")
Back to Journals » Drug Design, Development and Therapy » Volume 9
The investigational Aurora kinase A inhibitor alisertib (MLN8237) induces cell cycle G2/M arrest, apoptosis, and autophagy via p38 MAPK and Akt/mTOR signaling pathways in human breast cancer cells
Authors Li J, Yang Y, Liu Q, Pan S, He Z, Zhang X, Yang T, Chen X, Wang D, Qiu J, Zhou S
Received 3 October 2014
Accepted for publication 17 November 2014
Published 16 March 2015 Volume 2015:9 Pages 1627—1652
DOI https://doi.org/10.2147/DDDT.S75378
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Min Li
Jin-Ping Li,1,2 Yin-Xue Yang,3 Qi-Lun Liu,1 Shu-Ting Pan,1,4 Zhi-Xu He,5 Xueji Zhang,6 Tianxin Yang,7 Xiao-Wu Chen,8 Dong Wang,9 Jia-Xuan Qiu,4 Shu-Feng Zhou2,5
1Department of Surgical Oncology, General Hospital of Ningxia Medical University, Yinchuan, Ningxia, People’s Republic of China; 2Department of Pharmaceutical Sciences, College of Pharmacy, University of South Florida, Tampa, FL, USA; 3Department of Colorectal Surgery, General Hospital of Ningxia Medical University, Yinchuan, Ningxia; 4Department of Oral and Maxillofacial Surgery, The First Affiliated Hospital of Nanchang University, Nanchang, Jiangxi, 5Guizhou Provincial Key Laboratory for Regenerative Medicine, Stem Cell and Tissue Engineering Research Center and Sino-US Joint Laboratory for Medical Sciences, Guizhou Medical University, Guiyang, Guizhou, 6Research Center for Bioengineering and Sensing Technology, University of Science and Technology Beijing, Beijing, People’s Republic of China; 7Department of Internal Medicine, University of Utah and Salt Lake Veterans Affairs Medical Center, Salt Lake City, UT, USA; 8Department of General Surgery, The First People’s Hospital of Shunde, Southern Medical University, Shunde, Foshan, Guangdong, 9Cancer Center, Daping Hospital and Research Institute of Surgery, Third Military Medical University, Chongqing, People’s Republic of China
Abstract: Alisertib (ALS) is an investigational potent Aurora A kinase inhibitor currently undergoing clinical trials for the treatment of hematological and non-hematological malignancies. However, its antitumor activity has not been tested in human breast cancer. This study aimed to investigate the effect of ALS on the growth, apoptosis, and autophagy, and the underlying mechanisms in human breast cancer MCF7 and MDA-MB-231 cells. In the current study, we identified that ALS had potent growth-inhibitory, pro-apoptotic, and pro-autophagic effects in MCF7 and MDA-MB-231 cells. ALS arrested the cells in G2/M phase in MCF7 and MDA-MB-231 cells which was accompanied by the downregulation of cyclin-dependent kinase (CDK)1/cell division cycle (CDC) 2, CDK2, and cyclin B1 and upregulation of p21 Waf1/Cip1, p27 Kip1, and p53, suggesting that ALS induces G2/M arrest through modulation of p53/p21/CDC2/cyclin B1 pathways. ALS induced mitochondria-mediated apoptosis in MCF7 and MDA-MB-231 cells; ALS significantly decreased the expression of B-cell lymphoma 2 (Bcl-2), but increased the expression of B-cell lymphoma 2-associated X protein (Bax) and p53-upregulated modulator of apoptosis (PUMA), and increased the expression of cleaved caspases 3 and 9. ALS significantly increased the expression level of membrane-bound microtubule-associated protein 1 light chain 3 (LC3)-II and beclin 1 and induced inhibition of phosphatidylinositol 3-kinase (PI3K)/protein kinase B (Akt)/mammalian target of rapamycin (mTOR) and p38 mitogen-activated protein kinase (MAPK) pathways in MCF7 and MDA-MB-231 cells as indicated by their altered phosphorylation, contributing to the pro-autophagic activities of ALS. Furthermore, treatment with wortmannin markedly downregulated ALS-induced p38 MAPK activation and LC3 conversion. In addition, knockdown of the p38 MAPK gene by ribonucleic acid interference upregulated Akt activation and resulted in LC3-II accumulation. These findings indicate that ALS promotes cellular apoptosis and autophagy in breast cancer cells via modulation of p38 MAPK/Akt/ mTOR pathways. Further studies are warranted to further explore the molecular targets of ALS in the treatment of breast cancer.
Keywords: ALS, breast cancer, cell cycle, apoptosis, autophagy, p38 MAPK
Introduction
Breast cancer is the most lethal of the female-specific malignancies.1 The World Health Organization estimates that more than 1.2 million people are diagnosed with breast cancer each year.2 Standard breast cancer therapy generally combines surgery, multi-therapeutic agents, and ionizing radiation.3 The anticancer agents induce cell cycle arrest and/or cell death by apoptotic or non-apoptotic mechanisms including necrosis, senescence, autophagy, and mitotic catastrophe.4 Aurora A kinase (AURKA) plays an essential role in chromosome alignment, centrosome separation and maturation, mitotic spindle formation, and cytokinesis during mitosis.5–7 AURKA inhibition has been shown to induce aneuploidy, polyploidy, and mitotic catastrophe in cells.8
Currently, there are about 30 Aurora kinase inhibitors (AKIs) in different stages of pre-clinical and clinical development. Alisertib (ALS; also known as MLN8237) is one of the first AKIs to enter Phase I and II trials, showing great therapeutic potential in anticancer therapy in a wide range of types of cancer, including both advanced solid tumors and non-Hodgkin lymphoma.9–12 The clinical efficacy of ALS has largely been consistent with its cytostatic effects, with the best response so far being stable disease in about 23.7% of evaluable patients with advanced or metastatic solid tumor.12 However, data on the effect of ALS on breast cancer are few. In one study by Leontovich et al,13 ALS reduced cyclin A (cyclin-dependent kinase [CDK]2) expression in MCF-7 cells harboring abrogated p53 function (vMCF-7DNp53). ALS significantly enhanced the cytotoxicity of the microtubule inhibitor marchantin A from Marchantia polymorpha toward breast cancer cell lines A256, MCF7, and T47D.14 In addition, ALS augmented the antitumor efficacy of docetaxel or paclitaxel in in vivo models of triple-negative breast cancer grown in immunocompromised mice.15
The aims of the present study were to investigate the effects of ALS on the cell cycle, apoptosis, and autophagy and to elucidate the molecular mechanisms involved in human breast cancer MCF7 and MDA-MB-231 cells. We have demonstrated that ALS inhibits the proliferation and induced cell cycle G2/M arrest, apoptosis, and autophagy in MCF7 and MDA-MB-231 cells. We have found that p38 mitogen-activated protein kinase (MAPK) is required for ALS-induced autophagy at the sequestration step of autophagosome formation in MCF7 and MDA-MB-231 cells and we have confirmed that p38 MAPK and protein kinase B (Akt)/mammalian target of rapamycin (mTOR) signaling pathways play an important role in ALS-induced autophagy in MCF7 and MDA-MB-231 cells.
Materials and methods
Chemicals and reagents
ALS (MLN8237; 4-[[9-chloro-7-(2-fluoro-6-methoxyphenyl)-5H-pyrimido[5,4-d][2]benzazepin-2-yl]amino]-2-methoxybenzoic acid), thiazolyl blue tetrazolium bromide (MTT), Dulbecco’s phosphate buffered saline (PBS), 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES), ethylenediaminetetraacetic acid (EDTA), propidium iodide (PI), ribonuclease A, and fetal bovine serum (FBS) were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). 4′,6-Diamidino-2-phenylindole (DAPI) and 5-(and 6)-chloromethyl-2′,7′-dichlorodihy drofluorescein diacetate (CM-H2DCFDA) were bought from Thermo Fisher Scientific Inc. (Waltham, MA, USA). Dulbecco’s Modified Eagle’s Medium (DMEM/F12) and Roswell Park Memorial Institute (RPMI) 1640 medium were purchased from Corning Inc. (Corning, NY, USA). The annexinV:phycoerythrin (PE) apoptosis detection kit was purchased from BD Biosciences Inc. (San Jose, CA, USA). The autophagy detection kit containing the green fluorescent Cyto-ID® that can stain autophagic vacuoles was obtained from Enzo Life Sciences Inc. (Farmingdale, NY, USA). The polyvinylidene difluoride membrane was bought from Millipore Inc. (Bedford, MA, USA). Western blotting substrate was obtained from Thermo Fisher Scientific Inc. The autophagy inhibitors bafilomycin A1, wortmannin (WM, an irreversible and selective class I phosphatidylinositol 3-kinase [PI3K] inhibitor and a blocker of autophagosome formation), chloroquine, and Lipofectamine 2000 were purchased from Thermo Fisher Scientific Inc. SB202190 (an autophagy inducer and specific p38 MAP kinase inhibitor) was purchased from Santa Cruz Biotechnology Inc. (Dallas, TX, USA). The negative control small interfering ribonucleic acid (siRNA) and p38 MAPK siRNA were purchased from Cell Signaling Technology Inc. (Danvers, MA, USA). Primary antibodies against human p53, p21 Wafl/Cipl, p27 Kipl, cyclin B1, CDK1/CDC2, the p53-upregulated modulator of apoptosis (PUMA), B-cell lymphoma 2 (Bcl-2), Bcl-2-like protein4/Bcl-2-associated X protein (Bax), cleaved caspase 3, cleaved caspase 9, microtubule-associated protein 1 light chain 3 (LC3)-I (the cystolic form), LC3-II (the membrane-bound form), p38 MAPK, phosphorylated p38 MAPK (p-p38 MAPK at Thr180/Tyr182), PI3K, p-PI3K at Tyr458, Akt, p-Akt at Ser473, mTOR, p-mTOR at Ser2448, and beclin 1 were obtained from Cell Signaling Technology Inc. The primary antibody against human β-actin was bought from Santa Cruz Biotechnology Inc.
Cell lines and cell culture
MCF7 and MDA-MB-231 cells are the epithelial breast cancer cell lines, and MCF10A is the normal breast epithelial cell line. MCF7, MDA-MB-231, and MCF10A cells were all obtained from American Type Culture Collection (Manassas, VA, USA) and were cultured in different media. MCF7 and MDA-MB-231 cells were maintained in RPMI 1640 medium supplemented with heat-inactivated 10% FBS and 50 μg/mL penicillin. MCF10A cells were grown in DMEM/F12 (1/1) supplemented with 5% horse serum, 10 μg/mL insulin, 100 ng/mL cholera enterotoxin, 0.5 mg/mL hydrocortisone, and 20 ng/mL epidermal growth factor. All cells were maintained at 37°C in a 5% CO2/95% air humidified incubator. ALS was dissolved in dimethyl sulfoxide (DMSO) at a stock concentration of 50 mM, and was freshly diluted to the desired concentration with culture medium. The final concentration of DMSO was at 0.05% (v/v). The control cells received the vehicle only.
Cell viability assay
The MTT assay was performed to examine the effect of ALS on cell viability. Briefly, cells were seeded in 96-well culture plates at a density of 8×l03/well. After cells were attached, the media were changed to ALS at different concentrations (from 0.01–50 μM). The concentration of DMSO was at 0.05% (v/v). The control cells received the vehicle only. After 24 or 48 hour incubation, 10 μL MTT (5 g/L) was added to each well and cultured for another 4 hours. Then supernatant was removed and 150 μL DMSO was added. It was shaken for 5 minutes for the crystal dissolution. The absorbance at a wavelength 490 nm was measured with a Synergy™ H4 Hybrid microplate reader (BioTek Inc., Winooski, VT, USA). The half maximal inhibitory concentration (IC50) values were determined using the relative viability over ALS concentration curve.
Cell cycle analysis using flow cytometry
PI is commonly used as a DNA stain to determine DNA content in the cell cycle analysis. The effect of ALS on cell cycle distribution was determined by flow cytometry as described previously.16 Briefly, MCF7 and MDA-MB-231 cells were treated with ALS at different concentrations (0.1, 1.0, and 5.0 μM) for 24 hours. In separate experiments, MCF7 and MDA-MB-231 cells were treated with 1.0 μM ALS for 4, 8, 12, 24, 48, and 72 hours. Cells were suspended, washed with PBS, centrifuged, and fixed in 70% ethanol (v/v) at −20°C overnight. Then the cells were re-suspended in 1 mL of PBS containing 1 mg/mL ribonuclease A and 50 μg/mL PI. Cells were incubated in the dark for 30 minutes at room temperature. A total number of 1×104 events were subjected to cell cycle analysis using a flow cytometer (Becton Dickinson Immunocytometry Systems, San Jose, CA, USA).
Quantification of cellular apoptosis using flow cytometry
Apoptotic cells were quantitated using the annexinV:PE apoptosis detection kit (BD Biosciences Inc.) after cells were treated with ALS at different concentrations (0.1, 1.0, and 5.0 μM) for 24 hours as described previously.16 ALS was dissolved in DMSO at a final concentration of 0.05% (v/v). Annexin V is a 35 kDa Ca2+-dependent phospholipid-binding protein that has a high affinity for negatively charged phospholipid phosphatidylserine, and binds to cells that are actively undergoing apoptosis with exposed phospholipid phosphatidylserine. Briefly, cells were trypsinized and washed twice with cold PBS, and then cells were re-suspended in 1× binding buffer with 2.5 μL of annexin V:PE and 2.5 μL of 7-amino-actinomycin D (7-AAD, [used as a nucleic acid dye]) at 1×105 cells/mL in a total volume of 100 μL. The cells were gently mixed and incubated in the dark for 15 minutes at room temperature. A quota of 1× binding buffer (400 μL) was then added to a clean test tube and the number of apoptotic cells was quantified using a flow cytometer (Becton Dickinson Immunocytometry Systems) within 1 hour. Cells that stain positive for annexin V:PE and negative for 7-AAD are undergoing apoptosis; cells that stain positive for both annexin V:PE and 7-AAD are either in the end stage of apoptosis, are undergoing necrosis, or are already dead; and cells that stain negative for both annexin V:PE and 7-AAD are alive and not undergoing measurable apoptosis.
Quantification of cellular autophagy
For autophagy detection, each sample was washed by re-suspending the cell pellet in 1× assay buffer (Enzo Life Sciences Inc.; No: ENZ-51031-K200) and cells collected by centrifugation as described previously.16 After 24 hour incubation, the cells were treated with fresh medium alone, control vehicle alone (0.05% DMSO, v/v), or ALS (0.1, 1.0, and 5.0 μM) for 24 hours at 37°C. In separate experiments, MCF7 and MDA-MB-231 cells were treated with 1.0 μM ALS for 4, 8, 12, 24, 48, and 72 hours. To investigate the mechanisms for ALS-induced autophagy, cells were pre-treated with the 10 μM WM (a PI3K inhibitor and autophagy blocker), 10 μM bafilomycin A1 (an autophagy inhibitor), or 10 μM SB202190 (a selective inhibitor of p38 MAPK) for 30 minutes in MCF7 and MDA-MB-231 cells, then co-treated with 1.0 μM ALS for further 24 hours. Groups of cells treated with each of these compounds alone were also included. All inhibitors were dissolved in DMSO at a final concentration of 0.05% (v/v). Cells were re-suspended in 250 μL of phenol red-free culture medium (Thermo Fisher Scientific Inc.; No 1294895) containing 5% FBS, and 250 μL of the diluted Cyto-ID® Green stain solution (Enzo Life Sciences Inc.; No: ENZ-51031-K200) was added to each sample and mixed well. Cells were incubated for 30 minutes at 37°C in the dark, collected by centrifugation, washed with 1× assay buffer, and re-suspended in 250 μL fresh 1× assay buffer. Cells were analyzed using the green (FL1) channel of a flow cytometer (Becton Dickinson Immunocytometry Systems).
