP7+sEVs for CD9 and CD63. (E) Western blotting analysis of BMP7+hucMSCs cell and sEV for BMP7 and GAPDH. (F) Western blotting quantification. **P < 0.01. |
Tissue Distribution of BMP7+sEVs in vivo
In vivo imaging was used to demonstrate whether BMP7+sEVs could be efficiently and specifically delivered to the liver of normal C57BL/6J mice at various time points after intravenous injection with DiR-labeled PBS, blank-sEVs, and BMP7+sEVs. The fluorescent signals of PBS, blank-sEVs, and BMP7+sEVs were identified in the abdominal liver region, which peaked at 24 h and decreased thereafter (Figure 4A). Comparison of the temporal dispersion of sEVs in various organs, including the lungs, heart, spleen, liver, and kidneys, revealed that sEVs were primarily concentrated in the liver, implying that they could be efficiently administered to the liver in a targeted manner (Figure 4B). The same pattern was found after administration of sEVs to mice with CCl4-induced liver fibrosis (Figure 4C); hence, demonstrating the accumulation of sEVs mainly in the fibrotic liver, with the highest fluorescence signals detected in the livers of the BMP7+sEVs-treated animals (Figure 4D). Furthermore, 24 h after injection of DiI-labeled sEVs into the tail vein of fibrotic mice, immunofluorescence of liver tissues showed that sEVs mostly aggregated around α-SMA-expressing cells and BMP7+sEVs were more highly aggregated in liver tissues compared with the constructs used for the treatment of the other two groups of mice (Figure 4E). Fluorescence co-localization analysis showed that the fluorescence signals of BMP7+sEVs were more synchronized with α-SMA (Figure 4F). These results demonstrated that after systemic treatment, hucMSC-sEVs were mainly present in the liver and exhibited a certain degree of aHSC targeting, which was enhanced by loading BMP7.
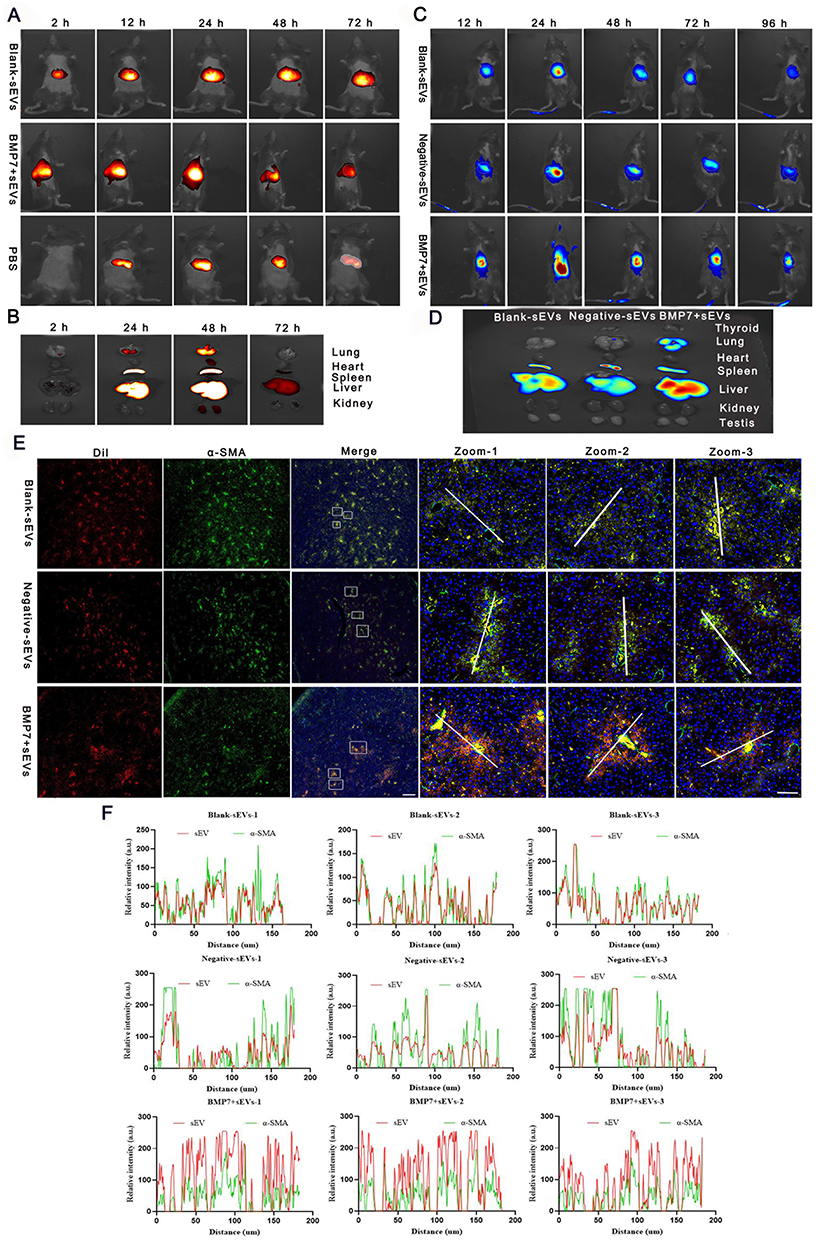 |
Figure 4 BMP7+sEVs in vivo biodistribution and ex vivo organ distribution. (A) In vivo biodistribution images of DiR (dissolved in PBS), blank-sEVs, and BMP7+sEVs in normal c57BL/6J mice. (B) In vivo biodistribution of blank-sEVs, negative-sEVs, and BMP7+sEVs in liver fibrosis C57BL/6J mice. (C) Major organs from healthy mice after intravenous injection with hucMSC-sEVs. (D) Fluorescence imaging of key organs in liver fibrosis mouse models 96 h following intravenous injection with DiR-labeled blank-sEVs, negative-sEVs, and BMP7+sEVs. (E) Immunofluorescence of α-SMA and sEVs in the liver tissues of C57BL/6J mice. Scale bars: 500 and 50 µm. (F) Co-quantification of hucMSC-sEVs and α-SMA fluorescence. |
Anti-Liver Fibrosis Effect of BMP7+sEVs in vivo
To verify the anti-liver fibrosis potential of BMP7+sEVs, the three groups of sEVs were injected into CCl4-induced hepatic fibrosis mice via the tail vein every two days for two weeks (Figure 5A). The HE staining showed severe chronic inflammation in the region of the portal vein of the liver of mice treated with CCl4, with disruption of the hepatic lobular structure and enlarged cellular interstitial space. Masson staining showed significant collagen fiber deposition in the livers of CCl4-treated mice. The IHC results demonstrated that the number of α-SMA-positive cells in the liver tissue of CCl4-treated mice was significantly higher than that in the normal control group. The immunofluorescence of liver tissue showed that collagen deposition in the BMP7+sEVs group was significantly lower than that in the other two groups of sEVs (Figure 5B). Quantification showed statistically significant differences in the collagen deposition area and α-SMA-positive area among groups (Figure 5C). These experimental results indicated that hucMSC-sEVs have a certain anti-liver fibrosis effect in vivo, with the strongest effect detected for BMP7+sEVs.
