")
Back to Journals » Drug Design, Development and Therapy » Volume 9
The relevance of piroxicam for the prevention and treatment of nonmelanoma skin cancer and its precursors
Authors Campione E , Paternò EJ, Candi E, Falconi M , Costanza G , Diluvio L , Terrinoni A, Bianchi L, Orlandi A
Received 26 April 2015
Accepted for publication 2 July 2015
Published 29 October 2015 Volume 2015:9 Pages 5843—5850
DOI https://doi.org/10.2147/DDDT.S84849
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Shu-Feng Zhou
Elena Campione,1 Evelin Jasmine Paternò,2 Eleonora Candi,3,4 Mattia Falconi,5 Gaetana Costanza,2 Laura Diluvio,1 Alessandro Terrinoni,4 Luca Bianchi,1 Augusto Orlandi2,6,7
1Department of Dermatology, 2Department of Biomedicine and Prevention, 3Department of Experimental Medicine and Surgery, University of Rome “Tor Vergata”, 4Biochemistry Laboratory IDI-IRCCS, Faculty of Medicine, University of Rome “Tor Vergata”, 5Department of Biology, University of Rome “Tor Vergata”, 6Institute of Anatomic Pathology, University of Rome “Tor Vergata”, 7Tor Vergata University-Policlinic of Rome, Rome, Italy
Abstract: Piroxicam (PXM), a nonsteroidal anti-inflammatory drug, is an enolic benzothiazine and a potent member of the oxicam series. The drug suppresses the synthesis of proinflammatory enzymes, such as cyclooxygenases-1 and -2 (COX-1 and 2), downregulates the production of prostaglandins (PGs) and tromboxanes, and inhibits polyamines production by blocking ornithine decarboxylase induction involved in nonmelanoma skin carcinogenesis. In addition, PXM is able to induce tumor cell apoptosis and suppresses metalloproteinase 2 activities. Skin carcinogenesis is a multistep process in which the accumulation of genetic events leads to a gradually dysplastic cellular expression, deregulation of cell growth, and carcinomatous progression. COX-1 upregulation plays a significant role in PG and vascular epidermal growth factor production supporting tumor growth. Increased level of PGs in premalignant and/or malignant cutaneous tumors is also favored by upregulation of COX-2 and downregulation of the tumor suppressor gene 15-hydroxy-prostaglandin dehydrogenase. Chemoprevention can be a hopeful approach to inhibit carcinoma occurrence before an invasive tumor develops. The chemopreventive effect of nonsteroidal anti-inflammatory drugs on nonmelanoma skin cancers has been established. In this study, we highlighted the different modalities of action of PXM on the pathogenesis of nonmelanoma skin cancer, analyzing and evaluating binding modes and energies between COX-1 or COX-2 and PXM by protein–ligand molecular docking. Our clinical experience about the local use of PXM on actinic keratoses and field cancerization is also reported, confirming its efficacy as target therapy.
Keywords: COXs inhibitor, actinic keratosis, tumor progression, binding mode
Introduction
Piroxicam (PXM), an enolic benzothiazine and potent member of the oxicam series, suppresses the synthesis of proinflammatory enzymes, reducing lipid mediators such as prostaglandins (PGs) and thromboxane (TXs), and also inhibits ornithine decarboxylase (ODC) induction,1 both pathways participating in carcinogenesis. Further effects of PXM are the inhibition of metalloproteinase-2 activity and induction of apoptosis, determined to have an antiaging effect on skin texture.2 PXM belongs to a class of nonsteroidal anti-inflammatory drugs (NSAIDs) indicated for the symptomatic treatment of inflammatory and degenerative rheumatic diseases (rheumatoid arthritis, osteoarthritis, and ankylosing spondylitis). On the basis of the documented effect of PXM in skin tumorigenesis, through the block of cyclooxygenases-1 and -2 (COX-1 and -2) activity,3 we reported preliminary studies of its usefulness in the treatment of actinic keratoses (AKs).4 COX inhibitors, diclofenac and celecoxib, have also been used for the treatment of AKs and squamous cell carcinomas (SCCs).5 The effectiveness of NSAIDs is attributable to the inhibitory effect on the increased COX-1 and -2 enzyme activity in skin tumors produced after chronic exposure to UVB (ultraviolet B) (accompanying skin inflammation). Aberrant overexpression of COX-2 in both AK and SCC upon UVB irradiation and UVB-induced p53 tumor suppressor gene mutations are early events responsible for the progression of sun-exposed nonmelanoma skin cancers (NMSCs).5 Topical NSAID treatment works as a nonspecific COX inhibitor and is an effective and well-tolerated treatment for AK. COX inhibitors have been examined with respect to their role in cancer prevention and cancer treatment in both animal models and humans.6 In humans, topical application of NSAIDs is also potent in inhibiting the UV light-induced acute sunburn reactions such as erythema and peeling of superficial layers, if applied immediately after UVB exposure.7,8 An exhaustive meta-analysis on PXM made by Richy et al9 on controlled clinical trials pointed out a more favorable efficacy and safety profile of PXM compared to other topical NSAIDs (diclofenac, naproxen, tenoxicam, indomethacin, etodolac, meloxicam, ibuprofen, salicylates, nabumetone, aceclofenac, droxicam, flurbiprofen, ketoprofen, nimesulide, and diflunisal).
Here, we described the different modalities of action of PXM on the pathogenesis of NMSC. The inhibitory effects of PXM on COX-1 and -2 activity have been investigated by analyzing its binding mode. We also reported our clinical experience about the effectiveness of local use of PXM as chemopreventive agent on AKs and field cancerization.