RNA interference
siRNA for downregulating gene expression was done by transfection of RNA oligonucleotides with Lipofectamine 2000 according to the manufacturer’s protocol. MCF7 cells were transfected with the negative control siRNA, p38 MAPK siRNA in Opti-MEM™ using Lipofectamine 2000. After incubation for 4 hours, the medium was changed with fresh complete culture medium. The cells were then incubated for an additional 48 hours and treated with ALS.
Western blotting analysis
The expression level of cellular proteins of interest was determined using Western blotting assays.16 MCF7 and MDA-MB-231 cells were washed with pre-cold PBS after 24 hour treatment with ALS at 0.1, 1.0, and 5.0 μM, lysed with the radioimmunoprecipitation assay buffer (50 mM HEPES at pH 7.5, 150 mM sodium chloride, 10% glycerol, 1.5 mM magnesium chloride, 1% Triton-X 100, 1 mM EDTA at pH 8.0, 10 mM sodium pyrophosphate, and 10 mM sodium fluoride) containing the protease and phosphatase inhibitor cocktails, and centrifuged at 3,000× g for 10 minutes at 4°C. Protein concentrations were measured using Pierce™ bicinchoninic acid protein assay kit (Thermo Fisher Scientific Inc.). An equal amount of protein sample (30 μg) was dissolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) sample loading buffer and electrophoresed on 10% SDS-PAGE mini-gel after thermal denaturation at 95°C for 5 minutes. Proteins were transferred onto Immobilon polyvinylidene difluoride membrane (EMD Millipore Inc., Billerica, MA, USA) at 400 mA for 2 hours at 4°C. Membranes were probed with indicated primary antibody overnight at 4°C and then blotted with respective secondary anti-mouse or anti-rabbit antibody. Visualization was performed using Bio-Rad ChemiDoc™ XRS system (BioRad Laboratories Inc., Hercules, CA, USA) with electrochemiluminescence substrate. Protein level was normalized to the matching densitometric value of the internal control β-actin.
Statistical analysis
Data are presented as the mean ± standard deviation (SD). Comparisons of multiple groups were evaluated by one-way analysis of variance (ANOVA) followed by Tukey’s multiple comparison procedure. Values of P<0.05 were considered statistically significant.
Results
ALS shows significantly higher cytotoxicity toward breast cancer cells than normal breast epithelial cells
In order to evaluate the cytotoxicity of ALS, we used the MTT assay to measure whether ALS inhibited the proliferation of malignant MCF7 and MDA-MB-231 cells and normal human breast epithelial MCF10A cells. The concentration-dependent inhibitory effects of ALS on the growth of MCF10A, MCF7, and MDA-MB-231 cells are shown in Figure 1A and B. When cells were treated with ALS at 0.01, 0.1, 1.0, 5.0, 20, and 50 μM for 24 hours, the percentage of cellular viability of MCF10A cells over the control cells (100%) was 97.3%, 89.2%, 85.8%, 64.9%, 58.4%, and 54.6%, respectively, and the IC50 value was 55.75 μM. The percentage of cellular viability of MCF7 cells was 91.5%, 82.9%, 65.3%, 58.7%, 45.2%, and 35.1%, respectively, and the IC50 was 17.13 μM; the percentage of cellular viability of MDA-MB-231 cells was 91.6%, 83.7%, 55.4%, 37.8%, 26.4%, and 26.3, respectively, and the IC50 was 12.43 μM (Figure 1A). When cells were treated with ALS at 0.01, 0.1, 1.0, 5.0, 20, and 50 μM for 48 hours, the percentage of cellular viability of MCF10A cells over the control cells (100%) was 95.2%, 87.4%, 81.1%, 68.1%, 53.1%, and 50.9%, respectively, and the IC50 value was 38.79 μM. the percentage of cellular viability of MCF7 cells over the control cells (100%) was 95.2%, 74.2%, 64.5%, 51.9%, 33.6%, and 24.6%, respectively, and the IC50 was 15.78 μM; the percentage of cellular viability of MDA-MB-231 cells over the control cells (100%) was 83.1%, 74.2%, 57.8%, 44.7%, 35.2%, and 25.5%, respectively, and the IC50 was 10.83 μM (Figure 1B). These results indicate that ALS induces a concentration-dependent inhibitory effect on the growth of MCF10A, MCF7, and MDA-MB-231 cells, but the cytotoxic effect of ALS is much lower in normal MCF10A cells than in both malignant cell lines.
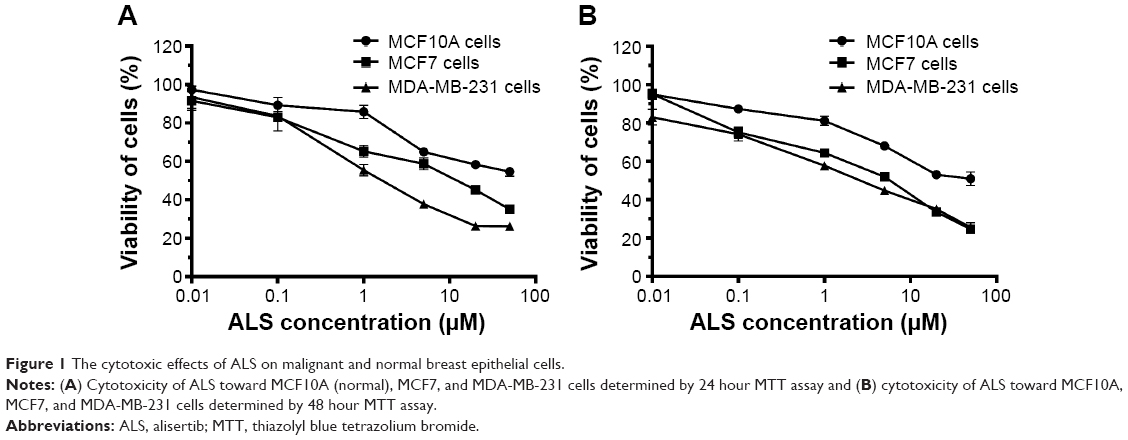 | Figure 1 The cytotoxic effects of ALS on malignant and normal breast epithelial cells. |
ALS induces G2/M arrest in MCF7 and MDA-MB-231 cells
To test whether ALS modulates the cell cycle in malignant breast cells, we assessed the cell cycle distribution of MCF7 and MDA-MB-231 cells exposed to ALS by flow cytometry. Treatment of MCF7 and MDA-MB-231 cells with ALS at different concentrations and different times arrested the cells at G2/M transition. Treatment with ALS led to an obvious G2/M arrest in concentration- and time-dependent manners in MCF7 and MDA-MB-231 cells. We treated MCF7 and MDA-MB-231 cells with ALS at 0.1, 1.0, and 5.0 μM for 24 hours, and then we measured the cell cycle distribution. At basal level, the percentage of MCF7 and MDA-MB-231 cells in G2/M phase was 18.6%, and 44.5%, respectively. As shown in Figure 2, a concentration-dependent increase in the cell number in G2/M phase in MCF7 and MDA-MB-231 cells was observed after incubation with ALS at 0.1, 1.0, and 5.0 μM for 24 hours. The percentage of cells arrested in G2/M phase was 42.5%, 78.9%, and 81.1% in MCF7 cells, respectively. This gave an increase of 2.3-, 4.2-, and 4.4-fold, respectively, compared to the control cells treated with DMSO only (P<0.001 by one-way ANOVA; Figure 2A and B). The percentage of cells arrested in G2/M phase was 55.2%, 79.6%, and 68.9% in MDA-MB-231 cells, respectively. This gave an increase of 1.2-, 1.8-, and 1.5-fold, respectively, compared to the control cells treated with DMSO only (P<0.01 by one-way ANOVA; Figure 2A and B). Treatment of MCF7 and MDA-MB-231 cells with ALS at 0.1, 1.0, and 5.0 μM also significantly affected the number of cells in G1 phase. The number of cells in G1 phase was significantly reduced by 32.0%, 77.6%, and 79.9% in MCF7 cells treated with ALS at 0.1, 1.0, and 5.0 μM, respectively. The number of cells in G1 phase was also significantly reduced by 25.0%, 69.9%, and 50.4% in MDA-MB-231 cells treated with ALS at 0.1, 1.0, and 5.0 μM, respectively (P<0.001 by one-way ANOVA; Figure 2B).
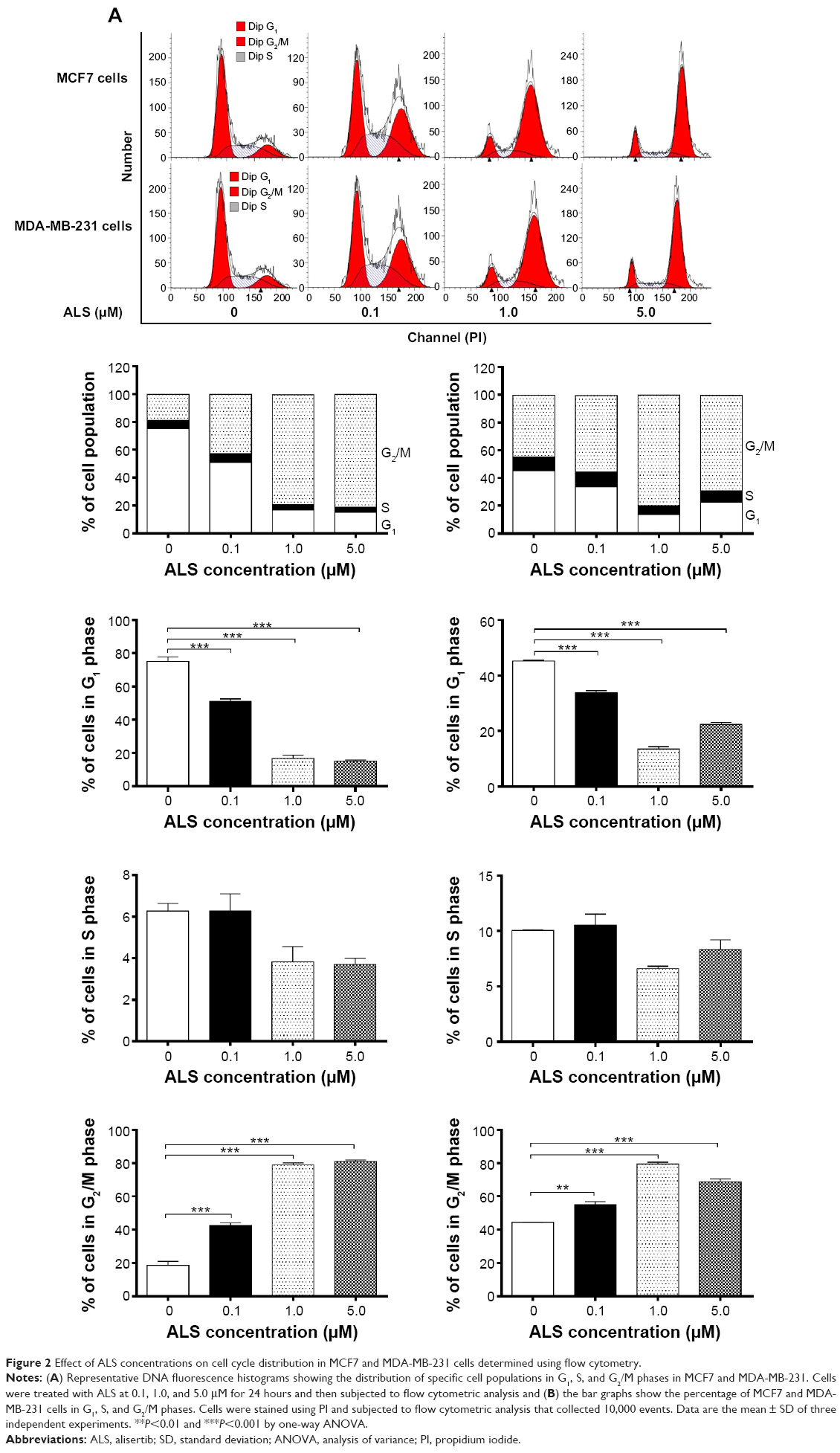 | Figure 2 Effect of ALS concentrations on cell cycle distribution in MCF7 and MDA-MB-231 cells determined using flow cytometry. |
We further conducted separate experiments to evaluate the effect of ALS treatment at 1.0 μM on cell cycle distribution in MCF7 and MDA-MB-231 cells over 72 hours. Compared to the control cells, the percentage of MCF7 cells in G2/M phase was increased from 12.8% at basal level to 66.6% and 55.3% after 24 and 48 hour treatment with 1.0 μM ALS, respectively. There was a significant increase in the percentage of MCF7 cells in G2/M phase after treatment with ALS for 24 and 48 hours (P<0.001 by one-way ANOVA; Figure 3A and B). The percentage of MDA-MB-231 cells in G2/M phase was increased from 12.7% at basal level to 26.5%, 31.9%, 39.9%, and 70.4% after 8, 12, 24, and 48 hour treatment with 1.0 μM ALS, respectively, but declined to 47.6% after 72 hour treatment of ALS. There was a significant increase in the percentage of MDA-MB-231 cells in G2/M phase after treatment with ALS for 8–72 hours (P<0.01 or P<0.001 by one-way ANOVA; Figure 3A and B). Taken together, the results show that ALS induces a remarkable cell cycle arrest in the MCF7 and MDA-MB-231 cells, contributing to its anticancer effect in the breast cancer treatment.
