")
Back to Journals » Advances and Applications in Bioinformatics and Chemistry » Volume 14
Antibacterial, Docking, DFT and ADMET Properties Evaluation of Chalcone-Sulfonamide Derivatives Prepared Using ZnO Nanoparticle Catalysis
Authors Weyesa A, Eswaramoorthy R , Melaku Y, Mulugeta E
Received 3 September 2021
Accepted for publication 18 November 2021
Published 11 December 2021 Volume 2021:14 Pages 133—144
DOI https://doi.org/10.2147/AABC.S336450
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Juan Fernandez-Recio
Abdanne Weyesa,1 Rajalakshmanan Eswaramoorthy,1,2 Yadessa Melaku,1 Endale Mulugeta1
1Department of Applied Chemistry, School of Applied Natural Science, Adama Science and Technology University, Adama, Ethiopia; 2Department of Biomaterials, Saveetha Dental College and Hospitals, Saveetha Institute of Medical and Technical Sciences, Saveetha University, Chennai, India
Correspondence: Endale Mulugeta Email [email protected]
Introduction: In the present work, two novel compounds were synthesized using zinc oxide nanoparticles through green synthesis protocol. The zinc oxide nanoparticles catalyzed reactions were afforded good to excellent yields of the target compounds 76.3– 98.6%.
Methodology: The synthesized compounds were characterized by UV-Vis, IR and NMR. The antibacterial activity of the synthesized compounds was screened against two Gram-positive bacteria (Bacillus cereus and Staphylococcus aureus) and two Gram-negative bacteria (Escherichia coli and Salmonella typhimurium).
Results and Discussion: The synthesized compounds displayed potent activity against the bacterial strains. Among them, compound 8 showed strong activity against Bacillus cereus relative to the standard drug. On the other hand, compound 9 exhibited strong activity against Escherichia coli. The molecular docking study of the synthesized compounds was conducted to investigate their binding pattern with DNA gyrase and E. coli dihydropteroate synthase and all of them were found to have minimum binding energy ranging from – 6.0 to − 7.3 kcal/mol, and the best result achieved with compound 8 and 9.
Conclusion: The findings of the in vitro antibacterial and molecular docking analysis demonstrated that the synthesized compounds have potential of antibacterial activity and can be further optimized to serve as a lead compound.
Keywords: ZnO nanoparticles, green synthesis, chalcone, sulfonamide, inhibition zone, molecular docking, ADMET and antibacterial
Introduction
Chalcone (Figure 1) is an aromatic ketone and an enone that serves as the central core for a variety of important biological compounds. For instance, chalcone derivatives have been synthesized and reported with respect to have various pharmacological activities including antimicrobial,1 antiinflammatory,2–4 antimalarial,5,6 anticancer,7 antiviral 1,8 and antifungal activities.9,10 Pharmacological properties of chalcone easily enhanced through modification of the core skeleton using introduction of various functional groups, substituents at different positions of the A or B ring of the skeleton or anchored other biologically active functional groups to the target compound. Moreover, they provide opportunity for developing inexpensive, easily synthetic protocol and therapeutic bioactive agents. As a result nowadays researchers focused on the synthesis of the title compound and its analogues through conventional methods. Still researchers are modified the synthesis protocol towards the title compound via ZnO nanoparticle to be greener.11 In the other trait the rapid expansion of resistance towards antibiotics, increases the need of developing new antibacterial agent. Hybridization of chalcone and arylsulfonamide moiety (Figure 2) has been investigated for their antiviral properties and the findings indicated that the synthesized derivatives displayed anti-TMV and anti-CMV activities in vivo.12 In this study through a new concise and facile green synthesis protocol chalcone-sulfonamide hybrids were synthesized using zinc oxide nanoparticles catalyzed reaction and investigated their antibacterial activities against two Gram-positive bacteria Staphylococcus aureus and Bacillus cereus and two Gram-negative bacteria Escherichia coli, and Salmonella typhimurium. Furthermore, the in silico molecular docking, ADMET, and DFT analysis of the synthesized chalcone-sulfonamide hybrids are incorporated.
 |
Figure 1 Chemical structure of chalcone 1 and chalcone-sulfonamide derivative 2. |
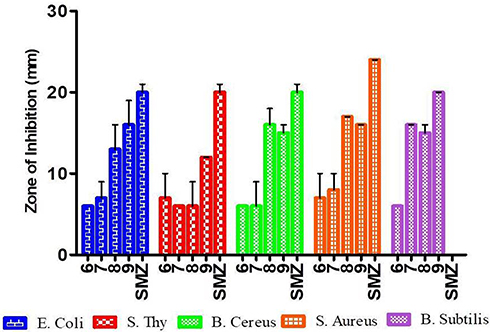 |
Figure 2 The inhibition zone of the synthetic compounds) at 300 μg/mL. |
Materials and Methods
All solvents and chemicals were obtained commercially from fine chemicals PLC (Addis Ababa) and were used as received without further purification. Melting points were determined in an open capillary using digital melting point apparatus, expressed in °C. Reaction progress was checked on pre-coated TLC plates and spots were visualized using UV light at 254 nm. Silica gel (60–120 mesh, Merck grade) has been used for column chromatography. The column was subjected to gradient elution by increasing ethyl acetate in n-hexane and spots were visualized under UV lamp (254 nm). The synthesized compounds were characterized on the basis of physical and spectral analysis. The UV-Vis spectra of some compounds were recorded on Double-beam UV-Vis spectrophotometer using DCM and MeOH as blank solvents and λmax values were expressed by nm. The 1H and 13C NMR spectra of the compounds were recorded on Bruker avance 400 MHz NMR spectrophotometer using Chloroform-d or Methanol-d4 as the solvent and the values are expressed in δ ppm.
