")
Back to Journals » Breast Cancer: Targets and Therapy » Volume 16
Amplifying Immune Responses: Microparticulate Vaccine Approach Against Breast Cancer
Authors Ubowski MM, VanSice R, Marriott M, Yacobucci MJ, Chablani L
Received 4 January 2024
Accepted for publication 16 March 2024
Published 28 March 2024 Volume 2024:16 Pages 149—162
DOI https://doi.org/10.2147/BCTT.S441368
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Robert Clarke
Michelle Marie Ubowski,1 Ryan VanSice,1 Morgan Marriott,1 Matthew J Yacobucci,2 Lipika Chablani1
1Wegmans School of Pharmacy, St. John Fisher University, Rochester, NY, 14618, USA; 2Albany College of Pharmacy and Health Sciences, Albany, NY, 12208, USA
Correspondence: Lipika Chablani, Department of Pharmaceutical Sciences, Wegmans School of Pharmacy, St. John Fisher University, 3690 East Ave, Rochester, NY, 14618, USA, Tel +1-585-899-3714, Fax +1-585-899-8453, Email [email protected]
Introduction: The study focuses on evaluating the immune responses generated by a novel microparticulate murine breast cancer vaccine.
Methods: The methodology included the use of a co-culture model of dendritic cells (DCs), and T-cells to evaluate the immunotherapeutic responses generated by the vaccine.
Results: The study observed that the dendritic cells expressed significantly higher levels of MHC I, MHC II, CD 40, and CD 80 cell surface markers in the presence of the vaccine microparticles than the controls (p< 0.05). This response was potentiated in the presence of an adjuvant, Poly (I:C). The study also demonstrated that the vaccine microparticles do not elicit inflammatory (TNF-alpha, IFN-gamma, IL-2, and IL-12) or immunosuppressive (IL-10) cytokine production when compared to the control.
Discussion: In conclusion, the study established the role of DCs in stimulating the cancer vaccine’s adaptive immune responses.
Keywords: dendritic cells, adjuvants, poly I:C, adaptive immune response, vaccine formulation
Introduction
As of 2022, breast cancer ranks as the most common malignancy occurring in women, regardless of race or ethnicity, in the United States. In 2023, it is estimated that there will be 297,790 Americans diagnosed with breast cancer, with 43,170 deaths attributed to breast cancer.1 Even more devastating, breast cancer causes most cancer deaths in Hispanic women and is the second most common cancer-related death in many other ethnicities.2 Immune dysfunction has emerged as a key player in breast cancer’s pathophysiology, suggesting that immunotherapy may be a viable component of a comprehensive treatment approach.3 Currently, there are two FDA-approved immunotherapies for breast cancer, including pembrolizumab and atezolizumab. Pembrolizumab is used to treat unresectable or metastatic, microsatellite instability-high (MSI-H) or mismatch repair-deficient (dMMR) solid tumors. These tumors have generally progressed following prior treatment or have no satisfactory alternative treatment options. Alternatively, atezolizumab is used (in combination with protein-bound paclitaxel) for patients with unresectable locally advanced or metastatic triple-negative breast cancer, whose tumors express programmed death-ligand 1 (PD-L1).
Vaccination is a method of immunotherapy that has been investigated against breast cancer. Most cancer vaccines to date have focused on stimulating a response from T lymphocytes in individuals with a preexisting disease by delivering tumor antigens to the patient’s immune system.3 Vaccines that fail to activate immune pathways beyond CD8+ T-cells typically result in transitory responses, leading to the development of vaccines capable of stimulating both CD8+ and CD4+ T-cells.3 One of the most well-studied breast cancer vaccines is NeuVax. NeuVax consists of two major components: nelipiminut-S (a HER2-derived peptide) and granulocyte-macrophage colony-stimulating factor (GM-CSF). HER2 is a well-studied tumor-associated antigen typically found in more aggressive forms of breast cancer. At the same time, GM-CSF is added to enhance antigen presentation and serves as an adjuvant/immunostimulant.4 The Phase III trial evaluating the efficacy and safety of NeuVax was stopped in 2016 due to futility. The disappointing results from the phase III trial led to the exploration of combination trials of NeuVax in conjunction with trastuzumab (Herceptin). A detailed description of the current status of NeuVax and further ongoing clinical studies is provided by Dillon et al.5
It is not uncommon for a vaccine candidate, such as NeuVax, to face clinical challenges. A major hurdle for immunotherapy is the innate ability of tumor cells to shut down the immune system. Tumor cells can secrete anti-inflammatory cytokines and increase regulatory T-cells to halt the antitumor immune responses.6 Another challenge is the constant dynamic nature of tumor cells leading to unknown mutations, which can pose issues for single-antigen vaccines if the cancer cells alter the tumor-associated antigens (TAAs) for which the vaccine was designed.6 One strategy for overcoming these barriers is to utilize tumor lysates in vaccine preparations to present the immune system with multiple tumor antigens at once.6 Tumor lysate-based vaccines are attractive options as they minimize the tumor’s ability to suppress the immune response, expand potential use by being unrestricted to tumors expressing specific antigens, and be less expensive and laborious produce. On similar lines, Nie et al have explored a prophylactic cancer vaccine using breast cancer tumor cellular membranes.7
