")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Roles of Ca2+/calmodulin-dependent protein kinase II in subcellular expression of striatal N-Methyl-D-aspartate receptors in L-3, 4-dihydroxyphenylalanine-induced dyskinetic rats
Received 6 September 2014
Accepted for publication 24 November 2014
Published 13 April 2015 Volume 2015:9 Pages 2119—2128
DOI https://doi.org/10.2147/DDDT.S73868
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Shu-Feng Zhou
Jing Gan, Chen Qi, Zhenguo Liu
Department of Neurology, Xinhua Hospital affiliated to Shanghai Jiao Tong University Medical School, Shanghai, People’s Republic of China
Background: The role of N-Methyl-D-aspartate (NMDA) receptors is critical to the development of l-3,4-dihydroxyphenylalanine (L-DOPA)-induced dyskinesia (LID) in Parkinson’s disease (PD). Ca2+/calmodulin-dependent protein kinase II (CaMKII) is thought to regulate the expression and activation of NMDA receptors in LID, but the interaction between LID and CaMKII-modulated NMDA receptor activity is not clear so far.
Methods: We used 6-hydroxydopamine-lesioned rats to create PD rat model, and at least 21 days of L-DOPA was administrated followed with or without microinjection of CaMKII inhibitor KN-93 into the lesioned striatum of all the PD rats and sham rats. A surface receptor cross-linking assay was used to distinguish expression of striatal NMDA receptors in surface and intracellular compartments.
Results: L-DOPA treatment enhanced surface levels of GluN1 expression and reduced its intracellular expression, but did not change total levels of GluN1 protein in the lesioned striatum. In contrast, L-DOPA decreased GluN2A surface expression but increased its intracellular expression. L-DOPA increased GluN2B expression preferentially in the surface compartment. We also found that L-DOPA increased CaMKII autophosphorylation at T286 in striatal neurons. The inhibition of CaMKII by microinjecting CaMKII inhibitor KN-93 into the lesioned striatum largely reversed the L-DOPA-induced changes in three subunits. In addition, dyskinetic behaviors of animals were observed alleviated after treatment of KN-93.
Conclusion: Our research indicates that long-term L-DOPA administration activates CaMKII in striatal neurons. Activated CaMKII is involved at least in part in mediating L-DOPA-induced changes of NMDA receptors surface/intracellular expression.
Keywords: glutamate, GluN1, GluN2A, GluN2B, dopamine, KN-93
Introduction
Parkinson’s disease (PD) is a degenerative disorder of the central nervous system, stemming from the progressive death of dopaminergic neurons along the substantia nigra projection to the striatum. The cause of this cell death is not clarified. L-3,4-dihydroxyphenylalanine (L-DOPA), as the standard medicament of dopamine replacement therapy, is still the most effective treatment of PD today. However, chronic L-DOPA treatment results in multiple side effects on motor activities, including L-DOPA induced dyskinesia (LID),1,2 which hampered the use of L-DOPA in PD treatment. Up to now, the mechanisms of LID are poorly understood. Several neurotransmitter systems in the local striatum have been implicated in the pathogenesis of LID. Central among non-dopaminergic systems is the glutamatergic transmission.3–5 N-Methyl-D-aspartate (NMDA) receptor, one subtype of the glutamate receptors, is rich in the striatum,6–8 composed of three major subunits, GluN1, GluN2A, and GluN2B (also known as NR1, NR2A, and NR2B). It has been demonstrated that NMDA receptor antagonists function as strong anti-dyskinetic agents for their significant suppression on LID.9 NMDA receptors were found involved in the occurrence of dyskinesia,10,11 but how expression and function of striatal NMDA receptors change cellularly and subcellularly stays unknown.
It is known that the properties of NMDA receptors depend on their subcellular localization, subunit composition, and also on NMDA receptor-associated proteins managing the response of signaling cascade, such as Ca2+/calmodulin-dependent protein kinase II (CaMKII).12,13 This kinase is activated by a transient Ca2+ rise with subsequent autophosphorylation at site of Thr286. After autophosphorylation, CaMKII can prolong its activity even after Ca2+ transients subside.14 A major set of direct substrates of CaMKII at local synaptic sites are glutamate receptors. By directly binding to the intracellular C-terminal tail of GluN2B,15 CaMKII phosphorylates GluN2B-C-terminal at a specific serine site (S1303) and thereby potentiates NMDA receptor function.16,17 Due to its tie to NMDA receptors, CaMKII is considered as the gateway of striatal NMDA- and DA-dependent functions.13 A selective inhibitor of CaMKII, N-[2-[[[3-(4-chlorophenyl)-2-propenyl]methylamino]methyl]phenyl]-N-(2-hydroxyethyl)-4-methoxybenzenesulfonamide (KN-93), like NMDA receptor antagonists, ameliorated LID in PD rat model after intrastriatal administration.18,19
Picconi et al13 reported that therapeutic effect of L-DOPA in short-term may be mediated by CaMKII activity in the striatum. The role of CaMKII-modulated NMDA receptor function involved in the development of dyskinesia after chronic L-DOPA treatment needs to be explored. In this study, we evaluated the influence of chronic L-DOPA administration on subcellular expression of striatal NMDA receptors in PD rats by a surface receptor cross-linking assay, and investigated the role of CaMKII in mediating NMDA receptor responses to L-DOPA by using KN-93.