Synthesis of p-Aminoacetophenone (4)
The compound 4 was prepared according to a previously described procedure with some modifications.13 A mixture of ethyl alcohol (30 mL) and 1.5 mL of distilled water were poured into three necked round bottom having p-nitro acetophenone (0.03 mol). To this suspension iron powder (0.09 mol), and CaCl2 (0.03 mol) were added and the reaction mixture was heated to 60°C with magnetic stirring on water bath. After completion of the reaction, the reaction mixture was filtered to remove the iron residues, which were washed with EtOAc (3 × 40 mL). The organic extracts were washed with H2O (3 × 30 mL), brine (2 × 30 mL), and dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The resulting compound 4 was obtained as yellow crystalline with melting point 102–104°C. Yield: 75.6%. 1H-NMR:(CDCl3) δ 2.48 (3H, CH3), 4.40 (2H, NH2), 7.76 (d, 2H, H-2, H-6, J= 8.6 Hz) and δ 6.64 (d,2H, H-3, H-5, J = 8.6 Hz).13C-NMR: (CDCl3) δ 25.7 (CH3), 113.4 (C-3, C-5), 126.4 (C-1), 130.8 (C-2, C-6) and 152.4 (C-4). UV-Vis (DCM): λmax at 314 nm and 273 nm.
Synthesis of Ketone Sulfonamide (5)
N-(4-acetyl phenyl) benzenesulfonamide (5a) and N-(4-acetyl phenyl)-4-methyl benzenesulfonamide (5b) were synthesized according to previously reported literature.14 To a solution of p-amino acetophenone (1 mmol) and benzenesulfonyl chloride (1 mmol) or 4-methyl benzene sulfonyl chloride (1 mmol) in methanol (50 mL) pyridine (5 mL) was added drop-wise. The reaction mixture was refluxed for 24 hrs. After completion the reaction, the mixture was poured into crushed ice and acidified with 10% HCl. The resulting solid was filtered, washed with 2% NaHCO3 and again with water.
Following the above procedure compound 5b obtained as white solid with m.p. of 188–190°C. Yield: 75.3% 1H-NMR: (400 MHz, CDCl3) δ 9.76 (N-H), 2.37 (CH3), 2.53 (COCH3), 7.19 (d, 2H, H-3, H-5 J= 8.0 Hz) and δ 7.25 (d, 2H, H-2, H-6, J=8.0 Hz), 7.74 (d, 2H, H-3ʹ, H-5ʹ, J= 8.2 Hz) and 7.82 (d, 2H, H-2ʹ, H-6ʹ, J=8.2 Hz). 13C-NMR: (101 MHz, CDCl3) δ 21.4 (CH3), 26.3 (COCH3), 118.4 (C-2, C-6), 127.1 (C-3, C-5), 129.8 (C-2ʹ, C-6ʹ) 130.0 (C-3ʹ, C-5ʹ), 132.3 (C-1), 136.3 (C-1ʹ), 142.4 (C-4ʹ) and 144.1 (C-4).
Chalcone and Chalcone-Sulfonamide Hybrids Synthesis Procedure Using ZnO NPs as a Catalyst
Equimolar quantities of substituted acetophenone (0.01 mol) and substituted benzaldehyde (0.01 mol) were mixed with ZnO nanoparticle (1 mmol, 0.081g), SnCl2.H2O (10%) and 5mL of distilled water. The reaction mixture was heated at 60°C on water bath for 4 hrs. The progress of the reaction was monitored using TLC in n-hexane: EtOAc (3:2) solvent system. After completion of the reaction, the mixture were cooled to room temperature and stirred with ethanol (50 mL) for 30 min and centrifuged for 10 min at 5000 rpm. The resulting liquid on the top solid was collected and concentrated under reduced pressure. The product was purified using column chromatography on silica gel using gradual increasing the polarity of solvent.
4ʹ-Amino-4-Hydroxychalcone (6)
Following the above procedure a yellow solid. M.p. 80–82°C. Yield: 98.6%. 1H-NMR: (400 MHz, MeOD) δ 7.87–7.94 (3H, m, H-β, H-3ʹ, H-5ʹ), 7.67 (1H, d, H-, J=15.5 Hz), 6.82–6.87 (4H, m, H-2, H-3, H-5, H-6), 4.20 (1H, s, O-H) and δ 3.31 (2H, s, N-H). 13C-NMR: (101 MHz, MeOD) δ 113.0 (C-3, C-5), 115.5 (C-3ʹ, C-5ʹ), 118.3 (C-α), 126.3 (C-1), 126.7 (C-1ʹ), 130.1 (C-2, C-6), 130.9 (C-2ʹ, C-6ʹ), 143.3 (C-β), 153.9 (C-4ʹ), 159.8 (C-4) and 188.9 (C=O).
4ʹ-Aminochalcone (7)
Following the above procedure yellow solid. M.p. 88–90ᵒC. Yield: 91.3%. 1H-NMR: (400 MHz, MeOD) δ 7.96 (2H, dd, H-2ʹ, H-6ʹ, J=11.3, 8.3 Hz), 7.85–7.72 (3H, m, H-2, H-6, H-β), 7.71 (1H, d, H-α, J=15.5 Hz), 7.24–7.12 (3H, m, H-3, H-4, H-5) and δ 6.65 (2H, d, H-3ʹ, H-5ʹ). 13C-NMR: (101 MHz, MeOD) δ 111.5 (C-3ʹ, C-5ʹ), 120.2 (H-α), 124.4 (C-1ʹ), 126.5 (C-2, C-2), 128.3 (C-3, C-5), 129.2 (C-4), 129.6 (C-2ʹ, C-6ʹ), 133.7 (C-1), 141.2 (C-β), 152.7 (C-4ʹ) and 187.02 (C=O).