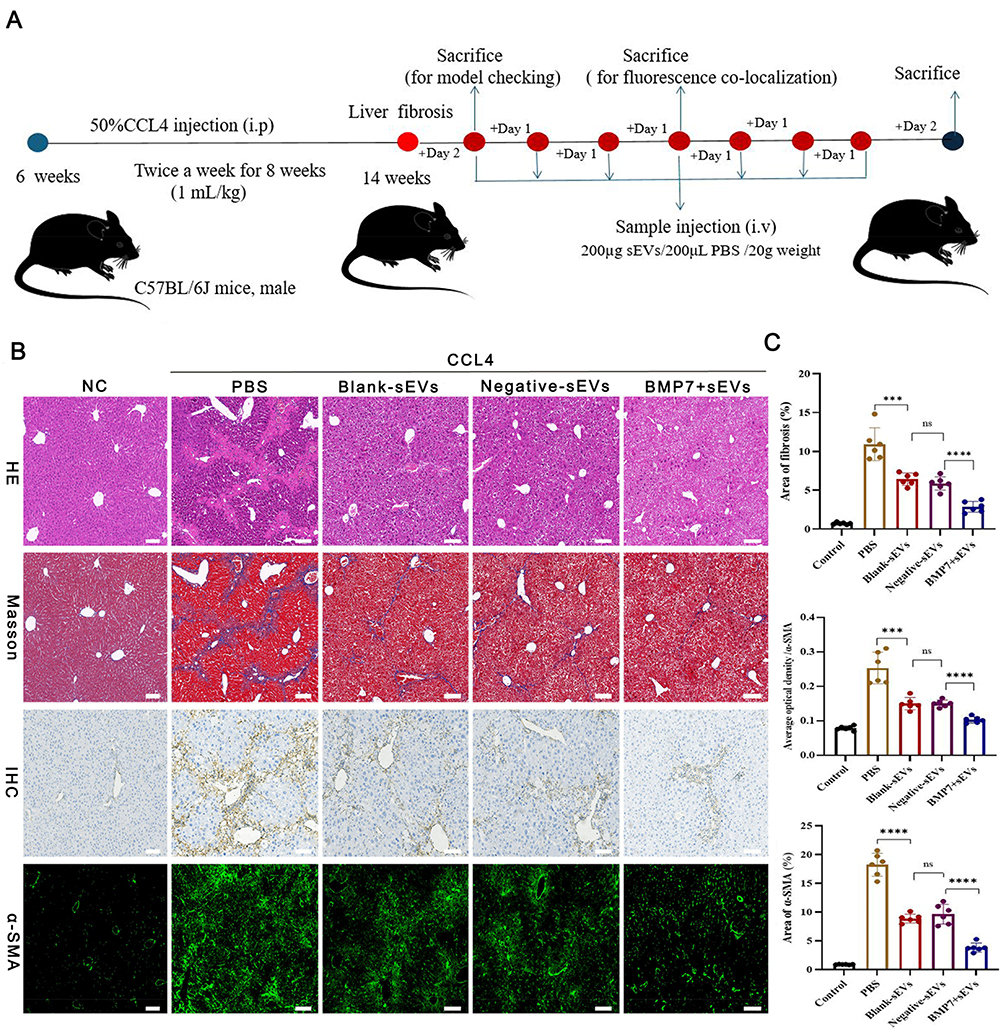 |
Figure 5 Therapeutic efficacy of BMP7+sEVs in liver fibrosis mice. (A) Treatment regimen using hucMSC-sEVs for liver fibrosis. i.p: intraperitoneal injection, i.v: tail vein injection. (B) HE staining, Masson staining, IHC, and immunofluorescence of α-SMA from liver tissues of normal control (NC) as well as PBS-, blank-sEVs-, negative-sEVs-, and BMP7+sEVs-treated mice. Scare bar: 100 µm. (C) Quantification of the fibrotic area using Masson staining. The bar graph depicts the quantification of α-SMA staining. ns P > 0.05, ***P < 0.001, ****P < 0.0001. |
BMP7+sEVs Attenuated Liver Fibrosis and Improved Liver Function
IHC showed that the number of COL1A1-positive cells in the liver tissue of the CCl4-treated mice was considerably higher than that in the control group. However, this effect was dramatically reduced by the sEV treatment, and COL1A1 expression in the BMP7+sEVs group was significantly lower than that in the blank-sEVs and negative-sEVs groups (Figure 6A and B). Furthermore, the ELISA results confirmed that α-SMA and COL1A1 levels in the liver tissues of CCl4-treated mice were significantly higher than those in the normal control group. Nevertheless, α-SMA and COL1A1 levels decreased after sEVs treatment, and the expression levels were significantly lower in the BMP7+sEVs group than that in the blank-sEVs and negative-sEVs groups (Figure 6C). Furthermore, the hucMSC-sEV treatment effectively reduced the serum levels of the liver function markers (ALT and AST) and liver fibrosis markers (HA, IV-C, and PC-III) compared with those of the CCl4-induced fibrosis model treated with PBS (Figure 6D and E). These results suggested that hucMSC-sEVs considerably improved liver function and attenuated fibrosis, with more dramatic effects detected in the BMP7+sEVs group than in the blank-sEVs and negative-sEVs groups.