Materials and methods
6-OHDA models
Animal experiments were executed according to the guidelines of the National Institutes of Health (publication no 80-23). All procedures were approved by the Institutional Review Board of Xinhua Hospital affiliated to Shanghai Jiao Tong University Medical School. Adult male rats (Sprague Dawley), weighing 180–220 g were used in this study.
This model was made as described previously.20 Briefly, ketamine (100 mg/kg) was used to anesthetize all rats by an intraperitoneal injection. After being placed onto a stereotaxic frame (Narishige, Tokyo, Japan), 4 μg/μL 6-hydroxydopamine (6-OHDA) (Sigma-Aldrich Co., St Louis, MO, USA) in a solution (in 0.9% saline with 0.02% ascorbic acid) was injected into the right medial forebrain bundle of rats. The total dosage of 6-OHDA was 32 μg/rat. Two coordinates were as follows: at anteroposterior (AP) −3.7 mm, mediolateral (ML) +1.7 mm, dorsoventral (DV) −7.8 mm; and at anteroposterior (AP) −4.4 mm, mediolateral (ML) +1.2 mm, dorsoventral (DV) −7.8 mm. The tooth bar was set to −2.4 mm.21 In control animals receiving sham surgery, the rats underwent the same procedure with an injection of a saline solution into the targeted sites.
Drug treatment and behavioral assessment
Twenty-one days after injection, the 6-OHDA-lesioned rats underwent a behavioral test for detecting contralateral rotations. The rats that exhibited rotational behaviors (at least seven turns per minute) following apomorphine injection (Sigma-Aldrich Co.) at a dose of 0.25 mg/kg were considered as successful PD rat models and could be used for the next experiment. The successful PD rats were divided into two groups randomly: Group 1 and Group 2. The two groups were both administrated with the L-DOPA solution (25 mg/kg L-DOPA plus 6.25 mg/kg benserazide per day, subcutaneously) for 21 consecutive days (twice daily). On day 22, pharmacological study with KN-93 was conducted in Group 1. KN-93 (Sigma-Aldrich Co.) (2 μg)19 was infused into the striatal side ipsilateral to the 6-OHDA lesion with the stereotaxic coordinates of AP +0.5 mm, ML +2.5 mm, DV −4.6 mm, as described in our previous study.18,19 Group 2 rats were not managed with KN-93 and followed the original procedures. The control group of sham-lesioned rats was given saline twice per day for 21 days instead of L-DOPA plus benserazide.
Abnormal involuntary movements (AIM) scores, axial, limb and orolingual (ALO) scores and the duration of rotation were used to assess the motor responses of PD rats treated with L-DOPA at the day 2, 7, 14, 21, and 22, as described elsewhere.18,22,23 Briefly, during 140 minutes after L-DOPA administration, rats were assessed every 20 minutes individually. Each rat was rated for appearance of abnormal movements in axial, limb, orolingual parts, and locomotor movements. For each AIM category, a severity score ranged from 0 to 4.18,22,23 The scores of each time point were added. The total scores of axial, limb and orolingual AIM were regarded as ALO scores.