Once the tumor lysate is prepared, antigen presentation largely depends on delivering these TAAs to the antigen-presenting cells. Antigen delivery in the form of a particulate vaccine has been studied as an effective vaccine delivery mechanism via various routes of administration.8–12 Microparticles less than five microns in size have the advantage of mimicking pathogens, resulting in more efficient uptake by antigen-presenting cells.13 Compared to soluble antigens, particulates are taken up more efficiently by antigen-presenting cells and may be more immunogenic.13 Another advantage of a microparticle vaccine is the co-delivery of antigen and adjuvant to the same dendritic cell. Adjuvants can be critical in providing a “danger signal” to ensure peripheral tolerance is overcome, and an immune response will be generated against the TAAs.6 Some adjuvants currently being used in cancer vaccines include granulocyte macrophage colony stimulating factor (GM-CSF), polyinosinic-polycytidylic acid (Poly (I:C)), pentaerythritol lipid A (PELA), and imiquimod.6,14–16 Poly (I:C), a ligand of Toll-like receptor 3 (TLR-3) is of particular interest as it is thought to have direct antitumor activity.14 Poly (I:C) can trigger both innate and adaptive immune pathways and has been explored as an immunostimulant for cancer vaccines.14 Poly (I:C), also commercially available as Hiltonol®, has been extensively evaluated as an adjuvant for cancer vaccines clinically, as observed in 111 clinical trials at various phases. Ammi et al provide a detailed review of Poly (I:C) as a cancer adjuvant, its role in activating the CD4+ and CD8+ T-cells, and its safety and efficacy as established by the clinical trials.17 While Poly (I:C) has been demonstrated to be an effective cancer adjuvant, studies have shown that its effects without the presence of an antigen are minimal to none. One study looking at Poly (I:C) alone showed no increase in cell surface markers or inflammatory cytokines.18
Previously, Chablani et al demonstrated the success of a novel spray-dried microparticulate murine breast cancer vaccine comprised whole-cell lysate and a mix of polymers in a murine model. The whole-cell lysate was derived from the murine breast cancer cell line 4T07. Two different routes of administration (oral and via skin) were evaluated to observe the immune responses generated by the microparticulate breast cancer vaccine. In vivo studies in Balb/c mice showed that unvaccinated/control animals developed significantly larger tumors than vaccinated animals when challenged with live murine breast cancer 4T07 cells. Flow cytometry analysis from the spleens of vaccinated animals revealed significantly higher levels of CD4+ T-cells, CD8+ T-cells, and B-cells in vaccinated animals compared to the controls (p<0.05).9,10
This study further aims to understand the immune mechanism followed by the above-mentioned novel spray-dried oral microparticulate breast cancer vaccine. Vaccine and placebo microparticles were formulated as per previously published studies, and in vitro studies using dendritic cells (DC 2.4) and T-cells (EL4.IL-2) were executed. Vaccine and placebo microparticles were compared to the negative control of cells alone without any treatments, and LPS treated cells were used as a positive control. LPS has been shown to be a potent immune activator, making it a good candidate for positive control.19 Dendritic cells have the capability to present TAAs to T-cells and lead to surface marker expressions. Dendritic cells have been recognized as professional antigen-presenting cells, and it provides them with a unique role in immunotherapy and in vitro studies to evaluate vaccine candidates.20
Dendritic cell maturation depends on the microenvironment of the surrounding cells. The presence of stimuli, cytokines, and chemokines released by neighboring cells dictates the activation of cell surface markers. A dendritic cell is capable of undergoing a maturation process as TAAs are presented to them, leading to further expression of processed antigens via the MHC (I or II) pathways. This antigen presentation is only complete when the co-stimulatory molecules, such as CD 40 and CD 80, are also expressed on the dendritic cell surface. The MHC (I or II) and their respective CD markers (80 or 40) interact with the T-cells via the T-cell receptor (TCR) and CD 28 co-stimulatory molecules to complete the communication between an antigen-presenting cell and an immune cell (T-cell). These can further lead to activation of CD4+ (via MHC II pathway) or CD8+ (via MHC I pathway) T-cells. However, activation of CD4+ T-cells will stimulate the T-helper cell mechanism to recruit B-cells and cause a robust antibody-based response against the cancer antigens presented via the MHC pathway through the vaccine particles. However, activation of CD8+ T-cells will cause the cytotoxic T-lymphocytes (CTLs) to stimulate a cytotoxic effect against the cells expressing cancer antigens in the body.
Further, these immune responses can be heightened in the presence of an adjuvant, such as Poly (I:C). The current study will examine the immune pathways utilized by the previously formulated and effective microparticulate breast cancer vaccine. In vitro studies involving flow cytometry to quantify the cell surface expression of the dendritic cells, ELISAs (enzyme-linked immunosorbent assays) to quantify the cytokine release, and confocal studies to qualitatively evaluate the dendritic cell expressions were performed to understand the immune pathways.
Materials
RPMI (11,875,119), DMEM (Catalog #6046), Murine IL-2 (RAB0287-1KT), TNF-alpha (RAB0477-1KT), IL-10 (RAB0245-1KT), IL-12 (RAB0255) and IFN-gamma (RAB0224-1KT) ELISA kits were purchased from Sigma Aldrich, USA. Flow cytometry cell surface markers for CD80 (50-967-2), CD40 (50-112-9392), MHC Class I (50-163-86), MHC Class II (50-982-8), propidium iodide (00-6690), and confocal cell surface markers for MHC Class I (MA511723), MHC Class II (14-5321-82), CD40 (MA5-32,619), and CD80 (PA5-85,913) primary antibodies were purchased from ThermoFisher, USA. Secondary antibody fluorophores (Alexa 555, A21137) were purchased from ThermoFisher, USA. NucBlue nuclei stain (Catalog # R37606) was purchased from Invitrogen, USA. Nunc 27 mm (150,682) glass bottom confocal dishes were purchased from Fisher Scientific, MA, USA. 4T07 murine breast cancer cell lines were obtained from Karmanos Cancer Institute, MI, USA, DC 2.4 cells (Catalog # SCC142) were purchased from EMD Millipore, MA, USA, and the EL4.IL-2 T-cells were a gift from the Rochester Institute of Technology, NY, USA. All other cell culture supplies were purchased from Fisher Scientific, MA, USA.