 |
Figure 6 BMP7+ sEVs improved live injury and attenuated liver fibrosis. (A) IHC of α-SMA and COL1A1 from liver tissues in normal control (NC) as well as PBS-, blank-sEVs-, negative-sEVs-, and BMP7+sEVs-treated mice. Scare bar: 100 µm. (B) The bar graph depicts the quantification of α-SMA and COL1A1 staining. (C) The expression level of α-SMA and COL1A1in liver tissue suspension. (D) The level of serum ALT and AST. (E) The level of serum HA, IV-C, and PC-III. ns P > 0.05, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. |
BMP7+sEVs Promoted the Phenotype Reversal of aHSCs
BMP7+sEVs had greater uptake by aHSCs (HSC-T6 and LX-2 cell lines) than blank-sEVs and negative-sEVs (Figure 7A). TEM observations and Oil Red O staining showed that hucMSC-sEVs increased the number of intracellular lipid droplets in aHSCs, which was most evident in the BMP7+sEVs group (Figure 7B). Transwell assays at 48 h after sEV treatment showed that the quantity of migrated cells in the BMP7+sEVs group was significantly decreased compared with that in the blank-sEV and negative-sEV groups (p < 0.001; Figure 8A and B). This suggested that hucMSC-sEVs can inhibit aHSC migration, with the largest effect detected for BMP7+sEVs. We also observed that the α-SMA expression of aHSCs was decreased after sEVs treatment, which was the lowest in the BMP7+sEVs group (Figure 8C and D). Collectively, these results suggest that hucMSC-sEVs can reverse the phenotype of aHSCs by direct targeting, with the greatest efficacy exhibited by BMP7+sEVs.
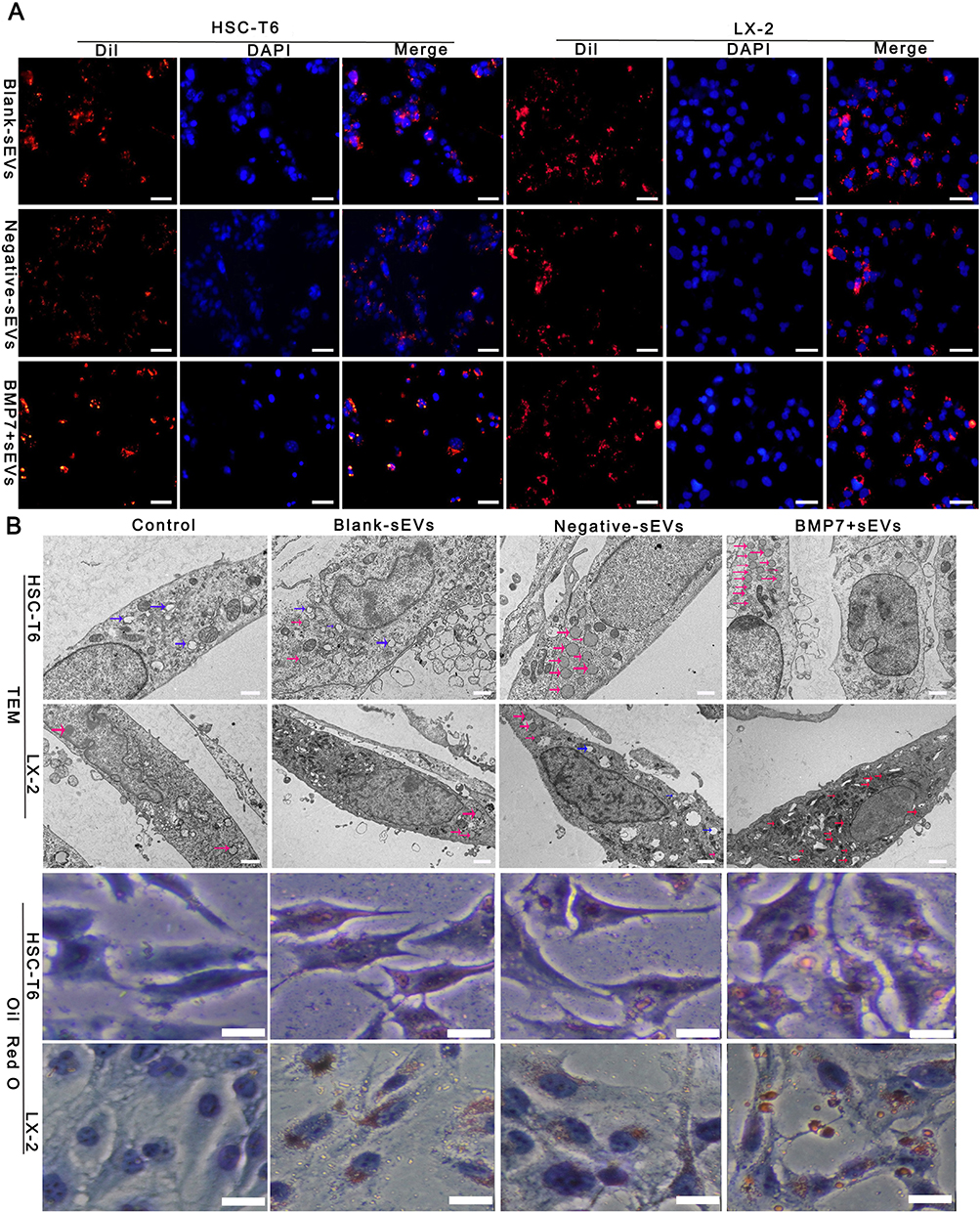 |
