")
Back to Journals » Drug Design, Development and Therapy » Volume 9
The clinicopathological significance and drug target potential of FHIT in breast cancer, a meta-analysis and literature review
Authors Su Y, Wang X, Li J, Xu J, Xu L
Received 4 June 2015
Accepted for publication 17 August 2015
Published 1 October 2015 Volume 2015:9 Pages 5439—5445
DOI https://doi.org/10.2147/DDDT.S89861
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Wei Duan
Yunshu Su,1 Xiaoli Wang,1,2 Jun Li,1 Junming Xu,3 Lijun Xu1
1Department of Cardiothoracic Surgery, 2Cancer Biology Research Center, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, Hubei, 3Department of General Surgery, Shanghai First People’s Hospital, Shanghai Jiaotong University, Shanghai, People’s Republic of China
Abstract: FHIT is a bona fide tumor-suppressor gene and its loss contributes to tumorigenesis of epithelial cancers including breast cancer (BC). However, the association and clinicopathological significance between FHIT promoter hypermethylation and BC remains unclear. The purpose of this study is to conduct a meta-analysis and literature review to investigate the clinicopathological significance of FHIT methylation in BC. A detailed literature search was performed in PubMed, EMBASE, Web of Science, and Google Scholar databases. The data were extracted and assessed by two reviewers independently. Odds ratios with 95% corresponding confidence intervals were calculated. A total of seven relevant articles were available for meta-analysis, which included 985 patients. The frequency of FHIT hypermethylation was significantly increased in invasive ductal carcinoma compared to benign breast disease, the pooled odds ratio was 8.43, P<0.00001. The rate of FHIT hypermethylation was not significantly different between stage I/II and stage III/IV, odds ratio was 2.98, P=0.06. In addition, FHIT hypermethylation was not significantly associated with ER and PR status. FHIT hypermethylation was not significantly correlated with premenopausal and postmenopausal patients with invasive ductal carcinoma. In summary, our meta-analysis indicated that the frequency of FHIT hypermethylation was significantly increased in BC compared to benign breast disease. The rate of FHIT hypermethylation in advanced stages of BC was higher than in earlier stages; however, the difference was not statistically significant. Our data suggested that FHIT methylation could be a diagnostic biomarker of BC carcinogenesis. FHIT is a potential drug target for development of demethylation treatment for patients with BC.
Keywords: FHIT, methylation, tumor suppressor gene, meta-analysis, odds ratio, drug target
Introduction
Breast cancer (BC) is the most frequently diagnosed cancer in women and the major cause of cancer-related female mortality in the USA.1 A series of epigenetic alterations and genetic abnormalities contribute to this process of BC onset and progression. Epigenetic alterations, which occur in transformed cells, involve changes in DNA methylation, including global hypomethylation, focal hypermethylation, histone modifications, and nucleosomal remodeling.2 Specifically, abnormal promoter region methylation in tumor suppressor genes results in loss of gene function which contributes to tumorigenesis of epithelial cancers.3 Therefore, it is critical to identify biomarkers for diagnosis and new molecular targets for development of personalized therapy. Common fragile sites are large chromosomal regions that are hot spots for alterations, especially in cancer cells. The three most frequently expressed common fragile site regions are FRA3B, FRA16D, and FRA6E which contain FHIT, WWOX, and PARK2.4,5 The FHIT gene is a bona fide tumor-suppressor gene present on the short arm of chromosome 3 and its loss of function has been evaluated in different types of cancers including BC.6,7 FHIT plays an important role in pro-apoptotic signaling, cell cycle control, and sensitivity to DNA damaging agents.8–10 Viral-mediated FHIT gene transfer to FHIT-deficient mice not only prevents but reverses the carcinogen-induced tumor development in vivo and restoration of FHIT protein induces tumor suppression in 50% of tumor cell lines tested in vitro.11 However, the association and clinical significance between FHIT promoter hypermethylation and BC remains under investigation. In this study, we systematically reviewed studies of FHIT promoter hypermethylation in this process of BC onset and progression, and quantified the association between FHIT promoter hypermethylation and BC by using meta-analysis methods. In addition, we summarize these findings and discuss the tumor suppressor function, as well as the clinical significance of FHIT in BC.