4ʹ-(Benzenesulfonamide)-4-Hydroxychalcone (8)
Following the above procedure a pale yellow solid. M.p. 204–206ᵒC. Yield: 89.8%. 1H-NMR: (400 MHz, MeOD) δ 7.93 (2H, d, H-2”, H-6”, J= 8.8 Hz), 7.88 (2H, d, H-2ʹ, H-6ʹ, J=8.6 Hz), 7.52 (6H, m, H-α, H-3”, H-4”, H-5”, H-2, H-6), 7.26 (2H, d, H-3, H-5, J=8.8), and 6.83 (2H, d, H-3ʹ, H-5ʹ, J=8.6 Hz). 13C-NMR: (101 MHz, MeOD) δ 115.6 (C-3, C-5), 117.8 (C-3ʹ, C-5ʹ), 118.5 (C-α), 126.3 (C-1), 126.8 (C-2”, C-6”), 128.9 (C-2, C-6), 129.8 (C-3”, C-5”), 130.6 (C-2ʹ, C-6ʹ), 132.9 (C-4”), 133.5 (C-1ʹ), 139.6 (C-1”), 142.3 (C-4ʹ), 145.2 (C-β), 160.3 (C-4) and 189.6 (C=O).
4ʹ-(p-Toluenesulfonamide)-4-Hydroxychalcone (9)
Following the above procedure a pale yellow solid. M.p. 104–106ᵒC. Yield: 76.3%. 1H-NMR: (400 MHz, MeOD) δ 9.76 (1H, s, N-H), δ 7.85 (2H, d, J= 8.2 Hz, H- 2”, H-6”), δ 7.75 (dd, J = 19.7, 8.0 Hz, H-2, H-6, H-α, H-β), 7.30 (2H, d, J = 7.9 Hz, H-2ʹ, H-6ʹ), 7.21 (2H, t, J = 9.0 Hz, H-3ʹ, H-5ʹ), δ 6.91 (2H, d, J=8.2 Hz, H-3”,H- 6”), δ 6.76 (2H, d, J = 8.1 Hz, H-3, H-5) and δ 2.36 (3H, s, CH3). 13C-NMR: (101 MHz, MeOD) δ 20.03 (CH3), 114.4 (C-3, C-5), 115.5 (C-3ʹ, C-5ʹ), 118.1 (C-α), 127.6 (C-2, C-6), 128.9 (C-1), 129.0 (C-1ʹ), 129.4 (C-3”, C-5”), 129.6 (C-2ʹ, C-6ʹ), 132.1 (C-β), 136.6 (C-1”), 142.8 (C-4”), 144.1 (C-4ʹ), 163.8 (C-4) and 191.4 (C=O).
Antibacterial Activity
Antibacterial activities of the synthesized compounds were tested using the disc-diffusion method. In the process, two-gram positive bacteria (Bacillus cereus ATCC6633 and Staphylococcus aureus ATCC25923) and two-gram negative bacteria (Escherichia coli ATCC 25922 and Salmonella typhimurium ATCC 27853) were used to evaluate the antibacterial activities. The medium was prepared from molten nutrient and Müeller-Hinton agar. Sulfamethoxazole was the standard drug used as positive control while DMSO was used as negative control. The four bacterial strains were tested with 300 and 500 µg/mL concentration using disc diffusion method. Each of the four compounds were dissolved in DMSO at concentration of 300 and 500 μg/mL and 6 mm diameter Whatman filter paper were soaked with 1 mL solution of the above two concentration for each compound then these saturated paper discs were inoculated at the center of each petri dish having bacterial lawn in triplicate. The plates were incubated at 37°C for 48 h and inhibition zone that appeared around the paper disc in each plate was determined by measuring the diameter of the inhibition zone.
Molecular Docking Study of the Synthesized Compounds
To study the interactions and binding affinity between the bacterial proteins and synthetic compounds in a 3D fashion, the compounds were docked within the binding site of the protein. AutoDock Vina with our recently reported protocol was used to dock the proteins (PDB ID:1AJ0) and compounds (6–9) into the active site of proteins.15,17–19 The chemical structures of the compounds were drawn using Chem Office tool (Chem Draw 16.0) assigned with proper orientation followed by the energy minimization of each molecule using ChemBio3D. The energy minimized ligand molecules were then used as input for AutoDock Vina, to carry out the docking simulation.20 The crystal structure of the receptor molecules crystal structure of a ternary complex of E. coli dihydropteroate synthase (PDB ID: 1AJ0) and E. Coli DNA gyrase B (PDB ID: 6F86) were downloaded from protein data bank. The protein preparation was done using the reported standard protocol by removing the co-crystallized ligand, deleting water molecules, adding polar hydrogens and cofactors, then the target protein file was prepared by leaving the associated residue with protein by using Auto preparation of target protein file Auto Dock 4.2 (MGL tools 1.5.7).15,20 The graphical user interface program was used to set the grid box for docking simulations. To surround the region of interest in the macromolecule, grid was used. The best docked conformation between the compounds and the protein was explored with the docking algorithm provided with Auto Dock Vina.20 During the docking process, a maximum of nine conformers were considered for each ligand. The conformations with the most favorable (least) free binding energy were selected for analyzing the interactions between the target receptor and ligands by Discovery studio visualizer. The ligands are represented in different color, H-bonds (distance range 2–3.5 Ǻ) and the interacting residues are represented in ball and stick model representation.
In-silico Drug-Likeness and Toxicity Predictions
In-silico drug-likeness is a prediction that concludes whether a particular pharmacological agent has properties consistent with being an orally active drug. This prediction is based on an already established concept by Lipinski et al, called Lipinski rule of five.21 The structures of synthesized compounds (6–9) were changed to their canonical simplified molecular input line entry system (SMILE) then submitted to SwissADME tool to estimate in silico pharmacokinetic parameters and other molecular properties based on the methodology reported by Amina et al (2016). SwissADME predictor gives information about the number of hydrogen donors, hydrogen acceptors and rotatable bonds, and total polar surface area of a compound. The compounds were also subjected to Lipinski et al, screened using SwissADME and PreADMET predictors. The organ toxicities and toxicological endpoints of the isolated compounds were predicted using PreADMET and OSIRIS Property (Table S1).22,24 The analyses of the compounds were compared with that of clinical drugs (sulfamethoxazole). The selection of compounds as drug candidates were determined by a parameter called drug score. The higher the drug score value, the higher the chance of the compound being considered as a drug candidate.
