")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Understanding the cross-resistance of oseltamivir to H1N1 and H5N1 influenza A neuraminidase mutations using multidimensional computational analyses
Received 31 January 2015
Accepted for publication 23 February 2015
Published 31 July 2015 Volume 2015:9 Pages 4137—4154
DOI https://doi.org/10.2147/DDDT.S81934
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Shu-Feng Zhou
Ashona Singh, Mahmoud E Soliman
School of Health Sciences, University of KwaZulu-Natal, Westville, Durban, South Africa
Abstract: This study embarks on a comprehensive description of the conformational contributions to resistance of neuraminidase (N1) in H1N1 and H5N1 to oseltamivir, using comparative multiple molecular dynamic simulations. The available data with regard to elucidation of the mechanism of resistance as a result of mutations in H1N1 and H5N1 neuraminidases is not well established. Enhanced post-dynamic analysis, such as principal component analysis, solvent accessible surface area, free binding energy calculations, and radius of gyration were performed to gain a precise insight into the binding mode and origin of resistance of oseltamivir in H1N1 and H5N1 mutants. Three significant features reflecting resistance in the presence of mutations H274Y and I222K, of the protein complexed with the inhibitor are: reduced flexibility of the a-carbon backbone; an improved ΔEele of ~15 (kcal/mol) for H1N1 coupled with an increase in ΔGsol (~13 kcal/mol) from wild-type to mutation; a low binding affinity in comparison with the wild-type of ~2 (kcal/mol) and ~7 (kcal/mol) with respect to each mutation for the H5N1 systems; and reduced hydrophobicity of the overall surface structure due to an impaired hydrogen bonding network. We believe the results of this study will ultimately provide a useful insight into the structural landscape of neuraminidase-associated binding of oseltamivir. Furthermore, the results can be used in the design and development of potent inhibitors of neuraminidases.
Keywords: neuraminidase, molecular dynamics, resistance, mutation, binding free energy
Introduction
The rapid evolution of highly pathogenic and resistant variants of influenza A viruses, H5N1 and H1N1, takes us to the precipice of a possible new-age influenza pandemic. The first human influenza pandemic documented in the 20th century was the 1918–1919 “Spanish flu”, which claimed the lives of approximately 50 million people worldwide.1–4 Since 1997, the highly pathogenic avian influenza A (H5N1) has had a detrimental effect on the socioeconomic development in countries across Africa, Asia, and the Middle East. Pharmacological advances in viable influenza chemotherapeutics and vaccines could not have predicted the influenza A (H1N1) virus outbreak in 2009–2010, which claimed an estimated 284,500 lives. The viruses H5N1 and H1N1 display similar pathogenesis of respiratory tract infection in humans. However, statistically H5N1 has a higher mortality rate.5
Influenza virus A is highly communicable. H1N1 is easily transmissible between humans, while avian influenza has not yet adapted sufficiently to facilitate human-human spread; the spread of H5N1 is largely dependent on close avian-human contact. Based on the research, the H1N1 and H5N1 viruses can both trace their lineage back to pigs as potential intermediate hosts. Both viruses can undergo reassortment in the intermediate host prior to infection in the new host.6,7 This raises the concern that more virulent and highly transmissible influenza virus strains may infect humans.8
Both the H1N1 and H5N1 viruses express two main surface antigens, H5/H1 (hemagglutinin type 5/1, respectively) and N1 (neuraminidase type 1).9–11 Neuraminidase has been the main target for potential pharmacological interventions.12 It plays a vital role in the spread of infection through cleavage of the viral receptor, sialic acid, from both viral and host proteins. This results in the release of newly replicated virus from an infected cell.12,13 The active site residues of the neuraminidase enzyme are highly conserved within all virus types and subtypes, ie, designed neuraminidase inhibitors may be effective against types A and B of the influenza virus.14–17
There are four effective neuraminidase inhibitors currently available: zanamivir,18 laninamivir,19 peramivir,20 and oseltamivir.21 Zanamivir is commercially known as Relenza® and is administered as an aerosol into the nasal cavity. Zanamivir has been approved in many countries since 1999/2000. Laninamivir is administered in the form of an inhaler and was made available to the Japanese market in 2010 (under the trade name Inavir®). Peramivir, commercially known as PeramiFlu®, is administered by intravenous drip and is approved in Japan, the Republic of Korea, and the People’s Republic of China. Oseltamivir commercially known as Tamiflu® (Figure 1), has long been the “drug of choice” due to ease of oral administration, improved bioavailability, and easy accessibility. There has been an increase in the number of reports describing oseltamivir resistance in H1N1 and H5N1, and the rapid development of resistance toward the drug poses a threat in the event of an outbreak.22,23
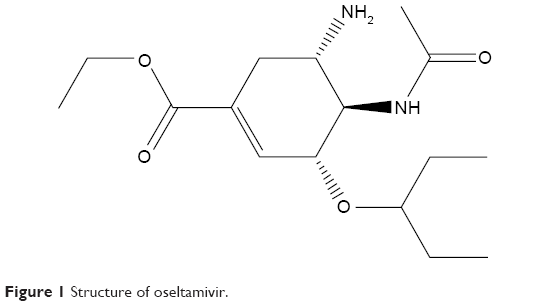 | Figure 1 Structure of oseltamivir. |
Research directed toward gaining an insight into the mechanism of resistance of influenza viruses is imperative in the future design of drugs as well as in the development of pre-emptive measures against possible mutations. The comparative multiple molecular dynamic (MD) simulations conducted in this study offer an atomistic perspective into the complex nature of protein motions as a function of time.24,25 Due to H5N1 virus sharing a 91.47% sequence identity and a conserved binding site with the 2009 H1N1, the results of molecular simulation may apply to both viruses.26
Several computational approaches have been used in an attempt to understand the impact of mutations on oseltamivir resistance to neuraminidase. Karthick et al initiated one of the first MD studies, which was based solely on the natural mutation H274Y of H1N1 isolated during the 2007–2008 Euroasia influenza season.27,28 Malaisree et al refined the source of oseltamivir resistance in avian influenza H5N1 virus with the H274Y mutation using MD simulation.29,30 Wang and Zheng performed a similar study that incorporated the mutations N294S and E119G.31 In the above studies, it was found that although residue 274 is a framework residue in the active site,21 the mutation of histidine (His) to tyrosine (Tyr) significantly impaired the binding of oseltamivir because the binding pocket could not accommodate the steric bulk of the drug. Further to this, the presence of additional mutations was found to amplify resistance of the neuraminidase enzyme to oseltamivir. A large database of crystallized glycoprotein of influenza virus A and B exists, each with a different mutation in the neuraminidase amino acid sequence.26,27,32–34 The MD studies for each mutation revealed enhanced surface expression of oseltamivir-resistant influenza neuraminidase.35 In many instances, these included zanamivir and peramivir resistance.36
Nguyen et al37 and Huang et al38 identified a mutation at position 222 from isoleucine to lysine (Lys) in strains of wild-type (WT) and H274Y mutants of H1N1 and H5N1. The point mutation from isoleucine to Lys exhibited increased resistance toward oseltamivir. Structural residue I222 of H1N1 and H5N1 neuraminidase offered stability to oseltamivir binding.39,40 Structure stability was reinforced by the presence of the hydrophobic pocket created by E276 and R224, which in turn supported a seven hydrogen bond interaction between the active site residues and the drug.30,36
Insufficient information about the origin of resistance of oseltamivir against neuraminidase H1N1 and H5N1 mutations, I222K and H274Y (Figure 2) prompted us to perform a comprehensive multidimensional analysis using a multiple MD approach. Multiple MD simulations have demonstrated better sampling efficiency.41 Multiple trajectories with different initial conditions improved the conformational sampling of MD simulations in proteins.42,43 Several post-dynamic analyses, such as root mean square fluctuation (RMSF), root mean square deviation (RMSD), free binding energy calculations, radius of gyration, solvent accessible surface area (SASA), and principal component analysis (PCA) were performed in order to gain a comprehensive understanding of the impact of mutations on binding and the conformational landscape of the oseltamivir–protein complex. Our research should contribute to understanding the effect of resistance and provide insight into the future development of innovative chemotherapeutics.
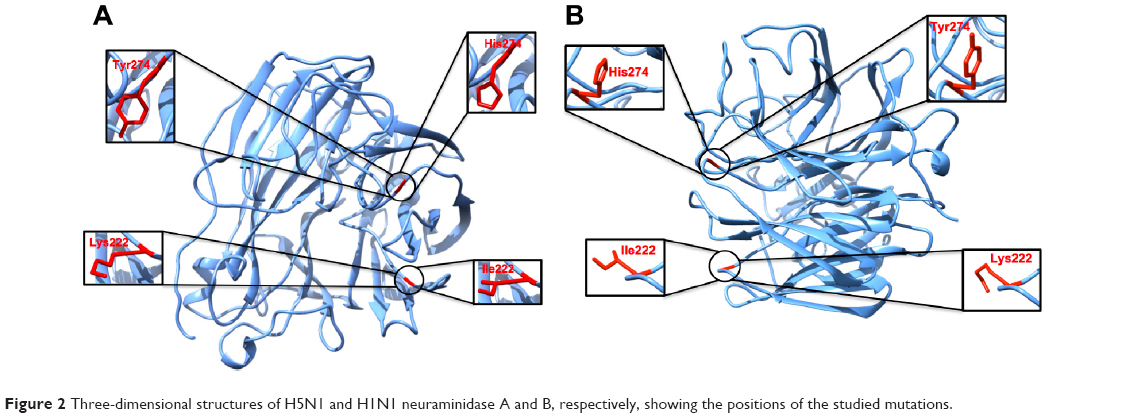 | Figure 2 Three-dimensional structures of H5N1 and H1N1 neuraminidase A and B, respectively, showing the positions of the studied mutations. |
Computational methods
System preparation
The X-ray crystal structures of H1N1 and H5N1 WT and mutant neuraminidase were extracted from Protein Data Bank (PDB) codes 4B7R (WT-H1N1),44 2HU4 (WT-H5N1),15 and 3CL0 (H274Y mutation-H5N1)45 complexed with oseltamivir, obtained from the Research Collaboratory for Structural Bioinformatics. Neuraminidase is a tetrameric enzyme, using the Chimera software package46 a single subunit was selected with the inclusion of an active site drug complex to reduce computational cost. Influenza virus, H1N1 PDB code 4B7R, formed the WT sequence with a His274 residue. Using Chimera, point mutations were introduced at positions 222 (from I to K) and 274 (from H to Y). Table 1 lists the three enzymes with relevant mutations resulting in eight simulations.
 | Table 1 Crystal structures of the simulated systems, PDB codes, and abbreviations |
MD simulation
Multiple MD simulations were performed to establish the impact of mutation I222K on the binding of oseltamivir to the WT enzyme and in the presence of mutation H274Y of H1N1 and H5N1. A long continuous MD trajectory may incur greater statistical errors as the protein denatures and evolves from one conformation to another during the time of the simulation.47 Thus, a multiple MD approach was undertaken. This method in turn reduced the force field artefacts, statistical bias, and computational time. A total simulation time of 100 ns partitioned in five distinct 20 ns MD runs was executed, with each trajectory having a different initial velocity. The multiple MD protocol applied in this study is described in Figure 3.
