")
Back to Journals » Drug Design, Development and Therapy » Volume 11
Spotlight on blisibimod and its potential in the treatment of systemic lupus erythematosus: evidence to date
Authors Lenert A, Niewold TB, Lenert P
Received 12 December 2016
Accepted for publication 10 February 2017
Published 13 March 2017 Volume 2017:11 Pages 747—757
DOI https://doi.org/10.2147/DDDT.S114552
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Frank Boeckler
Aleksander Lenert,1 Timothy B Niewold,2 Petar Lenert3
1Division of Rheumatology, University of Kentucky, Kentucky Clinic, Lexington, KY, 2Division of Rheumatology and Department of Immunology, Mayo Clinic, Rochester, MN, 3Division of Immunology, Department of Internal Medicine, The University of Iowa, Iowa City, IA, USA
Abstract: B cells in general and BAFF (B cell activating factor of the tumor necrosis factor [TNF] family) in particular have been primary targets of recent clinical trials in systemic lupus erythematosus (SLE). In 2011, belimumab, a monoclonal antibody against BAFF, became the first biologic agent approved for the treatment of SLE. Follow-up studies have shown excellent long-term safety and tolerability of belimumab. In this review, we critically analyze blisibimod, a novel BAFF-neutralizing agent. In contrast to belimumab that only blocks soluble BAFF trimer but not soluble 60-mer or membrane BAFF, blisibimod blocks with high affinity all three forms of BAFF. Furthermore, blisibimod has a unique structure built on four high-affinity BAFF-binding peptides fused to the IgG1-Fc carrier. It was tested in phase I and II trials in SLE where it showed safety and tolerability. While it failed to reach the primary endpoint in a recent phase II trial, post hoc analysis demonstrated its efficacy in SLE patients with higher disease activity. Based on these results, blisibimod is currently undergoing phase III trials targeting this responder subpopulation of SLE patients. The advantage of blisibimod, compared to its competitors, lies in its higher avidity for BAFF, but a possible drawback may come from its immunogenic potential and the anticipated loss of efficacy over time.
Keywords: BAFF, APRIL, lupus, B cells, blisibimod
Introduction
The primary role of B cells in initiating systemic autoimmune diseases has long been suspected. Production of autoantibodies, formation of circulating immune complexes with subsequent Fc-receptor engagement, and activation of the complement system have been considered to play a primary role in tissue inflammation in systemic lupus erythematosus (SLE).
It has been well established that B cells are excellent antigen-presenting cells for antigens that can bind to their surface Ig receptors.1 Direct evidence for the role of B cells apart from autoantibody production was first demonstrated in mice carrying the lpr/lpr mutation which develop a lupus-like disease.2 When MRL-lpr/lpr mice were crossed to JH knockouts, mice lacking B cells were generated. While their littermates with B cells developed nephritis and vasculitis and made autoantibodies, mice lacking B cells showed no evidence of renal disease or vasculitis. A similar effect was also observed in another lupus strain, NZM 2328, where absence of B cells completely protected mice from development of lupus.3 Subsequently, elegant experiments have shown that the requirement for B cells goes beyond their role as precursors of antibody-secreting cells and likely reflects their ability to serve as (auto)antigen-presenting cells.4 This autoantibody-independent role of B cells has been demonstrated in experiments where a mutant transgene encoding surface Ig was introduced into MRL-lpr/lpr mice. While these mice failed to secrete serum antibodies, they still had functional B cells expressing surface Ig receptors. In contrast to mice that completely lack B cells, mice carrying a mutant gene for surface Ig developed mononuclear cellular infiltrates in their kidneys, the characteristic of lupus nephritis in this strain, and had increased mortality compared to controls. These mice exhibited increased number of activated and memory CD4+ splenic T cells. Thus, this study showed that B cells themselves, independent of autoantibody secretion, likely play a primary pathogenic role in lupus.4–6
B cell processing of autoantigen can contribute to epitope spreading, a phenomenon in which initial reactivity to one epitope is followed by subsequent reactivity to additional epitopes expressed on the same or related autoantigens, a phenomenon commonly observed in lupus.7 B cells may additionally be a source of proinflammatory (ie, interleukin-6 [IL-6], tumor necrosis factor alpha [TNF-α]) and/or regulatory cytokines (ie, IL-10),8 and abnormalities in this cytokine-producing function have been observed in lupus mice.9 Remarkably, B cells that lack one of the innate Toll-like receptors, TLR9, may lose this regulatory function.10 Based on these observations, the Shlomchik lab was the first to suggest that B cell depletion, instead of bare mechanical removal of autoantibodies by plasma exchange, should be considered as a primary target for treating lupus.
However, the essential requirement for B cells early in the course of the disease does not rule out an important contribution from T cells, which serve downstream in the disease process as primary effector cells. For example, in autoimmune MRL-lpr/lpr mice, thymectomy or treatment with monoclonal T-cell-specific antibody could ameliorate lymphadenopathy and delay autoimmune-mediated inflammation.11,12
B cell hyperactivity has been recognized as an important characteristic of human SLE and animal models of lupus.13,14 It is associated with polyclonal hypergammaglobulinemia and production of numerous autoantibodies, particularly those recognizing components of the nuclear chromatin (ie, histones and dsDNA) and certain extractable nuclear antigens (ie, Smith antigen and U1-RNP). These antibodies (against Smith and dsDNA) are highly specific for lupus.15,16 Circulating levels of BAFF (B cell activating factor of the TNF family), a key B cell survival and activation factor, are elevated in SLE patients and in animal models of lupus.17–21 It is hypothesized that BAFF can be at least partially responsible for this activated B cell phenotype in lupus.