Methods
Whole Cell Lysate Preparation
The whole-cell lysate was prepared as per the protocol followed by Chablani et al.9 Briefly, the murine breast cancer cell line 4T07 was grown to confluence in T75 flasks with complete DMEM media supplemented with 10% fetal bovine serum, 50 U/mL penicillin, and 50 ug/mL of streptomycin. Cells were incubated at 37°C with 5% CO2. Upon confluence, each flask was gently scraped for cell removal in 3 mL of fresh media. Cells were pelleted by centrifugation at 1000 xg for ten minutes. After centrifugation, the supernatant was removed, and the cells were re-suspended in a hypotonic lysis buffer consisting of 10 mM Tris and 10 mM NaCl. The suspension was then subjected to five freeze–thaw cycles, between storage at −80°C and 37°C each for ten minutes. The freeze–thaw cycles resulted in the lysis of the whole cells, and the final whole cell lysate (WCL) preparation was stored at −80°C for further studies.
Quantification of the Whole Cell Lysate
The WCL was evaluated for the total protein content using the Quick Start Bradford Protein Assay kit by Bio-Rad (Catalog # 5000202) with bovine serum albumin (BSA) as a standard protein for the standard curve (1.25–20 µg/mL). Protein content was determined via the colorimetric assay as per the manufacturer’s protocol. Each sample or standard was tested in triplicates, and sample dilution was performed to remain in the linear range of the standard curve.
Formulation of the Vaccine Microparticle Using the Whole Cell Lysate
The 4T07 murine vaccine microparticles were formulated as per the protocol previously published by Chablani et al.11 Briefly, a suspension containing 1% w/v solid content comprised 4T07 WCL (5% w/w), hydroxyl propyl methylcellulose acetate succinate (HPMCAS) (32% w/w), and Eudragit FS 30 D aqueous suspension (Evonik Industries) (32% w/w), and bovine serum albumin (31% w/w) was prepared in deionized water. 1N sodium hydroxide was included to dissolve HPMCAS in water before it was added to the aqueous suspension. Aqueous suspension-based formulation for the placebo microparticles contained all the excipients as listed above except the WCL. The aqueous suspension of all the components, in each case, was spray-dried into microparticles using the Buchi B-290 mini spray dryer. Previously optimized parameters were used to spray-dry the microparticles.11 Briefly, the inlet temperature was maintained at 125°C, the outlet temperature was set at 80°C, the aspirator was at 50%, and a 5% peristaltic pump feed rate was used with a 0.7 mm spray drying nozzle.
Exposing DC2.4 Cells to the Microparticles and Co-Culture of Stimulated DC2.4 with EL4.IL2 Cells
DC2.4 cells were seeded in 24 well Corning plates at a density of 1 × 105 cells/well in complete RPMI media. The cells were allowed to adhere overnight under incubation. After 12 hours, the cells were exposed to the microparticles as follows: (a) vaccine microparticles (5 mg/well), (b) placebo microparticles (5 mg/well), (c) vaccine microparticles with the adjuvant Polyinosinic-polycytidylic acid [Poly (I:C)] (5 mg/well, 25 µg/mL respectively), (d) placebo microparticles with the adjuvant Poly (I:C) (5 mg/well, 25 µg/mL respectively), (e) LPS treated (positive control), and (e) Cells alone without any treatment (negative control). Each treatment was performed in triplicates and was allowed to incubate for four hours at 37°C/ 5% CO2. After the incubation period, EL4.IL2 T-cells were introduced directly to each well at a concentration of 1 × 105 cells/well. The plate was further incubated overnight.
Flow Cytometry Studies to Analyze DC2.4 Cell Surface Expression Upon Microparticle Treatments
For the flow cytometry studies, the aforementioned co-culture experiment was performed, and the supernatant from each well was removed, and 1 mL of fresh complete media was added to each well. The bottom of the wells was gently scrapped to harvest the attached DC2.4 cells. The cell suspension from each well was transferred to respectively labeled 2 mL microcentrifuge tubes. For the first experiment, each tube was stained with fluorescently labeled antibodies specific to MHC Class II (50-982-8) and CD40 (50-112-9392). In the second set of experiments, the cells were stained for MHC I (50-163-86) and CD80 (50-967-2) expression. An additional stain of propidium iodide (00-6690) was used to confirm the cell viability each time. The staining procedures were performed as per the manufacturer’s protocol for individual stains. The stained DC2.4 cell samples were analyzed using a BD Bioscience FacsCanto II fluorescence-activated cell sorter (Vaccinex Inc., Rochester, NY) to identify the proportion of live cell population expressing the cell surface markers for MHC I, MHC II, CD40, and CD80.