Striatal total protein extraction and immunoblot analysis
To prepare the total protein extraction of striatum, animals were sacrificed by decapitation. The method of extraction was described previously.20 Dissected striatal tissues were frozen in liquid nitrogen and were then homogenized by sonication in radioimmunoprecipitation assay buffer (50 Mm Tris, 150 mM sodium chloride, 0.1% sodium dodecyl sulfate, 1% Nonidet P-40, 1 mM ethylenediaminetetraacetic acid, a protease inhibitor cocktail and 2 mM phenylmethylsulfonyl fluoride). The homogenate was centrifuged at 4°C for 10 minutes at 700 g and the supernatant was collected. The pellet which contained large debris or nuclei was discarded. Protein concentrations were determined in the supernatant using a bicinchoninic acid assay kit (Pierce, Rockford, IL, USA). After boiling for 5 minutes in Laemmli sample buffer, samples were analyzed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) directly. Samples containing equivalent amounts of protein (30 μg) were electrophoresed on 4%–15% Tris-hydrochloride gradient gels (Bio-Rad Laboratories Inc., Hercules, CA, USA). Proteins were electrotransferred to polyvinylidene fluoride membrane (0.22 μm) for immunoblotting. After transfer, block membranes in blocking buffer with 5% non-fat dry milk in Tris-buffered saline (TBS)-Tween-20 (TBS-T) for an hour at room temperature. Membranes were incubated at 4°C overnight with antibody recognizing GluN1 (1:1,000), GluN2A (1:1,000), GluN2B (1:1,000) (EMD Millipore, Billerica, MA, USA); CaMKII (1:1,000; Cell Signaling Technology, Danvers, MA, USA); phospho-CaMKII-T286 (1:500; Cell Signaling Technology); α-actinin (1:1,000; EMD Millipore) or β-actin (1:5,000; Sigma-Aldrich Co.). The membranes were washed extensively with TBS-T and incubated with horseradish peroxidase-conjugated anti-rabbit or anti-mouse immunoglobulin G (1:2,000; Cell Signaling Technology) for an hour at room temperature. Visualization was achieved by using the enhanced chemiluminescence detection system (EMD Millipore). Bands of interest were analyzed quantitatively using the Image Lab Software (Bio-Rad Laboratories Inc.).
Surface receptor cross-linking assays
In this study, we performed surface receptor cross-linking assays with bis(sulfosuccinimidyl) suberate (BS3)24,25 to determine the changes in surface/intracellular expression of striatal NMDA receptors in response to L-DOPA or KN-93 administration. The protocol is similar to our previous study.20 Briefly, rats were decapitated after last behavior assessment. Brains were rapidly taken out and a coronal section containing the striatum was dissected. The striatum was cut manually into small pieces and transferred into a tube (Eppendorf) containing ice-cold oxygenated artificial cerebrospinal fluid (CSF) spiked with BS3 (Pierce) with a concentration of 2 mM. The samples were incubated at 4°C with gentle agitation for half an hour. To quench the cross-linking reaction, the samples were added with 20 mM glycine and incubated at 4°C for 10 minutes with gentle mixing. After being washed in cold artificial CSF four times, the tissue was centrifuged and homogenized. Samples were aliquoted and analyzed directly by 4%–15% Tris-hydrochloride gradient gels. Intracellular protein (α-actinin) was used as a loading control to quantify and normalize the expression levels of surface and intracellular proteins.20,24
Statistical analysis
Data were expressed as the mean ± standard error of the mean. Behavioral assessments and biochemical data analysis among different groups were performed using one-way analysis of variance (ANOVA) followed by Fisher’s Least Significant Difference statistics post hoc comparison tests. Statistical significance was set at P<0.05.
Results
Effects of KN-93 on L-DOPA-induced dyskinesia
The comparison of AIM score, ALO score and duration of rotation from day 2 through day 22 are shown in Figure 1. After 21 days of chronic L-DOPA treatment in PD rats, dyskinesia was observed in both Group 1 and 2 (displayed by the elevated AIM score [61.00±2.68 in Group 1, 63.20±2.33 in Group 2], the ALO score [45.83±2.76 in Group 1, 47.60±2.58 in Group 2] and the reduced duration of rotation [94.67±2.50 in Group 1, 96.00±3.85 in Group 2] on day 21 compared with that of day 2 respectively [P<0.05]). The AIM scores on day 2 were 50.00±3.22 in Group 1, 48.40±2.01 in Group 2; ALO scores were 34.67±2.76 in Group 1, 35.60±5.00 in Group 2; the duration of rotation was 130.33±3.02 in Group 1,130.40±3.54 in Group 2 (Figure 1). On day 22, we found that intrastriatal KN-93 injection in Group 1 (L-DOPA+/KN-93+) significantly reduced the ALO score to 37.67±1.52 and the AIM score to 53.83±2.15 compared to Group 2 (L-DOPA+/KN-93−) (ALO: 47.00±2.30, AIM: 61.80±2.80, respectively) (P<0.05). At the same time, the duration of rotation in Group 1 returned to 112.33±2.43 while in Group 2 that was 98.00±3.63 (P<0.01). In Group 1, on day 22, the ALO score was lower than that on day 21 (P<0.05) and the duration of rotation was longer than that on day 21 (P<0.05). The AIM score showed a tendency of decrease compared with day 21, though there was no statistical difference (Figure 1). Altogether, these data show that intrastriatal inhibition of CaMKII ameliorates LID in PD rats.