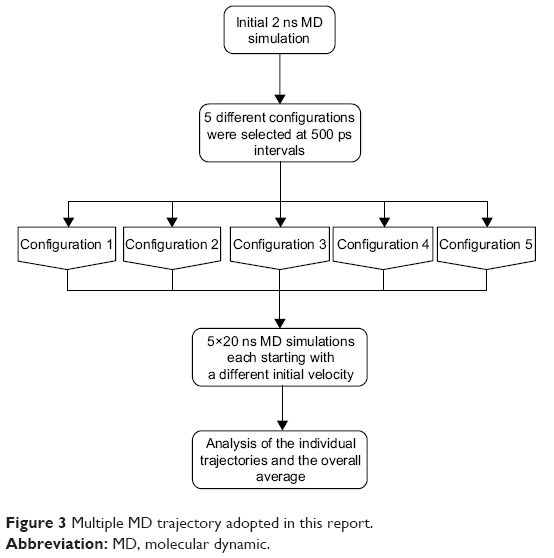 | Figure 3 Multiple MD trajectory adopted in this report. |
MD simulation set-up and parameters
The MD simulation was performed using the graphics processing unit version of the PMEMD engine provided with the Amber 12 and 14 package.48,49 The FF99SB variant of the Amber force field50 was used to describe the protein. The LEAP module of Amber 12/14 allowed for the addition of hydrogen atoms to the protein and three Na+ counter ions for neutralization. The system was suspended within a TIP3P51 water box such that all protein atoms were within 8 Å of any box edge. After energy minimization, a 1,000 step restraint gradient minimization was performed. Selective boundary conditions were enforced concurrently with the particle-mesh Ewald method,52 a component of Amber 12/14, with set parameters of direct space and a van der Waals cut-off of 12 Å. This would be conducive to the treatment of long-range electrostatic interactions. The system passed through an initial energy minimization with 2,500 steps of steepest descent and a restraint harmonic potential of 500 kcal/mol Å2 being applied to the solute. After this, an additional 1,000 step unrestrained conjugate gradient energy minimization was carried out on the complete system, ie, protein, ligand, solvent molecules, and added ions. Gradual heating of the MD simulation from 0 to 300 K was executed for 50 ps, such that the system maintained a fixed number of atoms and a fixed volume, ie, a canonical (NVT) ensemble. The solutes within the system were imposed with a potential harmonic restraint of 10 kcal/mol Å2 and a collision frequency of 1.0 ps−1. Superceding the heating input, a final equilibration estimating 500 ps of the systems was required. The operating temperature was kept constant at 300 K, accompanied by the number of atoms and pressure, mimicking an isobaric-isothermal (NPT) ensemble. The systems pressure was maintained at 1 bar using the Berendsen barostat.53 The total time for each MD simulation was 20 ns, and five trajectories for each of the eight systems were generated. In each simulation, the SHAKE algorithm54 was employed to constrict the bonds of the hydrogen atoms. The time step of each simulation was 2 fs and a single-precision floating-point precision model55 was used. The simulations coincided with randomized seeding of the NPT ensemble. It was accompanied by constant pressure of 1 bar maintained by the Berendsen barostat, a pressure coupling constant of 2 ps, with a temperature of 300 K and Langevin thermostat with a collision frequency of 1.0 ps−2. Co-ordinates were saved every 1 ps and the trajectories were analyzed every 1 ps using the PTRAJ module implemented in Amber 12.0 and CPPTRAJ module in Amber 14.0.
Post-dynamic analysis
Thermodynamic calculations
An implicit solvent model was employed to describe the thermodynamic free binding energies of oseltamivir bound to WT and mutant H1N1 and H5N1 neuraminidase in this study. The molecular mechanics/generalized Born surface area method was used to evaluate the ligand-protein complex binding affinities.56–59 To calculate the free binding energy contributions, 1,000 snapshots were extracted from each of the 20 ns trajectories. The following set of equations describes the calculation of the binding free energy:
| (1) |
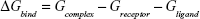
| (2) |
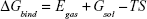
| (3) |
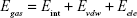
| (4) |
| (5) |
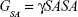
where Egas represents the gas-phase energy, Eint is the internal energy, Eele is the Coulomb energy, and EvdW is the van der Waals energy. The term Egas is directly measured from the FF99SB force field terms. The solvation energy (Gsol) is the summation of contributions from polar and nonpolar states. The polar solvation energy contribution, GGB, is derived from solving the GB equation. The term GSA corresponds to the non-polar solvation energy contribution, which is estimated from the SASA determined using a water probe radius of 1.4 Å. The temperature and total solute entropy are represented by T and S, respectively.60
Principal component analysis
PCA reveals the structure of atomic fluctuations, and describes the motion of the system in terms of eigenvectors (planar of motion) and eigenvalues (magnitude of motion).61 The individual MD trajectories were stripped of solvent and ions using the PTRAJ and CPPTRAJ modules in Amber 12.0/14.0. The resulting trajectories were aligned against a fully minimized structure. PCA was performed on a Cα backbone with 1,000 snapshots taken every 20 ps. The first two eigenvectors (PC1 and PC2) corresponding to the first two modes of PCA covariance matrices were generated using in-house scripts. An average of the PC1 and PC2 for the 5×20 ns trajectories of the H1N1 and H5N1 WT and mutant systems was generated. The corresponding PCA scatters were plotted using Origin software (http://www.originlab.com/) and structural postscript diagrams were created using visual MDs.62 Porcupine plots of the first and second modes developed by the normal mode wizard using the ProDy interface of visual MDs were sketched for each of the systems.63
Results and discussion
MD simulations and system stability
RMSD and potential energy plots in the Supplementary materials (Figures S1–12 of H5N1 and H1N1 neuraminidase, respectively) graphically monitor the convergence of the studied systems.64 All the systems of H5N1 and H1N1 influenza viruses converge at approximately 5,000 ps by both RMSD and potential energy calculations.
Post-dynamic analysis
RMSF and radius of gyration analysis was used to relate conformational changes and plasticity of the Cα backbone to mutation of each system.
Root mean square fluctuation
The RMSF captured the fluctuation of each residue, providing insight into the flexible regions of the protein that correspond to crystallographic β (temperature) factors.65 Figure 4 illustrates the fluctuations of systems WTH5N1 and H274YH5N1. The crystal structure of the WT neuraminidase consists of three α-helices and 24 β-strands. Evidence from the graph demonstrates an unfolding of the protein helix at residue 28 in H274YH5N1 in comparison with the WT, shortening the cardinal α-helix and β-strand by one and three residues, respectively. Closer inspection revealed a distinct change in fluctuation corresponding to region 108–144, possibly due to the presence of a hydrogen bond between Lys125 and Thr131. A similar effect was observed through β-strands 156–162 and 170–177 which share a hydrogen bond between amino acids Thr161 and Tyr171. The residue region 260–320 containing the compensatory mutation did not demonstrate large variations in fluctuations.
 | Figure 4 RMSF comparison of WTH5N1 and H247YH5N1: T1, T2, T3, T4, T5, and Tavg presenting the five individual 20 ns molecular dynamic trajectories and overall average, respectively. |
As depicted in Figure S13, by interacting with residues in the active site, the role of residue 222 appeared to shift from a structural one to a functional one. The Lys222 mutation contains a second amino group that acts as an electrophile at physiological pH; this prompts an interaction with the active site residues. Mutation I222KH5N1 caused a change in orientation of Gly332, promoting a disulfide link between parallel cysteine residues 339 and 364 and causing a shift in the β-strand orientation.66
Analysis of Figure S14 revealed a migration from a system of high flexibility (H274YH5N1) to one of low flexibility (H274Y-I222KH5N1). The change from a hydrophobic residue, His, to the aromatic Tyr274, which contains an ionizable phenolic group, introduced instability in a previously predominant hydrophobic region. Therefore, it is plausible to assume that the presence of mutation I222K in system H274Y-I222KH5N1 restricts interaction with the external solvent, thus minimizing flexibility throughout the protein.