In this review, we discuss new discoveries relevant to BAFF’s role in the pathogenesis of SLE. We also discuss available therapeutics that specifically target human BAFF focusing on blisibimod, a novel high-potency tetravalent BAFF inhibitor. While other B cell-targeted approaches in SLE, such as B cell depletion with the anti-CD20 antibody rituximab, were largely unsuccessful,22–24 BAFF neutralization with a monoclonal anti-BAFF antibody belimumab25 met primary endpoints in two large phase III clinical trials leading to its approval in 2011 by the US Food and Drug Administration (FDA) for the treatment of adult patients with SLE.26,27
BAFF and its receptors
BAFF also known as Blys (B lymphocyte stimulator), TALL-1,28 and THANK,29 were discovered in 1999.30,31 BAFF plays an essential role in B cell biology. Initial experiments suggested that BAFF may act as an important B cell growth factor by costimulating B cell proliferation induced by cross-linking of the B cell receptor for antigen. However, BAFF by itself fails to stimulate proliferation of resting B cells.30 It soon became clear that BAFF works by providing a critical B cell survival signal (rather than a proliferation signal). Consequently, mice deficient in BAFF have a marked reduction in B cell numbers, along with low serum Ig concentration.32 In contrast, excess production of BAFF causes an SLE-like disease in transgenic mice33,34 and likely plays a role in human SLE and other systemic autoimmune diseases.19–21,35
BAFF is produced by a variety of cell types including macrophages/monocytes, dendritic cells, and neutrophils.36 While initial reports failed to detect BAFF expression in freshly isolated or activated T- and B cells,31 these cells can also make BAFF upon certain conditions.37
Similar to other members of the TNF family, BAFF can exist as a type II cell membrane-bound protein (285 amino acids) and as a soluble form. The furin protease cleaves the surface BAFF and releases the soluble form of this cytokine.30,38 Soluble BAFF can adopt at least two different configurations: a typical homotrimer (3-mer) or a capsid-like structure that has 20 repeats of the 3-mer (60-mer).39 All three forms of BAFF are biologically active and can contribute to cell signaling39 (Figure 1). BAFF can also form heterotrimers with another B cell survival molecule APRIL (a proliferation-inducing ligand, CD256). However, it is not well understood under which conditions these heterotrimers are formed. Interestingly, the phenotype of mice that only express the membrane form of BAFF but lack the ability to make soluble BAFF is very similar to BAFF-deficient mice suggesting that membrane BAFF under normal conditions exerts a very limited role in B cell biology.40
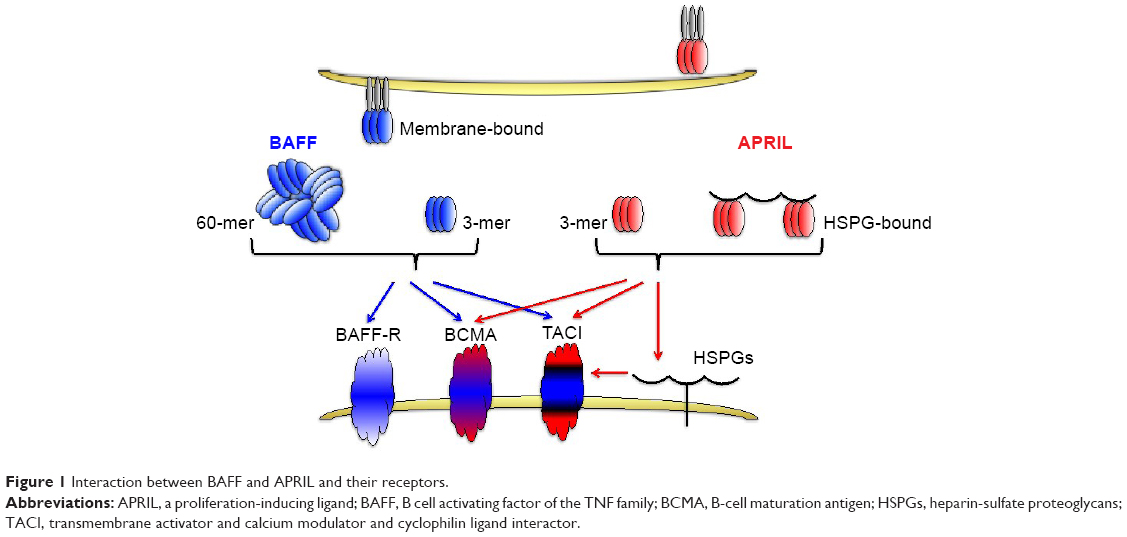 | Figure 1 Interaction between BAFF and APRIL and their receptors. |
BAFF binds to three receptors: transmembrane activator and calcium modulator and cyclophilin ligand interactor (TACI, CD267), B-cell maturation antigen (BCMA, CD269), and BAFF receptor (BAFF-R, also known as BR3, CD268).38,41 BAFF-R solely interacts with BAFF, whereas another TNF-family member, APRIL, can bind and signal through the other two BAFF receptors, TACI and BCMA, but not through BAFF-R.38 APRIL also binds to heparin-sulfate proteoglycans (HSPGs), with 3 mers and multimers bound to HSPGs, also contributing to signaling. BAFF-R-deficient mice have a phenotype similar to BAFF-deficient mice,42,43 while this is not the case with mice lacking either BCMA or TACI receptors.32,44
A very nice review by Mackay and Browning45 describes in much more detail the structure–function relationship between the existing ligands (BAFF and APRIL) and their respective receptors on B cells. Interestingly, the crystal structure of BAFF has revealed important differences between the BAFF and other members of the TNF family by revealing the existence of an extended loop in the BAFF, nonexistent in other TNF members, that may be involved in both receptor binding and viral-like assembly.
BAFF-R expression in B cells
Biotinylated and FLAG-tagged Blys/BAFF were initially used to search for BAFF-R receptor expression in different cell types. B cell tropism for BAFF was shown in peripheral CD20+ B cells, and in a variety of B cell tumor lines (ie, Raji, Namalwa, RPMI-8226), but not in bone marrow plasma cells, nor in T cells (resting or activated), monocytes, natural killer cells, granulocytes, or myeloid-derived cell lines.31 Furthermore, human BAFF/Blys acted across the species as it could bind and induce proliferation of murine B cells. BAFF-R recruits TNF receptor-associated factor 3 (TRAF3), which then leads to downstream signaling cascade resulting in activation of c-jun terminal kinase and nuclear factor kappaB (NF-κB) pathways.46
Rationale for targeting BAFF in SLE
In mice, autoreactive B cells, compared to non-autoreactive B cells, appear to be more dependent on BAFF for their survival.17,33,47 Evidence presented in the following paragraphs suggests that an environment rich in BAFF may allow survival and expansion of autoreactive B cells, ultimately leading to systemic autoimmune disease.
Animal studies
Administration of soluble recombinant BAFF to healthy BALB/c mice disrupted the spleen architecture and caused elevation of serum Ig concentration (particularly IgM and IgA).31
Transgenic mice overexpressing BAFF produced a variety of autoantibodies including rheumatoid factors and antibodies against dsDNA and had an expanded population of mature B cells. Interestingly, the marginal zone B cell subset and plasma cells, but not follicular B cells, were expanded in spleens of these transgenic mice. Intense germinal center formation was also observed. Peripheral blood B cells from these mice showed an increase in Major Histocompatibility Complex class II expression along with high expression of a survival (antiapoptotic) factor Bcl-2. Furthermore, these mice developed a clinical phenotype closely resembling SLE/Sjogren’s syndrome with hypergammaglobulinemia, proteinuria, and immune-complex-mediated glomerulonephritis. Additionally, peripheral blood T cells in these transgenic mice displayed an activated effector T-cell phenotype, and their number was expanded by at least twofold in the spleen and mesenteric lymph nodes.34
Treatment of NZB/NZW-F1 mice with a specific BAFF antagonist was able to slow down the disease progression in this strain of lupus mice and had a positive effect on overall survival.48,49
In NZM model of animal lupus, BAFF-deficient mice had a milder phenotype, and BAFF deficiency could prevent acceleration of clinical kidney disease in response to treatment with type I IFN.3,50
Treatment of NZBW-F1 mice and NZM 2410 mice with either BAFFR-Ig (which only neutralizes BAFF) or TACI-Ig (which neutralizes both BAFF and APRIL) showed that TACI-Ig was more immunosuppressive, causing a drop in serum IgM and IgG levels and Ig-secreting plasma cells in the spleen and bone marrow.49 However, both agents had a similar effect on clinical disease. When double-deficient (BAFF/APRIL) mice were compared to single-deficient BAFF mice, a similar effect on renal immunopathology was observed, despite pronounced differences in bone marrow plasma cells and serum autoantibodies.51 Taken together, these results suggest that APRIL may have a minor role in lupus pathogenesis.