Quantum Computational Studies
The density functional theory (DFT) analysis of synthesized compounds were performed using Gaussian 09 and visualized through Gauss view 6.0 (Figure 3).25 The structural coordinates of the lead compounds were optimized using B3LYP/6-31 G (d,p) level basis set without any symmetrical constraints. The molecular electrostatic potential (MEP) map and energies of the compounds were obtained from the optimized geometry. The Koopman’s approximation was used to estimate the HOMO-LUMO energy gap and related reactive parameters (electronegativity, chemical potential, hardness, softness, electrophilicity).25
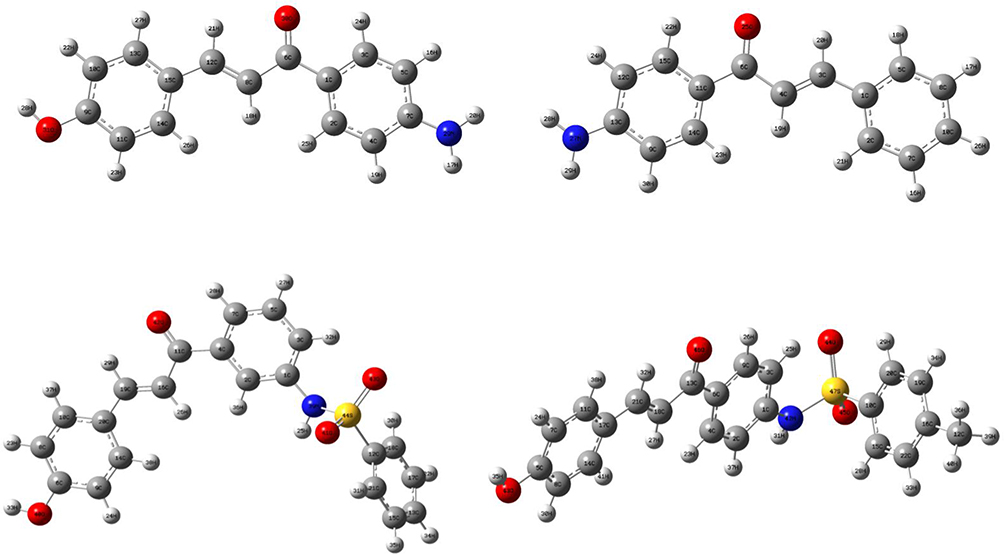 |
Figure 3 The Optimized structures of compounds (6–9). |
Statistics Data Analysis
The antimicrobial analysis data generated by triplicate measurements were reported as mean ± standard deviation. GraphPad Prism version 5.00 for Windows was used to perform the Analysis (GraphPad Software, San Diego California USA, www.graphpad.com”). Groups were analyzed for significant differences using a linear model of variance analysis (ANOVA) test for comparisons was performed, with significance accepted for p < 0.05 (supplementary information).
Results and Discussion
Chemistry
Through new concise and facile green synthesis methodology has been was explored to prepare chalcone derivatives containing benzenesulfonamide segment and their antibacterial properties were evaluated. In this work through a green synthesis protocol chalcone and chalcone-sulfonamide hybrids compounds were synthesized from commercially available materials. The starting material p-nitroacetophenone was reduced to p-aminoacetophenone using Fe powder/CaCl2 in EtOH-H2O mixture (Figures S1-S3). The resulting p-aminoacetophenone (4) reacted with substituted benzaldehyde to provide amino chalcone derivatives (Scheme 1). p-aminoacetophenone subsequently subjected to either benzenesulfonyl chloride or 4-methyl benzene sulfonyl chloride under reflux in methanol to afford the corresponding sulfonamide 5a and 5b (Figures S4 and S5). Compound 5a and 5b were used as a starting materials for ZnO NPs catalyzed Claisen-Schmidt type condensation reaction with substituted aromatic aldehydes to obtain chalcone-sulfonamide hybrids 8 and 9 (Figures S11-S14) and with the same reaction procedure compounds 6 and 7 synthesized, starting from p-aminoacetophenone (Figures S6-S10). The overall sequence of reactions used in the synthesis of various targeted chalcone derivatives are illustrated in Scheme 1. The structure elucidations of the synthesized compounds were accomplished using UV-Vis, FTIR, and NMR spectroscopic methods.
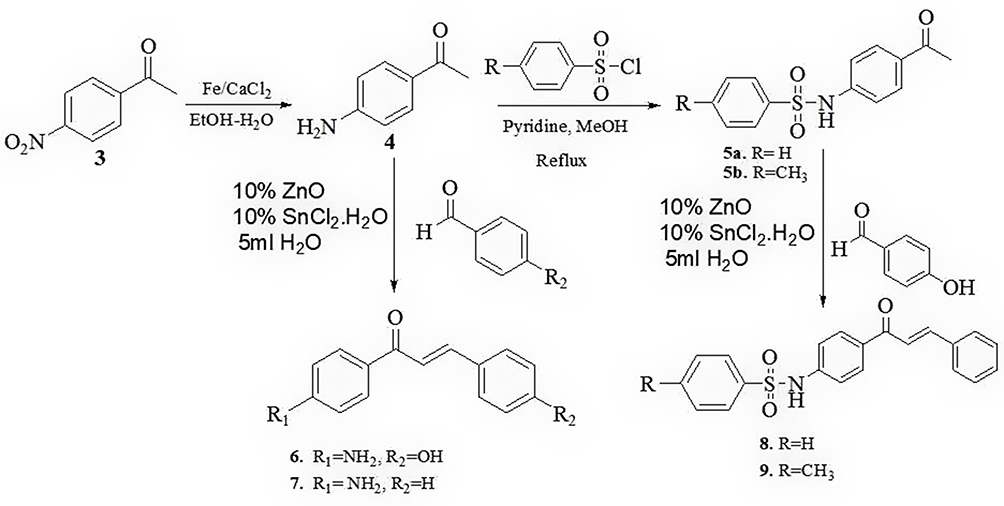 |
Scheme 1 Synthesis of chalcone and chalcone-sulfonamide hybrids using ZnO NPs catalyzed reaction. |
Antibacterial Activity
All synthesized target compounds were evaluated for their in vitro antibacterial activities against S. aureus, E. coli, B. cereus and S. typhimurium using disk diffusion method. Sulfamethoxazole was used as positive control. The zone of inhibition values indicated that all the compounds exhibited a varied range of 6–19 mm of antibacterial activities (Figure 2) against all the tested bacterial strains at 0.05, 0.075 and 0.1 mg/mL. Among synthesized compounds, compound 9 showed potent inhibitory activities against E. coli with 19 mm, B. cereus and S. aureus with 16 mm zone of inhibitions compared to standard drug Sulfamethoxazole (20 mm) at 0.1 mg/mL. Compound 8 showed active inhibition against B. cereus, S. aureus and E. coli with 18, 17, 16 mm zone of inhibitions respectively.