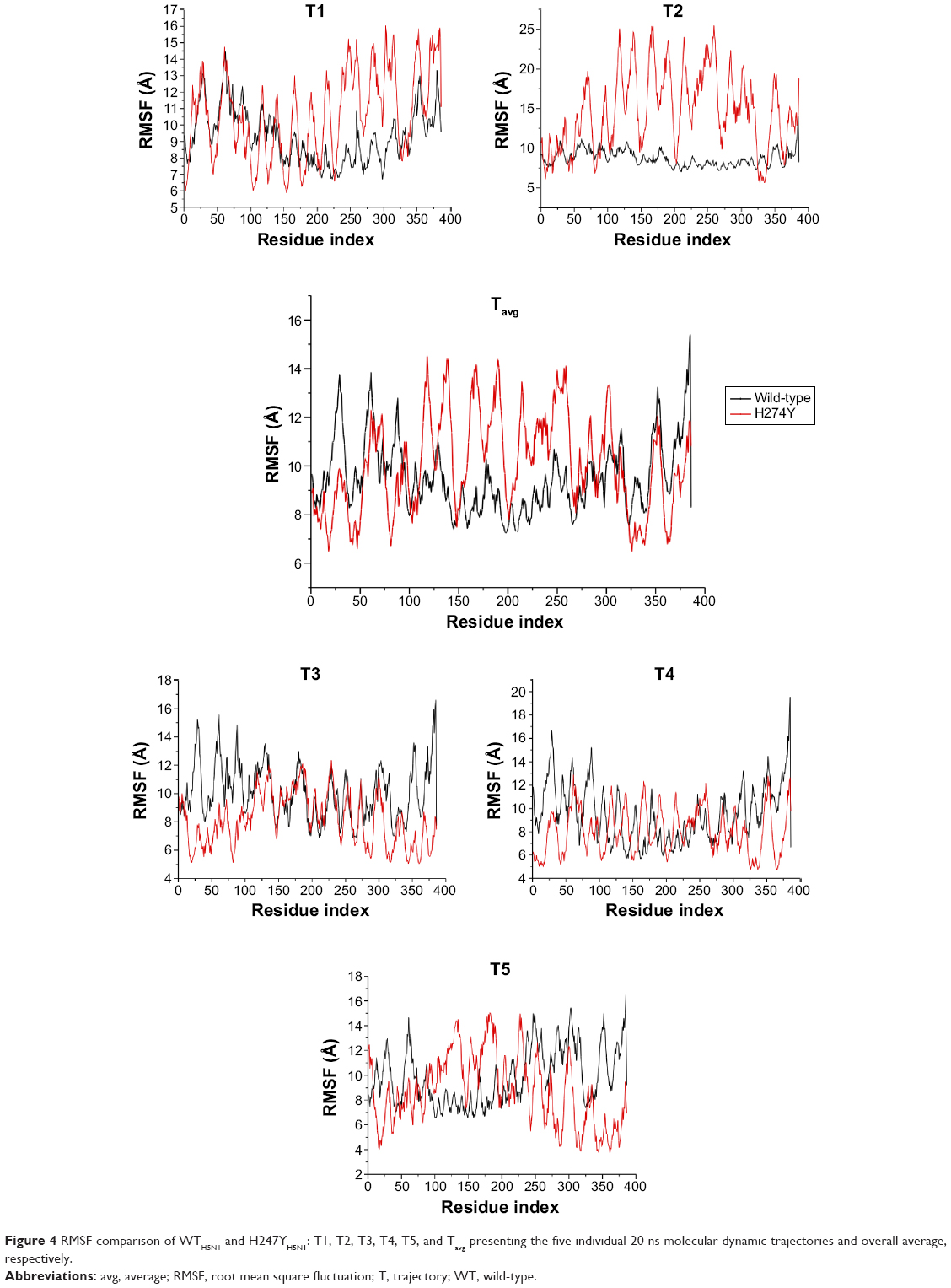
In the H274YH1N1 system (Figure 5), an increase in fluctuation at positions 58–65, 246–260, 284, and 301–305 occurred in comparison with WTH1N1. The H274Y mutation introduced a new hydrogen bond, and the aromatic group imposed a steric bulk disrupting previous hydrophobic interactions within that region. A considerable fluctuation was observed in the latter regions of the protein. There is only a single mutation in the I222KH1N1 system (Figure S15), which induced an increase in protein flexibility because Lys enhances solvent interaction. An overlay of the RMSF plots for systems H274Y-I222KH1N1 and H274YH1N1 (Figure S16) revealed no distinct difference between the two systems, indicating that no potential compensatory effect occurred to accommodate the double mutant species of protein H274Y-I222KH1N1. Thus, the conformation of the protein remains unchanged.
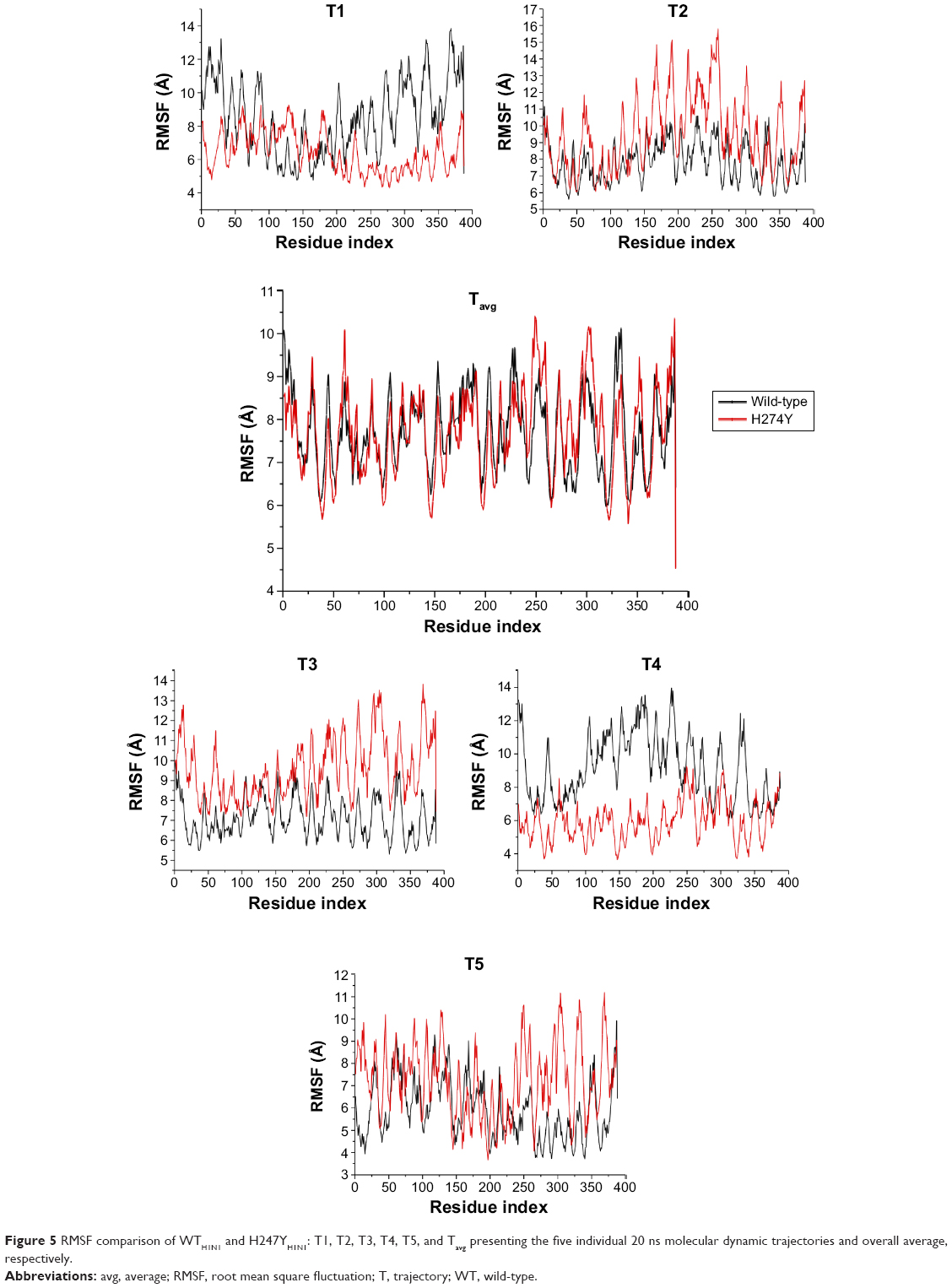 | Figure 5 RMSF comparison of WTH1N1 and H247YH1N1: T1, T2, T3, T4, T5, and Tavg presenting the five individual 20 ns molecular dynamic trajectories and overall average, respectively. |
Radius of gyration
Radius of gyration demonstrates the compactness of protein structures, providing insight into complex changes in the molecular shape.67,68 It is evident that systems WTH5N1 and H274YH5N1 (Figure 6) share a similar arrangement of amino acids in secondary and tertiary structures with an almost identical initial compactness. Although both systems share a close resemblance, the H274YH5N1 system showed a distinguishable increase in its radius of gyration over time, transmutating to a less compact structure.
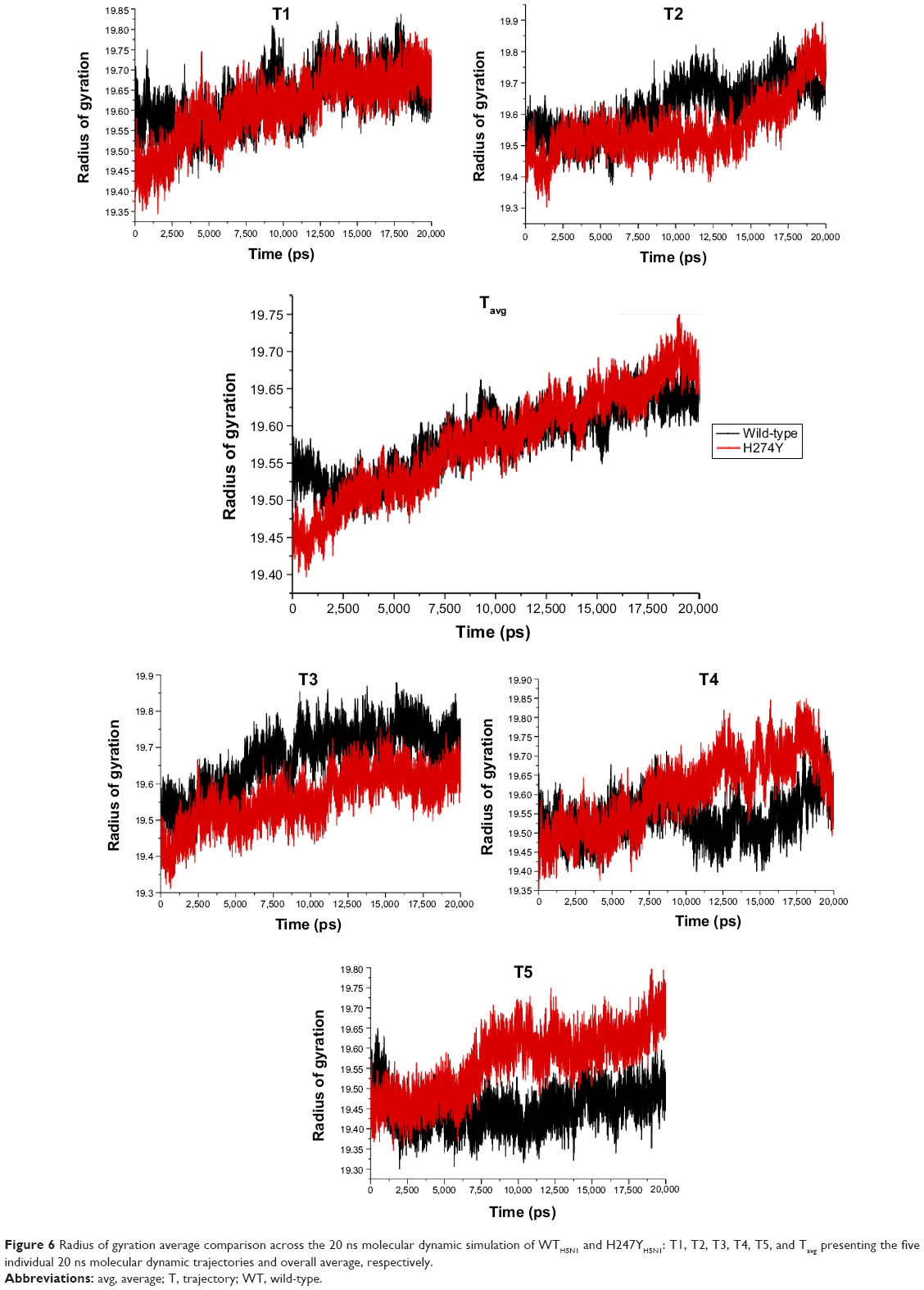 | Figure 6 Radius of gyration average comparison across the 20 ns molecular dynamic simulation of WTH5N1 and H247YH5N1: T1, T2, T3, T4, T5, and Tavg presenting the five individual 20 ns molecular dynamic trajectories and overall average, respectively. |
System comparison of WTH5N1 and I222KH5N1 (Figure S17) unveiled the I222K mutation as the more compact protein complex. Condensation of the structure created a semipermeable complex that prevented unwarranted solvent exposure of interior amino acid residues stabilizing the mutation I222KH5N1. Comparative analysis of systems H274YH5N1 and H274Y-I222KH5N1 (Figure S18) proposed an increase in flexibility of system H274Y-I222KH5N1. The compactness of system H274Y-I222KH5N1 attempts to preserve the enzymes bioactivity whilst inferring resistance against oseltamivir. Both systems H274YH5N1 and H274Y-I222KH5N1 share a similar radius of gyration profile, which suggests that both have a similar complex compactness.