Materials and methods
Search strategy and selection criteria
We performed comprehensive literature searches in PubMed, EMBASE, Web of Science, and Google Scholar databases in May 2015 with no limit set for date and language of publication using the search terms: “breast cancer or breast carcinoma”, “methylation”, and “FHIT, or Fragile histidine triad”. There were 103 articles identified from PubMed, 30 articles from EMBASE, 85 articles from Web of Science, and 16,600 articles from Google Scholar, first 400 of them were screened because the rest of them were not related to the present study. A total of 618 articles were screened by article titles and abstracts.
After screening by titles and abstracts, individual studies were screened using the inclusion and exclusion criteria. We included studies that met the following criteria: 1) studies that evaluated FHIT hypermethylation in the primary BC tissues, 2) research that revealed the relationship between FHIT hypermethylation and BC clinicopathological parameters, 3) FHIT hypermethylation examined by polymerase chain reaction. The exclusion criteria included the following: 1) reviews, case reports, letters, editorials, expert opinion, conference abstracts and 2) all studies using cell lines serum, human xenografts, and in vitro/ex vivo studies were also excluded. The search process was conducted independently by two reviewers (XW and JL), discrepancies were discussed and resolved by the third reviewer (JX). Forward and backward citation chasing of each included article was conducted. The most complete study was chosen to avoid duplication if the same patient populations were reported in several publications. Seven articles were eligible for inclusion in this meta-analysis.
Data extraction and methodological assessment
Two authors (YS, XW) independently reviewed and extracted the following data: last name of the first author, year of publication, country(ies) where the study was conducted, number of BC cases, clinicopathological parameters, cancer tumor-node-metastasis stage, methylation detection method, methylation rate and/or expression. The detailed information of seven relevant articles is listed in Table 1. Heterogeneity of investigation was evaluated to determine whether or not the data of various studies could be analyzed for a meta-analysis.
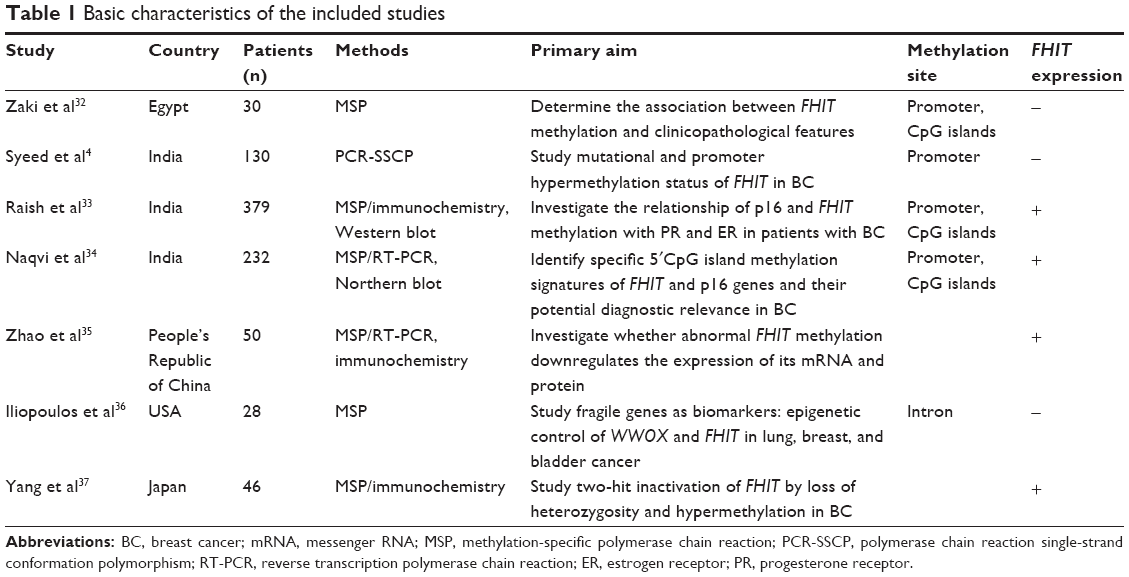 | Table 1 Basic characteristics of the included studies |
For the methodological evaluation of the studies, three investigators (JL, XW, and JX) read through each publication independently, and they assessed and scored them according to the Reporting Recommendations for Tumor Marker Prognostic Studies guidelines and European Lung Cancer Working Party quality scale.12,13 Three readers provided the quality scores and compared them, and then reached a consensus value for each item.