Steps in skin tumorigenesis and role of COX enzymes in cancer
Skin cancer is the most common cancer in humans. The skin of the head and neck accounts for less than 10% of the body’s surface area, but this region accounts for 70%–80% of skin cancer cases. Although mortality from NMSC is low, its high incidence leads to a significant public health burden, making them a suitable target for chemoprevention and long-lasting research. NMSC includes SCC and basal cell carcinoma (BCC).10 Carcinogenesis occurs in two steps: initiation and promotion.11 The promotion phase, an important target for the design of potential chemoprevention studies, is temporally prolonged and potentially reversible.11 A variety of model systems have demonstrated an increase in tissue polyamine levels, including putrescine, during tumor promotion.12 Mammalian polyamine biosynthesis is at least partially controlled via induction of ODC, which makes the inhibition of this enzyme a potential target for chemoprevention.13 α-Difluoromethylornithine is an enzyme-activated irreversible inhibitor of ODC that has been shown to prevent tumors in experimental animal systems.14 In addition, accumulation of genetic events within cells leads to a gradually dysplastic cellular manifestation, deregulated cell growth, and finally, carcinoma. An upregulation of COX-2 plays a significant role in PG and vascular epidermal growth factor (VEGF) production for the tumor proliferation.15 SCC of the head and neck showed little response to COX-2 inhibition. Therefore, a cotreatment of COX-1 and -2 determined a meaningful inhibition of VEGF, as observed by Park et al.15 Increased levels of prostaglandin E2 and F2 (PGE2 and PGF2) in premalignant and/or malignant cutaneous tumors are favored by upregulation of COX-2 and downregulation of tumor suppressor gene 15-hydroxy-prostaglandin dehydrogenase (15-PGDH).16 Chemoprevention can be a hopeful approach to inhibit carcinoma occurrence before an invasive tumor develops. The chemopreventive effect of NSAIDs on NMSC has been established in animals17–20 and in vitro studies.21 Experimental studies have proposed the topical or oral use of NSAIDs in human subjects for the regression of cutaneous tumors.22 Findings from observational cases studies or clinical trials documented a defensive effect of NSAIDs against colorectal cancer.23–25 The hypothesis that NSAIDs might prevent the occurrence of colorectal cancer arose from studies showing that PGE2 levels were higher in colorectal cancer than in surrounding normal tissue. On the other hand, few epidemiological studies have investigated the efficacy of NSAIDs in the prevention of skin cancer.26–28
PXM effects for the prevention of NMSC
It is well known that UV irradiation is the major cause of induction and development of AK and NMSC. Normally, sun-damaged cells undergo, through p53 activation, apoptosis for self-destruction.5 Skin cells can acquire tumorigenic mutations by UV irradiation or chemical stimulation and loss of the normal ability of cell differentiation and apoptosis. The stepwise evolution of AK with an increased expression of antiapoptotic Bcl-2 favors the progression to SCC.29 As a matter of fact, retinoid-induced modulation of apoptosis in NMSC is an efficacious therapeutic approach.30–32 In addition, the dermal response with inflammation, characterized by erythema and edema, mediated by PGs is a critical component of tumorigenesis, promoting tumor growth, tissue invasion, angiogenesis, and metastasis.29 The action of PXM plays a role in early stages of carcinogenesis by blocking the phases of inflammation and thus angiogenesis; the evidence of its effectiveness for the treatment of AKs has been described in our previous study.4
Also, prolonged use of PXM acts on photodamaged areas which is expression of field cancerization.4 To date, a higher occupational sun exposure of men compared with women justified the different incidence of NMSC.33 A study of Johannesdottir et al34 provides an alternative elucidation, which attributes the reduced incidence to the chronic use of analgesics for menstrual cycle–related symptoms. Several epidemiological studies confirm that aspirin can act as a chemopreventive agent and can decrease the incidence of various cancers, including skin tumors. The major inhibitor effect of aspirin on NMSC and precursors is related to its targeting elevated COX-2 levels in SCC. Most studies also examined the use of PXM in different disorders, especially for analyzing absorbance, tolerance, and efficacy in animal models,35–37 but the efficacy of PXM in humans was analyzed considering the general effects of NSAIDs, as in colon rectal carcinoma38,39 or in NMSC.40–42
Rathore et al43 recently reported that cotreatment of PXM with masitinib significantly inhibited cell proliferation of oral squamous cell carcinoma (OSCC) as compared to either drug alone, through the c-kit and AKT signaling pathways. PXM inhibited masitinib-induced COX-2 expression in all tested OSCCs. Therefore, targeting these two signaling pathways simultaneously was more efficient for inhibition of OSCCs across these species.43 A few studies evidenced the chemopreventive use of PXM for NMSC,4,44 but its interaction with COX-2 is well known;43 however, more studies provided evidences for efficacy of PXM in melanoma treatment.45–47
Cycloxygenase pathway
In vertebrates, two isoforms of active COX enzymes have been identified, which display similar enzymatic properties, but differ in their expression and regulation.48 The best-known mechanism of action of NSAIDs is the inhibition of COX isoforms, in particular, COX-1 and COX-2, both implicated in inflammation and promotion of tumorigenesis.16 Chronic inflammation correlates with the increased risk of developing cancer in the affected organ and can be caused by acute UV radiation and other factors.49,50 The metabolism of arachidonic acid, the predominant precursor of PGs and the major control point for their synthesis, can be triggered by UV effect on the skin cell membranes, thus starting the inflammatory process.51 Arachinoid acid metabolism and PG synthesis through the COX-related pathway promotes cancer progression. COX is the key enzyme in prostanoid biosynthesis, which catalyzes the conversion of arachidonic acid to prostaglandin H2 (PGH2), which is subsequently converted to biologically active lipids such as TX, PGE2, and prostacyclin (PGI2) by different enzymes.52 The skin is a major site of PG synthesis. All skin cells are PGs producers: keratinocytes, melanocytes, Langerhans cells, mast cells, fibroblasts, and endothelial cells.53 While COX-1 is constitutively expressed, COX-2 is induced by tumor promoters, growth factors, and cytokines.54 COX-1 is responsible for maintenance of the gastric and platelet functions, vascular homeostasis, as well as renal blood flow, and it also seems to contribute to PG synthesis during tumor progression in various tissues, including skin.55,56 The relative contribution of the two COX isoforms to skin inflammation is evaluated by combining studies with genetic knockout models and pharmacological intervention with isozyme-selective inhibitors.57,58 The carcinogenic actions of COX-2 should be distinguished from those of COX-1 in the application of PG-blocking therapy for cancer treatment.59 The proliferative effects of COX-2 are due primarily to the increased synthesis of PGs, which directly influence cell growth after binding to specific cell surface receptors, including PG E, F, and I classes of receptors.60,61 The protumorigenic effect of prostaciclins can be mediated by specific receptors as EP2, and the different forms of PGs mediated their biological effects through EP, FP, and IP receptors.62 PGs may also promote the retention of UV-damaged cells through the inhibition of apoptosis.63 A study of Kuzbicki et al64 demonstrated that differently from SCC, neither benign epithelial skin lesions nor BCC are associated with enhanced COX-2 expression. Both COX isoenzymes are strongly expressed in SCC, deriving from a more differentiated epidermal layer.64 A high COX-2 expression could be considered a risk factor of BCC recurrence, determining a role in BCC prognosis.65 In general, NSAIDs are effective chemopreventive agents for skin cancer, and they function by acting on COX pathway specifically during the treatment of AK and SCC.