Figure 7 Effect of BMP7+sEVs on aHSCs. (A) Co-localization of Dil-labeled blank-sEVs, negative-sEVs, and BMP7+sEVs (red), and cell nuclei stained with DAPI (blue) after sEVs treatment of aHSCs for 12 h. Scale bars: 50 µm. (B) Visualization of LDs using TEM and Oil Red O staining in control (negative control), blank-sEVs, negative-sEVs, and BMP7+sEVs after sEV treatment of aHSCs for 48 h. The three types of colored arrows in the TEM indicate the different types of LDs (blue: cholesterol LDs, red: triglycerides LDs, pink: mixed LDs). TEM scale bars: 5.0 µm, Oil Red O staining scale bars: 6 µm. |
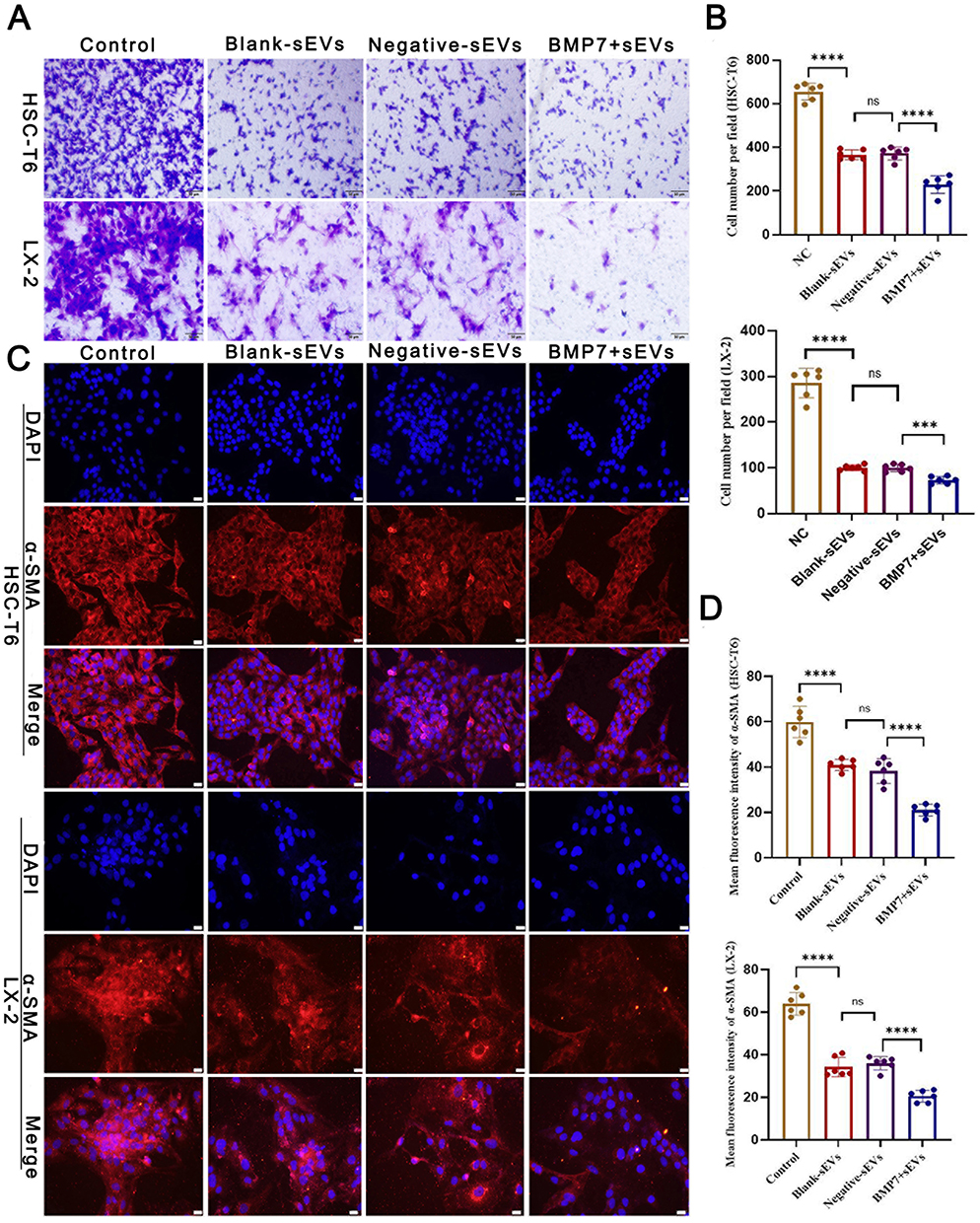 |
Figure 8 Anti-fibrosis effect of BMP7+sEVs in vitro. (A) Effect of hucMSC-sEVs on HSC-T6 and LX-2 cell migration abilities. Scale bars: 50 µm. (B) Bar graph shows the analysis results of the selected fields. (C) Immunofluorescence of α-SMA (red) in control (negative control), blank-sEVs-, negative-sEVs-, and BMP7+sEVs-treated aHSCs for 48 h. DAPI-stained nuclei (blue). Scale bars: 20 µm. (D) The bar graph shows the analysis results of the selected fields. ns P > 0.05, ***P < 0.001, ****P < 0.0001. |
BMP7+sEVs Inhibited the Proliferation of aHSCs
Finally, live-cell imaging of aHSCs for five days indicated that the proliferation rate of aHSCs in the hucMSC-sEV intervention groups was decreased relative to that of the control group, with the slowest rate representing with the BMP7+sEVs-treated cells (Figure 9A and B). The green fluorescence intensity of proliferating cell nuclear antigen in aHSCs was the weakest in the BMP7+sEVs group. These results indicated that BMP7+sEVs could also limit the proliferation of aHSCs to exert their anti-fibrotic effect in the liver (Figure 10A and B).