Statistical analysis
Analysis was conducted using Review Manager 5.2 (Cochrane Collaboration, Oxford, UK). The pooled odds ratios (ORs) with its 95% confidence intervals (CIs) were calculated. Heterogeneity among studies was estimated using the Cochran’s Q statistic and I2 tests.14 The I2 statistics were used to examine the difference for between-study variability due to heterogeneity rather than chance, with a range from 0% to 100%. When heterogeneity (I2) was less than 50%, a fixed effect model was used to calculate parameters. If there was substantial heterogeneity (I2 values ≥50%), a random-effects model was used to pool data and attempt to identify potential sources of heterogeneity based on subgroup analyses. The analysis was performed to compare the frequency of FHIT methylation between BC and nonmalignant breast tissue. The frequency of FHIT hypermethylation was compared in different tumor characteristics. The pooled ORs were estimated for the correlation between FHIT hypermethylation and clinicopathological features. P-values tailed less than 0.05 were considered statistically significant. Publication bias is what occurs whenever the research in the published literature is systematically unrepresentative of population of completed studies. Funnel plots were used for detection of publication bias.
Results
Identification of relevant studies
The initial database search resulted in a total of 618 citations. After review, seven articles were included for meta-analysis, as shown in Figure 1.
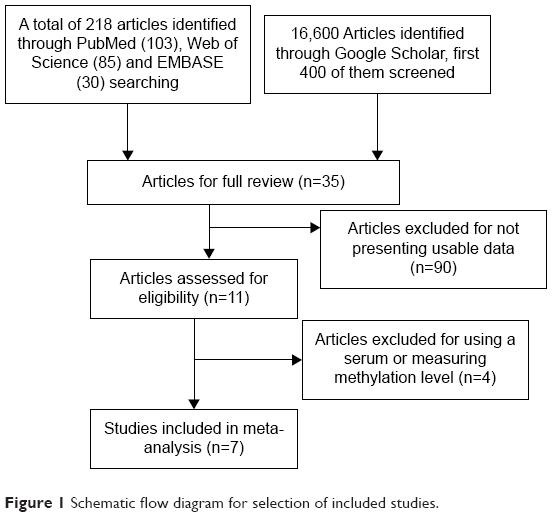 | Figure 1 Schematic flow diagram for selection of included studies. |
Study characteristics
Seven studies published from 2001 to 2011 were eligible for meta-analysis. A total of 985 BC patients from India, People’s Republic of China, Japan, Egypt, and the USA were enrolled. Their basic characteristics are summarized in Table 1.
The correlation of FHIT hypermethylation with clinicopathological features
The frequency of FHIT hypermethylation was significantly increased in invasive ductal carcinoma (IDC) compared to benign breast disease, the pooled OR was 8.43 with 95% CI 4.31–16.50, z=6.22, P<0.00001 (Figure 2). FHIT hypermethylation rate was not significantly correlated with stage, although FHIT was more frequently methylated in advanced stages (stage III/IV) than earlier stages (I/II), closely approaching statistical significance, OR was 2.98 with 95% CI 0.94–9.40, z=1.86, P=0.06 (Figure 3). FHIT hypermethylation rate was not associated with ER status, the rate of FHIT hypermethylation was not higher in ER-positive IDC than that in ER-negative IDC, OR was 1.25 with 95% CI 0.37–4.23, z=0.36, P=0.72 (Figure 4). FHIT hypermethylation rate was not associated with PR status, the rate of FHIT hypermethylation was similar between PR-positive and PR-negative IDC, OR was 0.66 with 95% CI 0.12–3.57, z=0.49, P=0.63 (Figure 5). FHIT hypermethylation was not related to premenopausal and postmenopausal patients with IDC. The frequency of FHIT hypermethylation was similar between postmenopausal and premenopausal IDC patients, OR was 0.87 with 95% CI 0.43–1.79, z=0.37, P=0.71 (Figure 6).