PXM binding mode
The inhibitory effects of PXM on COX-1 and -2 activity have been investigated by analyzing its binding mode. Protein–ligand molecular docking has been used to evaluate binding modes and energies between COX-1 or COX-2 and the PXM drug (Figures 1 and 2). The docking simulations have been performed by using the AutoDock Vina 1.1.2 program,66 through the AutoDock/Vina PyMOL plugin (http://wwwuser.gwdg.de/~dseelig/adplugin.html; the PyMOL Molecular Graphics System Version 1.5.0.4. Schrödinger, LLC). The PXM SDF file,67 downloaded from the PubChem compound database (https://pubchem.ncbi.nlm.nih.gov), has been converted into mol2 file and filled with hydrogens using the Open Babel program.68 In the absence of the human enzymes, the X-ray structures of Ovis aries COX-1, PDB ID code 1CQE,69 and Mus musculus COX-2, PDB ID code 1PXX,70 deposited in the Protein Data Bank (PDB, http://www.rcsb.org/pdb), have been selected as receptors since their sequence identity with human enzymes is very high (~95%) and the residues comprising the active sites fully conserved. In COX-1 and COX-2, the active sites are composed of the same group of amino acids with the exception of the residue in position 523, which is located at the border of the substrate pocket: that in COX-1 is an isoleucine, while in COX-2 is a valine.71 The cocrystallized ligands flurbiprofen (1CQE) and diclofenac (1PXX) have been removed before starting the simulations that have been performed using the Genetic Algorithm with local gradient optimization.66 The docking boxes (dimensions X=22.5; Y=22.5; Z=22.5 Å) have been centered according to the X-ray ligand positions and include all the residues forming the binding sites. As previously reported,4 the molecular docking analysis confirms that PXM is a nonselective drug that blocks the activity of COX-1 and -2, detecting the same binding energy (−7.6 kcal/mol) in both the enzymes, even if some slight differences in the binding mode are present. In COX-1 (Figures 1A and 2A), the Ser530 residue forms a hydrogen bond with the carboxamide group of the PXM, while a second hydrogen bond involves the Tyr355 hydroxyl group and the benzothiazine moiety of the drug. In COX-2 (Figures 1B and 2B), the same hydrogen bond occurs between Tyr355 and benzothiazine group, while the Ser530–carboxamide interaction is lost and a new hydrogen bond, involving the Arg120 side chain and the benzothiazine group, occurs. The remaining interacting residues show a similar distribution pattern, leading to a very comparable binding mode of the PXM molecule in the two binding sites. These results are in line with previous site-directed mutagenesis experiments that identified Arg120 as the key residue for selective inhibition of COX-2, while identifying Ser530 and Tyr355 as nonspecific binding determinants.72 Moreover, this structural analysis, determining the same binding energy, confirms that PXM is a nonselective COX-1 and COX-2 inhibitor.
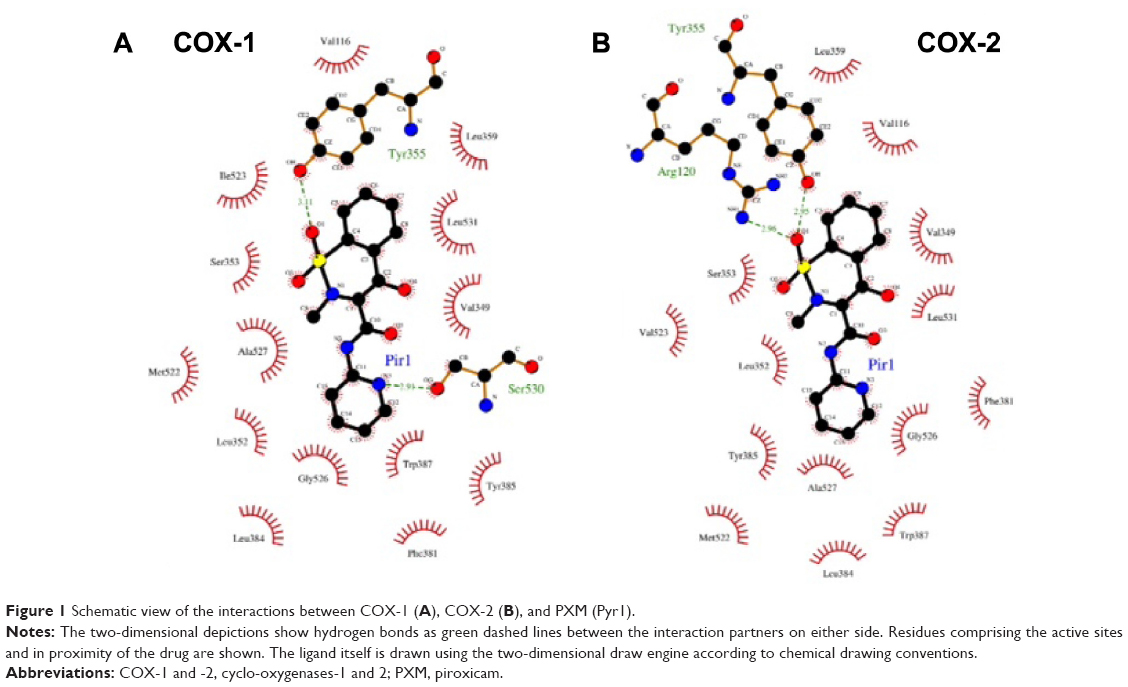 | Figure 1 Schematic view of the interactions between COX-1 (A), COX-2 (B), and PXM (Pyr1). |
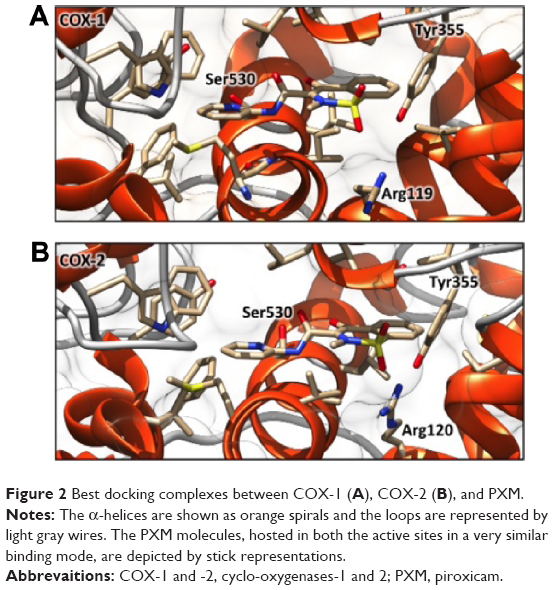 | Figure 2 Best docking complexes between COX-1 (A), COX-2 (B), and PXM. |
Pathogenesis of AKs as therapeutic target of PXM
AK is a chronic, progressive disease, which is expected to develop into SCC at a rate of 10% over 10 years,73 the latter potentially metastasizing in 5% of cases. Although it is not possible to predict which specific AKs will progress to SCC, histologic evidence indicates the majority (60%–70%) of SCCs arise from AKs lesions.74 The entire sun-damaged skin is a result of UV-induced field cancerization with multiple visible and subclinical lesions.75 AKs are typically seen on fair-skinned population on chronic sun-exposed areas such as the face, arms, dorsum of the hands, bald scalp, and upper back.76 UVB radiation, considered the most important factor implicated in AK pathogenesis, cause mutations in the telomerase gene and tumor suppressor gene p53.5 Increased telomerase activity delays programmed cell death, making the mutated cells immortal. The tumor suppressor gene p53 leads to the arrest of cell cycle, allowing the repair of damaged DNA. If mutated, the proliferation of damaged cells will give rise to potentially neoplastic cells.77 Several other factors