ELISA Experiments to Analyze Cytokine Expression Upon Microparticle Treatments
For the ELISA studies, the aforementioned co-culture experiment was performed, and the supernatant from each well was collected at varying time points of the co-culture. The time points evaluated included 4, 8, 12, and 24 hours. The supernatant from each well was collected and centrifuged to remove any cells or debris. All the supernatants were labeled and stored at −20°C for cytokine-specific ELISA studies. The cytokines evaluated via the ELISA studies included TNF-alpha (RAB0477-1KT), IFN-gamma (RAB0224-1KT), IL-2 (RAB0287-1KT), IL-12 (RAB0255), and IL-10 (RAB0245-1KT). Each time point sample was evaluated in triplicates. ELISAs were performed as per the manufacturer’s protocol for the kits associated with individual cytokine assay. The results were read using a Microtek UV plate reader at the UV wavelength respective to the ELISA protocol provided by the manufacturer.
Quantification of T-Cell Population Post Microparticle Treatments
After the aforementioned co-culture experiment, the cell supernatant containing the floating EL4.IL-2 (T-cells) was removed and collected in pre-labeled 2 mL centrifuge tubes. The tubes from all the treatment groups were centrifuged at 1000 xg for 10 min to collect the cell pellet. The cell pellet was then subjected to cell count analysis using trypan blue dye. Briefly, 20 µL of the cell suspension was mixed with 20 µL of sterile trypan blue dye. The stained sample was gently mixed and loaded into Bio-Rad’s TC20 cell counter slides as per the manufacturer’s protocol. The live T-cell population for each of the treatment groups post-co-culture experiment was determined in triplicates.
Confocal Microscopy Studies to Analyze DC2.4 Cell Surface Expression Upon Microparticle Treatments
The aforementioned co-culture experiment was identically performed in 27 mm Nunc glass-bottom dishes except for a total volume of 4 mL of complete RPMI media that was used for each dish. After 24-hours of the addition of EL4.IL2 cells in the co-culture experiment, the supernatant was removed from each dish. Each dish was then fixed with 1 mL of 10% formalin and incubated for 15 minutes. After fixation, each dish was washed three times with PBS (pH 7.4). Next, the cells were permeabilized with 1 mL of 0.5% Triton-X solution and incubated for 15 minutes. After permeabilization, each dish was washed three times with PBS (pH 7.4). Further, the dish background was blocked by adding 1 mL of 3% bovine serum albumin solution and incubation for 30 minutes. Consequentially, each dish was incubated with a primary antibody specific to MHC Class I (MA511723), MHC Class II (14–5321-82), CD40 (MA5-32,619), and CD80 (PA5-85,913) for 24 hours at 4°C. Upon incubation, each dish was stained with a fluorescently labeled antibody (Alexa 555, A21137) and incubated for 30 minutes. Samples were then stained with one drop of NucBlue (DAPI) (Invitrogen Catalog # R37606) and read immediately on a Leica TCS SP5 II confocal microscope (Rochester Institute of Technology, NY). Images were taken under a 40X water objective with 25% laser brightness. All the images were analyzed using Fiji for ImageJ software.
Results
Quantification of the Whole Cell Lysate
The WCL concentration of different batches of the murine 4T07 breast cancer cells ranged from 0.75 to 1.25 mg/mL. The individual batch concentration was used to determine the amount of WCL required to prepare the vaccine particles, keeping the concentration constant for each vaccine microparticle batch.
Formulation of the Vaccine Microparticle Using the Whole Cell Lysate
The vaccine and placebo microparticles were prepared and evaluated for their particle size as reported in previously published studies by Chablani et al.9 The formulated vaccine and placebo microparticles ranged from 1 to 5 µm in size and were stored in a tightly sealed clear glass vial at −20°C until further use.
Flow Cytometry Studies to Analyze DC2.4 Cell Surface Expression Upon Microparticle Treatments
Flow cytometry results for MHC class I, MHC class II, CD40, and CD80 are depicted in Figure 1. Co-cultured DC2.4 cells treated with vaccine + adjuvant [Poly(I:C)] demonstrated statistically significant increases in MHC class I, MHC class II, CD40, and CD80 expression compared to the negative cells only control (p<0.05). Vaccine treated wells showed statistically significant increases in MHC class I, MHC class II, CD40, and CD80 expression compared to the control (p<0.05), but numerically less expression than vaccine + adjuvant treatment group. No other groups differed significantly from the control group for MHC class I, MHC class II, or CD40 expression. Placebo and placebo + adjuvant groups exhibited significantly increased CD80 expression compared to the cells only control (p<0.05). Overall, the inclusion of Poly (I:C) resulted in statistically significant increased cell surface expression of MHC I, MHC II, CD 40, and CD80 for the cells treated with vaccine microparticles compared to the control (p<0.05). The role of Poly (I:C) as an immunostimulant was pronounced when used in combination with the 4T07 murine breast cancer vaccine microparticles when compared to the placebo microparticles. Poly (I:C) has been established as a cancer vaccine adjuvant as demonstrated in previous studies.17
 |
Figure 1 Flow cytometry analysis results to quantify dendritic cell surface marker expressions. (a) MHC I, (b) MHC II, (c) CD 40 costimulatory, and (d) CD 80 costimulatory. All results are expressed as mean ± SD (n=3). Statistical significance as observed by post-hoc analysis of ANOVA with cells alone treatment group as the negative control. *p<0.05, ***p<0.001, and ****p<0.0001. |
ELISA Experiments to Analyze Cytokine Expression Upon Microparticle Treatments
ELISA results showed the placebo, placebo + adjuvant, and LPS treatment groups led to statistically significantly higher levels of TNF-alpha, IFN-gamma, and IL-2 release at 24 hours compared to the cells-only control (p<0.05). The levels of cytokines released at prior time points (4, 8, and 12 hours) were not statistically different for all the treatment groups when compared to the controls. Further, the cytokine levels of IL-12 and IL-10 remained unchanged during the 24-hour study duration, and no difference was observed among the treatment and cells-only control group. Overall, increased concentrations of only TNF-alpha, IFN-gamma, and IL-2 cytokines were observed at 24 hours in the placebo, placebo + adjuvant, and LPS groups, as seen in Figures 2–4. The vaccine and vaccine + adjuvant treatment groups did not demonstrate any change in the cytokine release during the 24-hour duration. Further, no immunosuppression was observed, demonstrated by the lack of IL-10 release, by any of the groups tested for the 24-hour duration of the study.