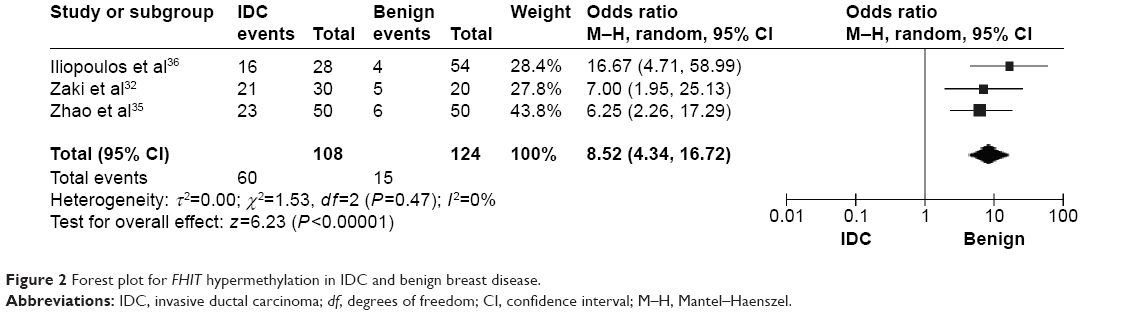 | Figure 2 Forest plot for FHIT hypermethylation in IDC and benign breast disease. |
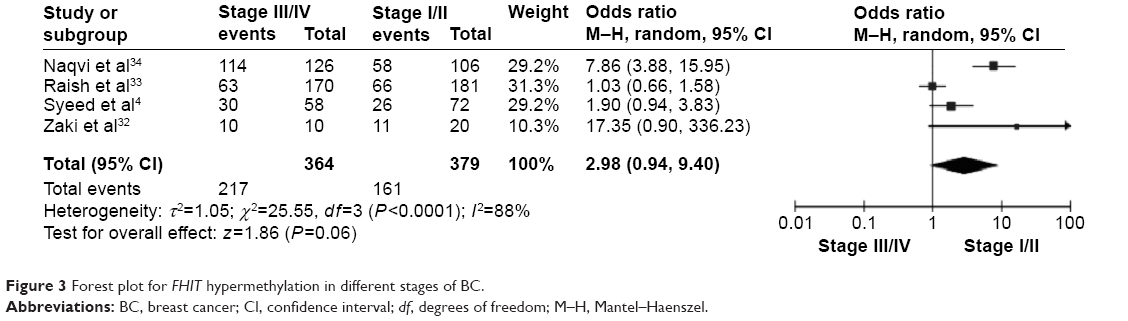 | Figure 3 Forest plot for FHIT hypermethylation in different stages of BC. |
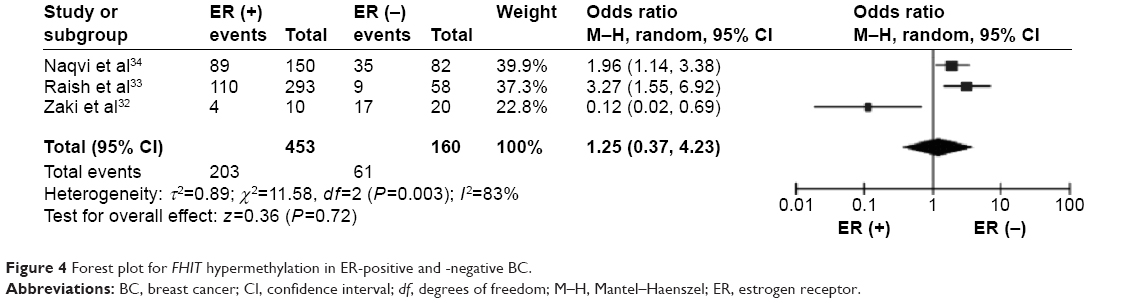 | Figure 4 Forest plot for FHIT hypermethylation in ER-positive and -negative BC. |
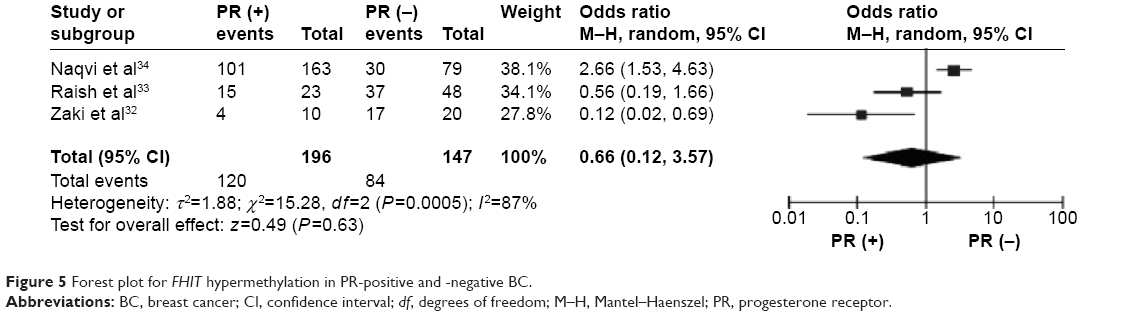 | Figure 5 Forest plot for FHIT hypermethylation in PR-positive and -negative BC. |
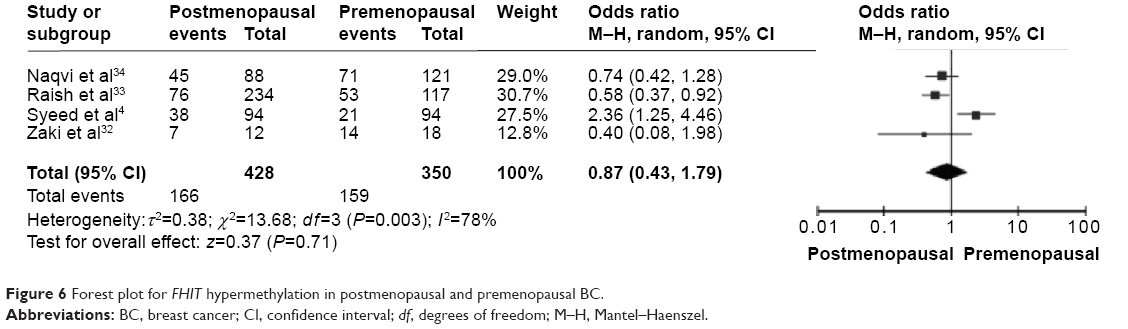 | Figure 6 Forest plot for FHIT hypermethylation in postmenopausal and premenopausal BC. |
Study quality, sensitivity analyses, and publication bias
The quality of each study was assessed with the Newcastle Ottawa Quality Assessment Scale. These scales were utilized to allocate a maximum of nine points for the quality of selection, comparability, exposure, and outcomes for study participants, and a score ≥7 indicates a good quality. Of the studies, four scored eight points, two scored seven points, and one scored six points. Hence, the studies were of a relatively high quality (data not shown). A sensitivity analysis was conducted by removing one study from the meta-analysis at a time; the overall results were not significantly affected. The pooled ORs were not significantly changed, indicating the stability of our analyses. The funnel plots were largely symmetric (Figure 7A–E), suggesting there were no publication biases in the meta-analysis of FHIT hypermethylation and clinicopathological features.
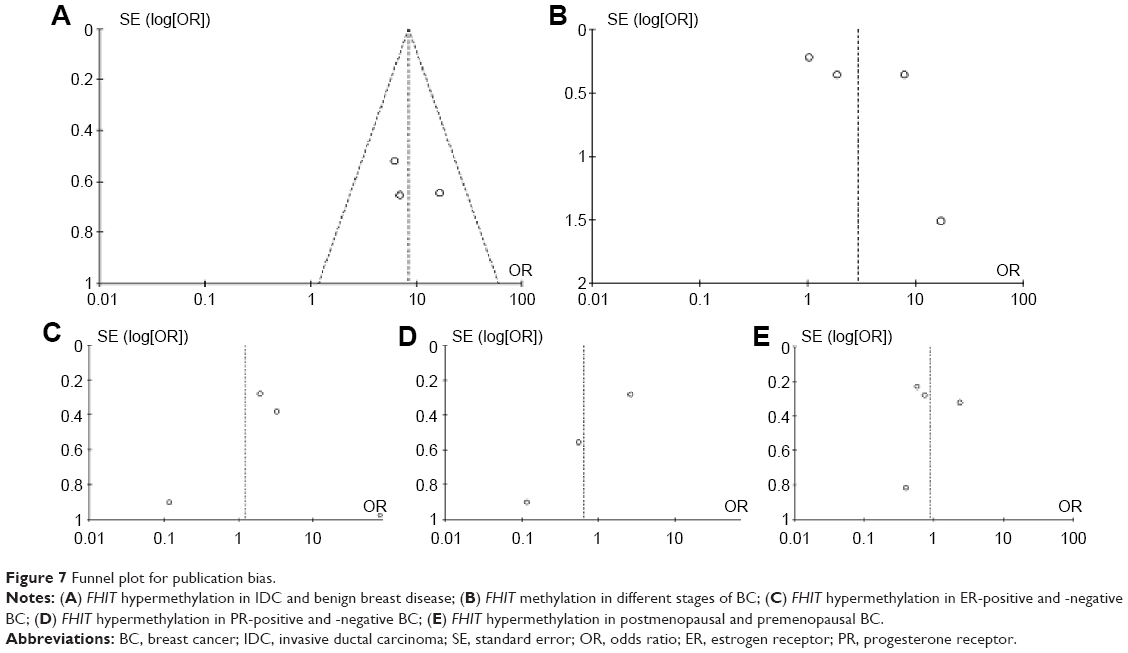 | Figure 7 Funnel plot for publication bias. |
Discussion
Aberrant DNA methylation has been recognized to contribute to breast carcinogenesis, and promoter hypermethylation of several tumor suppressor genes including APC, GSTP1, P14 (ARF), P16 (CDKN2A), P21 (CDKN1A), PTEN, and TIMP3 has been correlated with decreased gene expression.5,15–17 FHIT