Concurring with animal studies, treatment of human SLE patients (with or without lupus nephritis) with atacicept (TACI-Ig) was associated with safety issues related to ~30% reduction in serum IgG levels within 4 weeks of treatment. So, while atacicept may be an efficient agent for SLE, its potential toxicity may outweigh any gains in efficacy.52,53
Interestingly, NZM 2328 mice deficient in BAFF-R still develop full blown lupus despite having reduced numbers of B cells.54 This suggests that blockade of the BAFF-R alone may be therapeutically insufficient to control human SLE. At present, there are no clear plans of developing or using antibodies against BAFF-R, BCMA, or TACI for the treatment of lupus. However, a construct of two BAFF-R linked to the Fc domain of human IgG1 has been developed. This agent, named briobacept, binds BAFF (human, mouse, cynomolgus monkey) but not APRIL, and has been evaluated in phase I studies in rheumatoid arthritis (RA), but no plans for further development have been made.55
Human SLE
BAFF expression is increased in patients with SLE, and a clear correlation between BAFF levels and disease activity in SLE has been observed in some studies.19–21 Biologically active heterotrimers composed of BAFF and APRIL are elevated in peripheral blood of SLE patients.56,57 Interestingly, the membrane form of BAFF is abnormally expressed in B cells from patients with active SLE.37 In contrast to BAFF, the association between APRIL and SLE demonstrated an inverse relationship, suggesting a possible protective role for APRIL.58,59 Interestingly, BAFF levels are higher in African-American SLE patients as compared to European-American SLE patients,60 and a greater degree of B cell activation has been observed in African-American SLE patients as well.61
Treatment of SLE patients with the anti-BAFF antibody belimumab met its primary endpoint in two phase III clinical trials in 2011.26,27 Based on these studies, the FDA approved belimumab for the treatment of adult patients with SLE. It is worth mentioning that patients with active lupus nephritis or central nervous system (CNS) disease were excluded from these studies. While a study testing the efficacy of belimumab in lupus nephritis (BLISS-LN, NCT01639339) is ongoing, no similar trial in active CNS disease has been planned.
Interferons and BAFF
Type I and II IFN can regulate BAFF synthesis and thus play an important role in lupus pathogenesis. IFN-α can upregulate BAFF expression in mouse macrophages, human dendritic cells, and monocytes. Evidence of an IFN-signature in human SLE is found in about 50% of SLE patients and correlates with disease activity.62–66 Administration of type I IFN can lead to development of SLE-like syndromes in some patients.67,68 Therefore, type I IFN is a potential target for treatment of SLE, and several neutralizing agents are currently in the pipeline, with anifrolumab (anti-type I IFN receptor antibody) showing very promising effects in a recent phase II clinical trial in SLE.69
IFN-γ could induce a similar upregulation of BAFF in various cell types.70,71 For example, BAFF/Blys expression on human monocytes, both at the mRNA and protein level, was upregulated by IFN-γ up to fourfold.31 Not surprisingly, monocytes from SLE patients produced substantially more BAFF compared to healthy controls when stimulated with the same ligand.72
In SLE patients treated with a neutralizing anti-IFN-α antibody, BAFF mRNA expression was downregulated.73 Moreover, a relatively good correlation existed between BAFF and type I IFN levels in patients with SLE.60
Genetic deletion of the type I IFN receptor (Ifnar I) could prevent lupus development in NZB and NZM 2328 mice.74,75 Vice versa, the administration of type I IFN, or its upregulation, accelerated lupus nephritis, leading to death in several strains of lupus-prone mice. However, this was not the case with MRL-lpr/lpr mice as Ifnar-deficient MRL mice developed more severe disease compared to controls.75–78
Therefore, type I IFN and BAFF remain a legible target for treatment in SLE.79,80
B cell therapies in SLE
B cells are an important target for treatment of SLE patients. At first glimpse, it may look quite illogical why very similar treatment approaches aimed at targeting B cells result in quite different clinical outcomes in SLE. For example, rituximab, a CD20-directed B-cell-depleting antibody failed to show efficacy in two recent phase III clinical trials in SLE and lupus nephritis (NCT00137969 and NCT00282347), while belimumab, an anti-BAFF agent, became the first FDA approved drug for SLE in 50 years.26,27,81–83 A potential explanation for this controversy comes from the observation that circulating BAFF levels actually rise in rituximab-treated patients which may subsequently favor the selection and survival of autoreactive B cells and plasmablasts in an environment with very few remaining B cells. Furthermore, elevated BAFF levels could engage T follicular cells, promoting their interaction with autoreactive B cells.84–88
Current portfolio of BAFF (and APRIL) inhibitors
The portfolio of BAFF inhibitors includes belimumab (anti-BAFF antibody), blisibimod (anti-BAFF peptibody), tabalumab (anti-BAFF antibody), briobacept (BAFF receptor, inhibits only BAFF), atacicept (TACI receptor, inhibits both BAFF and APRIL), and anti-BAFF-R. In contrast, GSK2857916, an antibody drug conjugate, antagonizes only the BCMA receptor and is currently undergoing phase I study in multiple myeloma (NCT02064387) with no immediate plans for clinical studies in SLE.
Briobacept, consisting of an extracellular domain of BAFF-R fused to human IgG-Fc, has been evaluated in a phase I study in patients with RA. A report in the form of an abstract has been published in 2008, but since then further advancement to phase II or III clinical studies has been abandoned.55
Another BAFF-neutralizing agent, tabalumab, achieved its primary endpoint in a recent phase III study ILLUMINATE-2 (NCT01205438).89 However, important secondary endpoints such as time to severe flare, corticosteroid-sparing effect, and fatigue were not met. Therefore, the drug developer, Eli Lilly Co in 2014 decided to stop further clinical development of tabalumab in SLE.
Another interesting agent in development is atacicept, which neutralizes both BAFF and APRIL. While atacicept may still offer hopes for SLE, higher risk of serious infections and unsafe drop in serum IgG were observed in recent phase II/III trials.52,90
Several recent review articles have summarized clinical trials with the above agents, and thus will not be further discussed in this review.91–94
Development of A-623/AMG623/blisibimod as a novel high potency and selective BAFF-neutralizing agent
Dr Hsu et al95 at Amgen’s Department of Inflammation were looking for a new BAFF-neutralizing agent that would bind and neutralize both membrane-bound and soluble BAFF with high affinity. Their goal was to screen a 12-mer constrained phage library over Fc-BAFF protein immobilized on protein-A-coated magnetic beads. The next step involved fusing BAFF-binding peptides in tandem copies in-frame to the N-terminal part of the human IgG1-Fc region. The resulting construct (named peptibody) was then expressed in Escherichia coli. Purified peptibodies have a unique tetravalent structure, but since they are produced in E. coli, they lack the posttranslational glycosylation. A concern remains that BAFF-binding peptides in blisibimod may potentially become immunogenic in humans, causing the induction of neutralizing antibodies that will subsequently diminish blisibimod’s potency for BAFF.
In vitro B cell proliferation and affinity studies with A-623/AMG623/blisibimod
These novel peptibodies were first studied in an anti-IgM plus BAFF-mediated B cell proliferation assay. AMG623 showed high affinity for BAFF and inhibited BAFF (and anti-IgM)-mediated B cell proliferation at low nanomolar range (IC-50 at 6 ng/mL).