System H274YH1N1 showed a larger radius of gyration than the parent protein WTH1N1, indicating that H274YH1N1 is less tightly packed (Figure 7). Solvent interaction within the amino acid sequence perpetuated partial protein unfolding, facilitating a less compacted structure. The single mutation I222KH1N1 (Figure S19) shared a similar trend by way of possessing a large radius of gyration value as compared with the WTH1N1 system. However, isoleucine expels solvent, and when Lys was introduced, an ionizable species was generated, which, much like the Tyr residue at position 274, was capable of interacting with the surrounding solvent, relieving steric strain. The H274Y-I222KH1N1 system has a folding profile similar to that of H274YH1N1, which is indicative of an undisturbed compaction of structure. The H274Y-I222KH1N1 system (Figure S20) appeared unaffected by the mutation at position 222. This could be due to the presence of the solvent-interacting amino acid species Tyr having previously rendered the inner foldings of the protein susceptible to solvation. The introduction of an additional ionizable moiety better complemented the structure in this conformation.
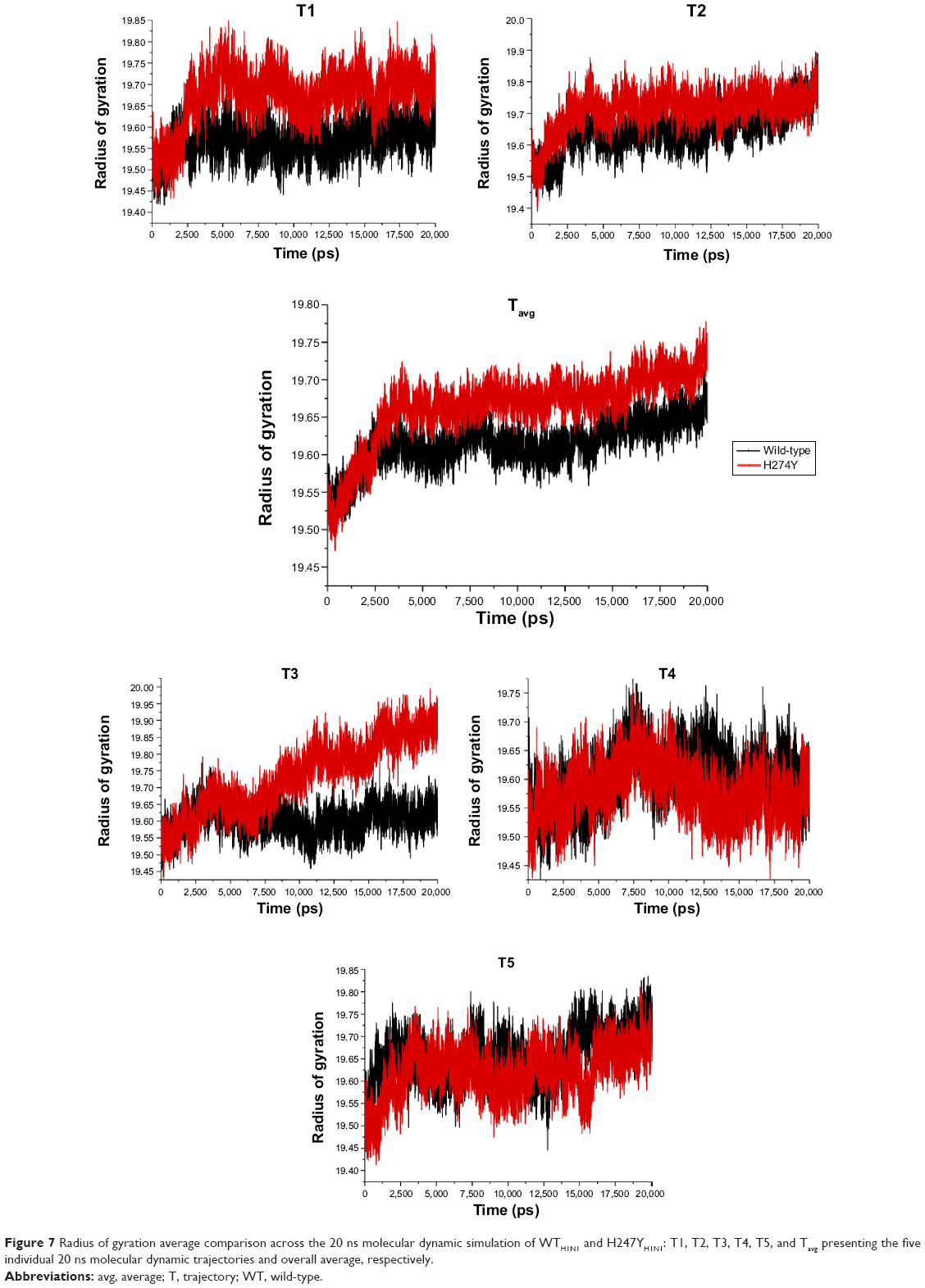 | Figure 7 Radius of gyration average comparison across the 20 ns molecular dynamic simulation of WTH1N1 and H247YH1N1: T1, T2, T3, T4, T5, and Tavg presenting the five individual 20 ns molecular dynamic trajectories and overall average, respectively. |
Calculation of MM/GBSA free binding energy
Evaluation of the free binding energies provides a thorough insight into the total energy of each of the systems. Table 2 summarizes the average molecular mechanics/generalized Born surface area (MM/GBSA) thermodynamic energy contributions elicited from the structural data of the five 20 ns MD simulations for the H5N1 and H1N1 systems.
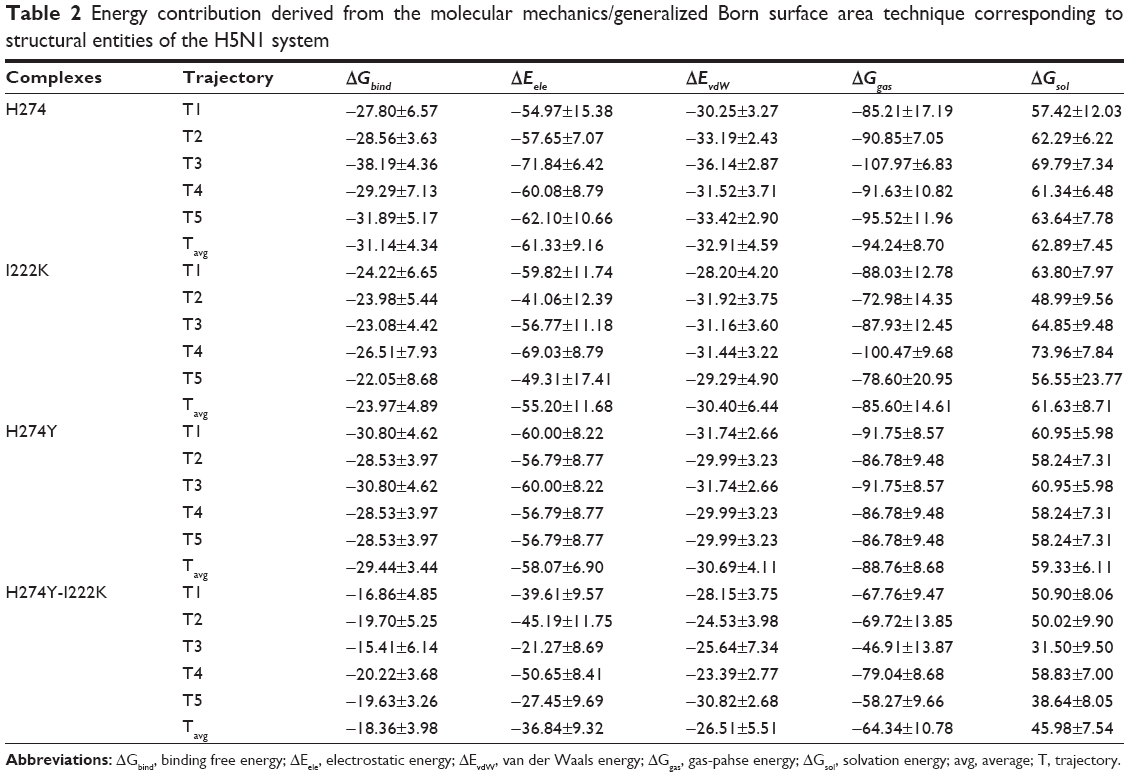 | Table 2 Energy contribution derived from the molecular mechanics/generalized Born surface area technique corresponding to structural entities of the H5N1 system |
The terms ΔEele and ΔEvdW represent the electrostatic and van der Waals intermolecular interacting components, respectively, between the protein and inhibitor.69–70 Systems WTH5N1 and I222KH5N1 showed a difference of −6.1302 kcal/mol (ΔEele) and −2.5055 kcal/mol (ΔEvdW). H274YH5N1 and H274Y-I222KH5N1 showed a difference of −21.2395 kcal/mol (ΔEele) and −4.1821 kcal/mol (ΔEvdW). The energy differences advocate a more stable protein–ligand interaction of WT and H274YH5N1. This was measured in terms of effective binding distance and positioning of amino acid residues for optimum interaction between charged entities. The ΔGsol, difference defined between systems WTH5N1 and I222KH5N1 was estimated to 1.2626 kcal/mol and for systems H274YH5N1 and H274Y-I222KH5N1 was estimated to be 13.3466 kcal/mol. The change to polar amino acids (eg from Ile to Lys and from His to Tyr) in systems I222KH5N1 and H274Y-I222KH5N1 contributed to an improved ΔGsol energy difference. Closer inspection of the free binding energy, ΔGbind, quantified the interaction between the protein and ligand. The difference in free binding energy between systems WTH5N1 and I222KH5N1 was −7.1732 kcal/mol, and that between systems H274YH5N1 and H274Y-I222KH5N1 was −11.0751 kcal/mol. The trend in binding free energy difference was observed to be as follows: WTH5N1<I222KH5N1~H274YH5N1<H274Y-I222KH5N1, indicating that the protein-ligand interaction became progressively thermodynamically unfavorable in the presence of mutations.