The above data shows all synthesized compounds displayed medium to good activity against the selected bacterial strains. Compound 9 showed good activities against Escherichia coli, and moderate activities on Bacillus cereus and Staphylococcus aureus. Whereas compound 8 revealed better activities on Bacillus cereus and Staphylococcus aureus and moderate activities on Escherichia coli with respect to the standard drug. Both compounds 8 and 9 can act as alternative drug to wards these bacteria strains. Among the synthetic compounds almost all displayed moderate to good activity against Staphylococcus aureus.
In silico Molecular Docking Binding Study of Synthesized Compounds Against E. coli Dihydropteroate Synthase (PDB ID: 1AJ0)
Sulfonamide and their derivatives were the first clinically used antibacterial agents.19 Among them, sulfamethoxazole has been the first-line treatment for pneumonia.28 Therefore in the present study, we have elucidate the molecular docking interaction between the synthesized compounds against E. Coli dihydropteroate synthase and compared with the clinical drugs (sulfamethoxazole). The synthesized compounds (6–9) were found to have minimum binding energy ranging from –6.6 to –8.2 kcal/mol (Table 3), with the best result achieved using compound 8 (–7.8 kcal/mol), and 9 (–8.2 kcal/mol). The binding affinity, H-bond and residual interaction of synthesized compounds and Clinical drugs (sulfamethoxazole) were summarized in Table 3. Compared to sulfamethoxazole, the synthesized compounds (6–9) shown similar residual interactions profile with amino acid residues Arg-63, Asn-22, Lys-221, Glu-60, Arg-220, and H-bond with Thr-62, Arg-255, and His-257. The compounds 9 (Ser-222), and 8 (Ser-222), 6 (Arg-220, Ser-219), and 7 (Arg-220) have shown additional hydrogen bonding interaction with amino acid residues. The synthesized compound 9 has shown similar residual amino acid interactions of clinical drugs (sulfamethoxazole) against human topoisomerase IIα (1AJ0). The in-silico analysis shown that the compound 8 (–7.8 kcal/mol), and 9 (–8.2 kcal/mol), revealed better activity. The compounds 6 (–6.6 kcal/mol), and 7 (–6.6 kcal/mol) docking results were partially matching the clinical drug interactions with amino acid residues. Based on the in silico molecular docking analysis results, compounds 8 and 9 shown high residual interactions and docking score than sulfamethoxazole. The in-silico results are in good agreement with the in vitro anti-microbial assay results. Therefore, compounds 9 and 8 might have better anti-microbial agents than the other compounds reported herein. The binding affinity, H-bond and residual interaction of ten compounds were summarized in Table 3 and with Figure S16.29
 |
Table 1 Molecular Docking Value of Synthetic Compounds (6–9) Against E. coli Dihydropteroate Synthase (PDB ID: 1AJ0) |
 |
Table 2 Molecular Docking Scores and Residual Amino Acid Interactions of Compounds 6–9 Against E. coli DNA GyraseB (PDB ID 6F86) |
 |
Table 3 Drug-Likeness Predictions of Isolated Compounds, Computed by SwissADME |
In silico Molecular Docking Binding Study of Synthesized Compounds Against E. coli DNA Gyrase B (PDB ID: 6F86)
Results obtained from the molecular docking study demonstrated that among the isolated compounds (6–9) have shown a comparable binding affinity against E. coli DNA gyrase B, the value ranged from –5.9 to –6.1 kcal/mol, with respect to ciprofloxacin (7.2 kcal/mol) and sulfamethoxazole (−6.6 kcal/mol) (Table 3 and Figure S17). Compounds (7, 8 and 9) showed two hydrogen bond interactions within active site of the protein (Ans-46, Thr-165) similar to clinical drug ciprofloxacin. Whereas compound (6) showed one hydrogen bond interaction with active site amino acid residue Asp-73.16 The hydrophobic interactions were observed with Glu-50, Ala-47, Gly-77, Ile-78, Pro-79, Ile-94, and Ile-78 (Table 1). The docking results showed comparable binding energy and amino acid interactions (–5.9 to –6.1 kcal/mol) compared to ciprofloxacin (−7.2 kcal/mol). This result supported by the experimental study of which showed in vitro a potential antimicrobial. Overall, the docking results indicate that the synthesized compounds may a potential antimicrobial agent against E. Coli strain.