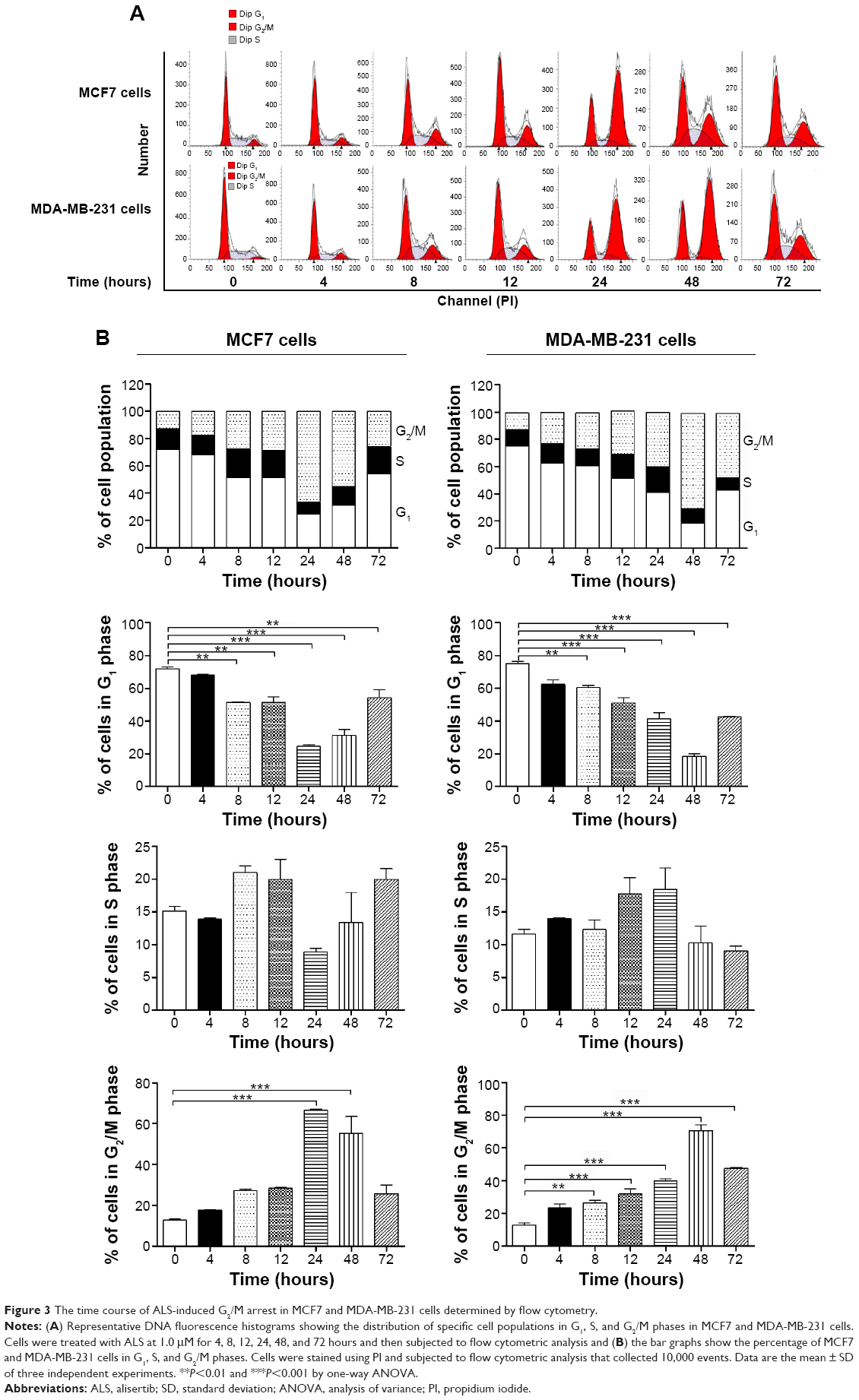 | Figure 3 The time course of ALS-induced G2/M arrest in MCF7 and MDA-MB-231 cells determined by flow cytometry. |
ALS regulates the expression of CDK1/CDC2, CDK2, cyclin B1, p21 Waf1/Cip1, p27 Kip1, and p53 in MCF7 and MDA-MB-231 cells
To explore the mechanism for ALS-induced cell cycle arrest, we examined the effect of ALS treatment on the expression levels of CDK1/CDC2, CDK2, cyclin B1, p21 Waf1/Cip1, p27 Kip1, and p53 in MCF7 and MDA-MB-231 cells using Western blotting assay. The expression level of CDK1/CDC2, CDK2, and cyclin B1 was remarkably decreased in MCF7 and MDA-MB-231 cells when treated with ALS, however, the expression level of p21 Waf1/Cip1, p27 Kip1, and p53 was markedly increased. In comparison to the control cells, the expression level of cyclin B1 was decreased 29.8% and 28.9% when MCF7 cells were treated with 1.0 and 5.0 μM ALS for 24 hours, respectively. There was a 43.4% and 20.5% reduction in the expression level of CDK1/CDC2 in MCF7 cells incubated with 1.0 and 5.0 μM ALS for 24 hours, respectively. Furthermore, the expression level of CDK2 was decreased 25.6% when treated with 1.0 μM ALS for 24 hours. Treatment of MCF7 cells with ALS at 0.1, 1.0, and 5.0 μM for 24 hours significantly increased the level of p21 Waf1/Cip1 2.8-, 2.9-, and 3.8-fold, respectively. Treatment of MCF7 cells with ALS at 1.0 and 5.0 μM for 24 hours also significantly increased the level of p27 Kip1 2.6- and 2.7-fold, respectively (P<0.01 or P<0.001 by one-way ANOVA; Figure 4A and B). Moreover, the expression level of p53 was increased when treated with ALS at 1.0 and 5.0 μM for 24 hours in MCF7 cells, although there was no statistical significance (Figure 4A and B).
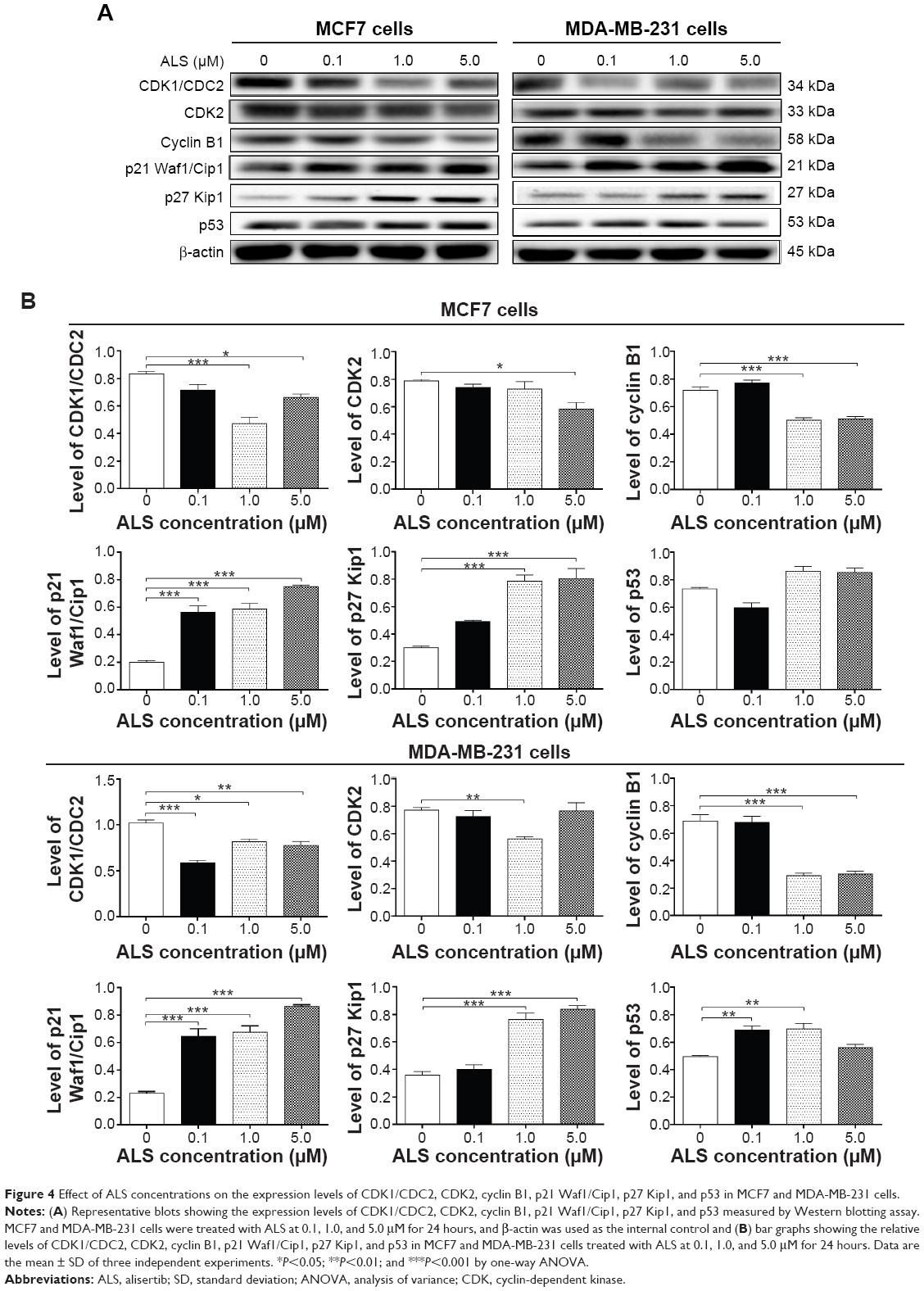 | Figure 4 Effect of ALS concentrations on the expression levels of CDK1/CDC2, CDK2, cyclin B1, p21 Waf1/Cip1, p27 Kip1, and p53 in MCF7 and MDA-MB-231 cells. |
Similarly, treatment of MDA-MB-231 cells with ALS at 0.1, 1.0, and 5.0 μM for 24 hours significantly decreased the level of CDK1/CDC2 by 42.2%, 19.6%, and 23.5%, respectively. Treatment of MDA-MB-231 cells with ALS at 1.0 μM for 24 hours significantly decreased the level of CDK2 by 27.2%. There was a 57.9% and 30.2% reduction in the expression level of cyclin B1 in MDA-MB-231 cells incubated with 1.0 and 5.0 μM ALS for 24 hours, respectively. Treatment of MDA-MB-231 cells with ALS at 0.1, 1.0, and 5.0 μM for 24 hours significantly increased the level of p21 Waf1/Cip1 2.8-, 2.9-, and 3.7-fold, respectively. Treatment of MDA-MB-231 cells with ALS at 1.0 and 5.0 μM for 24 hours significantly increased the level of p27 Kip1 2.1- and 2.3-fold, respectively. In addition, the expression level of p53 was increased 1.4- and 1.5-fold when treated with 0.1 and 1.0 μM ALS for 24 hours in MDA-MB-231 cells, respectively (P<0.01 by one-way ANOVA; Figure 4A and B).
To further investigate the molecular mechanism for ALS-induced cell cycle arrest, we conducted separate experiments to evaluate the expression levels of CDK1/CDC2, cyclin B1, p21 Waf1/Cip1, and p53 in MCF7 and MDA-MB-231 cells with the treatment of ALS over 72 hours using Western blotting analysis. Treatment of MCF7 cells with ALS at 1.0 μM for 8, 12, 48, and 72 hours significantly decreased the level of CDK1/CDC2 by 40.9%, 43.2%, 43.0%, and 52.3%, respectively. There was a 24.6%, 26.2%, 55.7%, and 65.6% reduction in the expression level of cyclin B1 in MCF7 cells incubated with 1.0 μM ALS for 4, 24, 48, and 72 hours, respectively. Treatment of MCF7 cells with ALS at 1.0 μM for 8, 12, 24, 48, and 72 hours significantly increased the level of p21 Waf1/Cip1 2.1-, 2.8-, 2.7-, 2.8-, and 2.4-fold, respectively. The expression level of p53 was increased 2.0-, 2.1-, and 2.7-fold when treated with 1.0 μM ALS for 24, 48, and 72 hours in MCF7 cells, respectively (P<0.01 or P<0.001 by one-way ANOVA; Figure 5A and B). Treatment of MDA-MB-231 cells with ALS at 1.0 μM for 4, 24, and 72 hours significantly decreased the level of CDK1/CDC2 by 25.0%, 34.5%, and 31.5%, respectively. There was a 41.2%, 25.0%, 46.3%, and 50.1% reduction in the expression level of cyclin B1 in MDA-MB-231 cells incubated with 1.0 μM ALS for 8, 12, 24, and 72 hours, respectively. Treatment of MDA-MB-231 cells with ALS at 1.0 μM for 8, 24, and 48 hours significantly increased the level of p21 Waf1/Cip1 2.1-, 1.7-, and 3.5-fold, respectively. In addition, the expression level of p53 was increased 1.9-, 2.0-, and 2.7-fold when treated with 1.0 μM ALS for 12, 24, and 72 hours in MDA-MB-231 cells, respectively (P<0.01 or P<0.001 by one-way ANOVA; Figure 5A and B).
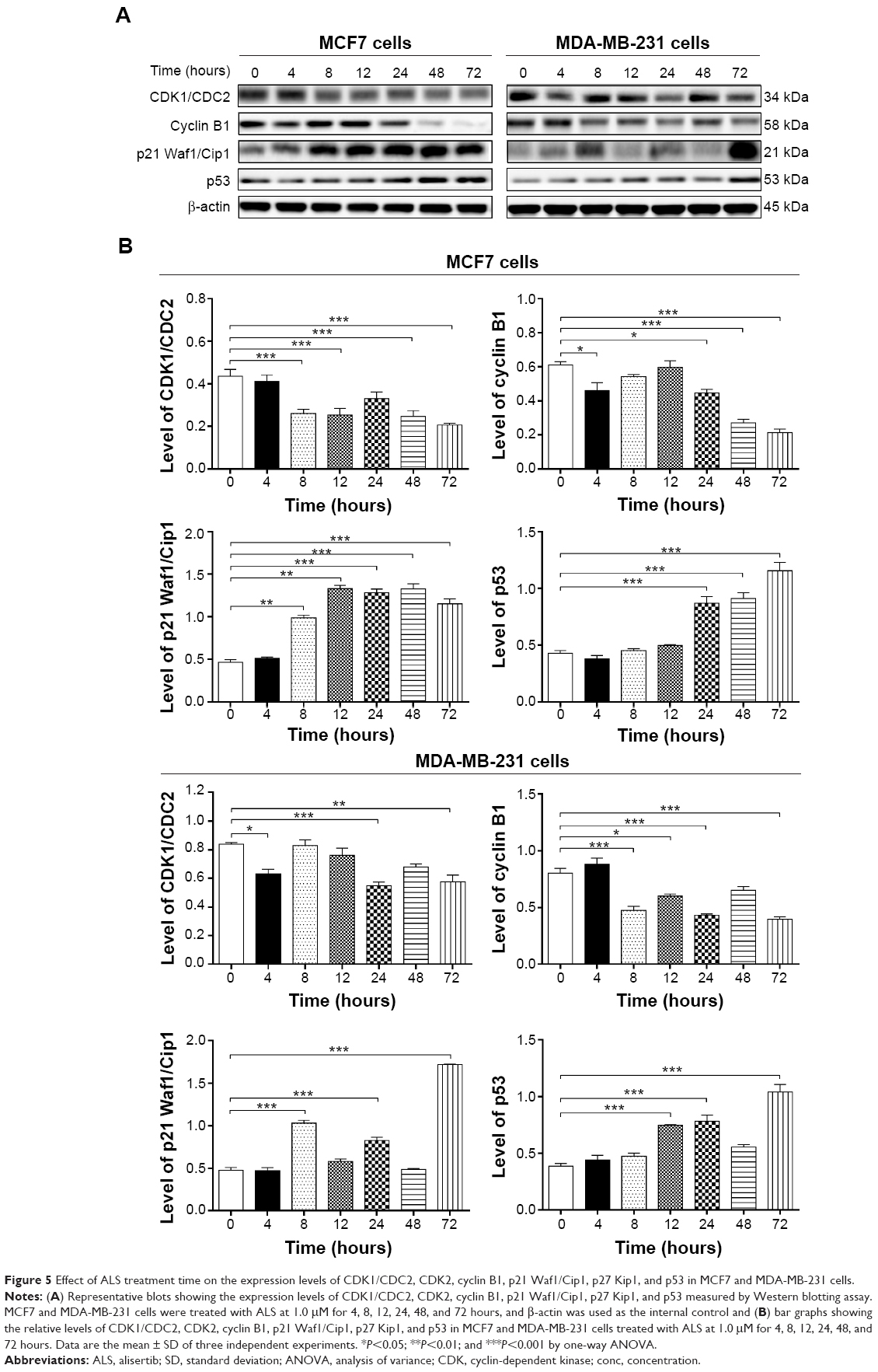 | Figure 5 Effect of ALS treatment time on the expression levels of CDK1/CDC2, CDK2, cyclin B1, p21 Waf1/Cip1, p27 Kip1, and p53 in MCF7 and MDA-MB-231 cells. |
ALS induces the apoptosis of MCF7 and MDA-MB-231 cells via activation of mitochondria-dependent pathway
In order to examine the apoptosis-inducing effect of ALS in MCF7 and MDA-MB-231 cells, the number of apoptotic cells was first quantified using flow cytometric analysis and the results are shown in Figure 6A. The number of apoptotic cells was 16.7% and 15.8% in MCF7 and MDA-MB-231 cells treated with the control vehicle only (0.05% DMSO, v/v), respectively. While MCF7 cells were treated with ALS at 0.1, 1.0, and 5.0 μM for 24 hours, the total percentage of apoptotic cells was increased 1.4-, 2.0-, and 2.6-fold compared to the control cells, respectively. Similarly, MDA-MB-231 cells were treated with ALS at 1.0 and 5.0 μM for 24 hours, the total percentage of apoptotic cells was increased 2.2- and 3.0-fold compared to the control cells, respectively (P<0.05 by one-way ANOVA; Figure 6A and B).