Table 3 corresponds to the calculated energies for the H1N1 neuraminidase systems. The amine group of oseltamivir was kept positively charged during the MD simulation. According to the literature, binding of drug in a positively charged state to the active site residues is falsely represented by an ameliorated free binding energy when comparing the mutant sequence with the WT sequence.71 Based on a study by Le et al it was proposed that a predominantly negatively charged column of residues at the binding pocket electrostatically funnels oseltamivir to the active site of N1 neuraminidases.1 This greatly impacts the binding pose, whereby a positively charged drug is drawn toward the epicenter of the active site. The energy contribution ΔGGB of H274YH1N1 and I222KH1N1 is testament to the proposed binding instruction as both systems highlighted an improved binding, with an energy difference from the WT of −4.1784 kcal/mol and −2.4447 kcal/mol, respectively. Surprisingly, the double mutation species H274Y-I222KH1N1 demonstrated a similar energy profile trend to WTH1N1. The electrostatic energy of the I222KH1N1 and H274YH1N1 species differed significantly from the WTH1N1 system by −8.3000 kcal/mol and –15.9730 kcal/mol, respectively. However, a remarkable drop in electrostatic energy was observed in the H274Y-I222KH1N1 system compared with H274YH1N1. This phenomenon could relate to the conformation of the double mutant system, as the residues interacting with the solvent direct themselves inwardly, interacting with neighboring amino acid residues. The ΔEvdW and ΔGsol differences between H274YH1N1 and double mutation H274Y-I222KH1N1 confirm the funneling mechanism, as both shared an improvement in the ΔEvdW value over the WT. However, a significant decline in ΔGsol of 37.4933 kcal/mol indicated a lack of solvent interaction. The van der Waals contributions for I222KH1N1 suggested a slight decline in hydrophobic interaction, with an energy difference from WT of −1.0594 kcal/mol. A ΔGsol difference between the systems of 4.7960 kcal/mol implied enhanced solvent interaction of the I222KH1N1 complex. The H274YH1N1 offered a similar van der Waals contribution to WTH1N1, as the aromatic group replaced a linear hydrocarbon chain. Despite this structural feature, a solvation energy difference of 13.1386 kcal/mol indicated that the hydroxyl group of Tyr imposed an increased solvent exposure.
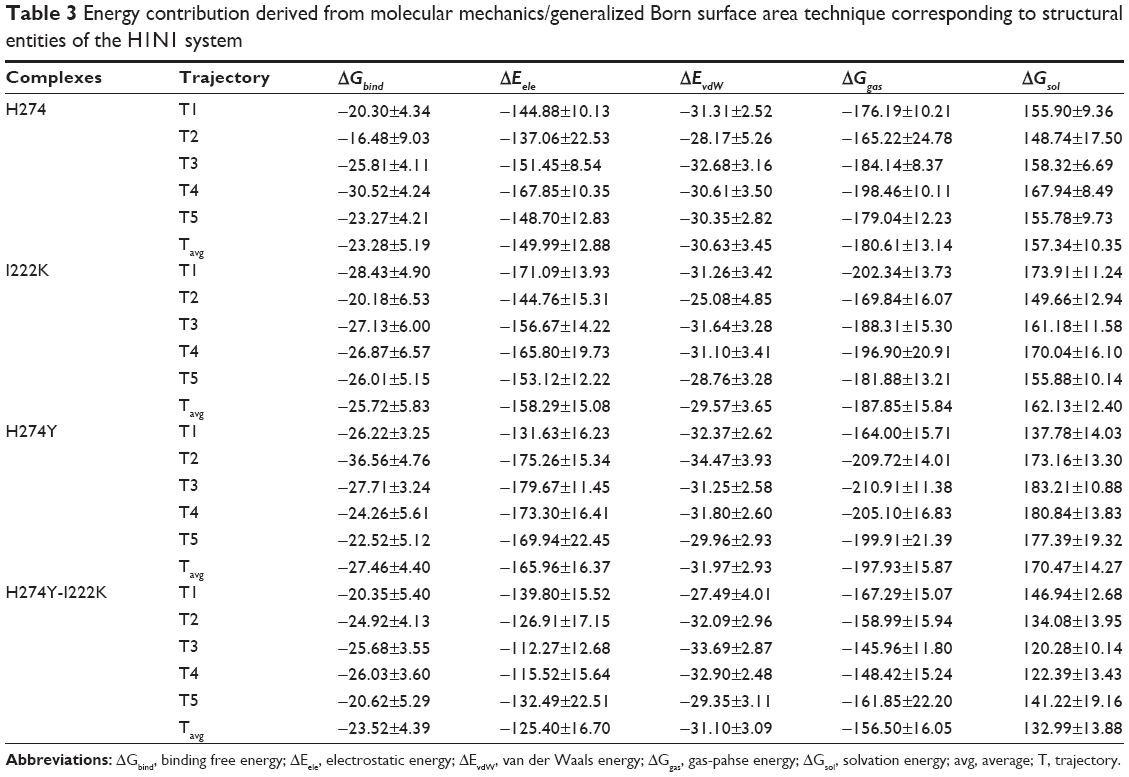 | Table 3 Energy contribution derived from molecular mechanics/generalized Born surface area technique corresponding to structural entities of the H1N1 system |
Hydrogen bond formation between amino acid residues
The dimensionality of a protein is a vector metric governed by the direction of interaction and number of hydrogen bonds. Comparative analysis of WTH5N1 and H274YH5N1 verified a decrease in the overall number of hydrogen bonds in the mutant species (Figure 8). This phenomenon was attributed to a reduction of hydrophobic interactions within the protein, evident from the van der Waals energy contribution. The residues of neuraminidase interact to a greater extent with the surrounding solvent, hence a depletion in the number of internal protein hydrogen bonds. System I222KH5N1 showed a similar trend to the H274YH5N1 system (Figure S21). The double mutation H274Y-I222KH5N1 showed an increase in the number of hydrogen bonds (Figure S22), offering a more compact structure that is supported by the radius of gyration.
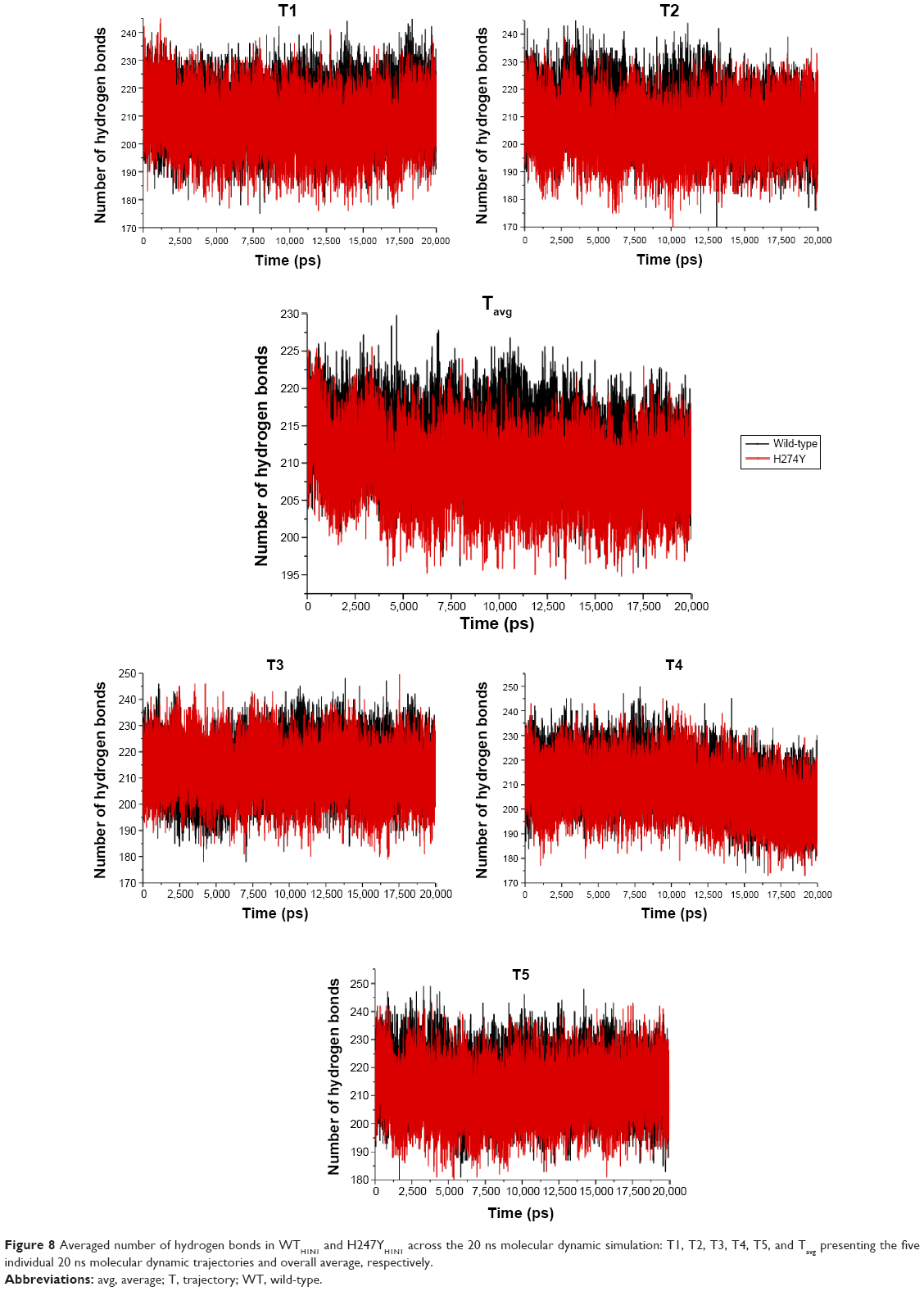 | Figure 8 Averaged number of hydrogen bonds in WTH1N1 and H247YH1N1 across the 20 ns molecular dynamic simulation: T1, T2, T3, T4, T5, and Tavg presenting the five individual 20 ns molecular dynamic trajectories and overall average, respectively. |
Mutation H274YH1N1 (Figure S23) showed a decline in the overall average of calculated hydrogen bonds as compared with the WTH1N1 system. This finding supports the idea of an unfolding of the protein due to depletion of interactions facilitating α-helices or β-strands capable of compacting the enzyme. Similarly, system I222KH1N1 (Figure S24), compared to the parent protein, WTH1N1, has a recorded decrease in the number of internal hydrogen bonds. A comparison of systems H274YH1N1 and H274Y-I222KH1N1 (Figure S25) revealed a near equivalent hydrogen bonding pattern. This could be due to torsional or steric stress of the macromolecule, such that thermodynamically no further unraveling of the protein can be accommodated before an irreversible deformation of the protein takes effect.