increase the prevalence of AK, like skin type I and II, advanced age, genetic predisposition, photodamage in outdoor workers, sun exposure in childhood, use of artificial tanning beds, PUVA (psoralens ultraviolet radiation) therapy, administration of X-rays, and immunosuppressive therapies.76 Human papilloma viruses are also implicated in etiopathogenesis.78 Many patients with epidermodysplasia verruciformis are immunosuppressed patients, ie, renal transplant recipients and develop HPV-associated AK and NMSCs. The viral E6 protein of HPV promotes proteolytic degradation of the proapoptotic BAK-protein, promoting skin cancerogenesis.79 Other molecular markers that may indicate an increased likelihood of malignancy include the expression of p16ink4, the CD95 ligand, tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and TRAIL receptors, and loss of heterozygosis.80,81 Skin inflammation is due to chronic UVB irradiation, which promotes the production of eicosanoids and COX-1 and -2 produce PGs from arachidonic acid. Inflammation is associated with the progression of AK to SCC. In fact, PGs promote tumor growth by stimulating cell proliferation, invasion, and angiogenesis.82,83 Increased levels of PGE2 and PGF2α in premalignant and/or malignant epithelial skin cancers are due to the constitutive upregulation of enzymes involved in PG biosynthesis, such as COX-2, and downregulation of tumor suppressor gene 15-PGDH.16 A schematic representation of PXM effect on AK after UVB radiation is shown in Figure 3: after chronic UVB exposure (290–320 nm), several genes underwent mutations, including p53, with alterations in the arrest of cell cycle. All the mutations and other factors determine the progression in AK. The local use of PXM blocked the action of COX, resulting in blocking the biosynthesis of PGs and in 15-PGDH increased expression. Finally, a reduction of proliferation, tumor progression, and angiogenesis occurred, whereas there was an increase in apoptosis. After topical treatment of AK with PXM, normal epidermal architecture was restored (Figure 3). In addition, COX-1 seems to contribute to tumor progression in various tissues including skin, and its inhibition lowers cell proliferation in some cancers, including ovarian and colon cancer.84,85 As recently reported, COX-1 and -2 are involved in VEGF production in SCC of head and neck.86 The coinhibition of COX-1 and -2 could block VEGF synthesis. Treg-inducing VEGF reduction counteracted pathological angiogenesis.87,88 The efficacy of PXM is related to its activity on both COX enzymes.89 In a preliminary open-label trial, we evaluated the efficacy and tolerability of PXM 1% gel in the treatment of patients affected by AKs. Ten patients affected by multiple AKs were treated twice daily for a period of 3 months.4
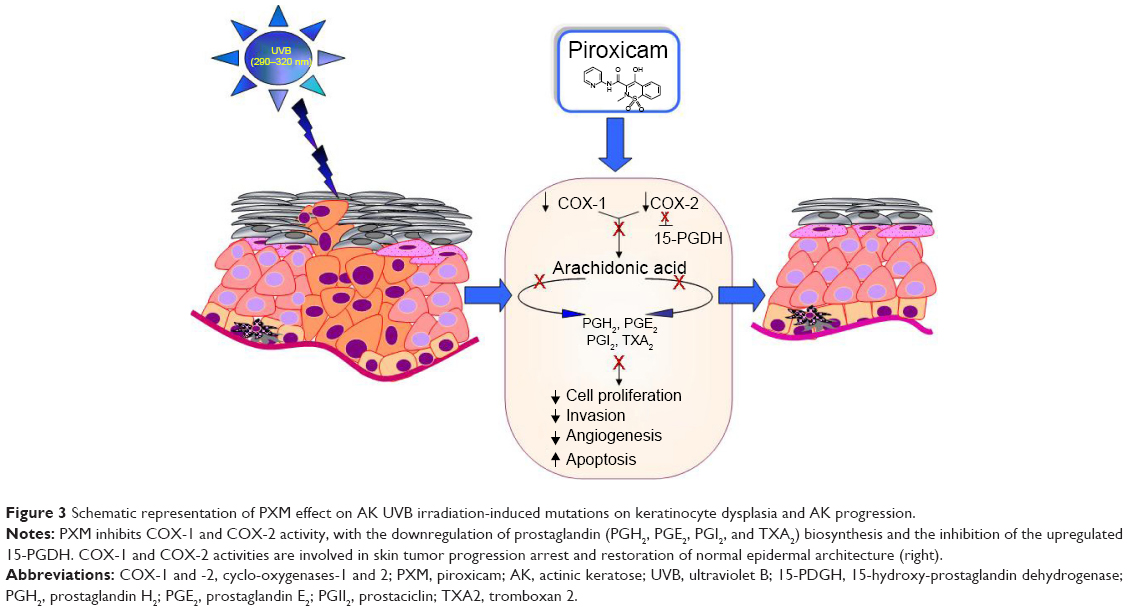 | Figure 3 Schematic representation of PXM effect on AK UVB irradiation-induced mutations on keratinocyte dysplasia and AK progression. |
To measure the efficacy of the therapy, we evaluated the lesions clinically and by means ± standard error of mean of dermoscopy analysis at an initial baseline visit, at intermediate visits, and after 3 months.4 The modifications during treatment were evaluated using a new scoring system, named AKESA, based on the clinical presence of erythema, scale, and atrophy on a target lesion.4
We observed improvement either in the typical features of the AKs, as confirmed by the AKESA score, or in the perilesional photodamaged skin area in terms of skin softening and a significant reduction in dermal vessels, also documented by dermoscopic investigation.4 On the basis of our clinical experience, we observed a healing response in more than 50% in AKs with the use of PXM, a reduction in the appearance of new lesions in patients with multiple AKs. In the preliminary trial, we showed that PXM exerts a significant antitumorigenic effect, and new studies with increased number of patients validated our results.4 AK is considered as a precursor to SCC, and its eradication is mandatory in affected patients. The drug with a nonspecific COX-1 and COX-2 inhibitor action works on the early stages of skin carcinogenesis, and for this reason it is eligible for the treatment of AKs and the field cancerization. The lack of side effects and its efficacy allow the use of PXM for a longer time even up to 1 year.