AMG623 was also tested for its ability to inhibit binding of human BAFF, murine BAFF, or murine APRIL to Biacore chips containing immobilized soluble receptors. AMG623 blocked binding equally well of both human and murine BAFF to BAFF-R, TACI, and BCMA, but had no effect on APRIL binding to its receptors TACI and BCMA.95
Animal models
Contrasting with belimumab, which lacks cross reactivity with murine BAFF,96,97 AMG623 could block murine BAFF activity in vitro. It was subsequently studied in healthy animals and in animal models of SLE and RA. Studies in healthy BALB/c mice showed the ability of AMG623 to reduce the number of B cells in peripheral blood and in spleens. Further animal testing showed the ability of AMG623, when injected intraperitoneally for 5 months, to delay the onset of proteinuria and prolong survival in lupus-prone NZB/NZW-F1 mice. However, this beneficial effect in lupus-prone mice was short-lived and disappeared 3 months after the last injection, suggesting that the defect in immune tolerance leading to disease induction in this model of lupus was not restored and that continuous BAFF inhibition is required.
Similar to animal models of lupus, AMG623 was also effective in collagen-induced arthritis, a mouse model of RA.95
Clinical trials with blisibimod
Early-phase trials
Stohl et al98 conducted early-phase clinical trials with blisibimod in SLE to characterize safety and tolerability. Adult patients with mild stable or inactive SLE (mean SLEDAI ~3) on standard treatment (including prednisone ≤10 mg daily) were enrolled in two early-phase trials with blisibimod. The primary endpoints of both trials were safety and tolerability; additional secondary endpoints were pharmacokinetic and pharmacodynamic parameters, and exploratory endpoints such as changes in peripheral B cell counts and expression of B cell surface markers.
In the phase Ia ascending single-dose study (NCT02443506), patients were sequentially enrolled into one of seven dose cohorts and randomized 3:1 to single dose of blisibimod, administered either subcutaneous (SC) or intravenous (IV), or placebo. A total of 56 participants were randomized, and 54 received the assigned treatment. Participants were followed up to 42 days postdose, and participants in the highest dose cohort (6.0 mg/kg IV) completed a 4-week extension after end-of-study visit. Comparable proportion of participants in the blisibimod (70%) and control (79%) arms experienced ≥1 adverse effect (AE). A total of two serious AEs (SAE) were reported, with one each in the blisibimod and control arms, related to the study drug.
In the phase Ib ascending multidose study (NCT02411136), patients were sequentially enrolled into one of four dose cohorts and randomized 4:1 to receive 4 weekly doses of blisibimod (SC or IV) or placebo. A total of 64 participants were randomized, while 63 received the assigned treatment; 4 participants (2 in each group) withdrew due to an AE. Similar proportion of participants in each group reported ≥1 AE (96% blisibimod vs 92% placebo); nine SAEs were reported in eight participants (5 in blisibimod, 3 in placebo). In the blisibimod arm, SAEs were lupus flare (polyarthritis), depression, chest pain with fever, and syncope; one participant experienced QTc prolongation possibly related to the study drug.
The overall results of these two early-phase clinical trials confirmed that blisibimod had favorable safety and tolerability compared with placebo, providing support for further study in phase II trials. However, a large proportion of participants tested positive for neutralizing antibodies, which could be of some concern. Additional exploratory analyses demonstrated blisibimod’s effect on decreasing peripheral B cell counts and changing B cell populations, with relative decrease in naïve B cells and increase in memory B cell compartments via BAFF-mediated differential B cell effects.
PEARL-SC (NCT01162681)
After promising phase I studies, PEARL-SC,99 a randomized, double-blind, placebo-controlled, phase II study was conducted to assess efficacy and safety of SC blisibimod in patients with active SLE (baseline SELENA-SLEDAI ≥6). The primary outcome was the proportion of responders in the blisibimod pooled group compared with the placebo pooled group meeting the composite endpoint SLE responder index (SRI-5) at 24 weeks. This multicenter international study enrolled 547 participants, randomized in parallel to receive blisibimod SC (either 100 mg once weekly, 200 mg once weekly, or 200 mg every 4 weeks) or placebo. The major exclusions were SLE patients with severe lupus nephritis, CNS lupus, and vasculitis.
Patient baseline characteristics were similar between groups, except for immunosuppressive use, which was higher in the placebo group (52.3%) compared with the control group (38.2%). In both groups, participants were predominantly female (94%), Hispanic (71%), and with active SLE (mean SELENA-SLEDAI ~10) with ≥3 organ domains involved (61%–65%). The most common SLE disease manifestations were mucocutaneous (91%), immunological (77%), and musculoskeletal (75%); renal manifestations were observed in 14%. Treatment with corticosteroids (~90%) and antimalarials (~70%) was common in both groups.
The primary efficacy endpoint for the study, the proportion of SRI-5 responders for blisibimod compared with placebo pooled groups, was not met. The proportion of SRI-5 responders in the highest blisibimod group (200 mg once weekly) was greater than pooled and regimen-matched placebo groups; however, this was not statistically significant. Similar trends were observed for modified SRI responses with blisibimod, ie, SRI-7 and SRI-8, compared with regimen-matched placebo. Additional subgroup analyses revealed favorable treatment response with blisibimod compared with placebo for severe SLE, defined as high baseline disease activity (SELENA-SLEDAI ≥10) and on corticosteroids. A higher difference in the responder rates (ΔSRI) between blisibimod and placebo was observed with increasing SRI responses, ie, from SRI-5 to SRI-8.
Compared to placebo, trends favoring blisibimod were seen for cumulative probability of severe flare and time to first severe flare, especially for the highest dose (200 mg once weekly). Among participants on prednisone >7.5 mg/d at baseline, more participants on the highest dose blisibimod (200 mg once weekly) were able to taper prednisone dose ≤7.5 mg/d compared with placebo (11.9% vs 9.5%). Subgroup analysis in participants with baseline proteinuria (urine protein to creatinine ratio of 1–6 g/g) achieved a greater mean reduction with blisibimod. Similar trends were seen for disease activity markers of SLE, with significant reduction of B cells and ds-DNA and increase in C3 and C4.
Further, Petri et al100 reported on the effects of blisibimod on patient-reported fatigue and disease activity in PEARL-SC. Improvements in fatigue measured by the FACIT-Fatigue score were seen in both groups, but were more profound with higher dose blisibimod (100 mg and 200 mg once weekly), exceeding the minimal clinically important difference in SLE (P <0.05). SRI-5 responders had significantly higher mean changes in fatigue compared with nonresponders. Improvements in fatigue, however, correlated poorly with disease activity. In terms of safety, a comparable proportion of participants in the blisibimod and placebo groups reported any AE (82.5% vs 85%), while fewer blisibimod-treated participants reported SAEs (11.1% vs 15.8%); overall, no differences were observed for severe infections, death, or malignancy. The presence of antidrug antibodies was not reported, given the high rates of assay positivity in blisibimod-naïve patients, which according to the authors needs further refinement. Thus, the possibility of immunogenicity to blisibimod with repeated exposure remains a concern for sustained clinical efficacy.
Despite not meeting its primary outcome, the PEARL-SC study redemonstrated safety and tolerability of blisibimod, and provided a hint of efficacy (especially for the highest blisibimod dose of 200 mg once weekly) in SLE patients with severe disease meeting a higher responder threshold (SRI-8). This subpopulation of SLE would benefit from further targeted study in phase III trials. On completion of PEARL-SC, 382 participants were enrolled in the open-label blisibimod extension study, PEARL-OLE, with preliminary reports demonstrating favorable long-term safety of blisibimod.101,102
Early-phase clinical trials of blisibimod in SLE are summarized in Table 1.