Principal component analysis
PCA is used to assess the impact of mutations on the conformational dynamics of neuraminidase to solicit an impartial resistance mechanism.72 Collation of systems WTH5N1 and H274YH5N1 (Figure 9) associated H274YH5N1 with restricted motion in relation to its α-carbon backbone. The displacements were reduced, minimizing the spatial occupancy of H274YH5N1 with a covariance of 0.3191. The magnitude of covariance for WTH5N1 was estimated to be −3.6367, advocating a more flexible framework with an inversely proportionate motion (Figure S26). System H274YH5N1 has a disproportionate motion of correlation (R2) value of 0.1814 in that the vectors commission dynamism in a single cooperative direction (Figure S27). The R2 value for WTH1N1 of −0.6526 indicated a more proportionate but antagonistic motion.
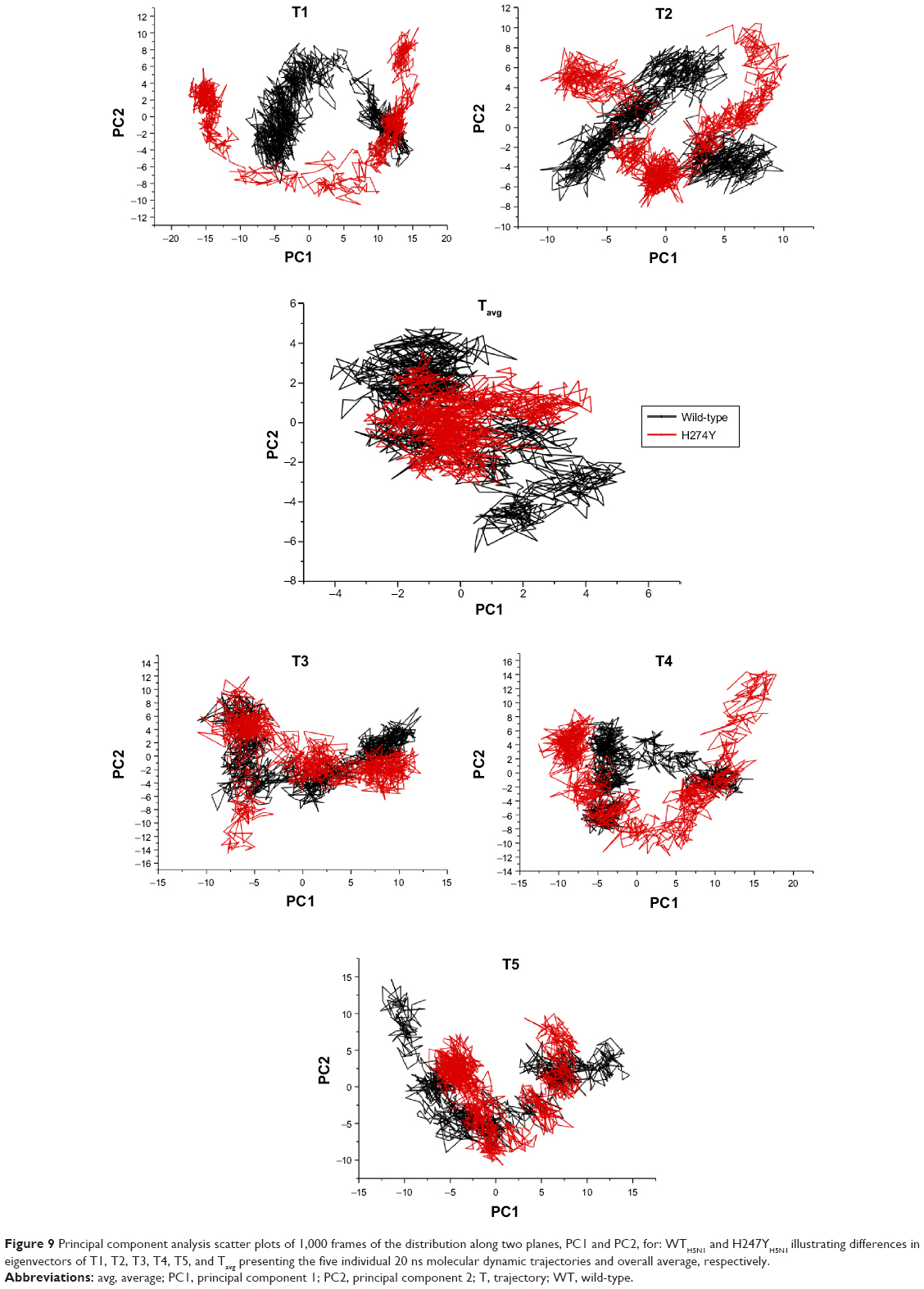 | Figure 9 Principal component analysis scatter plots of 1,000 frames of the distribution along two planes, PC1 and PC2, for: WTH5N1 and H247YH5N1 illustrating differences in eigenvectors of T1, T2, T3, T4, T5, and Tavg presenting the five individual 20 ns molecular dynamic trajectories and overall average, respectively. |
Examination of system I222KH5N1 (Figure S28) suggested that the neuraminidase enzyme tolerated an abridged flexibility of its α-carbon backbone in comparison with the WT enzyme. This corresponded with the determinations surmised from the RMSF values. The single mutation has a covariance of −3.1127, which is marginally smaller in magnitude than that for the WT, with an R2 of −0.3032. The direction of motion of I222KH5N1 is antiparallel, the associations predominate by an antonymous relationship of large and small variables (Figure S29). The double mutation, H274Y-I222KH5N1, has improved flexibility in the α-carbon backbone in comparison with H274YH5N1. This is demonstrated in the PCA scatter and covariance value estimated at 0.8148 (Figure S30). The improvement in flexibility coincided with the RMSD values, suggesting a greater number of conformational possibilities of the H274Y-I222KH5N1 system. An improved R2 of 0.2337, in contrast with H274YH5N1, described a unified direction of motion of disproportionate magnitude (Figure S31).
The WTH1N1 enzyme (Figure 10) appeared to have reduced internal motion of its α-carbon backbone in comparison with H274YH1N1, a restriction which corresponded to the compactness observed from calculation of the radius of gyration. The covariance and R2 values were estimated to be −1.2249 and −0.4616, respectively, suggesting a disproportionate antithetic motion of the enzyme (Figure S32). A similar effect was observed with the double mutation system H274Y-I222KH1N1, where the overall flexibility of the protein was rigid in comparison with H274YH1N1 (Figure S33). The covariance and R2 values of −0.4099 and −0.1539 support these findings (Figure S34). System H274YH1N1 demonstrated a more flexible α-carbon backbone which moved in a disorganized motion with a covariance of −0.7200 and an R2 value of −0.05218 (Figure S35). System I222KH1N1 exhibited a continuous linear motion along its α-carbon backbone, with a covariance of 0.6457 (Figure S36). This phenomenon was supported by an R2 of 0.3271 (Figure S37).
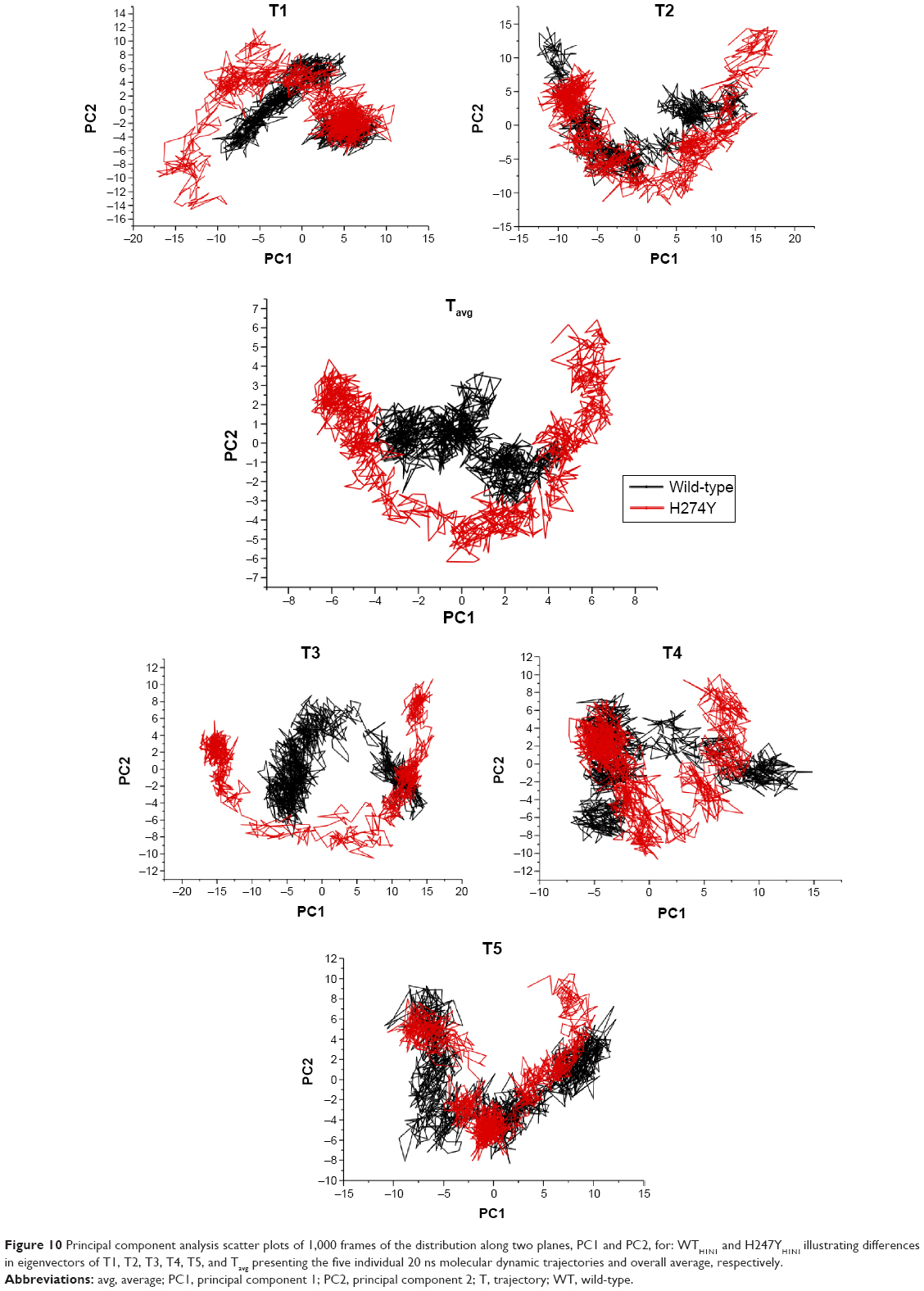 | Figure 10 Principal component analysis scatter plots of 1,000 frames of the distribution along two planes, PC1 and PC2, for: WTH1N1 and H247YH1N1 illustrating differences in eigenvectors of T1, T2, T3, T4, T5, and Tavg presenting the five individual 20 ns molecular dynamic trajectories and overall average, respectively. |
Solvent accessible surface area
The SASA imparts information relative to the compactness of the structure as well as the extent of hydrophobicity in the interior of the folded protein.73 System H274YH5N1 appeared to expand over time (Figure S38), whereas WTH5N1 remained unchanged. Both systems reflected a distinct and relatively stable complexed conformation, which is affirmed by the radius of gyration estimations. System I222KH5N1 (Figure S39) and H274Y-I222KH5N1 (Figure S40) demonstrated a nominally lower surface area than the WT and H274YH5N1, respectively. This implied that contact between the van der Waals partitions and the solvent was diminished. A dehydron species may be present in systems I222KH5N1 and H274Y-I222KH5N1 due to the contributions of ΔEvdW and ΔGsol, resulting in a reduced interaction between the solvent and van der Waals region.74 The preservation of the hydrophobic regions results in a loss in volume of the active site.