In-silico Drug-Likeness and Toxicity Predication
The SwissADME prediction outcome showed that all the synthesized compounds (6–9) satisfy Lipinski’s rule of five with zero violations (Table 2). The Kp values of all molecules are within the range of (–5.61 to –6.47 cm/s) with sulfamethoxazole (–7.21 cm/s) inferring low skin permeability. The predicted logP values revealed that they have optimal lipophilicity (ranging from 2.85 to 3.76). The SwissADME prediction parameters showed that all the compounds have high gastrointestinal (GI) absorption, and blood brain barrier (BBB) permeation. The compounds 6 and 7 are substrate of permeability glycoprotein (P-gp). These prediction results indicate that the synthesized compounds 8 and 9 can be active pharmacological agents. A range of cytochromes (CYP’s) regulates the drug metabolism, particularly the biotransformation of drug molecules are regulated by CYP1A2, CYP2C19, CYP2C9, CYP2D6 and CYP3A4.25 The prediction result exhibits that all the compounds (except 9 and clinical drugs) are found to be potential inhibitor for CYP1A2. The compounds 6, 7, and 9 are potential inhibitor for CYP2C19. All the compounds are inhibitors for CYP2D6. The in-silico predication results of Absorption, Distribution, Metabolism, and Excretion (ADME) for isolated compounds and clinical drugs are given in Table 3. Acute toxicity predictions result such as toxicity class classification and LD50 values indicates that none of the compound has shown acute toxicity and was found similar to sulfamethoxazole. Additionally, the compounds 8 and 9 are non-toxic (LD50> 5000) and compounds 6 and 7 are harmful if swallowed (300 < LD50 ≤ 5000).
The toxicological prediction gives results of endpoints such as Hepatotoxicity, Carcinogenicity, mutagenicity, and cytotoxicity. The isolated compounds and sulfamethoxazole were predicted to be non-cytotoxic, non-mutagenic, non-reproductive effective and non-irritant. However, all the compounds including clinical drugs have shown hepatotoxicity. The compounds 7, 8 and 9 were predicted to be carcinogenic. Pre-ADMET and OSIRIS property explorer prediction analysis have shown in Table S2. Hence, based on ADMET prediction analysis, the entire compounds may be a potential drug candidate in the investigation.
DFT Analysis for Optimized Structure and Quantum Chemical Parameters
DFT with the basis sets B3LYP/6-31 G (d,p) was used to compute the optimized geometries (Figure 4) 26 and MEP of synthesized compounds. The determination of the MEP region is the best fit for identifying sites for intra- and intermolecular interactions (Figure 5)
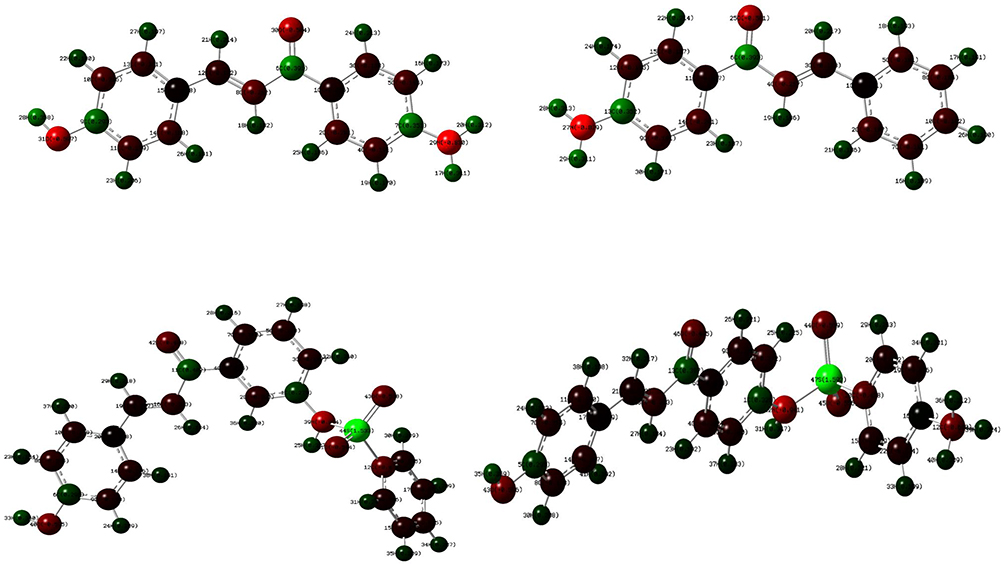 |