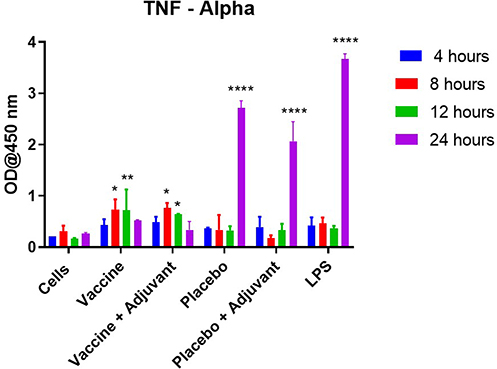 |
Figure 2 TNF-alpha cytokine release as observed in the DC/T-cell co-culture model for various time points (4–24 hours). All the results are expressed as mean ± SD (n=3). Statistical significance as observed by post-hoc analysis of ANOVA with cells alone treatment group, at the corresponding time interval, as the negative control. *p<0.05, **p<0.01, and ****p<0.0001. |
 |
Figure 3 IFN-gamma cytokine release as observed in the DC/T-cell co-culture model for various time points (4–24 hours). All the results are expressed as mean ± SD (n=3). Statistical significance as observed by post-hoc analysis of ANOVA with cells alone treatment group, at the corresponding time interval, as the negative control. ****p<0.0001. |
 |
Figure 4 IL-2 cytokine release as observed in the DC/T-cell co-culture model for various time points (4–24 hours. All the results are expressed as mean ± SD (n=3). Statistical significance as observed by post-hoc analysis of ANOVA with cells alone treatment group, at the corresponding time interval, as the negative control. ****p<0.0001. |
Quantification of T-Cell Population Post Microparticle Treatments
An expansion in the number of T-cells present after co-culture with dendritic cells was observed in both the vaccine and vaccine + adjuvant treatment groups. Figure 5 shows there was a significant increase in the number of live T-cells present when co-cultured with dendritic cells exposed to vaccine microparticles alone or in the presence of the adjuvant, Poly (I:C), (p<0.0001). No such changes were observed for the placebo and placebo + adjuvant treatment groups other than the cells alone control.
 |
Figure 5 Quantification of T-cells post microparticle treatment demonstrates a significantly higher live T-cell population when treated with vaccine microparticles (both in the absence and presence of the adjuvant). All the results are expressed as mean ± SD (n=3). Statistical significance as observed by post-hoc analysis of ANOVA with cells alone treatment group as the negative control. ****p<0.0001. |
Confocal Microscopy Studies to Analyze DC2.4 Cell Surface Expression Upon Microparticle Treatments
Figures 6–9 provide confocal images of cell surface expression of individual markers as observed on dendritic cells exposed to various treatment and control groups. The nuclei were stained with NucBlue (blue color), and the cell surface markers were fluorescently labeled with Alexa 555 antibody (red color) in individual studies. Confocal images qualitatively corroborated the results as observed with the flow cytometry studies. Higher cell surface expression of MHC Class I, MHC Class II, CD40, and CD80 were observed in the vaccine + adjuvant treatment group compared to the cells only control when analyzed with ImageJ. Treatments involving vaccine + adjuvant expressed more fluorescence for all four markers compared to other treatment groups. Also, placebo + adjuvant treated dendritic cells expressed higher fluorescence/expression of CD80 and MHC Class I compared to the vaccine, placebo, and cells-only groups as analyzed via ImageJ.
 |
Figure 6 Confocal images of DC2.4 cells after co-culture with T-cells. Images were processed with ImageJ and imaged under a 40X water objective. Cells were stained for 24 hours with a MHC I antibody. Cells were stained with fluorophores NucBlue (DAPI) and Alexa 555 secondary antibody for visualization.(a) Vaccine + Adjuvant (b) Placebo + Adjuvant (c) Vaccine only (d) Placebo only (e) Cells only. |
 |
Figure 7 Confocal images of DC2.4 cells after co-culture with T-cells. Images were processed with ImageJ and imaged under a 40X water objective. Cells were stained for 24 hours with a MHC Class II antibody. Cells were stained with fluorophores NucBlue (DAPI) and Alexa 555 secondary antibody for visualization. (a) Vaccine + Adjuvant (b) Placebo + Adjuvant (c) Vaccine only (d) Placebo only (e) Cells only. |
 |
Figure 8 Confocal images of DC2.4 cells after co-culture with T-cells. Images were processed with ImageJ and imaged under a 40X water objective. Cells were stained for 24 hours with a CD40 antibody. Cells were stained with fluorophores NucBlue (DAPI) and Alexa 555 secondary antibody for visualization. (a) Vaccine + Adjuvant (b) Placebo + Adjuvant (c) Vaccine only (d) Placebo only (e) Cells only. |
 |
Figure 9 Confocal images of DC2.4 cells after co-culture with T-cells. Images were processed with ImageJ and imaged under a 40X water objective. Cells were stained for 24 hours with a CD80 antibody. Cells were stained with fluorophores NucBlue (DAPI) and Alexa 555 secondary antibody for visualization. (a) Vaccine + Adjuvant (b) Placebo + Adjuvant (c) Vaccine only (d) Placebo only (e) Cells only. |
Statistical Analysis
All the statistical analyses were performed using GraphPad Prism 8. One-way ANOVA with Dunnett’s post-hoc test was used to determine the level of significance between the study groups. All the results are reported as mean ± SD (standard deviation). A p-value of 0.05 was considered significant for all the analyses.