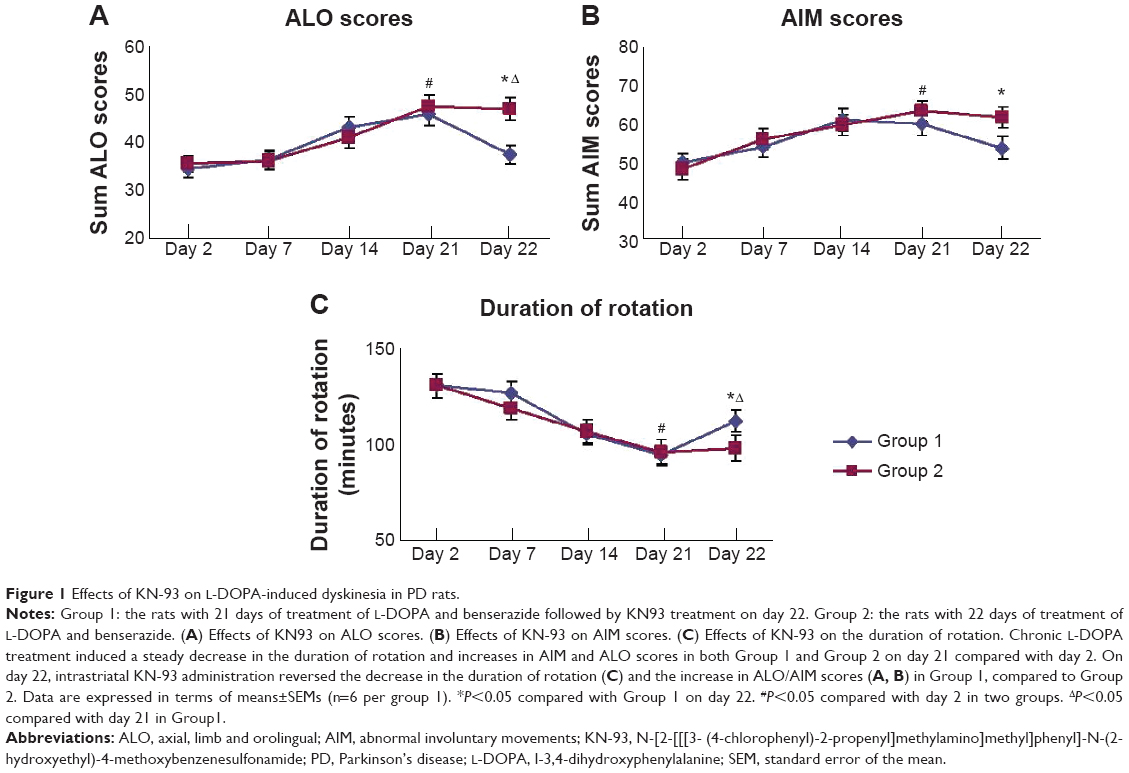 | Figure 1 Effects of KN-93 on L-DOPA-induced dyskinesia in PD rats. |
Phosphorylation of CaMKII at T286
T286 autophosphorylation indicates the activation of CaMKII. We found that L-DOPA administration in PD rats resulted in a robust increase in T286 phosphorylation (Figure 2). Quantitative analysis indicates that the levels of T286 phosphorylation were significantly elevated in both sides of LID rats compared to the sham rats, especially in the 6-OHDA-lesioned side. In the lesioned side of LID rats, the elevated level of T286 phosphorylation was significantly higher than un-lesioned side (P<0.05). In contrast, the total amount of CaMKII proteins remained unchanged (P>0.05). Intrastriatal KN-93 administration significantly suppressed the L-DOPA-induced T286 phosphorylation (Figure 2).