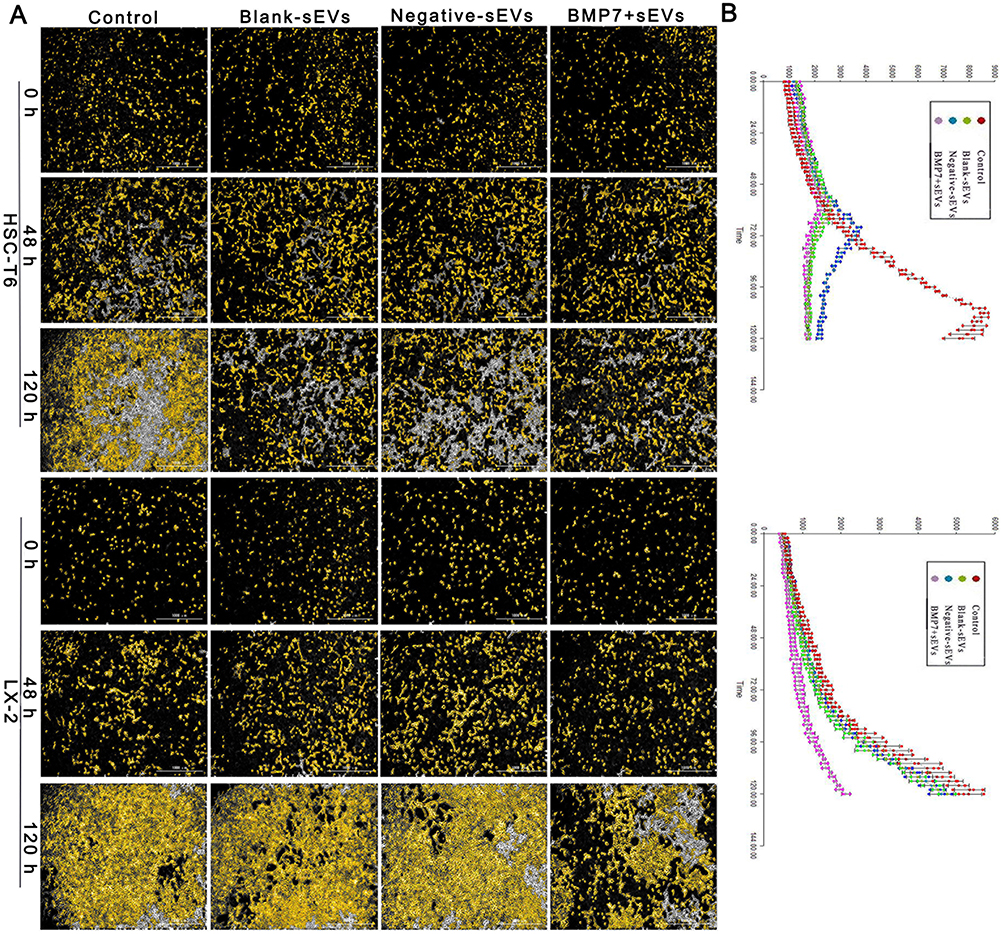 |
Figure 9 Effect of BMP7+sEVs on aHSC proliferation. (A) Cell count in the control (negative control), blank-sEVs, negative-sEVs, and BMP7+sEVs groups after sEV treatment of aHSCs for 0, 48, and 120 h. Scale bars: 1000 µm. (B) Line chart shows the growth curve in each group. |
 |
Figure 10 Effect of BMP7+sEVs on aHSC proliferation. (A) Proliferating cell nuclear antigen immunofluorescence (green) in control, blank-sEVs, negative-sEVs, and BMP7+sEVs after sEV treatment of aHSCs for 48 h. DAPI-stained nuclei (blue). Scale bars: 50 µm. (B) Bar graph shows the analysis results of the selected fields. ns P > 0.05, ***P < 0.001, ****P < 0.0001. |
Discussion
Liver fibrosis is a common pathological process in several chronic liver diseases and is a necessary step in the progression of various chronic liver injuries to cirrhosis or liver cancer. As liver fibrosis represents a major threat to human life and health, it is crucial to take proactive and effective treatment steps to prevent or even reverse disease progression. HSCs are attractive targets for liver fibrosis therapy because they play a critical role in the development of liver fibrosis.40 Currently, the mechanism of action of most drugs used in anti-liver fibrosis therapy mainly involves targeting aHSCs. However, the therapeutic effect is limited due to the drugs being susceptible to clearance by macrophages and uptake by other surrounding hepatocytes before reaching aHSCs. Additionally, their inherent toxicity and associated side effects further impede their effectiveness.41 MSCs and MSC-sEVs can alleviate liver fibrosis by acting on aHSCs through multiple mechanisms.42–44 However, their effect on anti-liver fibrosis is limited. To improve the anti-fibrotic activity of MSC-sEVs and their application in the clinic, our team first designed a conjugate of hucMSC-sEVs and BMP7 (BMP7+sEVs) by direct co-incubation of the sEVs with recombinant BMP7. However, the experimental result showed that this method was not only economically costly but also inefficient.
In this study, BMP7+sEVs were successfully generated through lentiviral transfection and led to BMP7 overexpression in hucMSCs. This genetic engineering technology, which generates sEVs loaded with the small molecule antifibrotic protein BMP7 by modifying the parental cells that produce BMP7+sEVs, is not only simple while reducing the costs associated with methods for the direct modification of sEVs but also does not disrupt the structure of sEVs. After BMP7+sEVs were injected into C57BL/6J mice via the tail vein, both in vivo and ex vivo imaging revealed that BMP7+sEVs accumulated mainly in the liver. Furthermore, immunofluorescence and fluorescence co-localization analyses showed that BMP7+sEVs were selectively taken up by aHSCs in the fibrotic liver tissue. These results showed that this targeting may result from receptor-mediated endocytosis. Compared to the BMP7+sEVs, blank-sEVs and negative-sEVs showed reduced accumulation in fibrotic liver tissue. Thus, engineered sEVs can modulate the fate and improve the microdistribution of sEVs within the mouse liver. Furthermore, the BMP7+sEVs resulted in a marked reduction in α-SMA and COLIA1 expression in fibrotic liver tissue and a significant decrease in serum levels of HA, PC-III, and IV-C in fibrotic liver mice. This reduction in biomarkers highlights the potential of BMP7+sEVs as a therapeutic approach for liver fibrosis. Liver function tests showed that all three types of hucMSC-sEVs improved liver injury, with BMP7+sEVs leading to the greatest improvement. Overall, these results suggested that BMP7+sEVs enhanced the efficacy of hucMSC-sEVs in curing fibrotic liver mice.
The key reported methods for targeting aHSCs to treat liver fibrosis include promoting apoptosis/senescence, reversing the aHSC phenotype, and suppressing the proliferation of aHSCs.45–47 P75NTR antagonists can promote the apoptosis of aHSCs.48 NF-κB inhibitors (PDTC) can also reduce oxidative stress and necroinflammation in the liver, which promotes the apoptosis of aHSCs.49 TRAIL agonists can also promote the apoptosis of aHSCs, thereby attenuating liver fibrosis.50 GATA4 inactivates HSCs by directly suppressing EPAS1 transcription.51 Praziquantel inactivates HSCs by decreasing TGF-β/Smad signaling and increasing Smad7 expression52 Recombinant human cytoglobin inhibits liver fibrosis by deactivating HSCs.53 However, these exogenous cytokines, recombinant proteins, and drugs are expensive, have numerous side effects, and most of them are cleared by macrophages after entering the body.