Figure 4 The DFT calculated Mullikan’s atomic charges of compounds (6–9). |
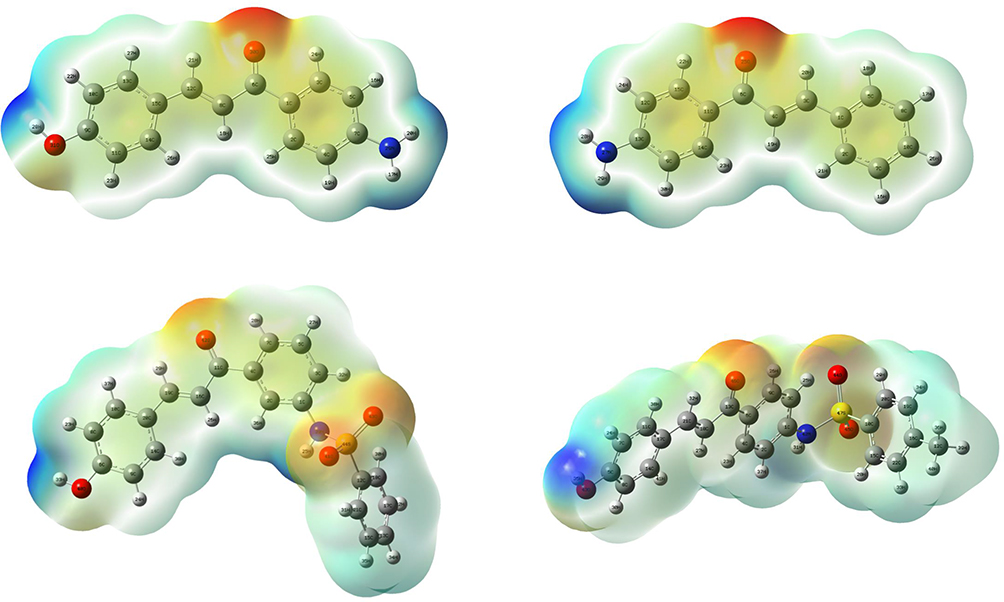 |
Figure 5 MEP surface of compounds. |
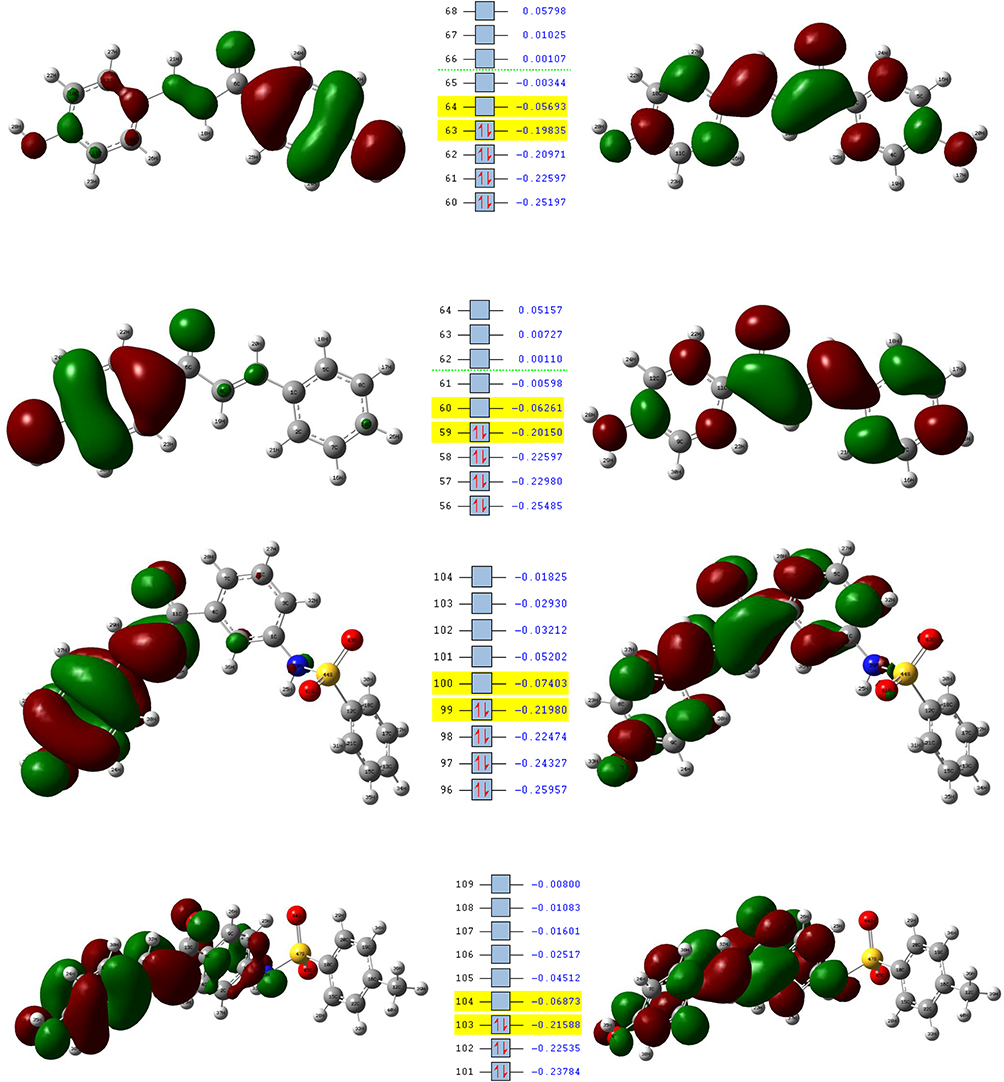 |
Figure 6 Molecular orbitals and energies for the HOMO and LUMO of compounds (6-9). |
Conclusion
Derivatives of chalcone and chalcone-sulfonamide hybrids were successfully synthesized using zinc oxide (ZnO) nanoparticles as green synthesis methodology. The ZnO nanoparticles catalyzed reaction proceeded smoothly to afford chalcone and chalcone-sulfonamide derivatives with good to excellent yield and shorter reaction time compared to conventional methodology. The synthesized compounds displayed potent activity against the bacterial strains. Among them, compound 8 showed maximum activity against Bacillus cereus with 20 mm zone of inhibition at 300 mg/mL relative to the standard drug. On the other hand, compound 9 exhibited maximum activity against Escherichia coli with 18 mm zone of inhibition at 300 mg/mL. The molecular docking study of the synthesized compounds was conducted to investigate their binding pattern with DNA gyrase and E. Coli dihydropteroate synthase and all of them were found to have minimum binding energy ranging from –6.0 to −7.3 kcal/mol and the best result achieved with compound 8 and 9.
Data Sharing Statement
The data used to support the findings of this study are included within the manuscript and also submitted as supporting information.
Acknowledgment
Thanks to Adama Science and Technology University for funded this research project under the grant number ASTU/SM-R/014/19. Thanks to Department of Chemistry, Addis Ababa University for access to NMR spectrometry. Thanks to Department of Applied Biology, Adama Science and Technology University for asses UV-Vis and screening antibacterial activities.
Disclosure
The authors assert that there is no conflict of interest.
References
1. Guo T, Xia R, Chen M, et al. Biological activity evaluation and action mechanism of chalcone derivatives containing thiophene sulfonate,”. RSC Adv. 2019;9(43):24942–24950.
2. Iqbal H, Prabhakar V, Sangith A, Chandrika B, Balasubramanian R. Synthesis, anti-inflammatory and antioxidant activity of ring-A-monosubstituted chalcone derivatives. Med Chem Res. 2014;23(10):4383–4394.
3. Jadhav SY, Bhosale RB, Shirame SP, et al. Synthesis and biological evaluation of fluoro-hydroxy substituted pyrazole chalcones as anti-inflammatory, antioxidant and antibacterial agents. Int J Pharm Bio Sci. 2013;4(2):309–397.
4. Vásquez-Martínez YA, Osorio ME, San Martín DA, et al. Antimicrobial, anti-inflammatory and antioxidant activities of polyoxygenated chalcones. J Braz Chem Soc. 2019;30(2):286–304.