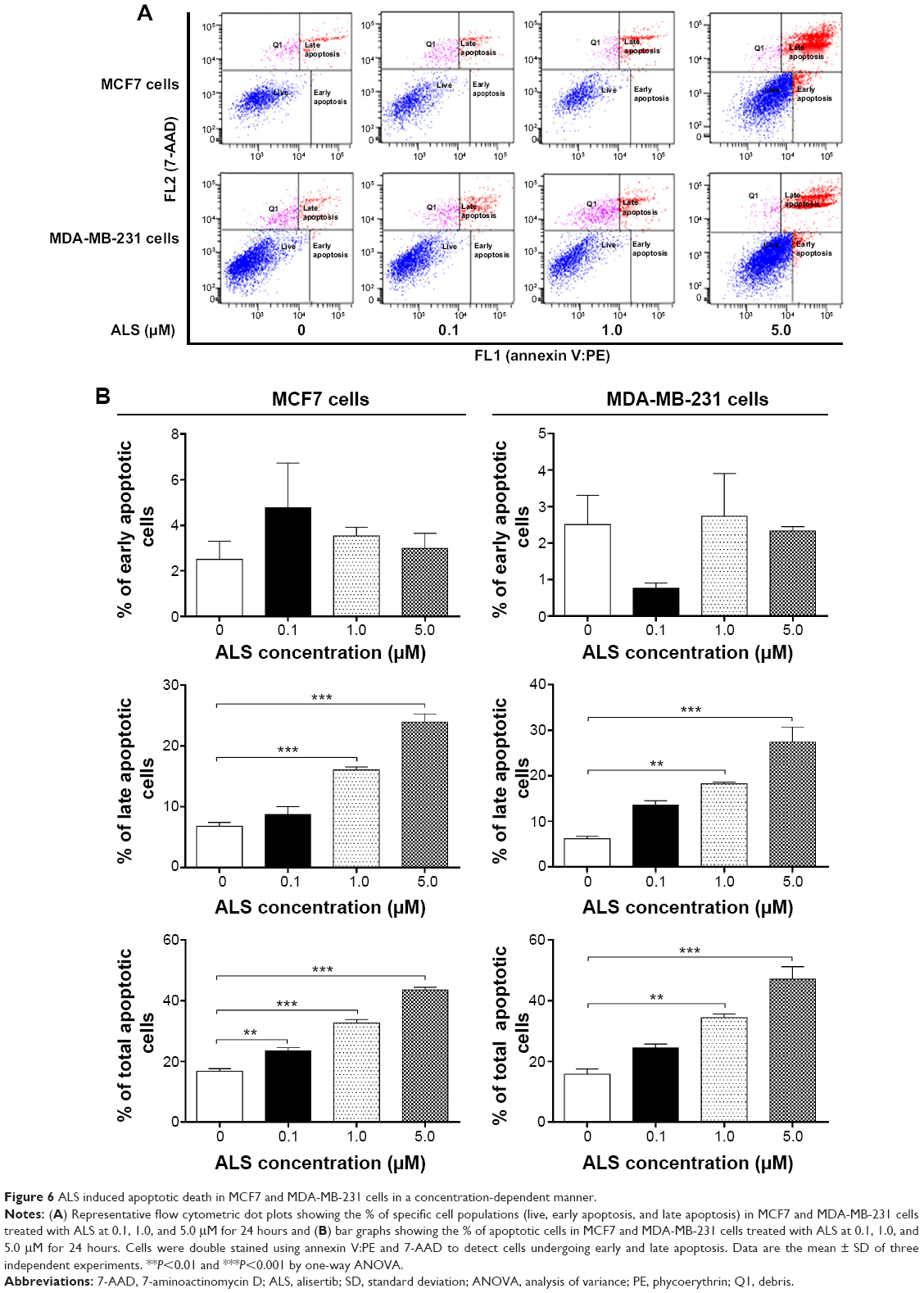 | Figure 6 ALS induced apoptotic death in MCF7 and MDA-MB-231 cells in a concentration-dependent manner. |
To investigate the mechanisms responsible for ALS-induced apoptosis in MCF7 and MDA-MB-231 cells, we evaluated the levels of Bcl-2, Bax, PUMA, cleaved caspase 3, and cleaved caspase 9 in above cells treated with ALS at 0.1, 1.0, and 5.0 μM for 24 hours. We first examined the effects of ALS treatment on the expression levels of the pro-apoptotic protein Bax and the anti-apoptotic protein Bcl-2. The expression level of Bax was concentration-dependently increased in both cell lines (P<0.05 by one-way ANOVA; Figure 7A and B). Incubation of MCF7 cells with 0.1, 1.0, and 5.0 μM ALS for 24 hours markedly increased Bax expression (1.4-, 2.8-, and 3.4-fold, respectively), and treatment of MDA-MB-231 cells with 0.1, 1.0, and 5.0 μM ALS for 24 hours remarkably increased the expression of Bax (1.7-, 1.5-, and 2.4-fold, respectively). In contrast, the expression level of Bcl-2 was decreased by 29.2%, 65.2%, and 53.4% in MCF7 cells, and 28.2%, 44.9%, and 53.8% in MDA-MB-231 cells when treated with ALS at 0.1, 1.0, and 5.0 μM, respectively. A significant increase in the expression level of Bax and reduction of Bcl-2 resulted in a significant decrease in Bcl-2/Bax ratio in both cell lines with a 50.4%, 87.5%, and 86.5% reduction in MCF7 cells and 58.3%, 64.6%, and 80.6% reduction in MDA-MB-231 cells when treated with ALS at 0.1, 1.0, and 5.0 μM for 24 hours, respectively (P<0.001 by one-way ANOVA; Figure 7A and B). Furthermore, the effect of ALS on the expression of PUMA was also examined due to its important role in the regulation of anti-apoptotic proteins. Incubation of MCF7 and MDA-MB-231 cells with ALS increased the expression level of PUMA in a concentration-dependent manner. Treatment of 0.1 and 5.0 μM ALS for 24 hours significantly increased the expression level of PUMA 1.5- and 2.4-fold in MCF7 cells, respectively. Treatment of 0.1, 1.0, and 5.0 μM ALS for 24 hours significantly increased the expression level of PUMA 1.5-, 1.8-, and 2.2-fold in MDA-MB-231 cells (P<0.05 by one-way ANOVA; Figure 7A and B). Subsequently, we observed a significant increase in the activation of caspases 9 and 3 in MCF7 and MDA-MB-231 cells. Incubation of MCF7 with ALS at 5.0 μM for 24 hours significantly increased the level of cleaved caspase 9 by 2.6-fold. Similarly, treatment of MDA-MB-231 cells with 1.0 and 5.0 μM ALS for 24 hours significantly increased the expression level of cleaved caspase 9 by 2.2- and 2.4-fold, respectively (P<0.001 by one-way ANOVA; Figure 7A and B). The level of cleaved caspase 3 was also significantly increased 1.5- and 1.6-fold when MCF7 cells were treated with 1.0 and 5.0 μM ALS for 24 hours, respectively. Moreover, treatment of MDA-MB-231 cells with ALS at 0.1, 1.0, and 5.0 μM for 24 hours increased the expression level of cleaved caspase 3 by 1.7-, 2.6-, and 2.7-fold, respectively (P<0.01 by one-way ANOVA; Figure 7A and B). These results indicate that ALS induces a remarkable activation of caspases 9 and 3, eventually leading to apoptotic cell death of MCF7 and MDA-MB-231 cells.
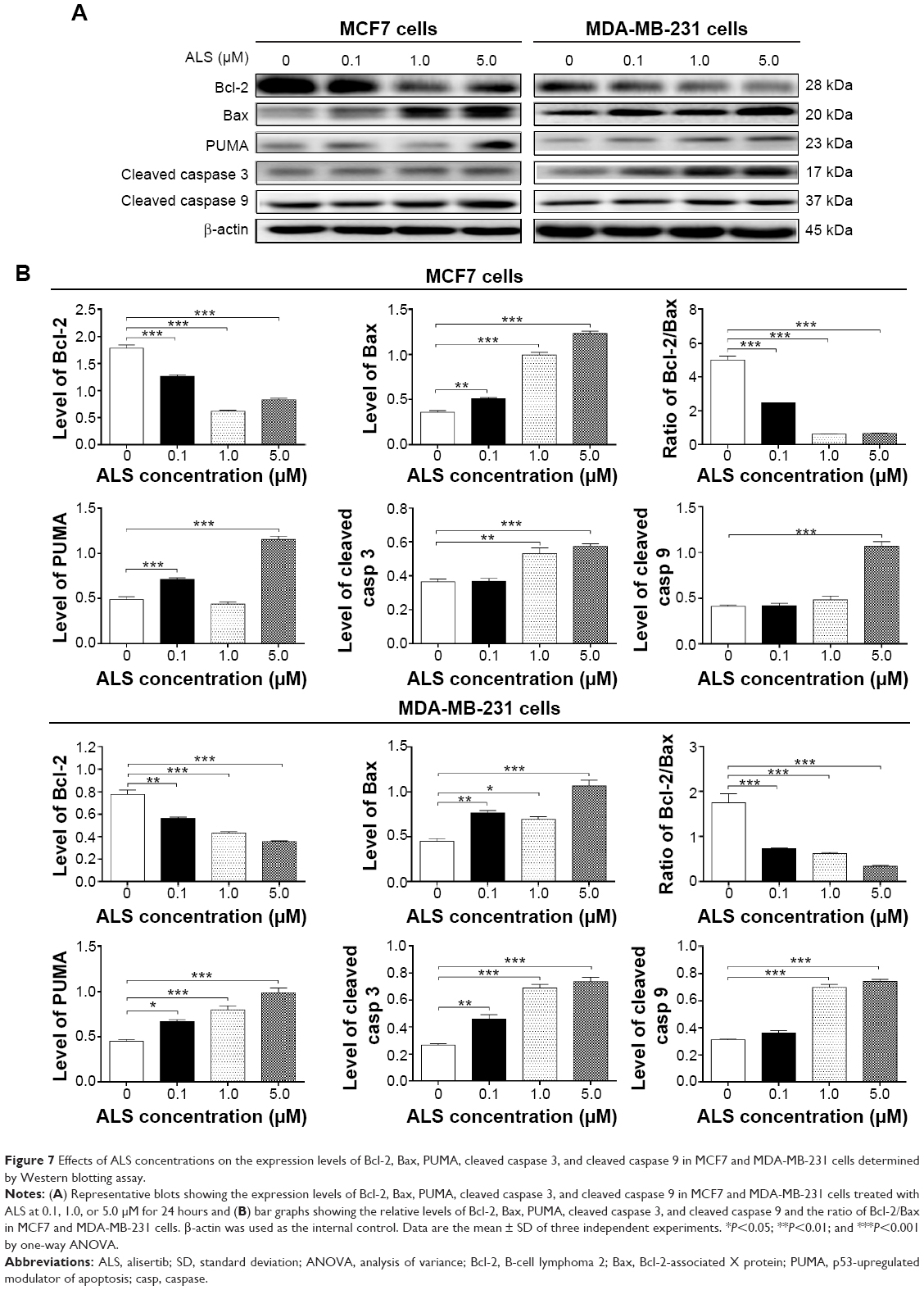 | Figure 7 Effects of ALS concentrations on the expression levels of Bcl-2, Bax, PUMA, cleaved caspase 3, and cleaved caspase 9 in MCF7 and MDA-MB-231 cells determined by Western blotting assay. |
ALS induces the autophagy of MCF7 and MDA-MB-231 cells
Autophagy is a cytoprotective mechanism against extracellular stress. To determine whether ALS induces autophagy, we examined the effect of ALS on the autophagy in MCF7 and MDA-MB-231 cells using flow cytometry and Western blotting analysis. ALS treatment of MCF7 and MDA-MB-231 cells induced remarkable autophagy in concentration- and time-dependent manners. In MCF7 cells, the percentage of autophagic cells at basal level was 3.6%, and incubation of the cells with ALS at 1.0 and 5.0 μM increased the autophagy 8.4- and 10.3-fold, respectively (P<0.001; Figure 8A and B). Treatment of MCF7 cells with 1.0 μM ALS for 24, 48, and 72 hours increased the autophagy 6.1-, 15.1-, and 34.3-fold, respectively (P<0.05 by one-way ANOVA; Figure 9A and B). Similarly, the percentage of autophagic cells at basal level was 1.3% in MDA-MB-231 cells, and incubation of the cells with ALS at 1.0 and 5.0 μM increased the autophagy 22.2- and 21.3-fold, respectively (P<0.01 by one-way ANOVA; Figure 8A and B). In addition, treatment of MDA-MB-231 cells with 1.0 μM ALS for 24, 48, and 72 hours increased the autophagy 5.6-, 14.9-, and 14.4-fold, respectively (P<0.01 by one-way ANOVA; Figure 9A and B).