Discussion
This study utilized the co-culture model of dendritic cells (DC 2.4) and T-cells (EL4.IL2) to evaluate the immune pathways followed by a microparticulate murine breast cancer vaccine. This vaccine has been successfully evaluated by the authors in an in vivo mouse model. Upon the oral and transdermal administration of the microparticulate breast cancer vaccine, we have observed that the vaccine can elicit both humoral (IgG) and cellular (CD4+, CD8+) mediated adaptive immune responses. Further, these immune responses led to protective immunity when the vaccinated animals were challenged with live murine breast cancer cells. We have utilized the in vitro co-culture (DC/T-Cell) model in this study further to understand the origin of such adaptive immune responses and included a cancer vaccine adjuvant [Poly (I:C)] to enhance them. The study immune responses obtained by the co-culture model were analyzed via a three-pronged approach: flow cytometry analysis of DC surface markers, cytokine analysis via ELISA, and confocal imaging to qualitatively evaluate the DC surface markers.
Flow cytometry studies confirmed that the dendritic cells co-cultured with EL4.IL2 T-cells undergo activation and maturation in the presence of vaccine microparticles and lead to significantly higher cell surface expression of MHC I, MHC II, CD40, and CD80 (p<0.05). These cell surface expressions were further potentiated in the presence of the adjuvant, Poly (I:C), as seen with the vaccine + adjuvant treatment group in comparison to the cells only control (p<0.05). The flow cytometry studies also demonstrated that the placebo particles are incapable of eliciting the same response. Further, this response does not change in the presence of an adjuvant for a placebo particle, as observed with the placebo + adjuvant treatment group. It is important to note that the vaccine and placebo particles are 95% w/w identical in composition. The only difference between them is the 5% w/w WCL present in the vaccine particles. These results confirm the significance of TAAs provided by the WCL to elicit the desired immune response mediated by the antigen presentation via the dendritic cells. Flow cytometry studies quantitatively demonstrate the significant levels of cell surface markers confirming the dendritic cells’ maturation, leading to immunostimulation of CD4+ and CD8+ T-cells. These results are further corroborated by the in vivo study results published previously.9,10 We have previously demonstrated that the spleens of vaccinated animals presented a significantly higher level of CD4+ T-cell, CD8+ T-cell, and B cell population when compared to the control animals (p<0.05). The current study also confirmed the role of Poly (I:C) as a potential adjuvant for this microparticulate breast cancer vaccine. The potentiation of cell surface markers in the presence of adjuvant for the vaccine treatment group demonstrates the role of Poly (I:C) as a cancer adjuvant capable of DC activation and T-cell stimulation. These results are in line with the studies published previously.21–24
Further, the ELISA results demonstrate that the vaccine and vaccine + adjuvant treatment groups do not lead to TNF-alpha, IFN-gamma, or IL-2 cytokine release. However, the placebo and placebo + adjuvant treatment groups expressed these cytokines at a significantly higher concentration during a 24-hour duration (p<0.05). These results indicate that the treatment groups, including vaccine microparticles, do not cause any inflammatory innate immune response versus the placebo microparticle counterpart, which leads to such immune responses. The lack of these cytokines in the presence of vaccine microparticles supports the safe application of these microparticles in vivo. Also, it indicates that the presence of the WCL shifts the immune response from innate to adaptive, as demonstrated by the flow cytometry studies. Our studies further support the understanding that cytokine release alone is not sufficient to cause dendritic cell maturation, as observed in the case of placebo and placebo + adjuvant treatment groups. Irrespective of the innate immune response observed by placebo microparticles, these groups failed to express significantly higher levels of cell surface markers, including MHC class I and II, and the co-stimulatory CD markers (40 and 80) as observed via the flow cytometry studies.
Additionally, the confocal studies performed with co-cultured dendritic cells supported the results observed via the FACS studies. The confocal studies qualitatively provided the visual confirmation of higher-level expression of cell surface markers including MHC I, MHC II, CD40, and CD80 in the vaccine microparticle containing treatment groups.
Lastly, the quantification of T-cells post-co-culture studies and the treatments with placebo and vaccine microparticles confirmed that the dendritic cells stimulated by the vaccine microparticles in the presence or absence of the adjuvant are capable of activating the T-cells leading to cell expansion and a statistically higher increased live T-cell count (p<0.0001). These results corroborate the results observed via the flow cytometry and confocal studies, confirming that the presence of cell surface markers of the dendritic cells allows them to effectively communicate with the T-cells, yielding the observed T-cell stimulation and expansion. Further, the placebo treatment groups did not experience any change in T-cell population when compared to cells alone as a negative control. Overall, the results confirm that the WCL/TAA source is vital to stimulate the dendritic cells and, eventually, the T-cells.