In this study, we demonstrated that hucMSC-sEVs could ameliorate liver fibrosis and reduce ECM deposition in vivo, with the greatest anti-liver fibrosis efficacy being associated with the BMP7+sEVs treatment. Cellular experiments further showed that BMP7+sEVs exerted antifibrotic effects by targeting HSCs. TEM, Oil Red O staining, and immunofluorescence staining showed that BMP7+sEVs promoted aHSC reversion to a relatively quiescent phenotype, whereas live cell imaging and immunofluorescence experiments showed that BMP7+sEVs inhibited aHSC proliferation. Therefore, hucMSC-sEVs effectively blocked the fibrotic cascade reaction by reversing profibrotic aHSCs to a relatively quiescent phenotype, while BMP7+sEVs further reversed the aHSC phenotype and inhibited their proliferation; thereby improving the anti-liver fibrosis efficacy of hucMSC-sEVs. In addition, we verified that the rotational speed of the ultracentrifugation method used to extract sEVs in previous studies was too high and resulted in breaking the structure of the sEVs. Therefore, we reduced the rotational speed from 120,000 × g to 100,000 × g and adjusted the rotational speed to 110,000 × g to wash and purify the sEVs, which generated not only pure sEVs but also structurally intact.
Despite our promising results, this study had some limitations. First, we only showed that BMP7+sEVs could enhance the antifibrotic effects of hucMSC-sEVs by targeting aHSCs; we did not investigate the target protein receptor of BMP7+sEVs nor have we clarified the specific mechanism through which they inhibit aHSC activation and proliferation. Second, MSC-sEVs contain genetic information from the parental cells and participate in several types of cellular activities such as antigen presentation, signaling, and immune responses. In this study, we only briefly explored whether hucMSC-sEVs could further promote the phenotypic reversal of aHSCs and inhibit their proliferation after reaching the target cells (aHSCs) through the transporter of BMP7. We have not explored whether other small molecules such as nucleic acids and proteins contained in the hucMSC-sEVs themselves synergistically interact with BMP7. We will further determine the exact intercellular messengers mediating these observed effects in a future study. Despite these limitations, this study clearly highlights BMP7+sEVs as promising, novel anti-fibrotic drugs; thus, demonstrating the precise application of nanomedicine to a single cell type in complex liver tissues as well as revealing that BMP7+sEVs strengthened the ability of hucMSC-sEVs to reverse liver fibrosis. This study further provides a general strategy for the use of nanoengineering technology to load hucMSC-sEVs with small molecules and overcome their current clinical limitations.
Conclusion
Our experimental results suggest that BMP7+sEVs generated following lentiviral transfection of parental hucMSCs can enhance the targeting of hucMSC-sEVs to aHSCs in the fibrotic mouse liver. BMP7+sEVs aggregated in fibrotic liver tissues after systemic administration to C57BL/6J mice and were preferentially taken up by aHSCs. BMP7+sEVs effectively modulated the liver fibrosis cascade response and thus enhanced the anti-liver fibrosis therapy of hucMSC-sEVs by promoting aHSC phenotypic reversion and suppressing aHSC proliferation. In conclusion, sEVs-based drugs developed through a simple engineering modification may serve as novel agents for anti-liver fibrosis therapy. Furthermore, modulation of the fibrotic cascade by sEVs based on engineered MSC-sEVs could be extended to a variety of fibroblast-accumulated diseases such as renal fibrosis, idiopathic pulmonary fibrosis, and cardiac fibrosis. In addition to stem cell-free therapies, the strategy described here provides a solid basis for the therapeutic use of diverse types of cell-derived sEVs for the refractory diseases therapy.
Abbreviations
HucMSC, human umbilical cord mesenchymal stem cell; sEVs, small extracellular vesicles; hucMSC-sEVs, human umbilical cord mesenchymal stem cell-derived small extracellular vesicles; BMP7, gene bone morphogenic protein 7; CCL4, carbon tetrachloride; IHC, immunohistochemistry; ELISA, enzyme-linked immunosorbent assay; aHSCs, activated hepatic stellate cells; ECM, excessive extracellular matrix; qHSCs, quiescent hepatic stellate cells; α-SMA, α-smooth muscle actin; COLI, collagen type I; LDs, lipid droplets; MSCs, mesenchymal stem cells; FITC, fluorescein isothiocyanate; PE, phycoerythrin; APC, allophycocyanin; CM: conditioned media; TEM, transmission electron microscopy; NTA, nanoparticle tracking analysis; PBS, phosphate-buffered saline; HE, Hematoxylin and eosin; ALT, alanine transaminase; AST, aspartate aminotransferase; HA, hyaluronic acid; IV-C, collagen type IV; PC-III, procollagen type III; PCNA, proliferating cell nuclear antigen.
Data Sharing Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Ethics Approval
This study was conducted in strict accordance with the ethical guidelines outlined in the Declaration of Helsinki. All study participants provided informed consent, and the study design and the research procedures were approved by Ethics Committee of LZU No. l Hospital (no. LDYYLL-2023-459).
Acknowledgments
We would like to thank the staff of the laboratory for their help with this project.
Author Contributions
All authors contributed to data analysis, drafting or revising the article, have agreed on the journal to which the article will be submitted, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Funding
This work was supported by the National Natural Science Foundation of China (Grant No. 82060119, 32171610) and Natural Science Foundation of Gansu province (Grant No.21JR1RA072, 20JR10FA661).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209–218. doi:10.1172/JCI24282
2. Kardashian A, Serper M, Terrault N, et al. Health disparities in chronic liver disease. Hepatology. 2023;77:1382–1403. doi:10.1002/hep.32743