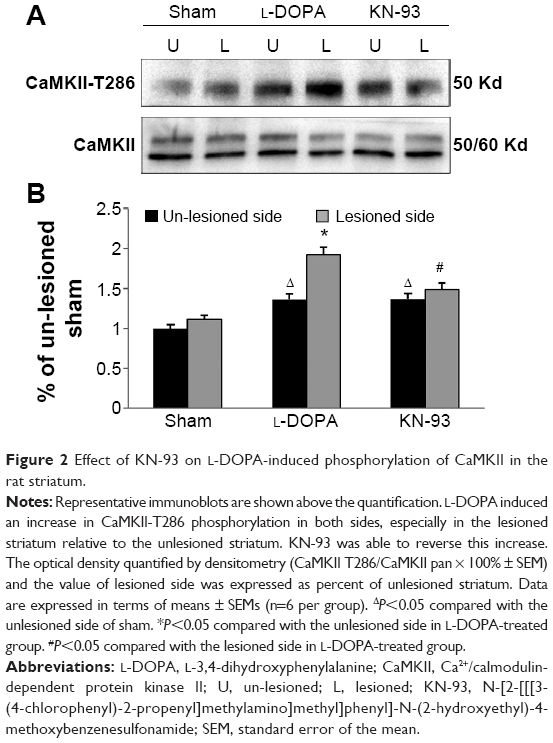 | Figure 2 Effect of KN-93 on L-DOPA-induced phosphorylation of CaMKII in the rat striatum. |
Changes in total protein expression of striatal NMDA receptors
We then detected the total cellular expression of key NMDA receptor subunits (GluN1, GluN2A, and GluN2B) in striatal neurons of PD rats with L-DOPA+/KN-93− versus PD rats with L-DOPA+/KN-93+. No significant difference of GluN1 level was found between the lesioned and un-lesioned side of striatum in either L-DOPA+/KN-93− group or L-DOPA+/KN-93+ group (Figure 3A and 3B). There was no difference between the two sides of striatum in sham rats after treatment with these drugs (Figure 3A and 3B). Similar results were obtained for GluN2A (Figure 3C and 3D). It was noted that the total expression of GluN2B was obviously higher in the lesioned side (152.02%±6.75% of the sham un-lesioned side) than that of the un-lesioned side (120.98%±1.66% of the sham un-lesioned side) in L-DOPA-treated models (P<0.001). KN-93 reduced this elevation of the lesioned side to 124.04%±8.09% of the sham un-lesioned side (P<0.001), while KN-93 caused no alteration in the GluN2B expression in the un-lesioned side of PD rats (121.02%±6.73% of the sham un-lesioned side). As compared to sham animals, GluN2B protein levels in all groups were increased in either lesioned or un-lesioned side (Figure 3E and 3F). Thus, chronic L-DOPA treatment that induces dyskinesia increases GluN2B expression in the striatum, which was reversed by local administration of a CaMKII inhibitor.
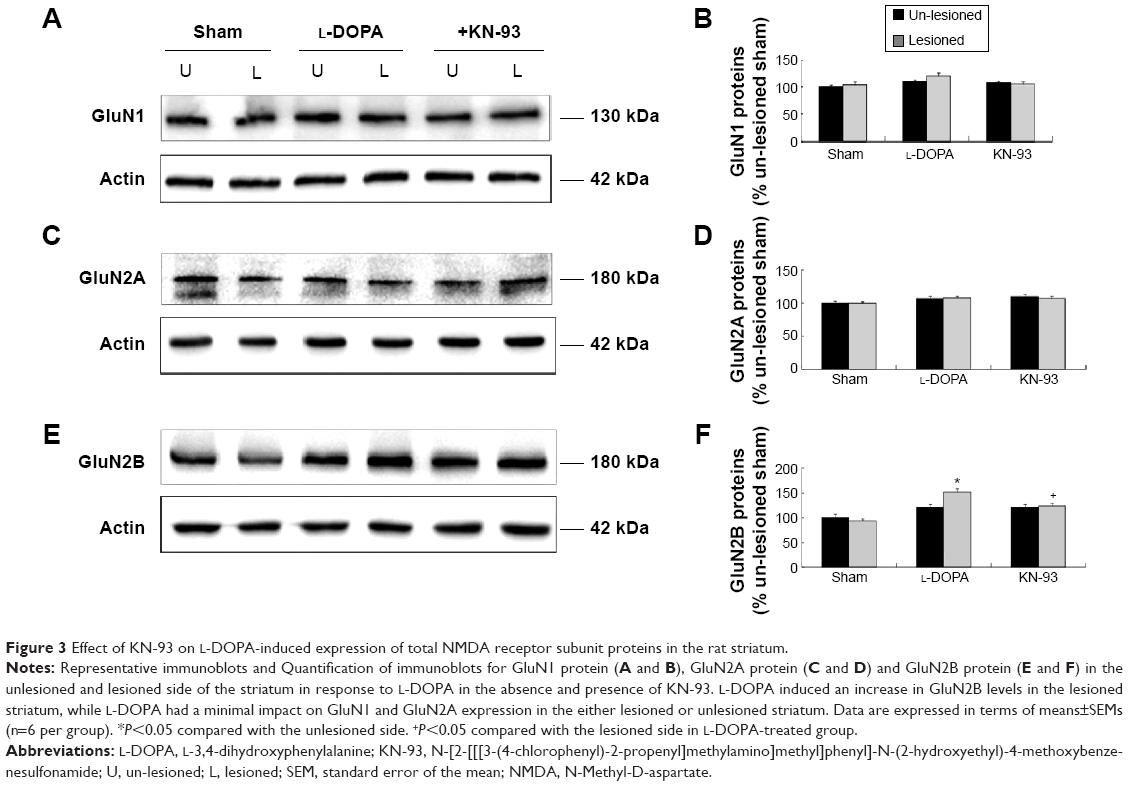 | Figure 3 Effect of KN-93 on L-DOPA-induced expression of total NMDA receptor subunit proteins in the rat striatum. |
Changes in subcellular expression of striatal NMDA receptors
According to BS3 assays, we demonstrated that the amount of GluN1 in the surface compartments increased in the lesioned side of L-DOPA-treated rats (179.26%±7.86% of the sham un-lesioned side) compared with the un-lesioned side (Figure 4A, B). On the contrary, intracellular GluN1 expressions dropped to 60.69%±2.98% of the sham un-lesioned side (P<0.01) (Figure 4A, C). Note that L-DOPA induced