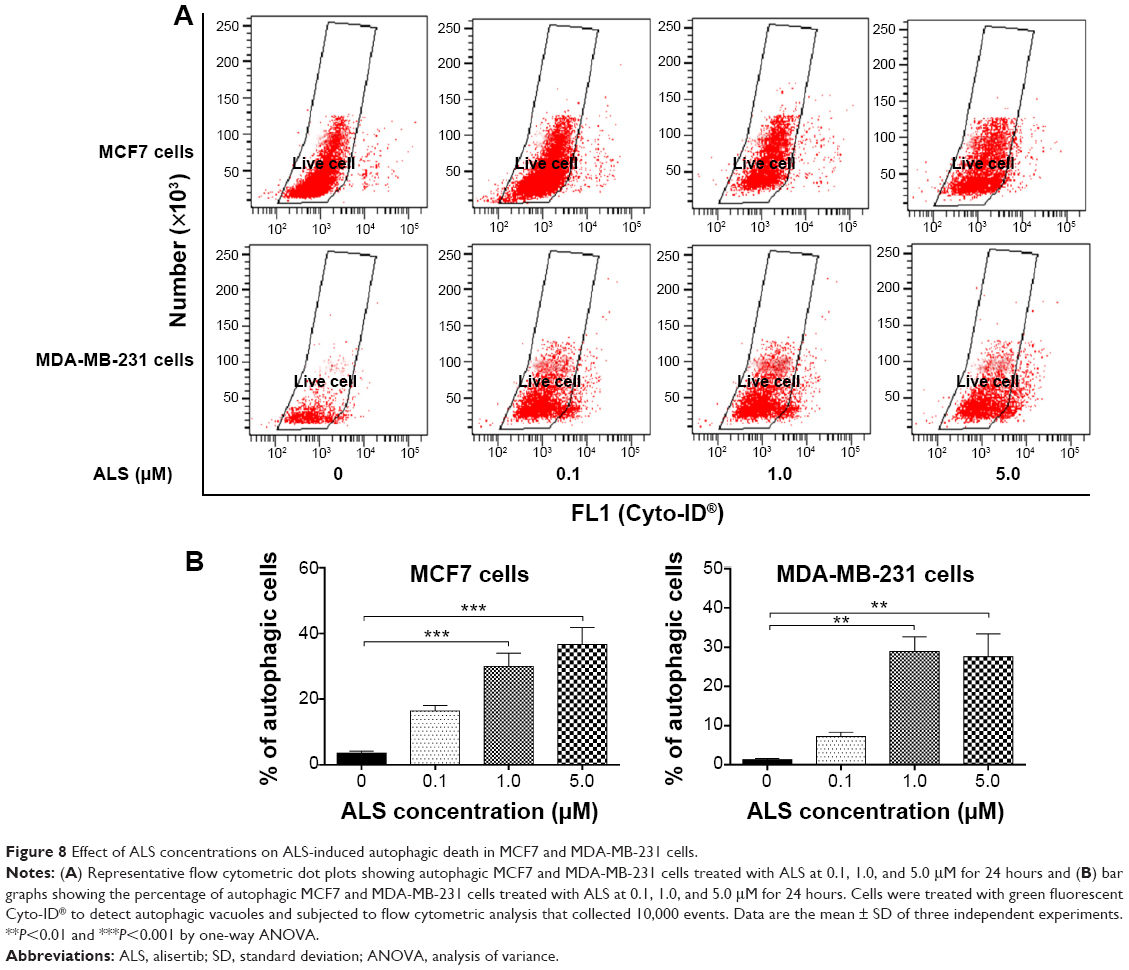 | Figure 8 Effect of ALS concentrations on ALS-induced autophagic death in MCF7 and MDA-MB-231 cells. |
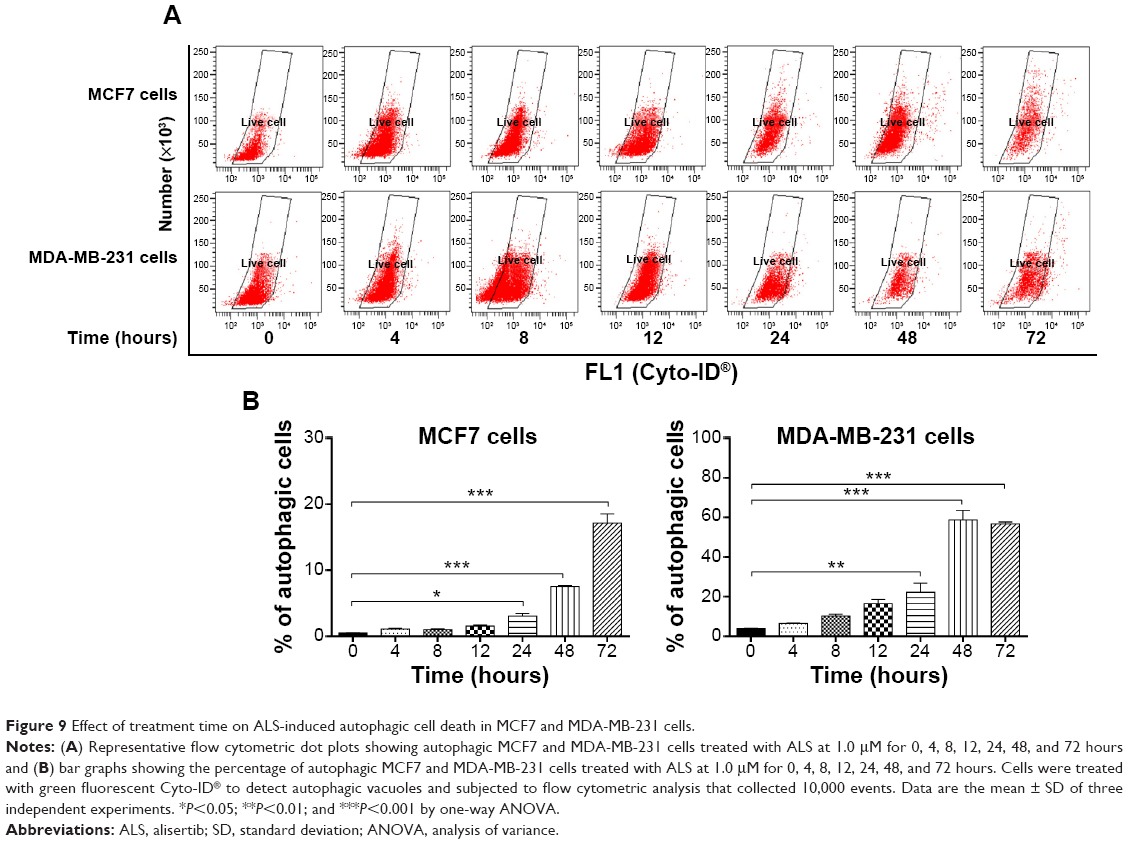 | Figure 9 Effect of treatment time on ALS-induced autophagic cell death in MCF7 and MDA-MB-231 cells. |
Next, we examined the effect of ALS on the expression level of beclin 1 and LC3-I/II. Autophagy is tightly regulated by beclin 1 (a mammalian homolog of yeast Atg6) that forms a complex with vacuolar protein sorting 34 (Vps34, also called class III PI3K), and serves as a platform for recruitment of other Atgs that are critical for autophagosome formation.17 Upon autophagy initiation, LC3 is cleaved at the C-terminus by Atg4 to form the cytosolic LC3-I.18 LC3-I is consequently proteolytically cleaved and lipidated by Atg3 and Atg7 to form LC3-II, which localizes to the autophagosome membrane. Treatment of MCF7 cells with ALS at 0.1, 1.0, and 5.0 μM for 24 hours increased the expression of beclin 1 1.5-, 1.4-, and 1.5-fold, respectively, compared to the control cells (P<0.001 by one-way ANOVA; Figure 10A and B). There was a 1.4- and 1.6-fold increase of beclin 1 in MDA-MB-231 cells treated with 1.0 and 5.0 μM ALS for 24 hours, respectively (P<0.01 by one-way ANOVA; Figure 10A and B). After 24 hour treatment with ALS at 0.1, 1.0, and 5.0 μM, our Western blotting analysis revealed two clear bands of LC3-I and II in MCF7 and MDA-MB-231 cells (Figure 10A). LC3-II migrated faster than LC3-I on SDS-PAGE, leading to the appearance of two bands after immunoblotting – LC3-I with an apparent mobility of about 18 kDa and LC3-II with an apparent mobility of 16 kDa. In both MCF7 and MDA-MB-231 cells, there was a concentration-dependent increase in the expression of LC3-II. Compared to the control cells, there was a 1.7-fold increase in the LC3-II level in MCF7 cells treated with ALS at 5.0 μM for 24 hours (P<0.01 by one-way ANOVA; Figure 10A and B). In MDA-MB-231 cells, 1.0 and 5.0 μM ALS resulted in a 1.8- and 2.8-fold increase in the expression of LC3-II, respectively, compared to the control cells (P<0.01 by one-way ANOVA; Figure 10A and B). In addition, treatment of MCF7 and MDA-MB-231 cells with ALS decreased the expression of LC3-I, although this was not statistically significant.
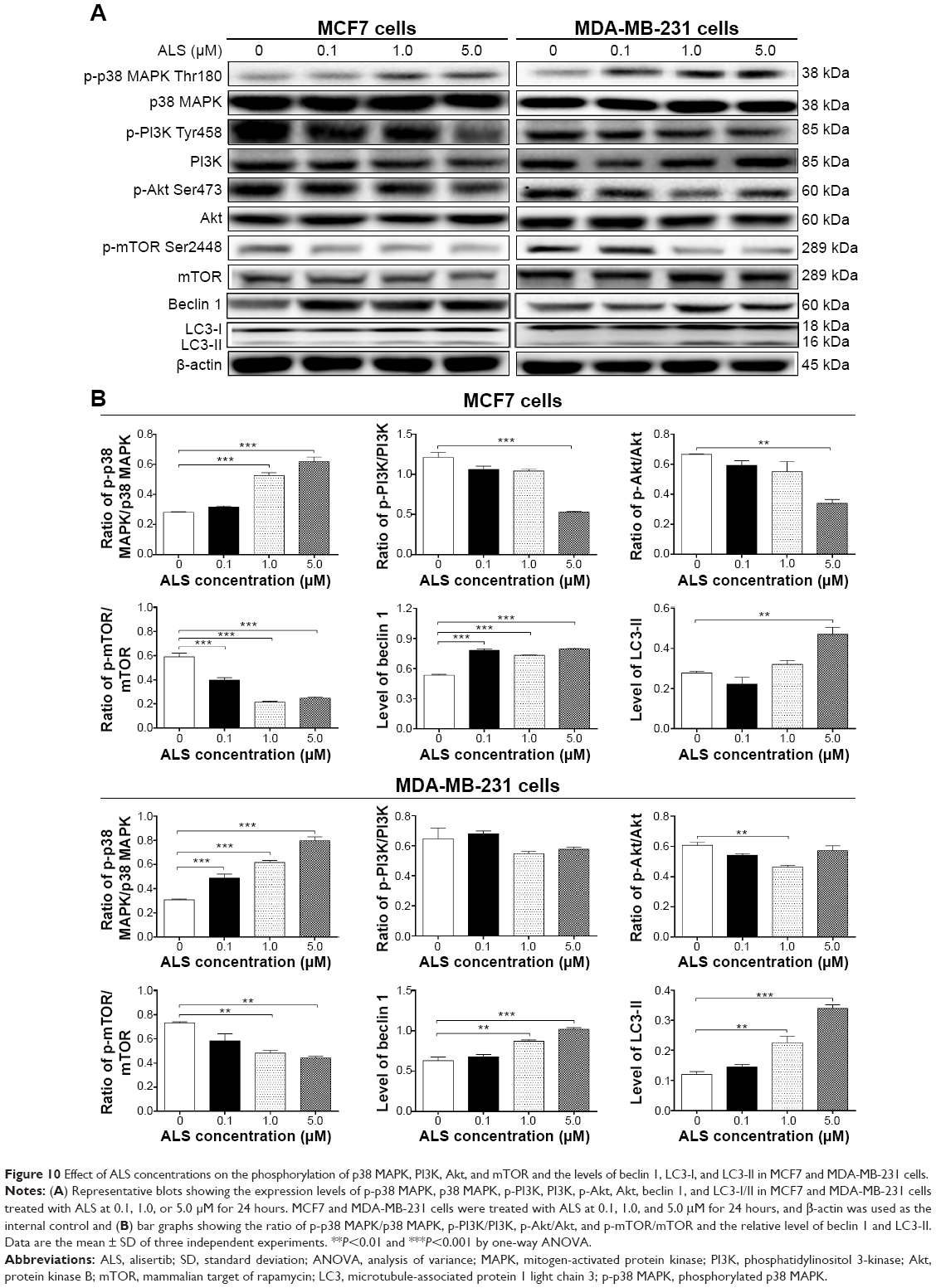 | Figure 10 Effect of ALS concentrations on the phosphorylation of p38 MAPK, PI3K, Akt, and mTOR and the levels of beclin 1, LC3-I, and LC3-II in MCF7 and MDA-MB-231 cells. |
To further elucidate the effect of ALS-induced autophagy in breast cancer cells, we examined the treatment time of ALS on the expression level of beclin 1 and LC3-I/II. Treatment of MCF7 cells with ALS at 1.0 μM for 12, 24, 48, and 72 hours increased the expression of beclin 1 by 1.9-, 2.3-, 3.0-, and 4.2-fold, respectively, compared to the control cells (P<0.001 by one-way ANOVA; Figure 11A and B). Similarly, there was a 1.7-, 2.0-, 3.3-, 4.2-, and 3.6-fold increase of beclin 1 in MDA-MB-231 cells treated with 1.0 μM ALS for 8, 12, 24, 48, and 72 hours, respectively (P<0.001 by one-way ANOVA; Figure 11A and B). In both MCF7 and MDA-MB-231 cells, there was also a time-dependent increase in the expression of LC3-II. Compared to the control cells, there was a 3.8-, 5.1-, 3.0-, and 6.1-fold increase in the LC3-II level in MCF7 cells treated with ALS at 1.0 μM for 12, 24, 48, and 72 hours, respectively (P<0.01 by one-way ANOVA; Figure 11B). In MDA-MB-231 cells, treatment with 1.0 μM ALS over 72 hours resulted in a 2.3- to 4.2-fold increase in the expression of LC3-II, compared to the control cells (P<0.001 by one-way ANOVA; Figure 11B). These results suggest ALS induces autophagy in breast cancer cells.
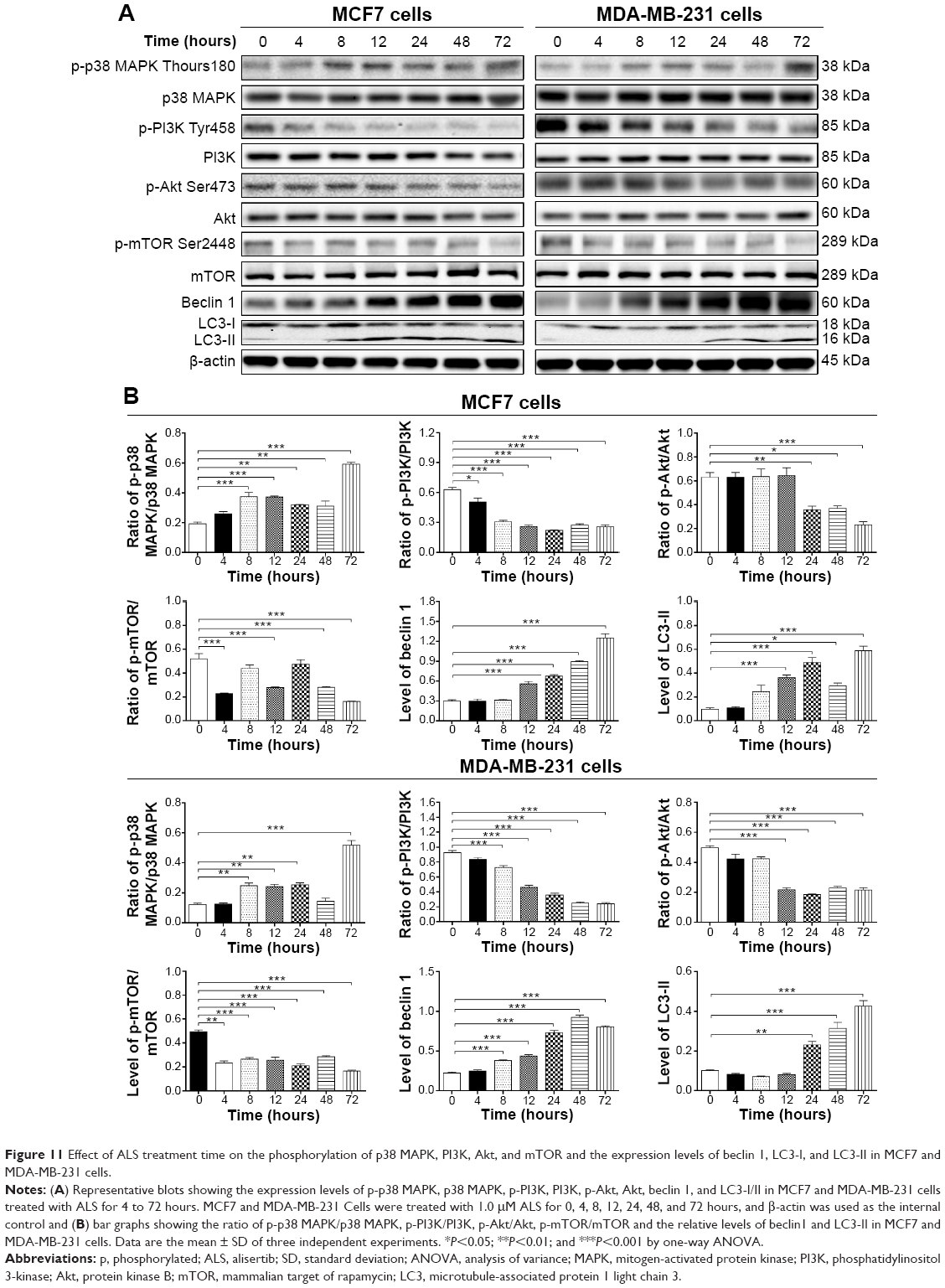 | Figure 11 Effect of ALS treatment time on the phosphorylation of p38 MAPK, PI3K, Akt, and mTOR and the expression levels of beclin 1, LC3-I, and LC3-II in MCF7 and MDA-MB-231 cells. |
ALS induces the activation of p38 MAPK in MCF7 and MDA-MB-231 cells
MAPK family members, including c-Jun N-terminal kinase, extracellular signal-regulated kinase, and p38 MAPK, have been reported to be involved in autophagy.19 To investigate whether the p38 MAPK signaling pathway was involved in ALS-induced autophagy, Western blotting assay was performed to detect the activated state of associated proteins. As shown in Figures 10 and 11, ALS markedly induced the phosphorylation of p38 MAPK.