3. Puche JE, Saiman Y, Friedman SL. Hepatic stellate cells and liver fibrosis. Compr Physiol. 2013;3:1473–1492.
4. Trivedi P, Wang S, Friedman SL. The power of plasticity-metabolic regulation of hepatic stellate cells. Cell Metab. 2021;33:242–257. doi:10.1016/j.cmet.2020.10.026
5. Kisseleva T, Brenner D. Molecular and cellular mechanisms of liver fibrosis and its regression. Nat Rev Gastroenterol Hepatol. 2021;18:151–166. doi:10.1038/s41575-020-00372-7
6. Parola M, Pinzani M. Liver fibrosis: pathophysiology, pathogenetic targets and clinical issues. Mol Aspects Med. 2019;65:37–55. doi:10.1016/j.mam.2018.09.002
7. Fu FU, Liu L, Halim H, Ju JU, Luo L, Song S. Mesenchymal stem cell migration and tissue repair. Cells. 2019;8(8):784. doi:10.3390/cells8080784
8. El Agha E, Kramann R, Schneider RK, et al. Mesenchymal stem cells in fibrotic disease. Cell Stem Cell. 2017;21(2):166–177. doi:10.1016/j.stem.2017.07.011
9. Tsuchiya A, Takeuchi S, Watanabe T, et al. Mesenchymal stem cell therapies for liver cirrhosis: mSCs as “conducting cells” for improvement of liver fibrosis and regeneration. Inflammation and Regeneration. 2019;39(1):18. doi:10.1186/s41232-019-0107-z
10. Yao Y, Xia Z, Cheng F, et al. Human placental mesenchymal stem cells ameliorate liver fibrosis in mice by upregulation of Caveolin 1 in hepatic stellate cells. Stem Cell Res Ther. 2021;12:294. doi:10.1186/s13287-021-02358-x
11. Trounson A, McDonald C. Stem cell therapies in clinical trials: progress and challenges. Cell Stem Cell. 2015;17:11–22. doi:10.1016/j.stem.2015.06.007
12. Li TT, Wang ZR, Yao WQ, Linghu EQ, Wang FS, Shi L. Stem cell therapies for chronic liver diseases: progress and challenges. Stem Cells Transl Med. 2022;11:900–911. doi:10.1093/stcltm/szac053
13. Yang X, Meng Y, Han Z, Ye F, Wei L, Zong C. Mesenchymal stem cell therapy for liver disease: full of chances and challenges. Cell Biosci. 2020;10:123. doi:10.1186/s13578-020-00480-6
14. Al-Azab M, Safi M, Idiiatullina E, Al-Shaebi F, Zaky MY. Aging of mesenchymal stem cell: machinery, markers, and strategies of fighting. Cell Mol Biol Lett. 2022;27:69. doi:10.1186/s11658-022-00366-0
15. Hwang NS, Zhang C, Hwang YS, Varghese S. Mesenchymal stem cell differentiation and roles in regenerative medicine. Wiley Interdiscip Rev Syst Biol Med. 2009;1:97–106. doi:10.1002/wsbm.26
16. Cheng W, Zeng Y, Wang D. Stem cell-based therapy for pulmonary fibrosis. Stem Cell Res Ther. 2022;13:492. doi:10.1186/s13287-022-03181-8
17. Liu P, Mao Y, Xie Y, Wei J, Yao J. Stem cells for treatment of liver fibrosis/cirrhosis: clinical progress and therapeutic potential. Stem Cell Res Ther. 2022;13:356. doi:10.1186/s13287-022-03041-5
18. Psaraki A, Ntari L, Karakostas C, Korrou-Karava D, Roubelakis MG. Extracellular vesicles derived from mesenchymal stem/stromal cells: the regenerative impact in liver diseases. Hepatology. 2022;75:1590–1603. doi:10.1002/hep.32129
19. Varderidou-Minasian S, Lorenowicz MJ. Mesenchymal stromal/stem cell-derived extracellular vesicles in tissue repair: challenges and opportunities. Theranostics. 2020;10:5979–5997. doi:10.7150/thno.40122
20. Cecchin R, Troyer Z, Witwer K, Morris K. Extracellular vesicles: the next generation in gene therapy delivery. Mol Ther. 2023;31:1225–1230. doi:10.1016/j.ymthe.2023.01.021
21. Hu C, Zhao L, Zhang L, Bao Q, Li L. Mesenchymal stem cell-based cell-free strategies: safe and effective treatments for liver injury. Stem Cell Res Ther. 2020;11:377. doi:10.1186/s13287-020-01895-1
22. Lou G, Chen Z, Zheng M, Liu Y. Mesenchymal stem cell-derived exosomes as a new therapeutic strategy for liver diseases. Exp Mol Med. 2017;49(e346):e346–e346. doi:10.1038/emm.2017.63
23. Ghafouri-Fard S, Niazi V, Hussen BM, Omrani MD, Taheri M, Basiri A. The emerging role of exosomes in the treatment of human disorders with a special focus on mesenchymal stem cells-derived exosomes. Front Cell Dev Biol. 2021;9:653296. doi:10.3389/fcell.2021.653296
24. Ceccotti E, Saccu G, Herrera Sanchez MB, Bruno S. Naïve or engineered extracellular vesicles from different cell sources: therapeutic tools for kidney diseases. Pharmaceutics. 2023;15(6):1715. doi:10.3390/pharmaceutics15061715
25. Hassanzadeh A, Rahman HS, Markov A, et al. Mesenchymal stem/stromal cell-derived exosomes in regenerative medicine and cancer; overview of development, challenges, and opportunities. Stem Cell Res Ther. 2021;12:297. doi:10.1186/s13287-021-02378-7
26. Fuloria S, Subramaniyan V, Dahiya R, et al. Mesenchymal stem cell-derived extracellular vesicles: regenerative potential and challenges. Biology (Basel). 2021;10:172. doi:10.3390/biology10030172
27. Wang X, Chen Y, Zhao Z, et al. Engineered exosomes with ischemic myocardium-targeting peptide for targeted therapy in myocardial infarction. J Am Heart Assoc. 2018;7:e008737. doi:10.1161/JAHA.118.008737
28. Lin Y, Yan M, Bai Z, et al. Huc-MSC-derived exosomes modified with the targeting peptide of aHSCs for liver fibrosis therapy. J Nanobiotechnology. 2022;20:432. doi:10.1186/s12951-022-01636-x
29. You DG, Oh BH, Nguyen VQ, et al. Vitamin A-coupled stem cell-derived extracellular vesicles regulate the fibrotic cascade by targeting activated hepatic stellate cells in vivo. J Control Release. 2021;336:285–295. doi:10.1016/j.jconrel.2021.06.031