Conclusion
PXM appears to be safe, effective, and well tolerated, although its use in AKs is still off-label. Further studies need to validate the use of PXM on NMSC, exploring its activity on different tumorigenesis pathways and on different histologic subtypes.
Acknowledgment
The authors thank Denis Mariano for editing assistance.
Disclosure
The authors report no conflicts of interest in this work.
References
Reddy BS, Nayini J, Tokumo K, Rigotty J, Zang E, Kelloff G. Chemoprevention of colon carcinogenesis by concurrent administration of piroxicam, a nonsteroidal anti-inflammatory drug with D, L-alpha-difluoromethylornithine, an ornithine decarboxylase inhibitor, in diet. Cancer Res. 1990;50(9):2562–2568. | ||
Mirshafiey A, Vaezzadeh F, Khorramizadeh MR, Saadat F. Effect of piroxicam on matrix metalloproteinase 2 and apoptosis. Int J Tissue React. 2004;26(1–2):1–7. | ||
Zhan H, Zheng H. The role of topical cyclo-oxygenase-2 inhibitors in skin cancer: treatment and prevention. Am J Clin Dermatol. 2007;8(4):195–200. | ||
Campione E, Diluvio L, Paterno EJ, Chimenti S. Topical treatment of actinic keratoses with piroxicam 1% gel: a preliminary open-label study utilizing a new clinical score. Am J Clin Dermatol. 2010;11(1):45–50. | ||
An KP, Athar M, Tang X, et al. Cyclooxygenase-2 expression in murine and human nonmelanoma skin cancers: implications for therapeutic approaches. Photochem Photobiol. 2002;76(1):73–80. | ||
Li H, Gao S, Ye J, et al. COX-2 is involved in ET-1-induced hypertrophy of neonatal rat cardiomyocytes: role of NFATc3. Mol Cell Endocrinol. 2014;382(2):998–1006. | ||
Wilgus TA, Koki AT, Zweifel BS, Kusewitt DF, Rubal PA, Oberyszyn TM. Inhibition of cutaneous ultraviolet light B-mediated inflammation and tumor formation with topical celecoxib treatment. Mol Carcinog. 2003;38(2):49–58. | ||
Black AK, Greaves MW, Hensby CN. The anti-inflammatory and pharmacological effects of topically applied flurbiprofen on human skin 24 hours after ultraviolet B irradiation. Prostaglandins Med. 1980;5(5):405–413. | ||
Richy F, Scarpignato C, Lanas A, Reginster JY. Efficacy and safety of piroxicam revisited. A global meta-analysis of randomised clinical trials. Pharmacol Res. 2009;60(4):254–263. | ||
Gloster HM Jr, Brodland DG. The epidemiology of skin cancer. Dermatol Surg. 1996;22(3):217–226. | ||
Becker K, Dosch J, Gregel CM, Martin BA, Kaina B. Targeted expression of human O(6)-methylguanine-DNA methyltransferase (MGMT) in transgenic mice protects against tumor initiation in two-stage skin carcinogenesis. Cancer Res. 1996;56(14):3244–3249. | ||
Verma AK, Duvick L, Ali M. Modulation of mouse skin tumor promotion by dietary 13-cis-retinoic acid and alpha-difluoromethylornithine. Carcinogenesis. 1986;7(6):1019–1023. | ||
Canizares F, Salinas J, de las Heras M, et al. Prognostic value of ornithine decarboxylase and polyamines in human breast cancer: correlation with clinicopathologic parameters. Clin Cancer Res. 1999;5(8): 2035–2041. | ||
Carbone PP, Douglas JA, Larson PO, et al. Phase I chemoprevention study of piroxicam and alpha-difluoromethylornithine. Cancer Epidemiol Biomarkers Prev. 1998;7(10):907–912. | ||
Park WS, Lee HK, Lee JY, Yoo NJ, Kim CS, Kim SH. p53 mutations in solar keratoses. Human Pathol. 1996;27(11):1180–1184. | ||
Muller-Decker K. Cyclooxygenase-dependent signaling is causally linked to non-melanoma skin carcinogenesis: pharmacological, genetic, and clinical evidence. Cancer Metastasis Rev. 2011;30(3–4):343–361. | ||
Huang C, Ma WY, Hanenberger D, Cleary MP, Bowden GT, Dong Z. Inhibition of ultraviolet B-induced activator protein-1 (AP-1) activity by aspirin in AP-1-luciferase transgenic mice. J Biol Chem. 1997;272(42):26325–26331. | ||
Bair WB 3rd, Hart N, Einspahr J, et al. Inhibitory effects of sodium salicylate and acetylsalicylic acid on UVB-induced mouse skin carcinogenesis. Cancer Epidemiol Biomarkers Prev. 2002;11(12):1645–1652. | ||
Marks F, Furstenberger G, Neufang G, Muller-Decker K. Mouse skin as a model for cancer chemoprevention by nonsteroidal anti-inflammatory drugs. Recent Results Cancer Res. 2003;163:46–57; discussion 264–266. | ||
Reeve VE, Matheson MJ, Bosnic M, Boehm-Wilcox C. The protective effect of indomethacin on photocarcinogenesis in hairless mice. Cancer Lett. 1995;95(1–2):213–219. | ||