an increase in surface expression of GluN1 and a decrease in intracellular expression of GluN1 in the lesioned striatum. As a result, the surface-to-intracellular ratio (S:I) ratio was substantially increased in L-DOPA-treated rats (Figure 4D). After intrastriatal KN-93 injection, the increased surface level of GluN1 was reduced (104.36%±3.47% in the sham un-lesioned side), while the decreased intracellular level of GluN1 was reversed (101.25%±6.81% in the sham un-lesioned side) relative to the L-DOPA-treated group (Figure 4A–C). The S:I ratio was also normalized. (Figure 4D). Our data reveal that a redistribution of GluN1 in striatal neurons from intracellular pools to cell surface exists in response to L-DOPA administration. CaMKII activation seems to be required for this redistribution.
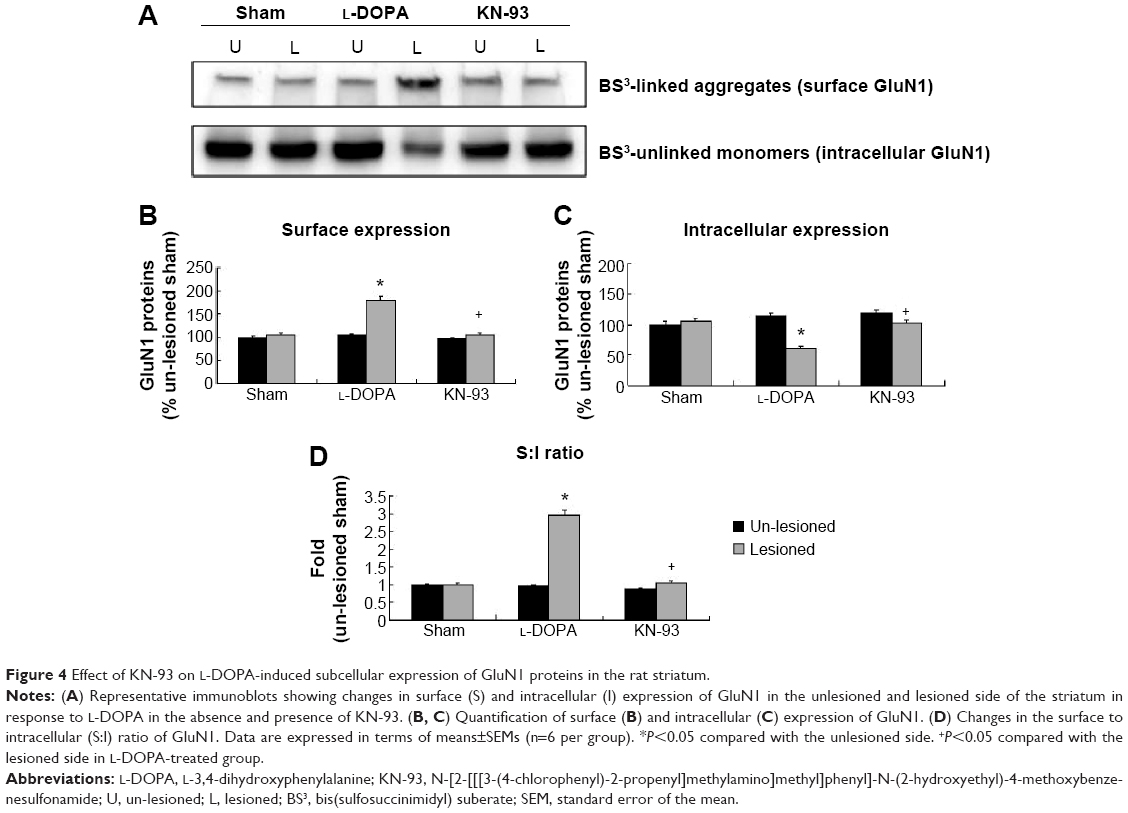 | Figure 4 Effect of KN-93 on L-DOPA-induced subcellular expression of GluN1 proteins in the rat striatum. |
Unlike GluN1, GluN2A expression in the two pools seemed to be regulated differentially. In the lesioned striatal side of L-DOPA-treated rats, the surface level of GluN2A was decreased (62.39%±4.64% of the sham un-lesioned side), while the intracellular level of GluN2A was concomitantly elevated (141.84%±13.87% of the sham un-lesioned side) (P<0.05) (Figure 5A–C). Note that L-DOPA induced a decrease in surface expression of GluN2A and an increase in intracellular expression of GluN2A in the lesioned striatum. As a result, the S:I ratio was reduced in L-DOPA-treated rats. KN-93 was able to completely reverse the decrease in surface GluN2A expression (P<0.05) and partially reduced the increase in the intracellular GluN2A expression (P=0.09). The S:I ratio was significantly decreased in the rats treated by L-DOPA and could be reversed by KN-93 (Figure 5D). These results indicate that GluN2A has redistribution from the cell surface to intracellular pools in striatal neurons after L-DOPA treatment. Local injection of KN-93 normalizes this redistribution.
 | Figure 5 Effect of KN-93 on L-DOPA-induced subcellular expression of GluN2A proteins in the rat striatum. |