30. Lu X, Guo H, Wei X, et al. Current status and prospect of delivery vehicle based on mesenchymal stem cell-derived exosomes in liver diseases. Int J Nanomed. 2023;18:2873–2890. doi:10.2147/IJN.S404925
31. Herrmann IK, Wood MJA, Fuhrmann G. Extracellular vesicles as a next-generation drug delivery platform. Nat Nanotechnol. 2021;16:748–759. doi:10.1038/s41565-021-00931-2
32. Zeisberg M, Hanai J, Sugimoto H, et al. BMP-7 counteracts TGF-beta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat Med. 2003;9:964–968. doi:10.1038/nm888
33. Yao H, Ge T, Zhang Y, et al. BMP7 antagonizes proliferative vitreoretinopathy through retinal pigment epithelial fibrosis in vivo and in vitro. FASEB J. 2019;33:3212–3224. doi:10.1096/fj.201800858RR
34. Weiskirchen R, Meurer SK, Gressner OA, Herrmann J, Borkham-Kamphorst E, Gressner AM. BMP-7 as antagonist of organ fibrosis. Front Biosci. 2009;14:4992–5012. doi:10.2741/3583
35. Zou GL, Zuo S, Lu S, et al. Bone morphogenetic protein-7 represses hepatic stellate cell activation and liver fibrosis via regulation of TGF-β/Smad signaling pathway. World J Gastroenterol. 2019;25:4222–4234. doi:10.3748/wjg.v25.i30.4222
36. Chen L, Zhou Q, Liu E, et al. rSjp40 inhibits activated hepatic stellate cells by promoting nuclear translocation of YB1 and inducing BMP-7/Smad1/5/8 pathway. Parasit Vectors. 2019;12:279. doi:10.1186/s13071-019-3539-z
37. Ji F, Wang K, Zhang Y, et al. MiR-542-3p controls hepatic stellate cell activation and fibrosis via targeting BMP-7. J Cell Biochem. 2019;120:4573–4581. doi:10.1002/jcb.27746
38. Mushahary D, Spittler A, Kasper C, Weber V, Charwat V. Isolation, cultivation, and characterization of human mesenchymal stem cells. Cytometry A. 2018;93:19–31. doi:10.1002/cyto.a.23242
39. Visan KS, Lobb RJ, Ham S, et al. Comparative analysis of tangential flow filtration and ultracentrifugation, both combined with subsequent size exclusion chromatography, for the isolation of small extracellular vesicles. J Extracell Vesicles. 2022;11:e12266.
40. Wang SS, Tang XT, Lin M, et al. Perivenous stellate cells are the main source of myofibroblasts and cancer-associated fibroblasts formed after chronic liver injuries. Hepatology. 2021;74:1578–1594. doi:10.1002/hep.31848
41. Schuppan D, Ashfaq-Khan M, Yang AT, Kim YO. Liver fibrosis: direct antifibrotic agents and targeted therapies. Matrix Biol. 2018;68–69:435–451. doi:10.1016/j.matbio.2018.04.006
42. Wang L, Bai G, Chen F. Human bone marrow mesenchymal stem cells suppress the proliferation of hepatic stellate cells by inhibiting the ubiquitination of p27. Biochem Cell Biol. 2017;95:628–633. doi:10.1139/bcb-2017-0127
43. Du Z, Wu T, Liu L, Luo B, Wei C. Extracellular vesicles-derived miR-150-5p secreted by adipose-derived mesenchymal stem cells inhibits CXCL1 expression to attenuate hepatic fibrosis. J Cell Mol Med. 2021;25:701–715. doi:10.1111/jcmm.16119
44. Kim J, Lee C, Shin Y, et al. sEVs from tonsil-derived mesenchymal stromal cells alleviate activation of hepatic stellate cells and liver fibrosis through miR-486-5p. Mol Ther. 2021;29:1471–1486. doi:10.1016/j.ymthe.2020.12.025
45. Zhang WS, Zhang R, Ge Y, et al. S100a16 deficiency prevents hepatic stellate cells activation and liver fibrosis via inhibiting CXCR4 expression. Metabolism. 2022;135:155271. doi:10.1016/j.metabol.2022.155271
46. Meng D, Li Z, Wang G, Ling L, Wu Y, Zhang C. Carvedilol attenuates liver fibrosis by suppressing autophagy and promoting apoptosis in hepatic stellate cells. Biomed Pharmacother. 2018;108:1617–1627. doi:10.1016/j.biopha.2018.10.005
47. Zhang M, Serna-Salas S, Damba T, Borghesan M, Demaria M, Moshage H. Hepatic stellate cell senescence in liver fibrosis: characteristics, mechanisms and perspectives. Mech Ageing Dev. 2021;199:111572. doi:10.1016/j.mad.2021.111572
48. Kendall TJ, Hennedige S, Aucott RL, et al. p75 neurotrophin receptor signaling regulates hepatic myofibroblast proliferation and apoptosis in recovery from rodent liver fibrosis. Hepatology. 2009;49:901–910. doi:10.1002/hep.22701
49. Bruck R, Schey R, Aeed H, Hochman A, Genina O, Pines M. A protective effect of pyrrolidine dithiocarbamate in a rat model of liver cirrhosis. Liver Int. 2004;24:169–176. doi:10.1111/j.1478-3231.2004.00900.x
50. Li R, Li Z, Feng Y, et al. PDGFRβ-targeted TRAIL specifically induces apoptosis of activated hepatic stellate cells and ameliorates liver fibrosis. Apoptosis. 2020;25(1–2):105–119. doi:10.1007/s10495-019-01583-3
51. Arroyo N, Villamayor L, Díaz I, et al. GATA4 induces liver fibrosis regression by deactivating hepatic stellate cells. JCI Insight. 2021;6(23):e150059. doi:10.1172/jci.insight150059
52. Liu J, Kong D, Qiu J, et al. Praziquantel ameliorates CCl(4)-induced liver fibrosis in mice by inhibiting TGF-β/Smad signalling via up-regulating Smad7 in hepatic stellate cells. Br J Pharmacol. 2019;176:4666–4680. doi:10.1111/bph.14831
53. Dat NQ, Thuy LTT, Hieu VN, et al. Hexa histidine-tagged recombinant human cytoglobin deactivates hepatic stellate cells and inhibits liver fibrosis by scavenging reactive oxygen species. Hepatology. 2021;73:2527–2545.
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.