5. Wanare G, Aher R, Kawathekar N, Ranjan R, Kaushik NK, Sahal D. Synthesis of novel α-pyranochalcones and pyrazoline derivatives as Plasmodium falciparum growth inhibitors. Bioorg Med Chem Lett. 2010;20(15):4675–4678.
6. Yadav N, Dixit SK, Bhattacharya A, et al. Antimalarial activity of newly synthesized chalcone derivatives in vitro. Chem Biol Drug Des. 2012;80(2):340–347.
7. Sissouma D, Ouattara M, Kone MW. Synthesis and antifungal activities of some benzimidazolyl-chalcones, analogues of chlormidazole. Afr J Pharm Pharmacol. 2015;9(12):418–423.
8. Wan Z, Hu D, Li P, Xie D, Gan X. Synthesis, antiviral bioactivity of novel 4-thioquinazoline derivatives containing chalcone moiety. Molecules. 2015;20(7):1186–11874.
9. Mellado M, Espinoza L, Madrid A, et al. Design, synthesis, antifungal activity, and structure–activity relationship studies of chalcones and hybrid dihydrochromane–chalcones. Mol Divers. 2019;24:1–13.
10. Geetha VM, Usha RU, Raghuveer P, Ramalingam P, Ravindra RJ, Padmanabha RY. New quinoxalinyl chalcone derivatives: search for potent antimicrobial agents. J Antimicrob Agents. 2017;3(1):1000137.
11. Saravanamurugan S, Palanichamy M, Arabindoo B, Murugesan V. Solvent free synthesis of chalcone and flavanone over zinc oxide supported metal oxide catalysts. Catal Commun. 2005;6:399–403.
12. Zhou D, Xie D, He F, Song B, Hu D. Antiviral properties and interaction of novel chalcone derivatives containing a purine and benzenesulfonamide moiety. Bioorg Med Chem Lett. 2018;28(11):2091–2097.
13. Chandrappa S, Vinaya K, Ramakrishnappa T, Rangappa KS. An efficient method for aryl nitro reduction and cleavage of azo compounds using iron powder/calcium chloride. Synlett. 2010;20:3019–3022.
14. Kobkeatthawin T, Chantrapromma S, Kumar CC, Fun HK. Synthesis, characterization, and crystal structure of sulfonamide chalcone:(E)-4-methoxy-N-(4-(3-(3, 4, 5-trimethoxyphenyl) acryloyl) phenyl)-benzenesulfonamide. Crystallography Rep. 2015;60(7):1058–1064.
15. Zeleke D, Eswaramoorthy R, Belay Z, Melaku Y. Synthesis and antibacterial, antioxidant, and molecular docking analysis of some novel quinoline derivatives. J Chem. 2020;(2020):1–16.
16. Carr SB, Makris G, Phillips SE, Thomas CD. Crystallization and preliminary X-ray diffraction analysis of two N-terminal fragments of the DNA-cleavage domain of topoisomerase IV from Staphylococcus aureus. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2006;62:1164–1167.
17. Narramore S, Stevenson CEM, Maxwell A, Lawson DM, Fishwick CWG. New insights into the binding mode of pyridine-3-carboxamide inhibitors of E. coli DNA gyrase. Bioorg Med Chem. 2019;27:3546–3550.
18. Wendorff TJ, Schmidt BH, Heslop P, Austin CA, Berger JM. The structure of DNA-bound human topoisomerase II alpha: conformational mechanisms for coordinating inter-subunit interactions with DNA cleavage. J Mol Biol. 2012;424:109–124.
19. Achari A, Somers DO, Champness JN, Bryant PK, Rosemond J, Stammers DK. Crystal structure of the anti-bacterial sulfonamide drug target dihydropteroate synthase. Nat Struct Biol. 1997;4:490–497.
20. Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455–461.
21. Lipinski CA. Rule of five in 2015 and beyond: target and ligand structural limitations, ligand chemistry structure and drug discovery project decisions. Adv Drug Deliv Rev. 2016;101:34–41.
22. Banerjee P, Eckert AO, Schrey AK, Preissner R. ProTox-II: a webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018;46:W257–W263.
23. Banerjee P, Dehnbostel FO, Preissner R. Prediction is a balancing act: importance of sampling methods to balance sensitivity and specificity of predictive models based on imbalanced chemical data sets. Front Chem. 2018;6:362.
24. Drwal MN, Banerjee P, Dunkel M, Wettig MR, Preissner R. ProTox: a web server for the in silico prediction of rodent oral toxicity. Nucleic Acids Res. 2014;42:W53–W58.
25. Sychev DA, Ashraf GM, Svistunov AA, et al. The cytochrome P450 isoenzyme and some new opportunities for the prediction of negative drug interaction in vivo. Drug Des Devel Ther. 2018;12:1147.
26. Mumit MA, Pal TK, Alam MA, Islam MA, Paul S, Sheikh MC. DFT studies on vibrational and electronic spectra, HOMO-LUMO, MEP, HOMA, NBO and molecular docking analysis of benzyl-3-N-(2,4,5-trimethoxyphenylmethylene)hydrazinecarbodithioate. J Mol Struct. 2020;1220:128715.
27. Abu-Melha S. Pyridyl thiosemicarbazide: synthesis, crystal structure, DFT/B3LYP, molecular docking studies and its biological investigations. Chem Cent J. 2018;12:101.
28. Mohamed MA, Kaur J, Wani F, Kichloo A, Bhanot R. Renal transplant recipient with concurrent COVID-19 and stenotrophomonas maltophilia pneumonia treated with trimethoprim/sulfamethoxazole leading to acute kidney injury: a therapeutic dilemma. Am J Case Rep. 2020;21:e926464.
29. Tamuly C., Saikia, I, Hazarika, M, et al. Bio-derived ZnO nanoflower: a highly efficient catalyst for the synthesis of chalcone derivatives. RSC Adv. 2015;5: 8604–8608.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.