First, we examined whether ALS induced autophagy of MCF7 and MDA-MB-231 cells in a concentration-dependent manner. Exposure of MCF7, and MDA-MB-231 cells to 0.1, 1.0, and 5.0 μM ALS for 24 hours increased the phosphorylation level of p38 MAPK; however, incubation of both cell lines with ALS did not significantly affect the expression of total p38 MAPK. The ratio of p-p38 MAPK over p38 MAPK was concentration-dependently increased by ALS in both cell lines compared to the control cells. In MCF7 cells, the p-p38 MAPK/p38 MAPK ratio was increased from 0.28 at basal level to 0.53 and 0.62, when MCF7 cells were treated with ALS at 1.0 and 5.0 μM, respectively (P<0.001 by one-way ANOVA; Figure 10A and B). In MDA-MB-231 cells, treatment with 0.1, 1.0, and 5.0 μM ALS significantly increased the ratio of p-p38 MAPK/p38 MAPK from 0.31 at basal level to 0.49, 0.62, and 0.80, respectively (P<0.001 by one-way ANOVA; Figure 10A and B).
We further investigated if the activation of p38 MAPK by ALS in breast cancer cell lines was in a time-dependent manner. A time-course study of p38 MAPK phosphorylation in MCF-7 and MDA-MB-231 cells showed that the activation of p38 MAPK was increased over treatment time. In MCF7 cells, the p-p38 MAPK/p38 MAPK ratio was increased from 0.19 at basal level to 0.37, 0.36, 0.32, 0.31, and 0.59, when MCF7 cells were treated with ALS at 1.0 μM for 8, 12, 24, 48, and 72 hours, respectively (P<0.01 by one-way ANOVA; Figure 11A and B). In MDA-MB-231 cells, 1.0 μM ALS for 8, 12, 24, and 72 hours increased the ratio of p-p38 MAPK/p38 MAPK from 0.12 at basal level to 0.25, 0.24, 0.25, and 0.52, respectively (P<0.01 by one-way ANOVA; Figure 11A and B). These observations indicate a possible involvement of p38 MAPK during ALS-induced autophagy.
Effects of p38 MAPK activation on ALS-induced autophagy in MCF7 and MDA-MB-231 cells
We have observed that treatment with ALS for 24 hours resulted in the sustained activation of p38 MAPK and significantly increased expression of LC3-II and beclin 1 in MCF7 and MDA-MB-231 cells. To further investigate how p38 MAPK was involved in ALS-induced autophagy, we co-incubated the cells with the autophagy inhibitors WM (an inhibitor for sequestration step of autophagosome) and bafilomycin A1 (an inhibitor for maturation step of autophagosome). First, we examined the effect of autophagy inhibitors on ALS-induced autophagy in MCF7 and MDA-MB-231 cells using flow cytometry. In MCF7 cells, WM at 10 μM significantly decreased ALS-induced autophagy by 31.4%; in contrast, bafilomycin A1 at 100 nM significantly increased ALS-induced autophagy 1.2-fold, compared to the control cells treated with ALS alone (P<0.01 by one-way ANOVA; Figure 12A and B). Similarly, in MDA-MB-231 cells, WM at 10 μM also markedly decreased ALS-induced autophagy by 45.5%, but bafilomycin A1 at 100 nM increased ALS-induced autophagy 1.3-fold, compared to the control cells treated with ALS alone (P<0.001 by one-way ANOVA; Figure 12A and B).
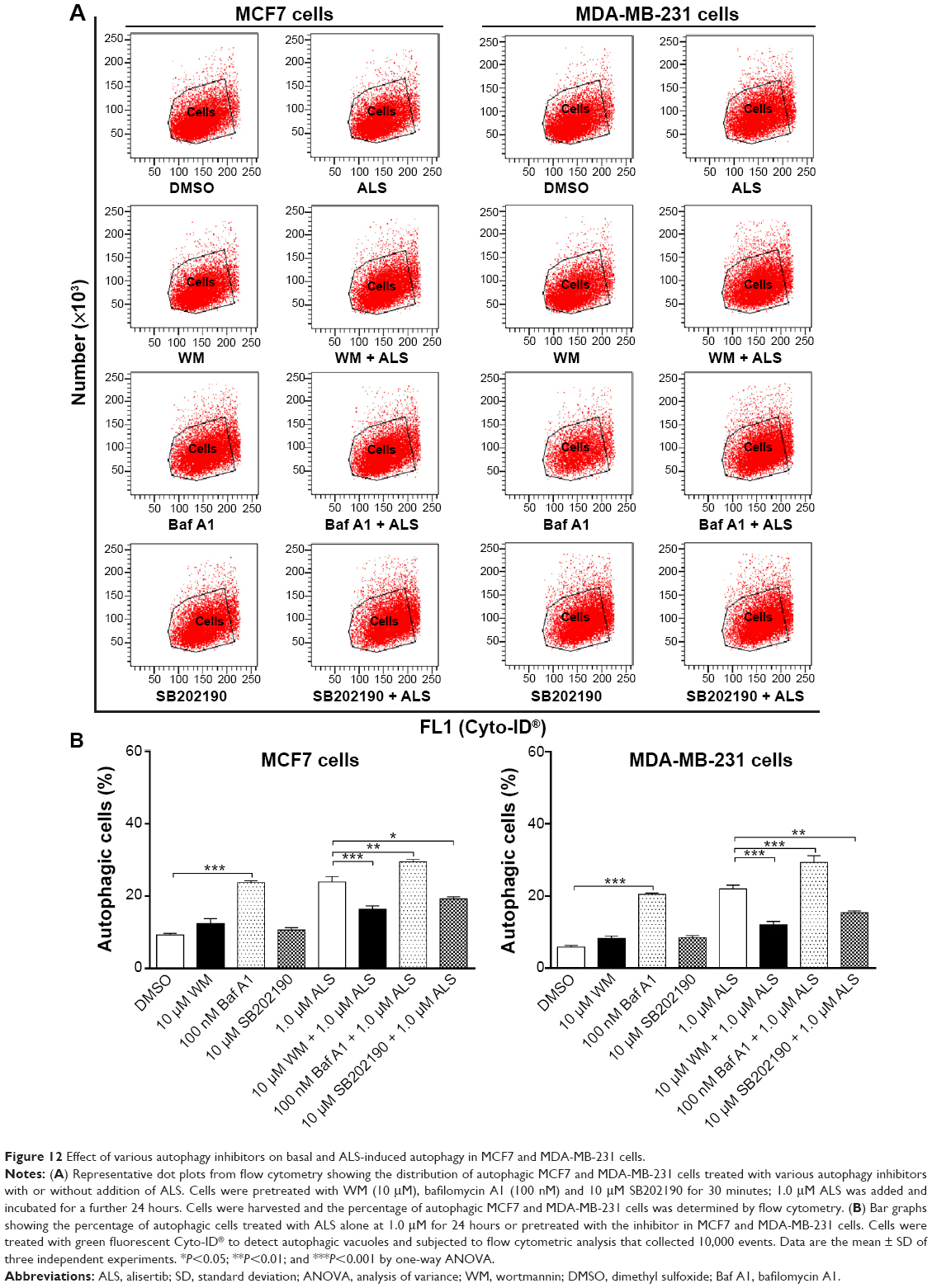 | Figure 12 Effect of various autophagy inhibitors on basal and ALS-induced autophagy in MCF7 and MDA-MB-231 cells. |
Furthermore, we examined the expression levels of beclin 1 and LC3-II when MCF7 and MDA-MB-231 cells were treated with ALS alone or combined with the autophagy inhibitors by Western blotting assay. Treatment with 1.0 μM ALS plus 10 μM WM or 100 nM bafilomycin A1 remarkably decreased the expression level of beclin 1 by 40.9% and 32.7% in MCF7 cells, respectively, compared to the control cells treated with ALS alone (P<0.001; Figure 13A and B). Similarly, treatment of MDA-MB-231 cells with 1.0 μM ALS plus 10 μM WM or 100 nM bafilomycin A1 markedly decreased the level of beclin 1 by 41.1% and 32.4%, respectively, compared to the control cells treated with ALS alone (P<0.001; Figure 13A and B). Moreover, treatment of cells with 1.0 μM ALS plus 10 μM WM decreased the level of LC3-II by 60.5% in MCF7 cells. In MDA-MB-231 cells, treatment of 1.0 μM ALS plus 10 μM WM decreased the level of LC3-II by 60.3%, compared to the control cells treated with ALS alone (P<0.05; Figure 13A and B). Treatment of cells with 1.0 μM ALS plus 100 nM bafilomycin A1 increased the expression level of LC3-II 2.2-fold in MCF7 and MDA-MB-231 cells, compared to the control cells treated with ALS alone (P<0.001; Figure 13A and B).
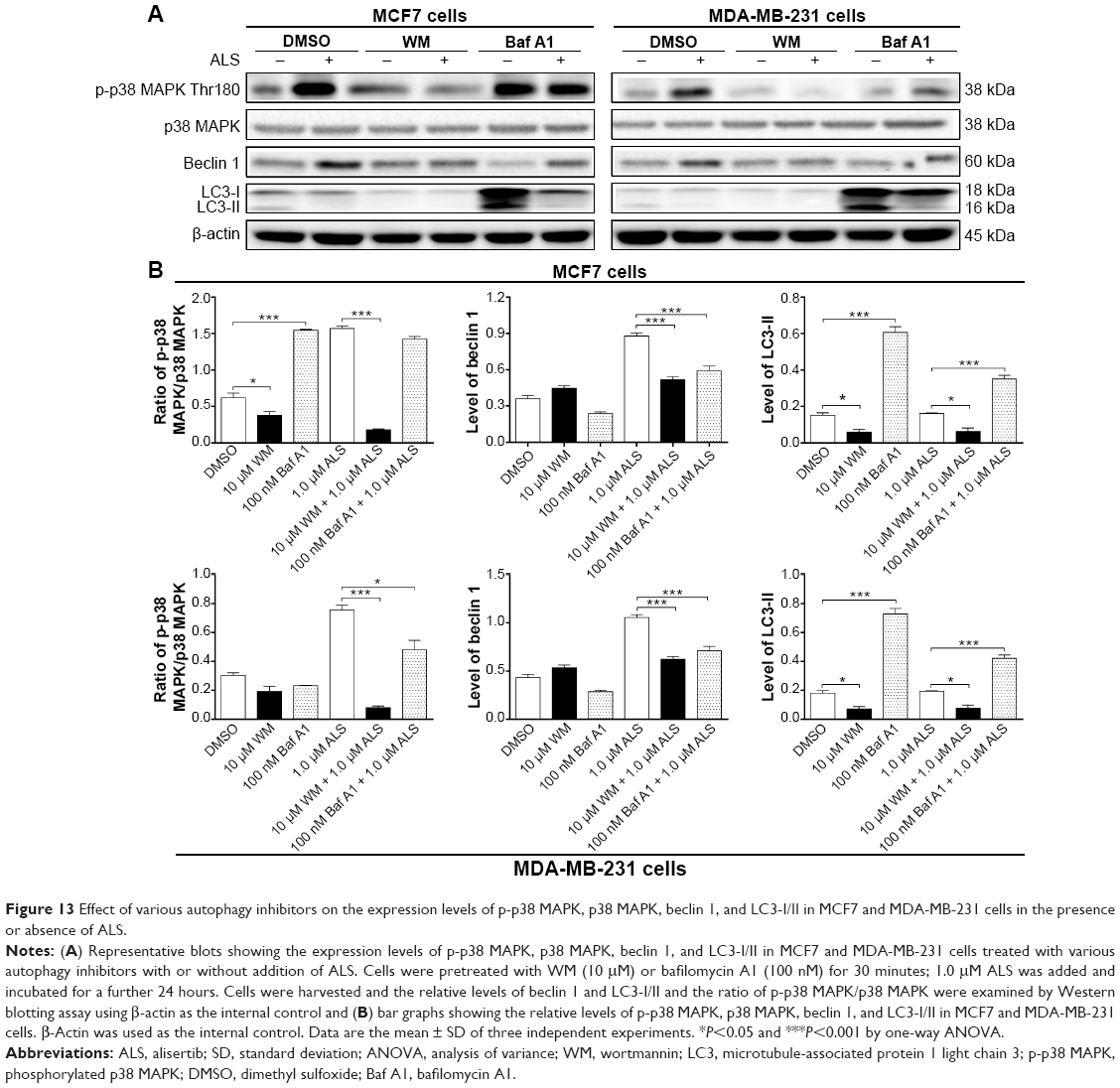 | Figure 13 Effect of various autophagy inhibitors on the expression levels of p-p38 MAPK, p38 MAPK, beclin 1, and LC3-I/II in MCF7 and MDA-MB-231 cells in the presence or absence of ALS. |
Next, we investigated the effects of p38 MAPK activation when cells were pretreated with WM or bafilomycin A1 before ALS treatment. In MCF7 and MDA-MB-231 cells, pretreatment with WM before ALS markedly attenuated p38 MAPK activation. Treatment of cells with 1.0 μM ALS plus 10 μM WM markedly decreased the ratio of p-p38 MAPK/p38 MAPK by 88.5% in MCF7 cells. In MDA-MB-231 cells, treatment with 1.0 μM ALS plus 10 μM WM remarkably decreased the ratio of p-p38 MAPK/p38 MAPK by 89.3%, compared to the control cells treated with ALS alone (P<0.001; Figure 13A and B). Moreover, pretreatment with bafilomycin A1 before ALS also decreased the ratio of p-p38 MAPK/p38 MAPK in MDA-MB-231 cells, compared to the control cells treated with ALS alone (P<0.05 by one-way ANOVA; Figure 13A and B). However, there was no significant difference in MDA-MB-231 cells (P>0.05 by one-way ANOVA; Figure 13A and B). Collectively, these results show that pretreatment with WM inhibits ALS-induced autophagy; whereas pretreatment with bafilomycin A1 does not markedly affect ALS-induced autophagy. These observations suggest that p38 MAPK regulates autophagosome formation at the sequestration step.
p38 MAPK regulates autophagy via Akt activation in MCF7 and MDA-MB-231 cells
To further determine the role of p38 MAPK in ALS-induced autophagy, we used pharmacological inhibitors. Cells were pretreated with SB202190 (a p38 inhibitor) for 30 minutes and then treated with ALS for 24 hours. First, we examined the effect of SB202190 on ALS-induced autophagy in MCF7 and MDA-MB-231 cells using flow cytometry. In MCF7 and MDA-MB-231 cells, treatment with 10 μM SB202190 decreased ALS-induced autophagy by 19.6% and 30.1%, respectively, compared to the control cells treated with ALS alone (P<0.05 by one-way ANOVA; Figure 12A and B). Next, we examined the expression levels of beclin 1 and LC3-II when MCF7 cells were treated with ALS alone or combined with SB202190 by Western blotting assay. Treatment of cells with 1.0 μM ALS plus 10 μM SB202190 did not significantly change the expression level of beclin 1 in MCF7 cells, compared to the control cells treated with ALS alone (P>0.05 by one-way ANOVA; Figure 14A and B). However, treatment of cells with 1.0 μM ALS plus 10 μM SB202190 markedly decreased the level of LC3-II by 84.6% in MCF7 cells, compared to the control cells treated with ALS alone (P<0.001 by one-way ANOVA; Figure 14A and B). In Figure 14, SB202190 partially blocked p38 MAPK activation. Treatment of cells with 1.0 μM ALS plus 10 μM SB202190 remarkably decreased the ratio of p-p38 MAPK/p38 MAPK by 65.4% in MCF7 cells, compared to the control cells treated with ALS alone (P<0.01 by one-way ANOVA; Figure 14A and B). Moreover, treatment of cells with 10 μM SB202190 alone decreased the ratio of p-Akt/Akt by 85.5% in MCF7 cells, compared to the control cells treated with DMSO only (P<0.001 by one-way ANOVA; Figure 14A and B). However, there was no significant change in the ratio of p-Akt/Akt when MCF7 cells were treated with 1.0 μM ALS plus 10 μM SB202190, compared to the control cells treated with ALS alone (P>0.05 by one-way ANOVA; Figure 14A and B). Treatment of cells with 10 μM SB202190 alone did not significantly change the ratio of p-mTOR/mTOR in MCF7 cells, compared to the control cells treated with DMSO only (P>0.05 by one-way ANOVA; Figure 14A and B). However, treatment of cells with 1.0 μM ALS plus 10 μM SB202190 remarkably decreased the ratio of p-mTOR/mTOR by 47.6% in MCF7 cells, compared to the control cells treated with ALS alone (P<0.001 by one-way ANOVA; Figure 14A and B).