The WTH1N1 system has a reduced exposed surface area when compared with H274YH1N1 (Figure S41). This observation corresponded to the PCA of H274YH1N1, facilitating a structure with greater potential for interaction with solvent. System I222KH1N1 exhibited a slightly different trend such that initially both the WT and single mutant shared similar SASA. Over time, at ~10,000 ps onward, a change in conformation occurred, with I222KH1N1 appearing to become more accessible to the solvent (Figure S42). A similar effect was found between H274YH1N1 and H274Y-I222KH1N1 (Figure S43). During the initial 10,000 ps of the simulation, there appeared to be a difference in the SASA of the two systems, with H274YH1N1 having greater solvent accessibility than the double mutant. In the later 10,000 ps, both systems begin to overlap. This anomaly suggests a conformational change in the presence of the double mutation H274Y and I222K, whereby these residues pointed outward, encouraging solvent interaction and disrupting the three-dimensional compactness.
Conclusion
Significant findings emerged in our endeavor to gain insight into the binding mode and origin of resistance of oseltamivir in H1N1 and H5N1. It was noted that the mechanism of resistance is entirely dependent on the type of mutation, relative positioning of such occurrences, and the repercussions thereof on the active site of the enzyme under analysis. Our results indicate that resistance of neuraminidase was inherited by the expression of H274Y and/or I222K mutations. The neuraminidase of H1N1 has known resistance against oseltamivir in the presence of mutations H274Y and/or I222K. Despite originating from the same host by reassortment, H5N1 has proven to be more susceptible to oseltamivir than H1N1 in the presence of mutations. Comparatively, neuraminidase of H5N1 appears to experience greater structural fluctuations in acquiring resistance against oseltamivir as opposed to the H1N1 system. H5N1 gains resistance by a loss in volume of the active site due to compaction of the neuraminidase, while H1N1 develops resistance by solvent exposure of active site residues. Subsidiary analyses of RMSD, RMSF and potential energy confirm the progressive development of resistance from WT through to mutants (for both H1N1 and H5N1).
The free binding energy established from the MM/GBSA algorithm offers the first sign of evidence of resistance. Based on the electrostatic funnel mechanism of neuraminidase (for H1N1), an increase in binding energy was observed. This was an artefact of the active site residues and drug interacting with solvent, rather than with one another. The presence of the H274Y mutation has a compensatory effect to ensure the survival of the viral species by inhibiting enzyme interaction with the drug, without compromising the integrity of the enzyme. Mutation I222K, through distance, transposes a putative constriction in the volume of the active site of H5N1, whereas in H1N1 the protein unfolds, promoting solvent interaction. As a result, the active site inherently cannot support the bulky 1-ethylpropoxy hydrophobic moiety of oseltamivir. It is reasonable to suppose that the I222K mutation has a potent effect in discriminating between binding of the substrate and binding of the drug oseltamivir. The H274Y-I222K double mutant exhibits even greater drug resistance. Overall resistance in systems H5N1 and H1N1 occurs as a result of loss of hydrophobicity within the binding pocket of the active site. The lack of hydrophobicity leads to structural collapse of the available active site, which in turn diminishes the integrity of the binding landscape. This poses a threat, given that H5N1/avian influenza has been found to have an increasing incidence of human transmissibility. The existence of a resistant strain is having a severe negative impact on human health. Therefore, it is imperative to design and identify unique, innovative, and selective chemotherapeutic agents to adapt to the newly defined binding pocket.
Acknowledgments
The authors acknowledge the School of Health Science, University of KwaZulu-Natal, Westville Campus, and the National Research Foundation for their financial assistance. We wish to further acknowledge the Center for High Performance Computing, Cape Town, Republic of South Africa, for high-performance computing resources. We acknowledge Sarentha Chetty for her computational and biological expertise throughout the project.
Disclosure
The authors report no conflicts of interest in this work.
References
Le L, Lee EH, Hardy DJ, Truong TN, Schulten K. Molecular dynamics simulations suggest that electrostatic funnel directs binding of Tamiflu to influenza N1 neuraminidases. PLoS Comput Biol. 2010;6(9):e1000939. | ||
World Health Organization. Pandemic preparedness. Available from: http://web.archive.org/web/20030202145905/http://www.who.int/csr/disease/influenza/pandemic/en. Accessed April 21, 2015. | ||
Cohen E. When a pandemic isn’t a pandemic. Available from: http://edition.cnn.com/2009/HEALTH/05/04/swine.flu.pandemic/index.html. Accessed April 22, 2015. | ||
Doshi P. The elusive definition of pandemic influenza. Bull World Health Organ. 2011;89(7):532–538. | ||
Patel RB, Mathur MB, Gould M, et al. Demographic and clinical predictors of mortality from highly pathogenic avian influenza A (H5N1) virus infection: CART analysis of international cases. PLoS One. 2014;9(3):e91630. | ||
Smith GJ, Vijaykrishna D, Bahl J, et al. Origins and evolutionary genomics of the 2009 swine – origin H1N1 influenza A epidemic. Nature. 2009; 459(7250):1122–1125. | ||
Yarris L. New biological route for swine flu to human infections. Berkeley Lab, 2009. Available from: https://newscenter.lbl.gov/2009/12/09/swine-flu-to-human/. Accessed June 12, 2015. | ||
Mostaço-Guidolin LC, Bowman CS, Greer AL, Fisman DN, Moghadas SM. Transmissibility of the 2009 H1N1 pandemic in remote and isolated Canadian communities: a modelling study. BMJ Open. 2012;2(5):e001614. | ||
Sifferlin A. H1N1’s death toll: 15 times higher than previously thought. Available from: http://healthland.time.com/2012/06/26/h1n1s-death-toll-15-times-higher-than-previously-thought/. Accessed April 23, 2015. | ||
Davis CP. Swine flu (swine influenza A [H1N1 and H3N2v] virus. Available from: http://www.medicinenet.com/swine_flu/article.htm. Accessed April 23, 2015. | ||
Baz M, Abed Y, Papenburg J, Bouhy X, Hamelin ME, Boivin G. Emergence of oseltamivir-resistant pandemic H1N1 virus during prophylaxis. N Engl J Med. 2009;361(23):2296–2297. | ||
Centers for Disease Control and Prevention. Influenza A (H3N2) variant virus. Available from: http://www.cdc.gov/flu/swineflu/h3n2v-cases.htm. Accessed April 25, 2015. | ||
Adabala PJ, LeGresley EB, Bance N, Niikura M, Pinto BM. Exploitation of the catalytic site and 150 cavity for design of influenza A neuraminidase inhibitors. J Org Chem. 2013;78(21):10867–10877. | ||
Sylte M, Suarez D. Influenza neuraminidase as a vaccine antigen. In: Compans RW, Orenstein WA, editors. Vaccines for Pandemic Influenza. Volume 333. Berlin, Germany: Springer; 2009. | ||
Russell RJ, Haire LF, Stevens DJ, et al. The structure of H5N1 avian influenza neuraminidase suggests new opportunities for drug design. Nature. 2006;443(7107):45–49. | ||
Li Q, Qi J, Zhang W, et al. The 2009 pandemic H1N1 neuraminidase N1 lacks the 150-cavity in its active site. Nat Struct Mol Biol. 2010;17(10):1266–1268. | ||
Woods CJ, Malaisree M, Long B, McIntosh-Smith S, Mulholland AJ. Analysis and assay of oseltamivir-resistant mutants of influenza neuraminidase via direct observation of drug unbinding and rebinding in simulation. Biochemistry. 2013;52(45):8150–8164. | ||
Han N, Liu X, Mu Y. Exploring the mechanism of zanamivir resistance in a neuraminidase mutant: a molecular dynamics study. PLoS One. 2012;7(9):e44057. | ||
Shobugawa Y, Saito R, Sato I, et al. Clinical effectiveness of neuraminidase inhibitors – oseltamivir, zanamivir, laninamivir, and peramivir – for treatment of influenza A(H3N2) and A(H1N1)pdm09 infection: an observational study in the 2010–2011 influenza season in Japan. J Infect Chemother. 2012;18(6):858–864. | ||
Takashita E, Ejima M, Itoh R, et al. A community cluster of influenza A(H1N1)pdm09 virus exhibiting cross-resistance to oseltamivir and peramivir in Japan, November to December 2013. Euro Surveill. 2014; 19(1):20666. | ||
Kim CU, Lew W, Williams MA, et al. Influenza neuraminidase inhibitors possessing a novel hydrophobic interaction in the enzyme active site: design, synthesis, and structural analysis of carbocyclic sialic acid analogues with potent anti-influenza activity. J Am Chem Soc. 1997; 119(4):681–690. | ||
[No authors listed]. Global monitoring of antiviral resistance in currently circulating human influenza viruses, November 2011. Wkly Epidemiol Rec. 2011;86(45):497–501. | ||
Meijer A, Rebelo-de-Andrade H, Correia V, et al. Global update on the susceptibility of human influenza viruses to neuraminidase inhibitors, 2012–2013. Antiviral Res. 2014;110:31–41. | ||
Karthick V, Shanthi V, Rajasekaran R, Ramanathan K. Exploring the cause of oseltamivir resistance against mutant H274Y neuraminidase by molecular simulation approach. Appl Biochem Biotechnol. 2012;167(2):237–249. | ||