gene, encompassing the FRA3B fragile site at chromosome 3p14.2, is a putative tumor suppressor gene involved in the pathogenesis of different tumor types including BC.18,19 FHIT messenger RNA (mRNA) is expressed in most normal human tissue, with highest level in epithelial cells and tissues.20 The FHIT gene is inactivated by genetic and epigenetic changes, ie, loss of heterozygosity or promoter hypermethylation, in numerous cancers.21 Originally, the absence of FHIT protein in tumors was examined as a loss of heterozygosity, mainly due to biallelic deletions of the gene. Recent evidence indicates that inactivation of FHIT by promoter hypermethylation plays an important role in tumorigenesis in several types of tumors including BC.22–27 However, the reported correlation between FHIT methylation and clinicopathological features in BC was inconsistent. We conducted the meta-analysis to determine the association between FHIT hypermethylation and clinicopathological characteristics in BC.
Our data revealed that the frequency of FHIT hypermethylation in BC was 8.43 times higher than that in benign breast tissues. Haroun et al observed that the methylation frequencies of the genes tested in non-small cell lung carcinoma specimens were 53.6% for FHIT.28 Jeong et al evaluated the methylation of FHIT in 60 BC samples; FHIT methylation was detected in 96.7% and the positive expression rate of FHIT in 87.3% of the patients.27 Our data and previous studies indicate that FHIT promoter hypermethylation is a useful diagnostic biomarker of BC. FHIT promoter hypermethylation is reversible, drug treatment through demethylation may be useful to delay tumorigenesis and progression, as well as improving prognosis. Inhibitors of DNA methylation such as 5-Aza-CdR and FdCyd were applied to human BC cells and induced apoptosis,16,28 and FdCyd is now in clinical trials for the treatment of BC and other solid tumors.29–31 More specifically, Stewart