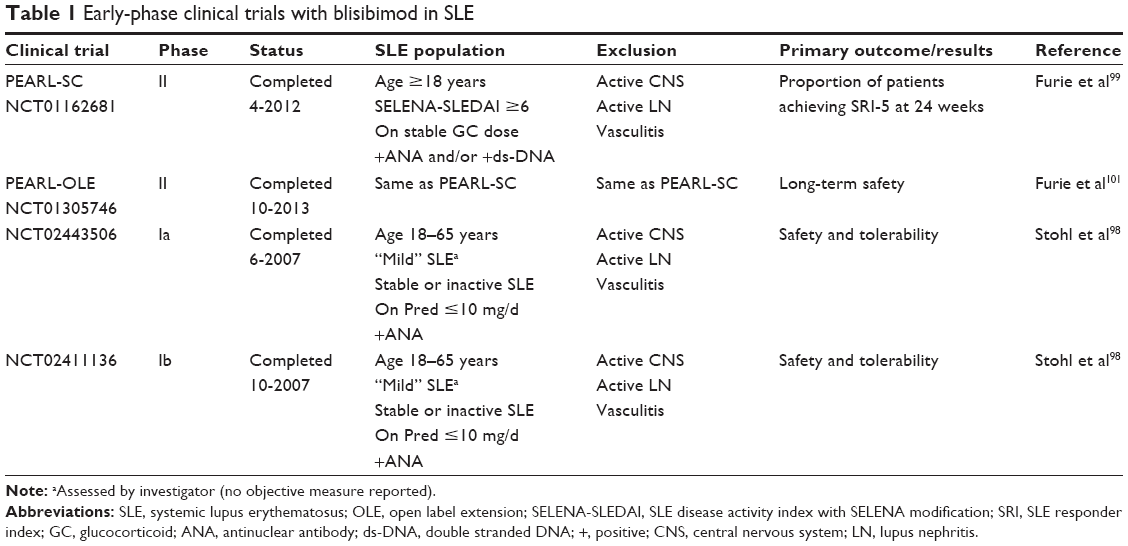 | Table 1 Early-phase clinical trials with blisibimod in SLE |
Late-phase trials
CHABLIS-SC1 (NCT01395745)
CHABLIS-SC1,102 a phase III randomized, double-blind, placebo-controlled trial, builds on prior successful early phase trials with blisibimod in SLE with the aim of targeting the responder populations identified in the PEARL-SC trial.102 This study enrolled 442 participants with active SLE (SELENA-SLEDAI ≥10) on systemic corticosteroids, randomized to blisibimod (200 mg SC once weekly) or placebo in addition to standard of care treatment. The primary endpoint was defined as SRI-6 at 52 weeks; additional secondary endpoints included time to first severe SLE flare, change in the number of tender and swollen joints, change in mucocutaneous disease activity, proportion of participants achieving prednisone ≤7.5 mg/d, and effect on biomarkers and safety. Due to the difficulty in evaluating new drug effect on background lupus treatment and high responder rates in placebo groups, this trial employed a strict requirement for early tapering of systemic corticosteroids after week 8 with the goal of achieving prednisone ≤7.5 mg/d.102 The study was completed in September 2016; however, no study results have been reported yet.
CHABLIS 7.5 (NCT02514967)
CHABLIS 7.5 is a phase III randomized, double-blind, placebo-controlled trial to evaluate the efficacy and safety of blisibimod in SLE participants with or without lupus nephritis. The study aims to enroll 350 patients with active SLE defined as SELENA-SLEDAI ≥10 despite systemic corticosteroids, who are positive for ds-DNA and have low complements. Participants will be randomized to blisibimod (200 mg SC once weekly) or placebo. The primary outcome is SRI-6 at 52 weeks, while secondary outcomes will be similar to those measured in CHABLIS-SC1. This trial is actively enrolling participants since June 2016 with the goal of completion in December 2018.
Late-phase clinical trials of blisibimod are summarized in Table 2.
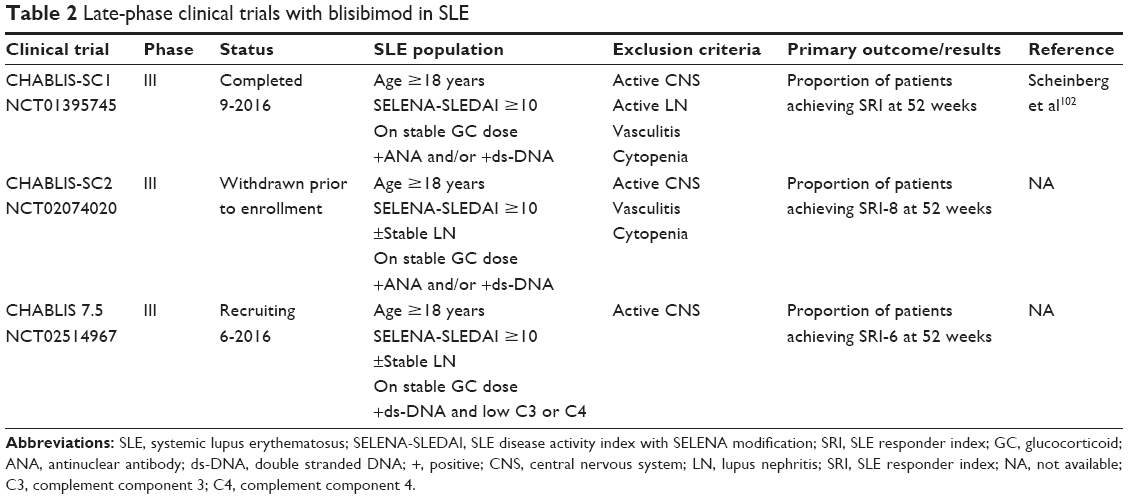 | Table 2 Late-phase clinical trials with blisibimod in SLE |
Conclusion
The identification of BAFF’s key role as a B cell survival, activation, and differentiation cytokine involved in SLE pathogenesis has led to the development of novel anti-BAFF agents with promising clinical results in SLE. The novel biologic agent blisibimod is building on the success of belimumab by targeting both soluble and membrane-bound BAFF. Early-phase clinical trials of blisibimod have proven its safety and tolerability and have provided a hint of efficacy, specifically in a subpopulation of SLE patients with higher disease activity. Blisibimod’s immunogenicity, which leads to the emergence of neutralizing antibodies reported in clinical trials, likely driven by its phage manufacturing process, remains a clinical concern. Despite this clinical hurdle, anti-BAFF therapy remains a promising new therapeutic option for the management of patients with SLE, and the results of late phase clinical trials are eagerly awaited.
This review has focused on the state of the art knowledge about the new anti-BAFF agent, named blisibimod, and its potential advantage over other BAFF-neutralizing agents for the treatment of lupus. However, numerous other pharmaceuticals, including small-molecular signaling pathway inhibitors (eg, Bruton tyrosine kinase inhibitors or proteasome inhibitors) are in various stages of preclinical and clinical development. It remains to be determined whether antibody-based neutralization of B cells may have any potential clinical advantages over these small-molecular inhibitors. Hopefully, we will not need to wait another 50 years for the next drug to be approved for the treatment of lupus.
Disclosure
The authors report no conflicts of interest in this work.