A significant increase in GluN2B in the surface pool was seen in the lesioned striatum (184.20%±13.48% of the sham un-lesioned side) of L-DOPA+/KN-93− rats (Figure 6A, B). KN-93 reduced this increase to 142.04%±8.12% (P<0.01) (Figure 6A, B). No significant change in the intracellular level of GluN2B was shown between the group L-DOPA+/KN-93− and the group L-DOPA+/KN-93+ (Figure 6A, C). The S:I ratio was significantly increased after L-DOPA administration, which was reduced after KN-93 treatment (Figure 6D). These data indicate that striatal GluN2B is preferentially upregulated in surface membranes in response to L-DOPA. This upregulation may also involve activation of CaMKII.
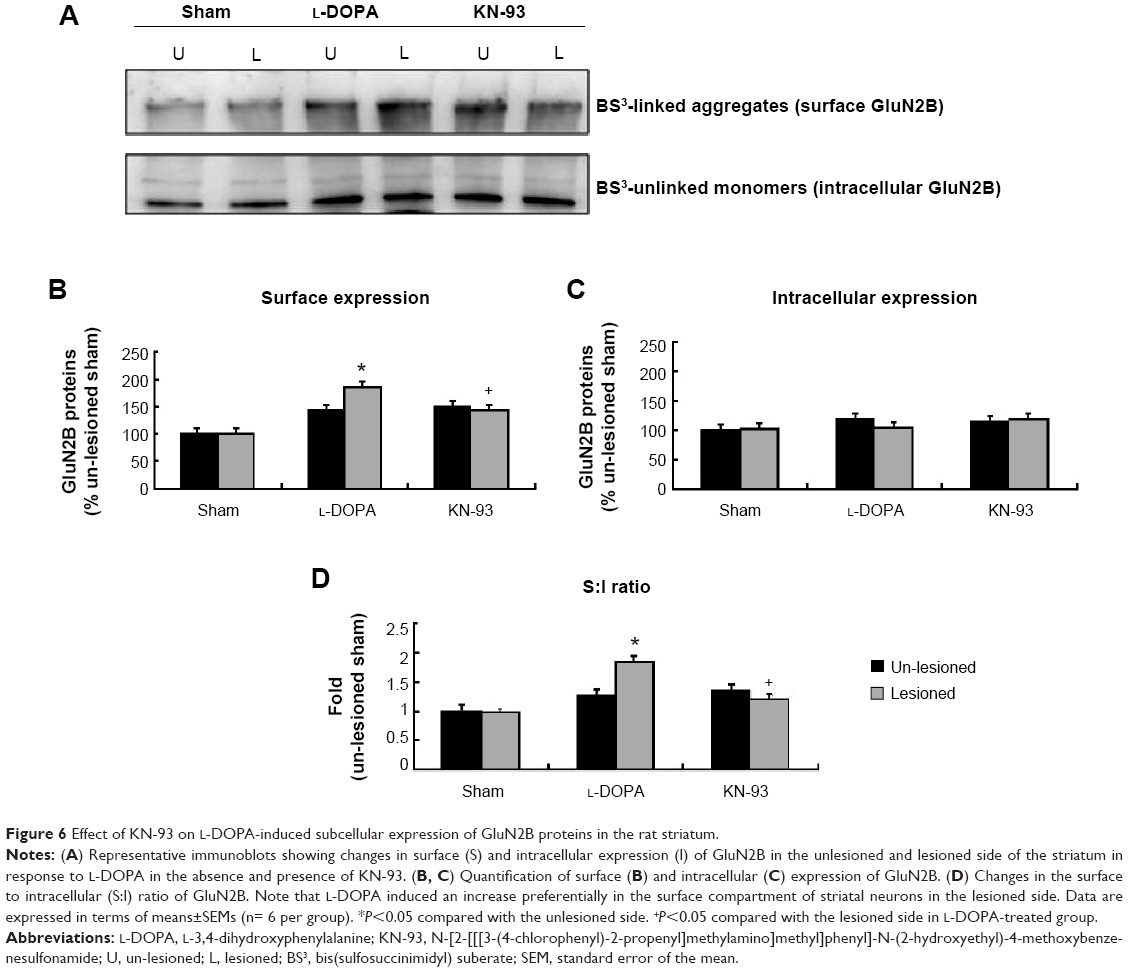 | Figure 6 Effect of KN-93 on L-DOPA-induced subcellular expression of GluN2B proteins in the rat striatum. |
In parallel, we monitored the response of an intracellular protein α-actinin to L-DOPA+/KN-93− group and to L-DOPA+/KN-93+ group. No significant change in α-actinin protein levels was observed in the lesioned side relative to the un-lesioned side in all groups (Figure 7).
 | Figure 7 Effects of L-DOPA and KN-93 on α-actinin expressions in the rat striatum. |
Discussion
In our present study, we investigated the effect of chronic L-DOPA treatment that caused typical dyskinesia on subcellular expression of striatal NMDA receptors in 6-OHDA-lesioned rats. We also evaluated the role of CaMKII in processing L-DOPA-induced changes on NMDA receptor expression.
First, we confirmed the effectiveness of CaMKII inhibitor suggested by the behavioral study that KN-93 alleviated dyskinetic behaviors. Parkinsonian rats and nonhuman primates treated with L-DOPA one or two times daily develop many abnormal movements with characteristics like human motor complication.18 In our study, PD rats displayed AIM after chronic L-DOPA treatment, with axial dyskinesia, forelimb dyskinesia, orolingual dyskinesia, and locomotive dyskinesia. The AIM scores and ALO scores increased correspondingly. Conversely, microinjection of KN-93 into striatum reduced dyskinetic scores and prolonged the duration of rotation, indicating alleviation of LID. This behavioral change was in line with previous studies.18,19