et al observed that there was a statistically significant increase in expression of FHIT as well as two other tumor suppressor genes, FUS1 and WWOX, in patients with tumor regression following decitabine, which is a demethylating agent.19 These preclinical studies and early clinical trials showed the potential to restore the activity of epigenetically silenced FHIT through epigenetic modulating agents and promise of reexpressed tumor suppressors as markers and effectors of responses. This approach provides a promising drug target for development of personalized therapy of BC in the future.
Our data showed the nonsignificant association of FHIT hypermethylation with ER, PR, and menopausal status of BC patients. Those data were different from previous studies which reported significant associations between FHIT hypermethylation and ER-negative as well as in PR-negative cases. Since the power of our analysis is small, further evaluation needs to be carried out in a larger sample study. Sensitivity analysis showed that no individual study significantly biased the pooled result and no evidence of publication bias was found. There are several potential limitations: 1) the search strategy was restricted to articles published in English and Chinese, some studies published in other languages were not included in current study; 2) the possibility of information and selection biases as well as unidentified confounders could not be completely excluded because all of the included studies were observational. Therefore, caution should be exercised when our findings are interpreted among the general population.
In conclusion, our meta-analysis indicated that the frequency of FHIT hypermethylation was significantly increased in BC compared to benign breast disease. Our data suggested FHIT could be a diagnostic biomarker. In addition, FHIT is a potential drug target for development of demethylation treatment for patients with BC.