The MHC Class I processing pathway is essential to immune surveillance by cytotoxic T-cells. Antitumor cytotoxic T-cell responses have been shown after cross-priming with dendritic cells.25 Cytotoxic T lymphocytes (CTLs) can recognize tumor antigens when presented on these MHC Class I molecules. Antigen recognition by CTLs allows these cells to lyse tumor cells.26 By having increased expression of MHC I molecules on dendritic cells’ surface, tumor cells are more susceptible to lysis by CTLs.26 The vaccine microparticles treatment groups (with and without the adjuvant) had significantly more MHC I expression than the control, indicating that in the presence of tumor cells, activated CTLs are likely to produce a significant antitumor response. The MHC class II pathway is regulated by dendritic cell stimuli as well. In normal dendritic cells, MHC class II is continuously recycled and degraded in the cell. Once the cell is matured, the MHC class II molecules are stabilized on the cell surface. The vaccine microparticle treatment groups had statistically higher MHC class II expression than the control, which indicates that these cells had matured; otherwise, degradation of this marker would be expected. These observations support that the microparticulate breast cancer vaccine activated the adaptive immune response, which was further boosted in the presence of Poly (I:C) adjuvant.
Further, it is established that the activation of T-cells is essential for the development of an adaptive immune response sufficient to eradicate tumor cells. Evaluating specific cell surface markers on antigen-presenting cells can help determine if the immune response generated is activating T-cells. For a T-cell to be fully activated, two signals are necessary. Signal 1 is the interaction of the T-cell receptor with the MHC molecule (I or II). Signal 2 occurs through the interaction of co-stimulatory molecules such as CD40 and CD80. These interactions between a T-cell and an antigen-presenting cell, such as a dendritic cell, allow the T-cell to become fully activated and elicit an adaptive immune response.27 Cell surface markers have been used to determine if cancer vaccines are promoting an immune response. Many studies have observed increased levels of MHC II, CD80, and CD86, but lack evaluation of immunotherapeutic effects of vaccines via MHC I and CD40.28 The current study fills the gap in evaluating the expression of such cell surface markers.
Additionally, the CD80/CD28 pathway has been shown to have the most vital activation of T-cells. Antigen presentation on MHC I can have improved effects on T-cell activation, specifically when looking at its ability to attack tumor cells by upregulation of the CD80 co-stimulatory molecule. When the molecules of CD80 bind to CD28, there is an activation of T-cell response.25
Conclusion
The co-culture model utilizing DC and T-cells confirmed that the vaccine microparticles containing 4T07 breast cancer whole cell lysate were capable of activating the dendritic cells to yield expression of MHC I, MHC II, CD40, and CD80 markers at statistically significant levels (p<0.05). Further, the role of Poly (I:C) as a successful adjuvant for the 4T07 microparticulate breast cancer vaccine was established. The inclusion of Poly (I:C) with the vaccine microparticles further boosted the cell surface marker expression. However, no such potentiation was observed in the case of placebo microparticles, confirming the significance of the whole cell lysate and the TAAs provided by the lysate to form the vaccine. Overall, the results confirm the role of DCs and MHC pathways utilized by the microparticulate breast cancer vaccine, which led to the protective adaptive immune responses observed previously in vivo.
Acknowledgments
The authors acknowledge the American Association of Colleges of Pharmacy (AACP) for the New Investigator Award grant funding that supported this project. We also acknowledge the American Foundation for Pharmaceutical Education (AFPE) for the Gateway to Research Scholarship to Michelle Ubowski regarding this research project. Additionally, we are thankful to Mr. Frank Murante and Dr Ernest Smith from Vaccinex Inc., Rochester, NY, for assisting with the flow cytometry studies. Lastly, we are grateful to Dr Hyla Sweet at Rochester Institute of Technology, Rochester, NY, for helping with the confocal studies.
Disclosure
Michelle Ubowski is employed by and is a shareholder of Pfizer, Inc., outside of the submitted work. The authors report no other conflicts of interest in this work.
References
1. Cancer stat facts: female Breast Cancer. 2023; Available from: https://seer.cancer.gov/statfacts/html/breast.html.
2. Control DoCPa. Breast Cancer Statistics. [Webpage]; 2020.
3. Ernst B, Anderson KS. Immunotherapy for the treatment of breast cancer. Curr Oncol Rep. 2015;17(2):5. doi:10.1007/s11912-014-0426-9