Bode AM, Dong Z. Signal transduction pathways: targets for chemoprevention of skin cancer. Lancet Oncol. 2000;1:181–188. | ||
Al-Waili NS. Indomethacin in basal cell carcinoma. J Pak Med Assoc. 1989;39(5):134–136. | ||
Benamouzig R, Chaussade S. Chemopreventive agents in colorectal cancer. La Revue du Praticien. 2003;53(20):2213–2214. | ||
Giardiello FM, Hamilton SR, Krush AJ, Offerhaus JA, Booker SV, Petersen GM. Nasopharyngeal angiofibroma in patients with familial adenomatous polyposis. Gastroenterology. 1993;105(5):1550–1552. | ||
Sandler RS, Halabi S, Baron JA, et al. A randomized trial of aspirin to prevent colorectal adenomas in patients with previous colorectal cancer. N Engl J Med. 2003;348(10):883–890. | ||
Sorensen HT, Friis S, Norgard B, et al. Risk of cancer in a large cohort of nonaspirin NSAID users: a population-based study. Br J Cancer. 2003;88(11):1687–1692. | ||
Arber N. Cyclooxygenase-2 inhibitors in colorectal cancer prevention: point. Cancer Epidemiol Biomarkers Prev. 2008;17(8):1852–1857. | ||
Friis S, Sorensen HT, McLaughlin JK, Johnsen SP, Blot WJ, Olsen JH. A population-based cohort study of the risk of colorectal and other cancers among users of low-dose aspirin. Br J Cancer. 2003;88(5):684–688. | ||
Boukamp P. Non-melanoma skin cancer: what drives tumor development and progression? Carcinogenesis. 2005;26(10):1657–1667. | ||
Gatti S, Carrozzo AM, Orlandi A, Nini G. Treatment of inflammatory linear verrucous epidermal naevus with calcipotriol. Br J Dermatol. 1995;132(5):837–839. | ||
Bianchi L, Orlandi A, Campione E, et al. Topical treatment of basal cell carcinoma with tazarotene: a clinicopathological study on a large series of cases. Br J Dermatol. 2004;151(1):148–156. | ||
Orlandi A, Bianchi L, Costanzo A, Campione E, Giusto Spagnoli L, Chimenti S. Evidence of increased apoptosis and reduced proliferation in basal cell carcinomas treated with tazarotene. J Invest Dermatol. 2004;122(4):1037–1041. | ||
Frost CA, Green AC, Williams GM. The prevalence and determinants of solar keratoses at a subtropical latitude (Queensland, Australia). Br J Dermatol. 1998;139(6):1033–1039. | ||
Johannesdottir SA, Chang ET, Mehnert F, Schmidt M, Olesen AB, Sorensen HT. Nonsteroidal anti-inflammatory drugs and the risk of skin cancer: a population-based case-control study. Cancer. 2012;118(19): 4768–4776. | ||
Santiago RM, Tonin FS, Barbiero J, et al. The nonsteroidal anti-inflammatory drug piroxicam reverses the onset of depressive-like behavior in 6-OHDA animal model of Parkinson’s disease. Neuroscience.2015;300:246–253. | ||
London CA, Gardner HL, Mathie T, et al. Impact of toceranib/piroxicam/cyclophosphamide maintenance therapy on outcome of dogs with appendicular osteosarcoma following amputation and carboplatin chemotherapy: a multi-institutional study. PloS One. 2015;10(4):e0124889. | ||
Dhawan B, Aggarwal G, Harikumar S. Enhanced transdermal permeability of piroxicam through novel nanoemulgel formulation. Int J Pharm Investig. 2014;4(2):65–76. | ||
Thun MJ, Henley SJ, Patrono C. Nonsteroidal anti-inflammatory drugs as anticancer agents: mechanistic, pharmacologic, and clinical issues. J Natl Cancer Inst. 2002;94(4):252–266. | ||
Clouser MC, Roe DJ, Foote JA, Harris RB. Effect of non-steroidal anti-inflammatory drugs on non-melanoma skin cancer incidence in the SKICAP-AK trial. Pharmacoepidemiol Drug Saf. 2009;18(4):276–283. | ||
Vad NM, Kudugunti SK, Wang H, Bhat GJ, Moridani MY. Efficacy of acetylsalicylic acid (aspirin) in skin B16-F0 melanoma tumor-bearing C57BL/6 mice. Tumour Biol. 2014;35(5):4967–4976. | ||
Dovizio M, Bruno A, Tacconelli S, Patrignani P. Mode of action of aspirin as a chemopreventive agent. Recent Results Cancer Res. 2013;191:39–65. | ||
Asgari M, White E, Chren MM. Nonsteroidal anti-inflammatory drug use in the prevention and treatment of squamous cell carcinoma. Dermatol Surg. 2004;30(10):1335–1342. | ||
Rathore K, Alexander M, Cekanova M. Piroxicam inhibits Masitinib-induced cyclooxygenase 2 expression in oral squamous cell carcinoma cells in vitro. Transl Res. 2014;164(2):158–168. | ||
Micali G, Lacarrubba F, Bhatt K, Nasca MR. Medical approaches to non-melanoma skin cancer. Expert Rev Anticancer Ther. 2013;13(12):1409–1421. | ||