Acknowledgments
This work is supported by the National Natural Sciences Foundation of China (30901364). The funding institution does not have any role in the study design, data collection, or analysis.
Disclosure
The authors declare that they have no competing interests, and have no financial disclosures relevant to this work.
References
Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63(1):11–30. | ||
Atalay C. Epigenetics in breast cancer. Exp Oncol. 2013;35(4):246–249. | ||
Wali A. FHIT: doubts are clear now. ScientificWorldJournal. 2010; 10:1142–1151. | ||
Syeed N, Husain SA, Sameer AS, Chowdhri NA, Siddiqi MA. Mutational and promoter hypermethylation status of FHIT gene in breast cancer patients of Kashmir. Mutat Res. 2011;707(1–2):1–8. | ||
Szyf M. DNA methylation signatures for breast cancer classification and prognosis. Genome Med. 2012;4(3):26. | ||
Pekarsky Y, Palamarchuk A, Huebner K, Croce CM. FHIT as tumor suppressor: mechanisms and therapeutic opportunities. Cancer Biol Ther. 2002;1(3):232–236. | ||
Hassan MI, Naiyer A, Ahmad F. Fragile histidine triad protein: structure, function, and its association with tumorogenesis. J Cancer Res Clin Oncol. 2010;136(3):333–350. | ||
Huang Q, Liu Z, Xie F, et al. Fragile histidine triad (FHIT) suppresses proliferation and promotes apoptosis in cholangiocarcinoma cells by blocking PI3K-Akt pathway. ScientificWorldJournal. 2014; 2014:179698. | ||
Rimessi A, Marchi S, Fotino C, et al. Intramitochondrial calcium regulation by the FHIT gene product sensitizes to apoptosis. Proc Natl Acad Sci U S A. 2009;106(31):12753–12758. | ||
Trapasso F, Pichiorri F, Gaspari M, et al. Fhit interaction with ferredoxin reductase triggers generation of reactive oxygen species and apoptosis of cancer cells. J Biol Chem. 2008;283(20):13736–13744. | ||
Ishii H, Vecchione A, Fong LY, et al. Cancer prevention and therapy in a preclinical mouse model: impact of FHIT viruses. Curr Gene Ther. 2004;4(1):53–63. | ||
McShane LM, Altman DG, Sauerbrei W, et al. Reporting recommendations for tumor marker prognostic studies (REMARK). J Natl Cancer Inst. 2005;97(16):1180–1184. | ||
Steels E, Paesmans M, Berghmans T, et al. Role of p53 as a prognostic factor for survival in lung cancer: a systematic review of the literature with a meta-analysis. Eur Respir J. 2001;18(4):705–719. | ||
Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta-analyses. BMJ. 2003;327(7414):557–560. | ||
Radpour R, Kohler C, Haghighi MM, Fan AX, Holzgreve W, Zhong XY. Methylation profiles of 22 candidate genes in breast cancer using high-throughput MALDI-TOF mass array. Oncogene. 2009;28(33):2969–2978. | ||
Davis NM, Sokolosky M, Stadelman K, et al. Deregulation of the EGFR/PI3K/PTEN/Akt/mTORC1 pathway in breast cancer: possibilities for therapeutic intervention. Oncotarget. 2014;5(13):4603–4650. | ||
Klarmann GJ, Decker A, Farrar WL. Epigenetic gene silencing in the Wnt pathway in breast cancer. Epigenetics. 2008;3(2):59–63. | ||
Ismail HM, Medhat AM, Karim AM, Zakhary NI. Multiple Patterns of FHIT Gene Homozygous Deletion in Egyptian Breast Cancer Patients. Int J Breast Cancer. 2011;2011:325947. | ||
Stewart DJ, Nunez MI, Jelinek J, et al. Impact of decitabine on immunohistochemistry expression of the putative tumor suppressor genes FHIT, WWOX, FUS1 and PTEN in clinical tumor samples. Clin Epigenetics. 2014;6(1):13. | ||
Druck T, Hadaczek P, Fu TB, et al. Structure and expression of the human FHIT gene in normal and tumor cells. Cancer Res. 1997;57(3):504–512. | ||