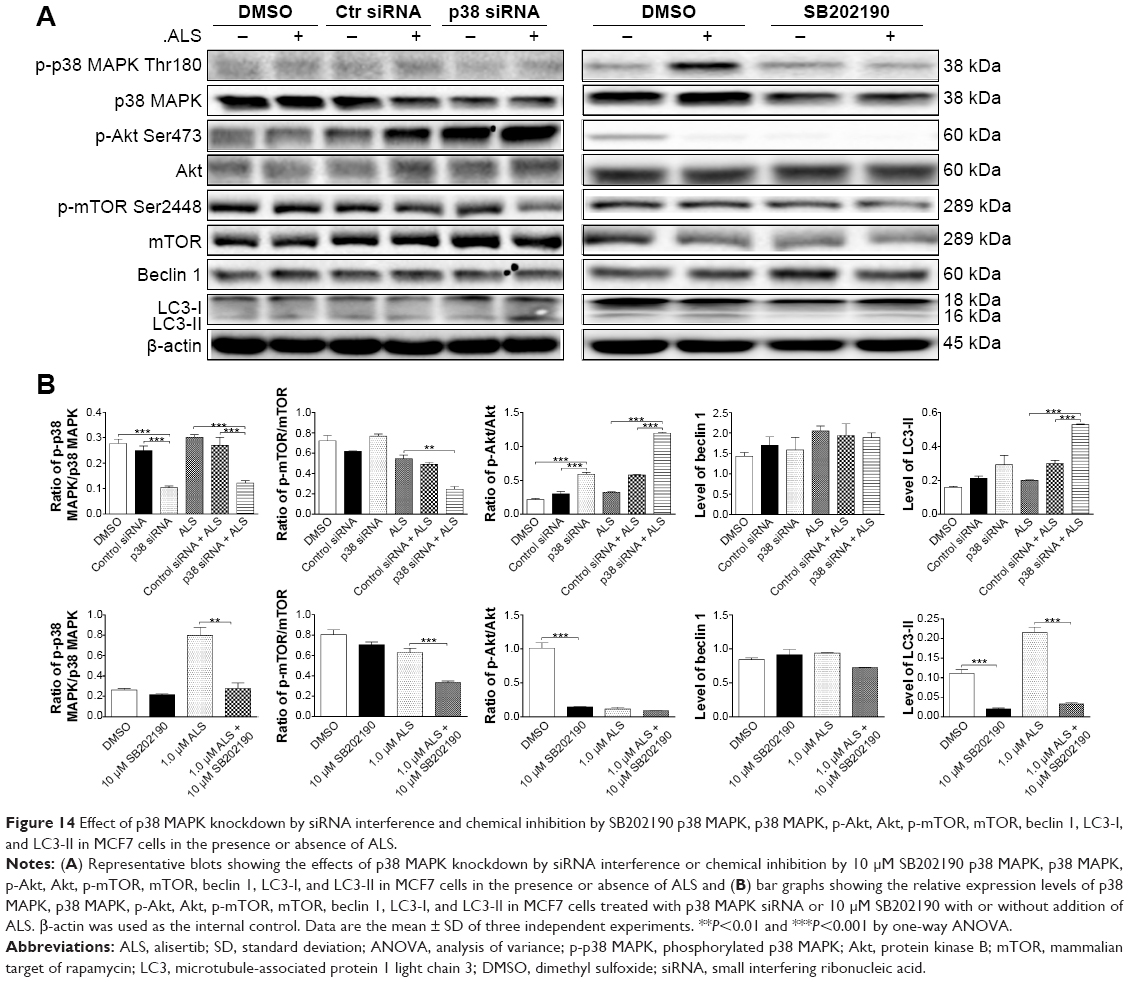 | Figure 14 Effect of p38 MAPK knockdown by siRNA interference and chemical inhibition by SB202190 p38 MAPK, p38 MAPK, p-Akt, Akt, p-mTOR, mTOR, beclin 1, LC3-I, and LC3-II in MCF7 cells in the presence or absence of ALS. |
To further investigate the role of p38 MAPK in the ALS-induced autophagy, p38 MAPK-specific siRNA was used to knock down the p38 MAPK gene. Transfection of MCF-7 cells with p38 MAPK siRNA downregulated the level of ALS-induced p-p38 and increased LC3-II conversion compared with parental or nonspecific siRNA-transfected control cells. Compared to the control cells treated with transfection of MCF-7 cells with control siRNA, transfecting p38 MAPK siRNA decreased the ratio of p-p38 MAPK/p38 MAPK by 58.4% (P<0.001 by one-way ANOVA; Figure 14A and B). After transfection of MCF-7 cells with p38 MAPK siRNA, the ratio of p-Akt/Akt was markedly increased 2.0-fold compared to cells transfected with the control siRNA (P<0.001 by one-way ANOVA; Figure 14A and B). However, there was no significant change in the ratio of p-mTOR/mTOR and the level of beclin 1 and LC3-II in MCF7 cells, compared to cells transfected with the control siRNA (P>0.05 by one-way ANOVA; Figure 14A and B).
Furthermore, we investigated the effect of knockdown of the p38 MAPK gene on ALS-induced autophagy. Compared to the control cells treated with transfection of MCF-7 cells with control siRNA plus 1.0 μM ALS, cells transfected with p38 MAPK siRNA showed a remarkable decrease in the ratio of p-p38 MAPK/p38 MAPK by 54.5% (P<0.001 by one-way ANOVA; Figure 14A and B). After transfection of MCF-7 cells with p38 MAPK siRNA plus 1.0 μM ALS, the ratio of p-Akt/Akt and the level of LC3-II were markedly increased 2.1- and 1.8-fold, compared to cells transfected with the control siRNA plus 1.0 μM ALS (P<0.001 by one-way ANOVA; Figure 14A and B). However, there was no significant change in the ratio of p-mTOR/mTOR and the level of beclin 1 compared to cells transfected with the control siRNA plus 1.0 μM ALS (P>0.05 by one-way ANOVA; Figure 14A and B). Taken together, these results confirm that p38 MAPK is involved in ALS-induced autophagy in MCF7 and MDA-MB-231 cells.
Discussion
Estimates of the worldwide incidence and mortality of cancer ranked breast cancer as the most frequent cancer among women with an estimated 1.67 million new cases diagnosed in 2012 (25% of all cancers) with slightly more cases in less developed than in more developed regions. Breast cancer mortality rates have declined in part due to therapeutic advances and it ranked as the fifth cause of death from cancer overall in 2012. However, it was still the most frequent cause of cancer death in women in less developed regions and the second in more developed regions after lung cancer,20 underscoring the need for better strategies in both prevention and therapy. Recent reports have shown that AURKA can induce chemotherapeutic resistance and regulate several key signaling pathways in cancer cells, suggesting its role as a central node in cancer cell signaling.21 Several AKIs have been tested in breast cancer pre-clinically in the last couple of years, indicating Aurora kinases are promising therapeutic targets for breast cancer treatment. In this study, we investigated the anti-proliferative and pro-apoptotic and pro-autophagic effects of ALS in breast cancer cell lines.
The Aurora kinases (AURKA, AURKB, and AURKC) are serine/threonine kinases that function as key regulators of mitosis.7 Abnormal expression or activation of these proteins leads to the development of cancer, and inhibiting their enzyme activity is potentially a good targeted cancer therapy. Currently more than 30 Aurora inhibitors (A-specific, B- specific, and pan-Aurora) are in early-phase clinical trials.22 ALS is a selective inhibitor of AURKA that is under clinical evaluation for the treatment of solid tumors and hematological malignancies. Our study shows that ALS inhibits proliferation of cells and induced G2/M arrest in breast cancer cell lines. Induced apoptosis and autophagy of cells was also observed in breast cancer cells on ALS exposure.
Cellular proliferation is regulated primarily by the regulation of cell cycle, which consists of four distinct sequential phases (G0/G1, S, G2, and M).23 In eukaryotes, the cell cycle is regulated by cyclins, CDKs, and cyclin-dependent kinases. In particular, cyclin B and CDK1 proteins are involved in the regulation of the progression of G2/M phase.24 It is widely known that cells are blocked in the G2/M phase during DNA damage, and cells are more susceptible to the cytotoxic effects of radiotherapy in the G2/M phase.25 Increasing induced G2/M phase arrest allows cell death which may be a useful strategy in cancer therapeutics.26 In the present study, we found that ALS concentration-dependently arrested MCF7 and MDA-MB-231 cells in G2/M phase. We further explored the effect of ALS on the key regulators in cell cycle checkpoints including CDK1/CDC2, CDK2, and cyclin B1 in MCF7 and MDA-MB-231 cells. The CDC2-cyclin B1 complex is pivotal in regulating the G2/M phase transition and mitosis. We observed a significant decrease in the expression level of cyclin B1 and CDC2 in MCF7 and MDA-MB-231 cells treated with ALS, providing an explanation for the inducing effect of ALS on G2/M phase arrest in MCF7 and MDA-MB-231 cells. The Cip/Kip family, including p21 Waf1/Cip1 and p27 Kip1, binds to cyclin-CDK complexes and prevents the kinase activation, subsequently blocking the progression of the cell cycle at the G2/M phase.27 p53 is a transcription factor that upregulates a number of important cell cycle-modulating genes such as p21 Waf1/Cip1. It has been reported that p21 Waf1/Cip1, a cyclin-dependent kinase inhibitor regulated by p 53, can bind to the CDK1/CDC2-cyclin B1 complex thereby inducing cell cycle arrest. We observed that the expression of p53, p27 Kip1, and p21 Waf1/Cip1 was remarkably increased in MCF7 and MDA-MB-231 cells treated with ALS, which probably contributes to the inhibitory effect of ALS on cell proliferation and the inducing effect on cell cycle arrest in breast cancer cells. These results indicate that upregulation of p53, p21 Waf1/Cip1, and p27 Kip1 expression and suppression of CDC2 and cyclin B1 by ALS may result in the G2/M phase arrest in human breast cancer cells.
Apoptosis is a very tightly programmed cell death with distinct biochemical and genetic pathways that play a critical role in the development and homeostasis in normal tissues.28 It contributes to elimination of unnecessary and unwanted cells to maintain the healthy balance between cell survival and cell death in metazoa.29,30 Apoptosis is caused by proteases, known as “caspases”, which specifically target cysteine aspartyl.31–33 Upon receiving specific signals instructing the cells to undergo apoptosis, a number of distinctive changes occur in the cell. A family of proteins known as caspases is typically activated in the early stages of apoptosis. These proteins cleave key cellular components that are required for normal cellular function including structural proteins in the cytoskeleton and nuclear proteins such as DNA repair enzymes. The caspases can also activate other degradative enzymes such as deoxyribonucleases, which begin to cleave the DNA in the nucleus. Cascade activation of caspases plays a critical role in both pathways, which are linked and both trigger the activation of caspase-3, 6 and 7.32 Tumor cells can acquire resistance to apoptosis by the overexpression of anti-apoptotic proteins such as Bcl-2 or by the downregulation or mutation of pro-apoptotic proteins such as Bax. The expression of both Bcl-2 and Bax is regulated by the p53 tumor suppressor gene.34 Anti-apoptotic members of Bcl-2 can be inhibited by post-translational modification and/or by increased expression of PUMA, which is an essential regulator of p53-mediated cell apoptosis.35 In our study, we found that the expression level of cleaved caspase-9 was significantly increased after ALS treatment, which subsequently activated caspase-3 in MCF7 and MDA-MB-231 cells. Activated caspase-3 in turn induced apoptosis with a decrease in Bcl-2 level. Moreover, we noted a concentration-dependent increase in the expression level of PUMA and Bax in MCF7 and MDA-MB-231 cells. These results indicate that ALS induces mitochondria-dependent apoptosis in breast cancer cells.
Chemotherapy in breast cancer often includes the use of anthracyclines, taxanes (docetaxel and paclitaxel), and DNA damaging agents like cyclophosphamide, fluorouracil or platinum-based compounds.36,37 Most of these agents are known to be good autophagy inducers in different types of cancer.38–41 In breast cancer cell lines, specifically in the MCF7 cell line, ALS was found to induce autophagy. Autophagy is a regulated lysosomal pathway involved in the degradation and recycling of long-lived proteins and organelles within cells. During autophagy, cytoplasmic constituents are sequestered into double-membraned autophagosomes, which then fuse with lysosomes to form autolysosomes, in which degradation occurs. There are many reports that show that autophagy may act as a tumor suppressor, in particular in the initiation of tumor.42,43 Keeping up with this concept, study has shown that induction of autophagy becomes an alternative strategy for cancer treatment.44 Many therapeutic drugs or natural compounds can trigger caspase-independent autophagic cell death by activating autophagy signaling pathways.44,45 Beclin 1 and LC3 are two specific markers of cell autophagy and both of them are strongly involved in autophagic process, especially in its early stage. Beclin 1 is a tumor suppressor which is particularly important for breast cancer since it was found to be deleted in 40%–75% of sporadic human breast and ovarian cancers. Beclin 1 expression is frequently low in human breast epithelial carcinoma cell lines and tissues and expressed at high levels in normal breast epithelia. Overexpression of beclin 1 in the MCF7 breast cancer cell line decreases proliferation, in vitro clonogenicity, and tumorigenesis.46 Beclin 1 promotes autophagy and inhibits proliferation of cancer cells by forming and activating the autophagy promoting complex beclin 1-hVps34, and the suppression of beclin 1 expression impairs autophagy.47 LC3, associated with the formation of the autophagic vacuole, is currently considered as a specific molecular marker for autophagosomes in mammals. LC3 proteins can be divided into two forms: LC3-I (18 kDa) and LC3-II (16 kDa). The amount of LC3-II correlates with the extent of autophagosome formation and is an autophagosomal marker.48 In our study, the induction of autophagy was supported by the following observations. First, we found that ALS treatment of MCF7 and MDA-MB-231 cells induced remarkable autophagy in concentration- and time-dependent manners using flow cytometric analysis. Second, there was a striking increase in the expression level of beclin 1 and LC3-II in MCF7 and MDA-MB-231 cells treated with ALS using Western blotting assay. These results indicate ALS can induce autophagy in breast cancer cells.
Furthermore, we found that ALS significantly induced p38 MAPK and subsequently suppressed the activation of PI3K, Akt, and mTOR. PI3K/Akt/mTOR is a major intracellular signaling pathway, which has received much attention in recent years given its potential role in cancer.49,50 This critical pathway acts as a convergence point for a multitude of upstream signals and in turn stimulates the activity of numerous downstream effectors, thereby mediating enhanced cellular survival, growth, protein synthesis, motility, and other functions of pro-tumorigenic impact.49 As shown by our Western blotting analysis, ALS significantly decreased the activation of PI3K, Akt, and mTOR, but increased LC3-II levels in MCF7 and MDA-MB-231 cells. These results are consistent with the role of PI3K/Akt/mTOR in ALS-induced autophagy.51,52 Our results suggest that the loss of p-mTOR protein is a consequence of the ALS aggregation, and multiple upstream signal inputs to mTOR could lead to such an event. First, we found the classical upstream kinase p-Akt as an essential player in regulating p-mTOR. Accumulation of ALS led to a significant decrease in p-Akt level; low levels of p-Akt produced by the ALS correlated with low levels of p-mTOR and high levels of LC3-II. In our study, ALS significantly reduced the phosphorylation of mTOR at Ser2448 and Akt at Ser473 in both MCF7 and MDA-MB-231 cells. With increasing concentrations of ALS, a decreasing phosphorylation of mTOR and Akt was observed. Similar data were obtained in MCF7 and MDA-MB-231 cells. However, the total Akt and mTOR levels were not changed when MCF7 and MDA-MB-231 cells were treated with ALS. These results suggest that ALS accumulation causes a downregulation of Akt and mTOR function, and reduced Akt and mTOR function might exert a role, at least in part, on the regulation of autophagy.