References
Lanzavecchia A. Antigen-specific interaction between T and B cells. Nature. 1985;314(6011):537–539. | ||
Shlomchik MJ, Madaio MP, Ni D, Trounstein M, Huszar D. The role of B cells in lpr/lpr-induced autoimmunity. J Exp Med. 1994;180(4):1295–1306. | ||
Jacob N, Guo S, Mathian A, et al. B Cell and BAFF dependence of IFN-α-exaggerated disease in systemic lupus erythematosus-prone NZM 2328 mice. J Immunol. 2011;186(8):4984–4993. | ||
Chan OT, Hannum LG, Haberman AM, Madaio MP, Shlomchik MJ. A novel mouse with B cells but lacking serum antibody reveals an antibody-independent role for B cells in murine lupus. J Exp Med. 1999;189(10):1639–1648. | ||
Mamula MJ, Lin RH, Janeway CA Jr, Hardin JA. Breaking T cell tolerance with foreign and self co-immunogens. A study of autoimmune B and T cell epitopes of cytochrome c. J Immunol. 1992;149(3):789–795. | ||
Mamula MJ, Fatenejad S, Craft J. B cells process and present lupus autoantigens that initiate autoimmune T cell responses. J Immunol. 1994;152(3):1453–1461. | ||
James JA, Harley JB. B-cell epitope spreading in autoimmunity. Immunol Rev. 1998;164:185–200. | ||
Fillatreau S, Sweenie CH, McGeachy MJ, Gray D, Anderton SM. B cells regulate autoimmunity by provision of IL-10. Nat Immunol. 2002;3(10):944–950. | ||
Lenert P, Brummel R, Field EH, Ashman RF. TLR-9 activation of marginal zone B cells in lupus mice regulates immunity through increased IL-10 production. J Clin Immunol. 2005;25(1):29–40. | ||
Tilstra JS, John S, Nickerson K, Bastacky S, Shlomchik MJ. B cell specific TLR9 modulates disease in murine lupus. Presented at: ACR meeting; 2016; Washington DC. | ||
Steinberg AD, Roths JB, Murphy ED, Steinberg RT, Raveche ES. Effects of thymectomy or androgen administration upon the autoimmune disease of MRL/Mp-lpr/lpr mice. J Immunol. 1980;125(2):871–873. | ||
Wofsy D, Ledbetter JA, Hendler PL, Seaman WE. Treatment of murine lupus with monoclonal anti-T cell antibody. J Immunol. 1985;134(2):852–857. | ||
Klinman DM, Steinberg AD. Systemic autoimmune disease arises from polyclonal B cell activation. J Exp Med. 1987;165(6):1755–1760. | ||
Izui S, McConahey PJ, Dixon FJ. Increased spontaneous polyclonal activation of B lymphocytes in mice with spontaneous autoimmune disease. J Immunol. 1978;121(6):2213–2219. | ||
Holman HR, Kunkel HG. Affinity between the lupus erythematosus serum factor and cell nuclei and nucleoprotein. Science. 1957;126(3265):162–163. | ||
Tan EM, Schur PH, Carr RI, Kunkel HG. Deoxybonucleic acid (DNA) and antibodies to DNA in the serum of patients with systemic lupus erythematosus. J Clin Invest. 1966;45(11):1732–1740. | ||
Ota M, Duong BH, Torkamani A, et al. Regulation of the B cell receptor repertoire and self-reactivity by BAFF. J Immunol. 2010;185(7):4128–4136. | ||
Stohl W, Metyas S, Tan SM, et al. B lymphocyte stimulator overexpression in patients with systemic lupus erythematosus: longitudinal observations. Arthritis Rheum. 2003;48(12):3475–3486. | ||
Zhang J, Roschke V, Baker KP, et al. Cutting edge: a role for B lymphocyte stimulator in systemic lupus erythematosus. J Immunol. 2001;166(1):6–10. | ||
Cheema GS, Roschke V, Hilbert DM, Stohl W. Elevated serum B lymphocyte stimulator levels in patients with systemic immune-based rheumatic diseases. Arthritis Rheum. 2001;44(6):1313–1319. | ||
Petri M, Stohl W, Chatham W, et al. Association of plasma B lymphocyte stimulator levels and disease activity in systemic lupus erythematosus. Arthritis Rheum. 2008;58(8):2453–2459. | ||
Merrill JT, Neuwelt CM, Wallace DJ, et al. Efficacy and safety of rituximab in moderately-to-severely active systemic lupus erythematosus: the randomized, double-blind, phase II/III systemic lupus erythematosus evaluation of rituximab trial. Arthritis Rheum. 2010;62(1):222–233. | ||
Rovin BH, Furie R, Latinis K, et al. Efficacy and safety of rituximab in patients with active proliferative lupus nephritis: the Lupus Nephritis Assessment with Rituximab study. Arthritis Rheum. 2012; 64(4):1215–1226. | ||
Reddy V, Jayne D, Close D, Isenberg D. B-cell depletion in SLE: clinical and trial experience with rituximab and ocrelizumab and implications for study design. Arthritis Res Ther. 2015;15(Suppl 1):S2. | ||
Baker KP, Edwards BM, Main SH, et al. Generation and characterization of LymphoStat-B, a human monoclonal antibody that antagonizes the bioactivities of B lymphocyte stimulator. Arthritis Rheum. 2003;48(11):3253–3265. | ||
Navarra SV, Guzmán RM, Gallacher AE, et al. Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: a randomised, placebo-controlled, phase 3 trial. Lancet. 2011;377(9767):721–731. | ||
Furie R, Petri M, Zamani O, et al. A phase III, randomized, placebo-controlled study of belimumab, a monoclonal antibody that inhibits B lymphocyte stimulator, in patients with systemic lupus erythematosus. Arthritis Rheum. 2011;63(12):3918–3930. | ||
Shu HB, Hu WH, Johnson H. TALL-1 is a novel member of the TNF family that is down-regulated by mitogens. J Leukoc Biol. 1999;65(5):680–683. | ||
Mukhopadhyay A, Ni J, Zhai Y, Yu GL, Aggarwal BB. Identification and characterization of a novel cytokine, THANK, a TNF homologue that activates apoptosis, nuclear factor-kappaB, and c-Jun NH2-terminal kinase. J Biol Chem. 1999;274(23):15978–15981. | ||
Schneider P, MacKay F, Steiner V, et al. BAFF, a novel ligand of the tumor necrosis factor family, stimulates B cell growth. J Exp Med. 1999;189(11):1747–1756. | ||
Moore PA, Belvedere O, Orr A, et al. BLyS: member of the tumor necrosis factor family and B lymphocyte stimulator. Science. 1999;285(5425):260–263. | ||
Schiemann B, Gommerman JL, Vora K, et al. An essential role for BAFF in the normal development of B cells through a BCMA-independent pathway. Science. 2001;293(5537):2111–2114. | ||
Thien M, Phan TG, Gardam S, et al. Excess BAFF rescues self-reactive B cells from peripheral deletion and allows them to enter forbidden follicular and marginal zone niches. Immunity. 2004;20(6):785–798. | ||
Mackay F, Woodcock SA, Lawton P, et al. Mice transgenic for BAFF develop lymphocytic disorders along with autoimmune manifestations. J Exp Med. 1999;190(11):1697–1710. | ||
Mariette X, Roux S, Zhang J, et al. The level of BLyS (BAFF) correlates with the titre of autoantibodies in human Sjögren’s syndrome. Ann Rheum Dis. 2003;62(2):168–171. | ||
Scapini P, Nardelli B, Nadali G, et al. G-CSF-stimulated neutrophils are a prominent source of functional BLyS. J Exp Med. 2003;197(3):297–302. | ||