Bianchi F, Tagliabue E, Menard S, Campiglio M. Fhit expression protects against HER2–driven breast tumor development: unraveling the molecular interconnections. Cell Cycle. 2007;6(6):643–646. | ||
Yanagawa N, Osakabe M, Hayashi M, Tamura G, Motoyama T. Frequent epigenetic silencing of the FHIT gene in penile squamous cell carcinomas. Virchows Arch. 2008;452(2):377–382. | ||
Lee WT, Akst LM, Adelstein DJ, et al. Risk factors for hypopharyngeal/upper esophageal stricture formation after concurrent chemoradiation. Head Neck. 2006;28(9):808–812. | ||
Zheng S, Ma X, Zhang L, et al. Hypermethylation of the 5′ CpG island of the FHIT gene is associated with hyperdiploid and translocation-negative subtypes of pediatric leukemia. Cancer Res. 2004;64(6):2000–2006. | ||
Cecener G, Tunca B, Egeli U, et al. The promoter hypermethylation status of GATA6, MGMT, and FHIT in glioblastoma. Cell Mol Neurobiol. 2012;32(2):237–244. | ||
Banzai C, Nishino K, Quan J, et al. Promoter methylation of DAPK1, FHIT, MGMT, and CDKN2A genes in cervical carcinoma. Int J Clin Oncol. 2014;19(1):127–132. | ||
Jeong YJ, Jeong HY, Lee SM, Bong JG, Park SH, Oh HK. Promoter methylation status of the FHIT gene and Fhit expression: association with HER2/neu status in breast cancer patients. Oncol Rep. 2013;30(5):2270–2278. | ||
Haroun RA, Zakhary NI, Mohamed MR, Abdelrahman AM, Kandil EI, Shalaby KA. Assessment of the prognostic value of methylation status and expression levels of FHIT, GSTP1 and p16 in non-small cell lung cancer in Egyptian patients. Asian Pac J Cancer Prev. 2014;15(10):4281–4287. | ||
Beumer JH, Parise RA, Newman EM, et al. Concentrations of the DNA methyltransferase inhibitor 5-fluoro-2′-deoxycytidine (FdCyd) and its cytotoxic metabolites in plasma of patients treated with FdCyd and tetrahydrouridine (THU). Cancer Chemother Pharmacol. 2008;62(2):363–368. | ||
Gowher H, Jeltsch A. Mechanism of inhibition of DNA methyltransferases by cytidine analogs in cancer therapy. Cancer Biol Ther. 2004;3(11):1062–1068. | ||
Newman EM, Morgan RJ, Kummar S, et al. A phase I, pharmacokinetic, and pharmacodynamic evaluation of the DNA methyltransferase inhibitor 5-fluoro-2′-deoxycytidine, administered with tetrahydrouridine. Cancer Chemother Pharmacol. 2015;75(3):537–546. | ||
Zaki SM, Abdel-Azeez HA, El Nagar MR, Metwally KA, S Ahmeed MM. Analysis of FHIT gene methylation in egyptian breast cancer women: association with clinicopathological features. Asian Pac J Cancer Prev. 2015;16(3):1235–1239. | ||
Raish M, Dhillon VS, Ahmad A, et al. Promoter Hypermethylation in Tumor Suppressing Genes p16 and FHIT and Their Relationship with Estrogen Receptor and Progesterone Receptor Status in Breast Cancer Patients from Northern India. Transl Oncol. 2009;2(4):264–270. | ||
Naqvi RA, Hussain A, Raish M, et al. Specific 50′CpG island methylation signatures of FHIT and p16 genes and their potential diagnostic relevance in Indian breast cancer patients. DNA Cell Biol. 2008; 27(9):517–525. | ||
D Zhao, LJ, G Liu, S Li, A Shen. Breast fragile histidine triad abnormal methylation can down-regulate the expression of its mRNA and protein. Chinese Journal of Breast Disease. 2012. Accessed from: http://www.cqvip.com/qk/88763x/201201/41530095.html | ||
Iliopoulos D, Guler G, Han SY, et al. Fragile genes as biomarkers: epigenetic control of WWOX and FHIT in lung, breast and bladder cancer. Oncogene. 2005;24(9):1625–1633. | ||
Yang Q, Nakamura M, Nakamura Y, et al. Two-hit inactivation of FHIT by loss of heterozygosity and hypermethylation in breast cancer. Clin Cancer Res. 2002;8(9):2890–2893. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.