4. Cimino-Mathews A, Foote JB, Emens LA. Immune targeting in breast cancer. Oncology. 2015;29(5):375–385.
5. Dillon PM, Brenin CM, Slingluff CL Jr. Evaluating nelipepimut-s in the treatment of Breast Cancer: a short report on the emerging data. Breast Cancer. 2020;12:69–75. doi:10.2147/BCTT.S224758
6. Mittendorf EA, Clifton GT, Holmes JP, et al. Final report of the phase I/II clinical trial of the E75 (nelipepimut-S) vaccine with booster inoculations to prevent disease recurrence in high-risk breast cancer patients. Ann Oncol. 2014;25(9):1735–1742. doi:10.1093/annonc/mdu211
7. Nie X, Shi C, Chen X, et al. A single-shot prophylactic tumor vaccine enabled by an injectable biomembrane hydrogel. Acta Biomater. 2023;169:306–316. doi:10.1016/j.actbio.2023.08.010
8. Akalkotkar A, Chablani L, Tawde SA, D’Souza C, D’Souza MJ. Development of a microparticulate prostate cancer vaccine and evaluating the effect of route of administration on its efficacy via the skin. J Microencapsul. 2015;32(3):281–289. doi:10.3109/02652048.2015.1017615
9. Chablani L, Tawde SA, Akalkotkar A, D’Souza C, Selvaraj P, D’Souza MJ. Formulation and evaluation of a particulate oral breast cancer vaccine. J Pharm Sci. 2012;101(10):3661–3671. doi:10.1002/jps.23275
10. Chablani L, Tawde SA, Akalkotkar A, D’Souza MJ. Evaluation of a particulate breast cancer vaccine delivered via skin. Aaps j. 2019;21(2):12. doi:10.1208/s12248-018-0285-7
11. Chablani L, Tawde SA, D’Souza MJ. Spray-dried microparticles: a potential vehicle for oral delivery of vaccines. J Microencapsul. 2012;29(4):388–397. doi:10.3109/02652048.2011.651503
12. Tawde SA, Chablani L, Akalkotkar A, D’Souza MJ. Evaluation of microparticulate ovarian cancer vaccine via transdermal route of delivery. J Control Release. 2016;235:147–154. doi:10.1016/j.jconrel.2016.05.058
13. Joshi VB, Geary SM, Gross BP, Wongrakpanich A, Norian LA, Salem AK. Tumor lysate-loaded biodegradable microparticles as cancer vaccines. Expert Rev Vaccines. 2014;13(1):9–15. doi:10.1586/14760584.2014.851606
14. Forghani P, Waller EK. Poly (I: c) modulates the immunosuppressive activity of myeloid-derived suppressor cells in a murine model of breast cancer. Breast Cancer Res Treat. 2015;153(1):21–30. doi:10.1007/s10549-015-3508-y
15. Wafa EI, Geary SM, Ross KA, Goodman JT, Narasimhan B, Salem AK. Pentaerythritol-based lipid A bolsters the antitumor efficacy of a polyanhydride particle-based cancer vaccine. Nanomedicine. 2019;21:102055. doi:10.1016/j.nano.2019.102055
16. Falke J, Hulsbergen-van de Kaa CA, Maj R, Oosterwijk E, Witjes JA. A placebo-controlled efficacy study of the intravesical immunomodulators TMX-101 and TMX-202 in an orthotopic bladder cancer rat model. World J Urol. 2018;36(11):1719–1725. doi:10.1007/s00345-018-2334-3
17. Ammi R, De Waele J, Willemen Y, et al. Poly(I:C) as cancer vaccine adjuvant: knocking on the door of medical breakthroughs. Pharmacol Ther. 2015;146:120–131. doi:10.1016/j.pharmthera.2014.09.010
18. Saxena M, Sabado RL, La Mar M, et al. Poly-ICLC, a TLR3 agonist, induces transient innate immune responses in patients with treated HIV-infection: a randomized double-blinded placebo controlled trial. Front Immunol. 2019;10:725. doi:10.3389/fimmu.2019.00725
19. El-Hussein A, Lam SSK, Raker J, Chen WR, Hamblin MR. N-dihydrogalactochitosan as a potent immune activator for dendritic cells. J Biomed Mater Res A. 2017;105(4):963–972. doi:10.1002/jbm.a.35991
20. El-Murr T, Patel A, Sedlak C, D’Souza-Lobo B. Evaluating dendritic cells as an in vitro screening tool for immunotherapeutic formulations. J Immunol Methods. 2018;459:55–62. doi:10.1016/j.jim.2018.05.005
21. Verdijk RM, Mutis T, Esendam B, et al. Polyriboinosinic polyribocytidylic acid (poly(I:C)) induces stable maturation of functionally active human dendritic cells. J Immunol. 1999;163(1):57–61. doi:10.4049/jimmunol.163.1.57
22. Smits EL, Ponsaerts P, Van de Velde AL, et al. Proinflammatory response of human leukemic cells to dsRNA transfection linked to activation of dendritic cells. Leukemia. 2007;21(8):1691–1699. doi:10.1038/sj.leu.2404763
23. Benwell RK, Hruska JE, Fritsche KL, Lee DR. Double stranded RNA- relative to other TLR ligand-activated dendritic cells induce extremely polarized human Th1 responses. Cell Immunol. 2010;264(2):119–126. doi:10.1016/j.cellimm.2010.05.008
24. Salem ML, Diaz-Montero CM, El-Naggar SA, Chen Y, Moussa O, Cole DJ. The TLR3 agonist poly(I:C) targets CD8+ T cells and augments their antigen-specific responses upon their adoptive transfer into naïve recipient mice. Vaccine. 2009;27(4):549–557. doi:10.1016/j.vaccine.2008.11.013
25. Driessens G, Kline J, Gajewski TF. Costimulatory and coinhibitory receptors in anti-tumor immunity. Immunol Rev. 2009;229(1):126–144. doi:10.1111/j.1600-065X.2009.00771.x
26. Zheng Y, Manzotti CN, Liu M, Burke F, Mead KI, Sansom DM. CD86 and CD80 differentially modulate the suppressive function of human regulatory T cells. J Immunol. 2004;172(5):2778–2784. doi:10.4049/jimmunol.172.5.2778
27. den Haan JM, Arens R, van Zelm MC. The activation of the adaptive immune system: cross-talk between antigen-presenting cells, T cells and B cells. Immunol Lett. 2014;162(2):Pt B):103–112. doi:10.1016/j.imlet.2014.10.011
28. Rojas-Sepúlveda D, Tittarelli A, Gleisner MA, et al. Tumor lysate-based vaccines: on the road to immunotherapy for gallbladder cancer. Cancer Immunol Immunother. 2018;67(12):1897–1910. doi:10.1007/s00262-018-2157-5
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.