Chu VT, Enghard P, Schürer S, et al. Systemic activation of the immune system induces aberrant BAFF and APRIL expression in B cells in patients with systemic lupus erythematosus. Arthritis Rheum. 2009;60(7):2083–2093. | ||
Mackay F, Schneider P, Rennert P, Browning J. BAFF AND APRIL: a tutorial on B cell survival. Annu Rev Immunol. 2003;21:231–264. | ||
Liu Y, Hong X, Kappler J, et al. Ligand-receptor binding revealed by the TNF family member TALL-1. Nature. 2003;423(6935):49–56. | ||
Bossen C, Tardivel A, Willen L, et al. Mutation of the BAFF furin cleavage site impairs B-cell homeostasis and antibody responses. Eur J Immunol. 2011;41(3):787–797. | ||
Gross JA, Johnston J, Mudri S, et al. TACI and BCMA are receptors for a TNF homologue implicated in B-cell autoimmune disease. Nature. 2000;404(6781):995–999. | ||
Sasaki Y, Casola S, Kutok JL, Rajewsky K, Schmidt-Supprian M. TNF family member B cell-activating factor (BAFF) receptor-dependent and -independent roles for BAFF in B cell physiology. J Immunol. 2004;173(4):2245–2252. | ||
Shulga-Morskaya S, Dobles M, Walsh ME, et al. B cell-activating factor belonging to the TNF family acts through separate receptors to support B cell survival and T cell-independent antibody formation. J Immunol. 2004;173(4):2331–2341. | ||
Yan M, Wang H, Chan B, et al. Activation and accumulation of B cells in TACI-deficient mice. Nat Immunol. 2001;2(7):638–643. | ||
Mackay F, Browning JL. BAFF: a fundamental survival factor for B cells. Nat Rev Immunol. 2002;2(7):465–475. | ||
Rauch M, Tussiwand R, Bosco N, Rolink AG. Crucial role for BAFF-BAFF-R signaling in the survival and maintenance of mature B cells. PLoS One. 2009;4(5):e5456. | ||
Lesley R, Xu Y, Kalled SL, et al. Reduced competitiveness of autoantigen-engaged B cells due to increased dependence on BAFF. Immunity. 2004;20(4):441–453. | ||
Kayagaki N, Yan M, Seshasayee D, et al. BAFF/BLyS receptor 3 binds the B cell survival factor BAFF ligand through a discrete surface loop and promotes processing of NF-kappaB2. Immunity. 2002;17(4):515–524. | ||
Ramanujam M, Wang X, Huang W, et al. Similarities and differences between selective and nonselective BAFF blockade in murine SLE. J Clin Invest. 2006;116(3):724–734. | ||
Jacob CO, Pricop L, Putterman C, et al. Paucity of clinical disease despite serological autoimmunity and kidney pathology in lupus-prone New Zealand mixed 2328 mice deficient in BAFF. J Immunol. 2006;177(4):2671–2680. | ||
Jacob CO, Guo S, Jacob N, et al. Dispensability of APRIL to the development of systemic lupus erythematosus in NZM 2328 mice. Arthritis Rheum. 2012;64(5):1610–1619. | ||
Dall’Era M, Chakravarty E, Wallace D, et al. Reduced B lymphocyte and immunoglobulin levels after atacicept treatment in patients with systemic lupus erythematosus: results of a multicenter, phase Ib, double-blind, placebo-controlled, dose-escalating trial. Arthritis Rheum. 2007;56(12):4142–4150. | ||
Ginzler EM, Wax S, Rajeswaran A, et al. Atacicept in combination with MMF and corticosteroids in lupus nephritis: results of a prematurely terminated trial. Arthritis Res Ther. 2012;14(1):R33. | ||
Jacob CO, Yu N, Guo S, et al. Development of systemic lupus erythematosus in NZM 2328 mice in the absence of any single BAFF receptor. Arthritis Rheum. 2013;65(4):1043–1054. | ||
Shaw M, Trapp R, Del Giudice J, et al. The effects of repeated doses of briobacept (BR3-FC) in patients with rheumatoid arthritis. Presented at: EULAR 2008; June 11–14; 2008; Paris, France. Presentation OP-0123. | ||
Roschke V, Sosnovtseva S, Ward CD, et al. BLyS and APRIL form biologically active heterotrimers that are expressed in patients with systemic immune-based rheumatic diseases. J Immunol. 2002;169(8):4314–4321. | ||
Dillon SR, Harder B, Lewis KB, et al. B-lymphocyte stimulator/a proliferation-inducing ligand heterotrimers are elevated in the sera of patients with autoimmune disease and are neutralized by atacicept and B-cell maturation antigen-immunoglobulin. Arthritis Res Ther. 2010;12(2):R48. | ||
Stohl W, Metyas S, Tan SM, et al. Inverse association between circulating APRIL levels and serological and clinical disease activity in patients with systemic lupus erythematosus. Ann Rheum Dis. 2004;63(9):1096–1103. | ||
Morel J, Roubille C, Planelles L, et al. Serum levels of tumour necrosis factor family members a proliferation-inducing ligand (APRIL) and B lymphocyte stimulator (BLyS) are inversely correlated in systemic lupus erythematosus. Ann Rheum Dis. 2009;68(6):997–1002. | ||
Ritterhouse LL, Crowe SR, Niewold TB, et al. B lymphocyte stimulator levels in systemic lupus erythematosus: higher circulating levels in African American patients and increased production after influenza vaccination in patients with low baseline levels. Arthritis Rheum. 2011;63(12):3931–3941. | ||
Menard LC, Habte S, Gonsiorek W, et al. B cells from African American lupus patients exhibit an activated phenotype. JCI Insight. 2016;1(9):e87310. | ||
Weckerle CE, Franek BS, Kelly JA, et al. Network analysis of associations between serum interferon-α activity, autoantibodies, and clinical features in systemic lupus erythematosus. Arthritis Rheum. 2011;63(4):1044–1053. | ||
Niewold TB, Hua J, Lehman TJ, Harley JB, Crow MK. High serum IFN-alpha activity is a heritable risk factor for systemic lupus erythematosus. Genes Immun. 2007;8(6):492–502. | ||
Baechler EC, Batliwalla FM, Karypis G, et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci U S A. 2003;100(5):2610–2615. | ||
Bennett L, Palucka AK, Arce E, et al. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med. 2003;197(6):711–723. | ||
Crow MK, Kirou KA, Wohlgemuth J. Microarray analysis of interferon-regulated genes in SLE. Autoimmunity. 2003;36(8):481–490. | ||
Rönnblom LE, Alm GV, Oberg KE. Possible induction of systemic lupus erythematosus by interferon-alpha treatment in a patient with a malignant carcinoid tumour. J Intern Med. 1990;227(3):207–210. | ||
Niewold TB, Swedler WI. Systemic lupus erythematosus arising during interferon-alpha therapy for cryoglobulinemic vasculitis associated with hepatitis C. Clin Rheumatol. 2005;24(2):178–181. | ||
Narain S, Furie R. Update on clinical trials in systemic lupus erythematosus. Curr Opin Rheumatol. 2016;28(5):477–487. | ||
Panchanathan R, Choubey D. Murine BAFF expression is up-regulated by estrogen and interferons: implications for sex bias in the development of autoimmunity. Mol Immunol. 2013;53(1–2):15–23. | ||
Litinskiy MB, Nardelli B, Hilbert DM, et al. DCs induce CD40-independent immunoglobulin class switching through BLyS and APRIL. Nat Immunol. 2002;3(9):822–829. | ||