Harris J, Bines S, Das Gupta T. Therapy of disseminated malignant melanoma with recombinant alpha 2b-interferon and piroxicam: clinical results with a report of an unusual response-associated feature (vitiligo) and unusual toxicity (diffuse pulmonary interstitial fibrosis). Med Pediatr Oncol. 1994;22(2):103–106. | ||
Chiu LC, Tong KF, Ooi VE. Cytostatic and cytotoxic effects of cyclooxygenase inhibitors and their synergy with docosahexaenoic acid on the growth of human skin melanoma A-375 cells. Biomed Pharmacother. 2005;59(Suppl 2):S293–S297. | ||
Brockley LK, Cooper MA, Bennett PF. Malignant melanoma in 63 dogs (2001–2011): the effect of carboplatin chemotherapy on survival. N Z Vet J. 2013;61(1):25–31. | ||
Salvemini D, Kim SF, Mollace V. Reciprocal regulation of the nitric oxide and cyclooxygenase pathway in pathophysiology: relevance and clinical implications. Am J Physiol Regul Integr Comp Physiol. 2013;304(7):R473–R487. | ||
Fischer SM, Conti CJ, Viner J, Aldaz CM, Lubet RA. Celecoxib and difluoromethylornithine in combination have strong therapeutic activity against UV-induced skin tumors in mice. Carcinogenesis. 2003;24(5):945–952. | ||
Steele VE, Hawk ET, Viner JL, Lubet RA. Mechanisms and applications of non-steroidal anti-inflammatory drugs in the chemoprevention of cancer. Mutat Res. 2003;523–524:137–144. | ||
Moison RM, Beijersbergen van Henegouwen GM. Topical antioxidant vitamins C and E prevent UVB-radiation-induced peroxidation of eicosapentaenoic acid in pig skin. Radiat Res. 2002;157(4):402–409. | ||
Smith WL. The eicosanoids and their biochemical mechanisms of action. Biochem J. 1989;259(2):315–324. | ||
Xu W, Jia S, Xie P, et al. The expression of proinflammatory genes in epidermal keratinocytes is regulated by hydration status. J Invest Dermatol. 2014;134(4):1044–1055; quiz e1015. | ||
Vezza R, Habib A, Li H, Lawson JA, FitzGerald GA. Regulation of cyclooxygenases by protein kinase C. Evidence against the importance of direct enzyme phosphorylation. J Biol Chem. 1996;271(47):30028–30033. | ||
Loftin CD, Tiano HF, Langenbach R. Phenotypes of the COX-deficient mice indicate physiological and pathophysiological roles for COX-1 and COX-2. Prostaglandins Other Lipid Mediat. 2002;68–69:177–185. | ||
Cha YI, DuBois RN. NSAIDs and cancer prevention: targets downstream of COX-2. Annu Rev Med. 2007;58:239–252. | ||
Morgan GJ, Poland M, DeLapp RE. Efficacy and safety of nabumetone versus diclofenac, naproxen, ibuprofen, and piroxicam in the elderly. Am J Med. 1993;95(2A):19S–27S. | ||
Moser U, Waldburger H, Schwarz HA, Gobelet CA. A double-blind randomised multicentre study with tenoxicam, piroxicam and diclofenac sodium retard in the treatment of ambulant patients with osteoarthritis and extra-articular rheumatism. Scand J Rheumatol Suppl. 1989;80:71–80. | ||
Park SW, Kim HS, Choi MS, et al. The influence of cyclooxygenase-1 expression on the efficacy of cyclooxygenase-2 inhibition in head and neck squamous cell carcinoma cell lines. Anticancer Drugs. 2011;22(5):416–423. | ||
Hata AN, Breyer RM. Pharmacology and signaling of prostaglandin receptors: multiple roles in inflammation and immune modulation. Pharmacol Ther. 2004;103(2):147–166. | ||
Tober KL, Thomas-Ahner JM, Maruyama T, Oberyszyn TM. Possible cross-regulation of the E prostanoid receptors. Mol Carcinog. 2007;46(8):711–715. | ||
Sung YM, He G, Fischer SM. Lack of expression of the EP2 but not EP3 receptor for prostaglandin E2 results in suppression of skin tumor development. Cancer Res. 2005;65(20):9304–9311. | ||
Friedman ES, LaNatra N, Stiller MJ. NSAIDs in dermatologic therapy: review and preview. J Cutan Med Surg. 2002;6(5):449–459. | ||
Kuzbicki L, Lange D, Stanek-Widera A, Chwirot BW. Different expression of cyclooxygenase-2 (COX-2) in selected nonmelanocytic human cutaneous lesions. Folia Histochem Cytobiol. 2011;49(3):381–388. | ||
El-Khalawany MA, Abou-Bakr AA. Role of cyclooxygenase-2, ezrin and matrix metalloproteinase-9 as predictive markers for recurrence of basal cell carcinoma. J Cancer Res Ther. 2013;9(4):613–617. | ||
Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455–461. | ||
Seeliger D, de Groot BL. Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J Comput Aided Mol Des. 2010;24(5):417–422. | ||
O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. Open Babel: an open chemical toolbox. J Cheminform. 2011;3:33. | ||