Notably, there may be crosstalk between apoptosis and autophagy which is induced by ALS in cancer cells.53 The interplay of apoptosis and autophagy is very perplexing.54 Autophagy has been shown to exhibit cytoprotective effects and negatively regulate apoptosis under certain conditions.55 On the contrary, apoptosis suppresses the process of autophagy. It has been shown that mitochondria play a pivotal role in the process of apoptosis and autophagy. Mitochondrial dysfunction and loss of mitochondrial membrane potential lead to the release of pro-apoptotic molecules, which in turn initiates the process of apoptosis. However, autophagy can, to some extent, eliminate the dysfunctional mitochondria, eliciting a cytoprotective effect with a number of cellular signaling molecules and pathways.56–58
Moreover, we also provided evidence about the existence of crosstalk between the p38 MAPK and Akt/mTOR signaling pathways in MCF7 and MDA-MB-231 cells exposed to ALS. The p38 MAPK is an important stress kinase that is involved in regulation of inflammation, cell growth and differentiation, cell cycle, and cell death. Activation of p38 MAPK can lead to an inhibition or induction of autophagy, depending on the cellular context and cell type.59–61 However, the molecular mechanisms that link p38 MAPK to autophagy are not yet fully understood. In the present study, we have observed that ALS treatment significantly increases the phosphorylation of p38 MAPK in MCF7 and MDA-MB-231 cells. With increasing concentration or sustained time of ALS, an increasing amount of p-p38 MAPK and highly accumulated LC3-II was observed. Thus, we speculate that the activation of p38 MAPK by ALS contributes to autophagy induction. We further consolidated this point by pharmacological inhibitors of autophagy. WM, an inhibitor of the sequestration step of autophagosome, completely blocked ALS-induced p38 MAPK activation in MCF7 and MDA-MB-231 cells. These observations suggest that p38 MAPK regulates autophagosome formation at the sequestration step. On the contrary, knockdown of the p38 gene using p38 MAPK siRNA caused accumulation of LC3-II. These observations further confirm that p38 MAPK plays an important role in ALS-induced autophagy.
Our previous studies have demonstrated that ALS induced the activation of p38 MAPK and decreased the activation of Akt and mTOR. To confirm the role of p38 MAPK in ALS-induced autophagy via Akt/mTOR signaling pathway, we knocked down the p38 MAPK gene using p38 MAPK siRNA in MCF7 cells and investigated the change of the phosphorylation of Akt. We found that transfection of MCF7 cells with p38 MAPK siRNA downregulated the level of ALS-induced p-p38 MAPK, and in contrast, upregulated the activation of Akt and led to LC3-II accumulation. Furthermore, we observed a similar effect of the p38 MAPK specific inhibitor SB202190 on ALS-inhibited p-Akt in MCF7 cells. SB 202190 completely inhibited the activation of Akt in MCF7 cells. These results suggest that p38 MAPK is involved in the regulation of Akt activation and autophagy process induced by ALS.
In summary, the present study shows that ALS inhibits cell proliferation, arrests cells in G2/M phase, and induces apoptosis via mitochondria-dependent pathway and autophagy via modulation of p38 MAPK and Akt/mTOR signaling pathways in MCF7 and MDA-MB-231 cells. The crosstalk between apoptosis and autophagy contributes, at least in part, to the breast cancer killing effect of ALS, and the interplay between p38 MAPK and Akt/mTOR signaling pathways contributes to ALS-induced autophagy of MCF7 and MDA-MB-231 cells. This study suggests that ALS may represent a promising therapeutic agent for targeted therapy in breast cancer. More studies are needed to reveal the underlying mechanisms for the anticancer effect of ALS and to verify the efficacy and safety of ALS in the treatment of breast cancer.
Acknowledgment
The authors appreciate the financial support from the Startup Fund of the College of Pharmacy, University of South Florida, Tampa, FL 33612, USA.
Disclosure
There is no conflict of interest in this work.
References
DeSantis C, Ma J, Bryan L, Jemal A. Breast cancer statistics, 2013. CA Cancer J Clin. 2014;64(1):52–62. | ||
Kasami M, Uematsu T, Honda M, et al. Sugimura, Comparison of estrogen receptor, progesterone receptor and Her-2 status in breast cancer pre- and post-neoadjuvant chemotherapy. Breast. 2008;17(5):523–527. | ||
Morgan G, Ward R, Barton M. The contribution of cytotoxic chemotherapy to 5-year survival in adult malignancies. Clin Oncol (R Coll Radiol). 2004;16(8):549–560. | ||
Okada H, Mak TW. Pathways of apoptotic and non-apoptotic death in tumour cells. Nat Rev Cancer. 2004;4(8):592–603. | ||
Fu J, Bian M, Jiang Q, Zhang C. Roles of Aurora kinases in mitosis and tumorigenesis. Mol Cancer Res. 2007;5(1):1–10. | ||
Barr AR, Gergely F. Aurora-A: the maker and breaker of spindle poles. J Cell Sci. 2007;120(Pt 17):2987–2996. | ||
Marumoto T, Honda S, Hara T, et al. Aurora-A kinase maintains the fidelity of early and late mitotic events in HeLa cells. J Biol Chem. 2003;278(51):51786–51795. | ||
Qi W, Cooke LS, Liu X, et al. Mahadevan, Aurora inhibitor MLN8237 in combination with docetaxel enhances apoptosis and anti-tumor activity in mantle cell lymphoma. Biochem Pharmacol. 2011;81(7):881–890. | ||
Falchook G, Kurzrock R, Gouw L, et al. Investigational Aurora A kinase inhibitor alisertib (MLN8237) as an enteric-coated tablet formulation in non-hematologic malignancies: Phase 1 dose-escalation study. Invest New Drugs. 2014;32(6):1181–1187. | ||
Friedberg JW, Mahadevan D, Cebula E, et al. Phase II study of alisertib, a selective Aurora A kinase inhibitor, in relapsed and refractory aggressive B- and T-cell non-Hodgkin lymphomas. J Clin Oncol. 2014;32(1):44–50. | ||
Goldberg SL, Fenaux P, Craig MD, et al. An exploratory phase 2 study of investigational Aurora A kinase inhibitor alisertib (MLN8237) in acute myelogenous leukemia and myelodysplastic syndromes. Leuk Res Rep. 2014;3(2):58–61. | ||
Matulonis UA, Sharma S, Ghamande S, et al. Phase II study of MLN8237 (alisertib), an investigational Aurora A kinase inhibitor, in patients with platinum-resistant or -refractory epithelial ovarian, fallopian tube, or primary peritoneal carcinoma. Gynecol Oncol. 2012;127(1):63–69. | ||
Leontovich AA, Salisbury JL, Veroux M, et al. Inhibition of Cdk2 activity decreases Aurora-A kinase centrosomal localization and prevents centrosome amplification in breast cancer cells. Oncol Rep. 2013;29(5):1785–1788. | ||
Jensen JS, Omarsdottir S, Thorsteinsdottir JB, Ogmundsdottir HM, Olafsdottir ES. Synergistic cytotoxic effect of the microtubule inhibitor marchantin A from Marchantia polymorpha and the Aurora kinase inhibitor MLN8237 on breast cancer cells in vitro. Planta Med. 2012;78(5):448–454. | ||
Huck JJ, Zhang M, Mettetal J, et al. Translational exposure-efficacy modeling to optimize the dose and schedule of taxanes combined with the investigational Aurora A kinase inhibitor MLN8237 (alisertib). Mol Cancer Ther. 2014;13(9):2170–2183. | ||
Li YC, He SM, He ZX, et al. Plumbagin induces apoptotic and autophagic cell death through inhibition of the PI3K/Akt/mTOR pathway in human non-small cell lung cancer cells. Cancer Lett. 2014;344(2):239–259. | ||
Kang R, Zeh HJ, Lotze MT, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011;18(4):571–580. | ||
Kabeya Y, Mizushima N, Ueno T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19(21):5720–5728. | ||
Zhang C, Kawauchi J, Adachi MT, et al. Activation of JNK and transcriptional repressor ATF3/LRF1 through the IRE1/TRAF2 pathway is implicated in human vascular endothelial cell death by homocysteine. Biochem Biophys Res Commun. 2001;289(3):718–724. | ||
Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359–E386. | ||
Dar AA, Goff LW, Majid S, Berlin J, El-Rifai W. Aurora kinase inhibitors – rising stars in cancer therapeutics? Mol Cancer Ther. 2010;9(2):268–278. | ||
Cheung CH, Sarvagalla S, Lee JY, Huang YC, Coumar MS. Aurora kinase inhibitor patents and agents in clinical testing: an update (2011–2013). Expert Opin Ther Pat. 2014;24(9):1021–1038. | ||
Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407(6805):770–776. | ||
Cho HJ, Oh YJ, Han SH, et al. Cdk1 protein-mediated phosphorylation of receptor-associated protein 80 (RAP80) serine 677 modulates DNA damage-induced G2/M checkpoint and cell survival. J Biol Chem. 2013;288(6):3768–3776. | ||
Pawlik TM, Keyomarsi K. Role of cell cycle in mediating sensitivity to radiotherapy. Int J Radiat Oncol Biol Phys. 2004;59(4):928–942. | ||
Montenegro MF, Sanchez-Del-Campo L, Fernandez-Perez MP, Saez-Ayala M, Cabezas-Herrera J, Rodriguez-Lopez JN. Targeting the epigenetic machinery of cancer cells. Oncogene. 2015;34(2):135–143. | ||
Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 2004;73:39–85. | ||
Galluzzi L, Bravo-San Pedro JM, Vitale I, et al. Essential versus accessory aspects of cell death: recommendations of the NCCD 2015. Cell Death Differ. 2015;22(1):58–73. | ||
Cotter TG. Apoptosis and cancer: the genesis of a research field. Nat Rev Cancer. 2009;9(7):501–507. | ||
Lee MJ, Ye AS, Gardino AK, et al. Sequential application of anticancer drugs enhances cell death by rewiring apoptotic signaling networks. Cell. 2012;149(4):780–794. | ||
Thornberry NA, Lazebnik Y. Caspases: enemies within. Science. 1998;281(5381):1312–1316. | ||
Lakhani SA, Masud A, Kuida K. Caspases 3 and 7: key mediators of mitochondrial events of apoptosis. Science. 2006;311(5762):847–851. | ||
Thornberry NA. Caspases: key mediators of apoptosis. Chem Biol. 1998;5(5):R97–R103. | ||
Miyashita T, Krajewski S, Krajewska M, et al. Tumor suppressor p53 is a regulator of bcl-2 and bax gene expression in vitro and in vivo. Oncogene. 1994;9(6):1799–1805. | ||
Taylor RC, Cullen SP, Martin SJ. Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol. 2008;9(3):231–241. | ||
Bese NS. Radiochemotherapy in the treatment of breast cancer. Clin Oncol (R Coll Radiol). 2009;21(7):532–535. | ||
Oakman C, Viale G, Di Leo A. Management of triple negative breast cancer. Breast. 2010;19(5):312–321. | ||
Kung CP, Budina A, Balaburski G, Bergenstock MK, Murphy M. Autophagy in tumor suppression and cancer therapy. Crit Rev Eukaryot Gene Expr. 2011;21(1):71–100. | ||
Li JL, Han SL, Fan X. Modulating autophagy: a strategy for cancer therapy. Chin J Cancer. 2011;30(10):655–668. | ||
Yang ZJ, Chee CE, Huang S, Sinicrope F. Autophagy modulation for cancer therapy. Cancer Biol Ther. 2011;11(2):169–176. | ||
Maycotte P, Thorburn A. Autophagy and cancer therapy. Cancer Biol Ther. 2011;11(2):127–137. | ||
Liang C, Feng P, Ku B, et al. Autophagic and tumour suppressor activity of a novel Beclin 1-binding protein UVRAG. Nat Cell Biol. 2006;8(7):688–699. | ||
Mathew R, Karp CM, Beaudoin B, et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009;137(6):1062–1075. | ||
Kondo Y, Kanzawa T, Sawaya R, Kondo S. The role of autophagy in cancer development and response to therapy. Nat Rev Cancer. 2005;5(9):726–734. | ||
Xu ZX, Liang J, Haridas V, et al. A plant triterpenoid, avicin D, induces autophagy by activation of AMP-activated protein kinase. Cell Death Differ. 2007;14(11):1948–1957. | ||
Liang XH, Jackson S, Seamanet M, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402(6762):672–676. | ||
Wang W, Fan H, Zhou Y, Duan P, Zhao G, Wu G. Knockdown of autophagy-related gene BECLIN1 promotes cell growth and inhibits apoptosis in the A549 human lung cancer cell line. Mol Med Rep. 2013;7(5):1501–1505. | ||
Lee YJ, Hah YJ, Kang YN, et al. The autophagy-related marker LC3 can predict prognosis in human hepatocellular carcinoma. PLoS One. 2013;8(11):e81540. | ||
Vivanco I, Sawyers CL. The phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2(7):489–501. | ||
Luo J, Manning BD, Cantley LC. Targeting the PI3K-Akt pathway in human cancer: rationale and promise. Cancer Cell. 2003;4(4):257–262. | ||
Ravikumar B, Vacher C, Berger Z, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36(6):585–595. | ||
Yamamoto A, Cremona ML, Rothman JE. Autophagy-mediated clearance of huntingtin aggregates triggered by the insulin-signaling pathway. J Cell Biol. 2006;172(5):719–731. | ||
Ding YH, Zhou ZW, Ha CF, et al. The Aurora kinase A inhibitor alisertib induces apoptosis and autophagy but inhibits epithelial to mesenchymal transition in human epithelial ovarian cancer cells. Drug Des Devel Ther. 2015;9:425–464. | ||
Su M, Mei Y, Sinha S. Role of the crosstalk between autophagy and apoptosis in cancer. J Oncol. 2013;2013:102735. | ||
Bhogal RH, Weston CJ, Curbishley SM, Adams DH, Afford SC. Autophagy: a cyto-protective mechanism which prevents primary human hepatocyte apoptosis during oxidative stress. Autophagy. 2012;8(4):545–558. | ||
Levine B, Sinha S, Kroemer G. Bcl-2 family members: dual regulators of apoptosis and autophagy. Autophagy. 2008;4(5):600–606. | ||
Yousefi S, Simon HU. Apoptosis regulation by autophagy gene 5. Crit Rev Oncol Hematol. 2007;63(3):241–244. | ||
Marquez RT, Xu L. Bcl-2: Beclin 1 complex: multiple, mechanisms regulating autophagy/apoptosis toggle switch. Am J Cancer Res. 2012;2(2):214–221. | ||
Brancho D, Tanaka N, Jaeschke A, et al. Mechanism of p38 MAP kinase activation in vivo. Genes Dev. 2003;17(16):1969–1978. | ||
Brancho D, Ventura JJ, Jaeschke A, Doran B, Flavell RA, Davis RJ. Role of MLK3 in the regulation of mitogen-activated protein kinase signaling cascades. Mol Cell Biol. 2005;25(9):3670–3681. | ||
Qin Y, Zhou ZW, Pan ST, et al. Graphene quantum dots induce apoptosis, autophagy, and inflammatory response via p38 mitogen-activated protein kinase and nuclear factor-kB mediated signaling pathways in activated THP-1 macrophages. Toxicology. 2015;327:62–76. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.