Harigai M, Kawamoto M, Hara M, Kubota T, Kamatani N, Miyasaka N. Excessive production of IFN-gamma in patients with systemic lupus erythematosus and its contribution to induction of B lymphocyte stimulator/B cell-activating factor/TNF ligand superfamily-13B. J Immunol. 2008;181(3):2211–2219. | ||
Yao Y, Richman L, Higgs BW, et al. Neutralization of interferon-alpha/beta-inducible genes and downstream effect in a phase I trial of an anti-interferon-alpha monoclonal antibody in systemic lupus erythematosus. Arthritis Rheum. 2009;60(6):1785–1796. | ||
Santiago-Raber ML, Baccala R, Haraldsson KM, et al. Type-I interferon receptor deficiency reduces lupus-like disease in NZB mice. J Exp Med. 2003;197(6):777–788. | ||
Agrawal H, Jacob N, Carreras E, et al. Deficiency of type I IFN receptor in lupus-prone New Zealand mixed 2328 mice decreases dendritic cell numbers and activation and protects from disease. J Immunol. 2009;183(9):6021–6029. | ||
Mathian A, Weinberg A, Gallegos M, Banchereau J, Koutouzov S. IFN-alpha induces early lethal lupus in preautoimmune (New Zealand Black x New Zealand White) F1 but not in BALB/c mice. J Immunol. 2005;174(5):2499–2506. | ||
Fairhurst AM, Mathian A, Connolly JE, et al. Systemic IFN-alpha drives kidney nephritis in B6.Sle123 mice. Eur J Immunol. 2008;38(7):1948–1960. | ||
Hron JD, Peng SL. Type I IFN protects against murine lupus. J Immunol. 2004;173(3):2134–2142. | ||
Postal M, Sinicato NA, Appenzeller S, Niewold TB. Drugs in early clinical development for systemic lupus erythematosus. Expert Opin Investig Drugs. 2016;25(5):573–583. | ||
Singh N, Kumar B, Aluri V, Lenert P. Interfering with baffled B cells at the lupus tollway: promises, successes, and failed expectations. J Allergy Clin Immunol. 2016;137(5):1325–1333. | ||
Wallace DJ, Navarra S, Petri MA, et al. Safety profile of belimumab: pooled data from placebo-controlled phase 2 and 3 studies in patients with systemic lupus erythematosus. Lupus. 2013;22(2):144–154. | ||
Stohl W, Hiepe F, Latinis KM, et al. Belimumab reduces autoantibodies, normalizes low complement levels, and reduces select B cell populations in patients with systemic lupus erythematosus. Arthritis Rheum. 2012;64(7):2328–2337. | ||
Askanase AD, Yazdany J, Molta CT. Post-marketing experiences with belimumab in the treatment of SLE patients. Rheum Dis Clin North Am. 2014;40(3):507–517. | ||
Lenert A, Lenert P. Current and emerging treatment options for ANCA-associated vasculitis: potential role of belimumab and other BAFF/APRIL targeting agents. Drug Des Devel Ther. 2015;9:333–347. | ||
Ehrenstein MR, Wing C. The BAFFling effects of rituximab in lupus: danger ahead? Nat Rev Rheumatol. 2016;12(6):367–372. | ||
Kreuzaler M, Rauch M, Salzer U, et al. Soluble BAFF levels inversely correlate with peripheral B cell numbers and the expression of BAFF receptors. J Immunol. 2012;188(1):497–503. | ||
Pollard RP, Abdulahad WH, Vissink A, et al. Serum levels of BAFF, but not APRIL, are increased after rituximab treatment in patients with primary Sjogren’s syndrome: data from a placebo-controlled clinical trial. Ann Rheum Dis. 2013;72(1):146–148. | ||
Wardemann H, Yurasov S, Schaefer A, et al. Predominant autoantibody production by early human B cell precursors. Science. 2003(5638);301:1374–1377. | ||
Merrill JT, van Vollenhoven RF, Buyon JP, et al. Efficacy and safety of subcutaneous tabalumab, a monoclonal antibody to B-cell activating factor, in patients with systemic lupus erythematosus: results from ILLUMINATE-2, a 52-week, phase III, multicentre, randomised, double-blind, placebo-controlled study. Ann Rheum Dis. 2016;75(2):332–340. | ||
Isenberg D, Gordon C, Licu D, Copt S, Rossi CP, Wofsy D. Efficacy and safety of atacicept for prevention of flares in patients with moderate-to-severe systemic lupus erythematosus (SLE): 52-week data (APRIL-SLE randomised trial). Ann Rheum Dis. 2015;74(11):2006–2015. | ||
Lazaro E, Scherlinger M, Truchetet ME, et al. Biotherapies in systemic lupus erythematosus: new targets. Joint Bone Spine. Epub 2016 Sep 20. | ||
Morais SA, Vilas-Boas A, Isenberg DA. B-cell survival factors in autoimmune rheumatic disorders. Ther Adv Musculoskelet Dis. 2015;7(4):122–151. | ||
Mok CC. Emerging biological therapies for systemic lupus erythematosus. Expert Opin Emerg Drugs. 2014;19(2):303–322. | ||
Stohl W. Therapeutic targeting of the BAFF/APRIL axis in systemic lupus erythematosus. Expert Opin Ther Targets. 2014;18(4):473–489. | ||
Hsu H, Khare SD, Lee F, et al. A novel modality of BAFF-specific inhibitor AMG623 peptibody reduces B-cell number and improves outcomes in murine models of autoimmune disease. Clin Exp Rheumatol. 2012;30(2):197–201. | ||
Stohl W. Biologic differences between various inhibitors of the BLyS/BAFF pathway: should we expect differences between belimumab and other inhibitors in development? Curr Rheumatol Rep. 2012;14(4):303–309. | ||
Nicoletti AM, Kenny CH, Khalil AM, et al. Unexpected potency differences between B-Cell-Activating Factor (BAFF) antagonist antibodies against various forms of BAFF: Trimer, 60-Mer, and Membrane-Bound. J Pharmacol Exp Ther. 2016;359(1):37–44. | ||
Stohl W, Merrill JT, Looney RJ, et al. Treatment of systemic lupus erythematosus patients with the BAFF antagonist “peptibody” blisibimod (AMG 623/A-623): results from randomized, double-blind phase 1a and phase 1b trials. Arthritis Res Ther. 2015;17:215. | ||
Furie RA, Leon G, Thomas M, et al. PEARL-SC Study. A phase 2, randomised, placebo-controlled clinical trial of blisibimod, an inhibitor of B cell activating factor, in patients with moderate-to-severe systemic lupus erythematosus, the PEARL-SC study. Ann Rheum Dis. 2015;74(9):1667–1675. | ||
Petri MA, Martin RS, Scheinberg MA, Furie RA. Assessments of fatigue and disease activity in patients with systemic lupus erythematosus enrolled in the Phase 2 clinical trial with blisibimod. Lupus. Epub 2016 Jun 26. | ||
Furie RA, Thomas C, Chu A, et al. Effects of blisibimod, and inhibitor of B cell activating factor, on serum immunoglobulins and infection risk in patients with systemic lupus erythematosus: observations from the placebo-controlled PEARL_SC and open-label extension studies. Arthritis Rheum. 2013;65(Suppl 10):1742. | ||
Scheinberg MA, Hislop CM, Martin RS. Blisibimod for treatment of systemic lupus erythematosus: with trials you become wiser. Expert Opin Biol Ther. 2016;16(5):723–733. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.