Picot D, Loll PJ, Garavito RM. The X-ray crystal structure of the membrane protein prostaglandin H2 synthase-1. Nature. 1994;367(6460):243–249. | ||
Rowlinson SW, Kiefer JR, Prusakiewicz JJ, et al. A novel mechanism of cyclooxygenase-2 inhibition involving interactions with Ser-530 and Tyr-385. J Biol Chem. 2003;278(46):45763–45769. | ||
Gierse JK, McDonald JJ, Hauser SD, Rangwala SH, Koboldt CM, Seibert K. A single amino acid difference between cyclooxygenase-1 (COX-1) and -2 (COX-2) reverses the selectivity of COX-2 specific inhibitors. J Biol Chem. 1996;271(26):15810–15814. | ||
Duggan KC, Hermanson DJ, Musee J, et al. (R)-Profens are substrate-selective inhibitors of endocannabinoid oxygenation by COX-2. Nat Chemical Biol. 2011;7(11):803–809. | ||
Dodson JM, DeSpain J, Hewett JE, Clark DP. Malignant potential of actinic keratoses and the controversy over treatment. A patient-oriented perspective. Arch Dermatol. 1991;127(7):1029–1031. | ||
Czarnecki D, Meehan CJ, Bruce F, Culjak G. The majority of cutaneous squamous cell carcinomas arise in actinic keratoses. J Cutan Med Surg. 2002;6(3):207–209. | ||
Braakhuis BJ, Tabor MP, Kummer JA, Leemans CR, Brakenhoff RH. A genetic explanation of Slaughter’s concept of field cancerization: evidence and clinical implications. Cancer Res. 2003;63(8):1727–1730. | ||
Philipp-Dormston WG. Field cancerization: from molecular basis to selective field-directed management of actinic keratosis. Curr Probl Dermatol. 2015;46:115–121. | ||
Ouhtit A, Nakazawa H, Armstrong BK, et al. UV-radiation-specific p53 mutation frequency in normal skin as a predictor of risk of basal cell carcinoma. J Natl Cancer Inst. 1998;90(7):523–531. | ||
Harwood CA, McGregor JM, Proby CM, Breuer J. Human papillomavirus and the development of non-melanoma skin cancer. J Clin Pathol. 1999;52(4):249–253. | ||
Harwood CA, Proby CM. Human papillomaviruses and non-melanoma skin cancer. Curr Opin Infect Dis. 2002;15(2):101–114. | ||
Kushida Y, Miki H, Ohmori M. Loss of heterozygosity in actinic keratosis, squamous cell carcinoma and sun-exposed normal-appearing skin in Japanese: difference between Japanese and Caucasians. Cancer Lett. 1999;140(1–2):169–175. | ||
Mortier L, Marchetti P, Delaporte E, et al. Progression of actinic keratosis to squamous cell carcinoma of the skin correlates with deletion of the 9p21 region encoding the p16(INK4a) tumor suppressor. Cancer Lett. 2002;176(2):205–214. | ||
Berhane T, Halliday GM, Cooke B, Barnetson RS. Inflammation is associated with progression of actinic keratoses to squamous cell carcinomas in humans. Br J Dermatol. 2002;146(5):810–815. | ||
Nakagawa K, Yamamura K, Maeda S, Ichihashi M. Bcl-2 expression in epidermal keratinocytic diseases. Cancer. 1994;74(6):1720–1724. | ||
Daikoku T, Wang D, Tranguch S, et al. Cyclooxygenase-1 is a potential target for prevention and treatment of ovarian epithelial cancer. Cancer Res. 2005;65(9):3735–3744. | ||
Farivar-Mohseni H, Kandzari SJ, Zaslau S, Riggs DR, Jackson BJ, McFadden DW. Synergistic effects of Cox-1 and -2 inhibition on bladder and prostate cancer in vitro. Am J Surg. 2004;188(5):505–510. | ||
Park SW, Kim HS, Hah JH, Kim KH, Heo DS, Sung MW. Differential effects between cyclooxygenase-2 inhibitors and siRNA on vascular endothelial growth factor production in head and neck squamous cell carcinoma cell lines. Head Neck. 2010;32(11):1534–1543. | ||
Stasi MA, Scioli MG, Arcuri G, et al. Propionyl-L-carnitine improves postischemic blood flow recovery and arteriogenetic revascularization and reduces endothelial NADPH-oxidase 4-mediated superoxide production. Arterioscler Thromb Vasc Biol. 2010;30(3):426–435. | ||
Tarallo V, Vesci L, Capasso O, et al. A placental growth factor variant unable to recognize vascular endothelial growth factor (VEGF) receptor-1 inhibits VEGF-dependent tumor angiogenesis via heterodimerization. Cancer Res. 2010;70(5):1804–1813. | ||
de Miranda AS, Bispo Junior W, da Silva YK, et al. Design, synthesis, antinociceptive and anti-inflammatory activities of novel piroxicam analogues. Molecules. 2012;17(12):14126–14145. | ||
Laskowski RA, Swindells MB. LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J Chem Inf Model. 2011;51(10):2778–2786. | ||
Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera – a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–1612. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.