Karplus M, McCammon JA. Molecular dynamics simulationa of biomolecules. Nature. 2002;9(9):646. | ||
Li L, Li Y, Zhang L, Hou T. Theoretical studies on the susceptibility of oseltamivir against variants of 2009 A/H1N1 influenza neuraminidase. J Chem Inf Model. 2012;52(10):2715–2729. | ||
Karthick V, Ramanathan K. Insight into the oseltamivir resistance R292K mutation in H5N1 influenza virus: a molecular docking and molecular dynamics approach. Cell Biochem Biophys. 2014;68(2):291–299. | ||
Operario DJ, Moser MJ, St George K. Highly sensitive and quantitative detection of the H274Y oseltamivir resistance mutation in seasonal A/H1N1 influenza virus. J Clin Microbiol. 2010;48(10):3517–3524. | ||
Malaisree M, Rungrotmongkol T, Nunthaboot N, et al. Source of oseltamivir resistance in avian influenza H5N1 virus with the H274Y mutation. Amino Acids. 2009;37(4):725–732. | ||
Rungrotmongkol T, Intharathep P, Malaisree M, et al. Susceptibility of antiviral drugs against 2009 influenza A (H1N1) virus. Biochem Biophys Res Commun. 2009;385(3):390–394. | ||
Wang NX, Zheng JJ. Computational studies of H5N1 influenza virus resistance to oseltamivir. Protein Sci. 2009;18(4):707–715. | ||
Yang Z, Yang G, Zhou L. Mutation effects of neuraminidases and their docking with ligands: a molecular dynamics and free energy calculation study. J Comput Aided Mol Des. 2013;27(11):935–950. | ||
Pan D, Sun H, Bai C, et al. Prediction of zanamivir efficiency over the possible 2009 influenza A (H1N1) mutants by multiple molecular dynamics simulations and free energy calculations. J Mol Model. 2011; 17(10):2465–2473. | ||
Takano R, Kiso M, Igarashi M, et al. Molecular mechanisms underlying oseltamivir resistance mediated by an I117V substitution in the neuraminidase of subtype H5N1 avian influenza A viruses. J Infect Dis. 2013;207(1):89–97. | ||
Bloom JD, Nayak JS, Baltimore D. A computational-experimental approach identifies mutations that enhance surface expression of an oseltamivir-resistant influenza neuraminidase. PLoS One. 2011;6(7):e22201. | ||
McKimm-Breschkin JL, Rootes C, Mohr PG, Barrett S, Streltsov VA. In vitro passaging of a pandemic H1N1/09 virus selects for viruses with neuraminidase mutations conferring high-level resistance to oseltamivir and peramivir, but not to zanamivir. J Antimicrob Chemother. 2012; 67(8):1874–1883. | ||
Nguyen HT, Fry AM, Gubareva LV. Neuraminidase inhibitor resistance in influenza viruses and laboratory testing methods. Antivir Ther. 2012; 17(1 Pt B):159–173. | ||
Huang L, Cao Y, Zhou J, et al. A conformational restriction in the influenza A virus neuraminidase binding site by R152 results in a combinational effect of I222T and H274Y on oseltamivir resistance. Antimicrob Agents Chemother. 2014;58(3):1639–1645. | ||
Hayden FG, de Jong MD. Emerging influenza antiviral resistance threats. J Infect Dis. 2011;203(1):6–10. | ||
Wagh K, Bhatia A, Greenbaum BD, Bhanot G. Bird to human transmission biases and vaccine escape mutants in H5N1 infections. PLoS One. 2014;9(7):e100754. | ||
Mirjalili V, Noyes K, Feig M. Physics-based protein structure refinement through multiple molecular dynamics trajectories and structure averaging. Proteins. 2014;82 Suppl 2:196–207. | ||
Altis A, Nguyen PH, Hegger R, Stock G. Dihedral angle principal component analysis of molecular dynamics simulations. J Chem Phys. 2007;126(24):244111. | ||
Shlens J. A Tutorial on Principal Component Analysis. San Diego, CA, USA: University of California; 2005. | ||
van der Vries E, Collins PJ, Vachieri SG, et al. H1N1 2009 pandemic influenza virus: resistance of the I223R neuraminidase mutant explained by kinetic and structural analysis. PLoS Pathog. 2012;8(9):e1002914. | ||
Collins PJ, Haire LF, Lin YP, et al. Crystal structures of oseltamivir- resistant influenza virus neuraminidase mutants. Nature. 2008;453(7199):1258–1261. | ||
UCSF Chimera – an Extensible Molecular Modeling System. Resource for Biocomputing, Visualization, and Informatics, University of California, San Francisco; National Institute of General Medical Sciences; National Institutes of Health. Available from: http://www.cgl.ucsf.edu/chimera/. Accessed January 11, 2015. | ||
Kanibolotsky DS, Novosyl’na OV, Abbott CM, Negrutskii BS, El’skaya AV. Multiple molecular dynamics simulation of the isoforms of human translation elongation factor 1A reveals reversible fluctuations between “open” and “closed” conformations and suggests specific for eEF1A1 affinity for Ca2+-calmodulin. BMC Struct Biol. 2008;8:4. | ||
Götz AW, Williamson MJ, Xu D, Poole D, Le Grand S, Walker RC. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 1. Generalized Born. J Chem Theory Comput. 2012;8(5):1542–1555. | ||
Case DA, Berryman JT, Betz RM, et al. AMBER 12, University of California, 2012. Available from: http://ambermd.org/doc12/Amber12.pdf. Accessed June 11, 2015. | ||
Salomon-Ferrer R, Götz AW, Poole D, Le Grand S, Walker RC. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 2. Explicit solvent particle mesh Ewald. J Chem Theory Comput. 2013; 9(9):3878–3888. | ||
Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79(2):926. | ||
Essmann U, Perera L, Berkowitz M, Darden T, Lee H, Pedersen L. A smooth particle mesh Ewald method. J Chem Phys. 1995;103:8577. | ||
Berendsen HJC, Postma JPM, Van Gunsteren WF, DiNola A, Haak JR. Molecular dynamics with coupling to an external bath. J Chem Phys. 1984; 81:3684–3690. | ||
Ryckaert JPC, Berendsen HJC. Numerical integration of the Cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Phys. 1977;23:327–341. | ||
Le Grand S, Gotz AW, Walker RC. SPFP: speed without compromise – a mixed precision model for GPU accelerated molecular dynamics simulations. Comput Phys Commun. 2013;184:374–380. | ||
Sun H, Li Y, Tian S, Xu L, Hou T. Assessing the performance of MM/PBSA and MM/GBSA methods. 4. Accuracies of MM/PBSA and MM/GBSA methodologies evaluated by various simulation protocols using PDBbind data set. Phys Chem Chem Phys. 2014;16(31): 16719–16729. | ||
Greenidge PA, Kramer C, Mozziconacci JC, Wolf RM. MM/GBSA binding energy prediction on the PDBbind data set: successes, failures, and directions for further improvement. J Chem Inf Model. 2013;53(1):201–209. | ||
Godschalk F, Genheden S, Söderhjelm P, Ryde U. Comparison of MM/GBSA calculations based on explicit and implicit solvent simulations. Phys Chem Chem Phys. 2013;15(20):7731–7739. | ||
Mitomo D, Fukunishi Y, Higo J, Nakamura H. Calculation of protein-ligand binding free energy using smooth reaction path generation (SRPG) method: a comparison of the explicit water model, GB/SA model and docking score function. Genome Inform. 2009;23(1):85–97. | ||
Hou T, Wang J, Li Y, Wang W. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J Chem Inf Model. 2011;51(1):69–82. | ||
Cocco S, Monasson R, Weigt M. From principal component to direct coupling analysis of coevolution in proteins: low-eigenvalue modes are needed for structure prediction. PLoS Comput Biol. 2013;9(8):e1003176. | ||
Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14(1):33–38. | ||
Bakan A, Meireles LM, Bahar I. ProDy: protein dynamics inferred from theory and experiments. Bioinformatics. 2011;27(11):1575–1577. | ||
Hess B. Convergence of sampling in protein simulations. Phys Rev E Stat Nonlin Soft Matter Phys. 2002;65(3 Pt 1):031910. | ||
Wassenaar T. A molecular dynamics hands-on session II. Available from: http://www.nmr.chem.uu.nl/~tsjerk/course/md-tutorial/analysis.html. Accessed October 31, 2014. | ||
[No authors listed]. From sequence to structure. New Science Press Ltd; 2004. Available from: http://www3.uah.es/farmamol/New_Science_Press/nsp-protein-1.pdf. Accessed June 11, 2015. | ||
Lobanov MI, Bogatyreva NS, Galzitskaia OV. [Radius of gyration as an indicator of protein structure compactness]. Mol Biol (Mosk). 2008; 42(4):623–628. Russian. | ||
Caves LS, Evanseck JD, Karplus M. Locally accessible conformations of proteins: multiple molecular dynamics simulations of crambin. Protein Sci. 1998;7(3):649–666. | ||
Caravella JA, Carbeck JD, Duffy DC, Whitesides GM, Tidor B. Long-range electrostatic contributions to protein-ligand binding estimated using protein charge ladders, affinity capillary electrophoresis, and continuum electrostatic theory. J Am Chem Soc. 1999;121(18):4340–4347. | ||
Cottrell D, Tupper PF. Energy drift in molecular dynamics simulations. BIT Numerical Mathematics. 2007;47(3):507–523. | ||
Rungrotmongkol T, Intharathep P, Malaisree M, et al. Susceptibility of antiviral drugs against 2009 influenza A (H1N1) virus. Biochem Biophys Res Commun. 2009;385(3):390–394. | ||
Jolliffe IT. Principal Component Analysis. New York, NY, USA: Springer; 2002. | ||
Richmond TJ. Solvent accessible surface area and excluded volume in proteins: analytical equations for overlapping spheres and implications for the hydrophobic effect. J Mol Biol. 1984;178(1):63–89. | ||
Chaplin M. Protein folding and denaturation. Available from: http://www1.lsbu.ac.uk/water/protein_denatured.html. Accessed October 31, 2014. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.