Daily administration of L-DOPA for 21 days could induce a unique change in subcellular expression of striatal GluN1. L-DOPA increased surface expression of GluN1, but lowered intracellular expression of the subunit with no change in total protein abundance. In contrast to GluN1, GluN2A exhibited an opposite response to L-DOPA. A decrease in surface pool of striatal GluN2A and an increase in intracellular pool simultaneously occurred in response to L-DOPA without changes in total protein levels. The expression of GluN2B was only upregulated in the surface pool. The L-DOPA-induced changes in expression of all three subunits seem to involve activation of synapse-enriched CaMKII. The CaMKII inhibitor KN-93 which was injected into the striatum reversed the aforementioned changes induced by L-DOPA. Our data demonstrated compartment-specific responses of key NMDA receptor subunits to L-DOPA treatment, and these L-DOPA-sensitive responses, to some extent, seem to be mediated by the signaling pathway involving CaMKII.
A number of early studies performed in humans or animals with dyskinetic conditions have found an increase in GluN2B expression in the striatum.3–5 In this study, we confirmed that total cellular levels of GluN2B protein in striatal neurons were elevated in L-DOPA-treated rats. We also found that the increase in GluN2B expression primarily occurred in the surface membrane. This may lead to an increase in activity of GluN2B-containing NMDA receptor after long-term application of L-DOPA. In addition to GluN2B, GluN1 and GluN2A were also sensitive to L-DOPA, although they were regulated differentially. GluN1 expression was increased in its amount in the striatal surface and decreased in the intracellular location, but GluN2A expression was modified in an opposite way. It may imply a redistribution of GluN1 and GluN2A between the two subcellular pools, probably due to altered exocytosis or endocytosis. The concomitant increases in surface GluN1 and GluN2B and decreases in surface GluN2A seem to suggest the existence of a higher level of GluN1/GluN2B receptors than GluN1/GluN2A receptors in striatal neurons during dyskinesia. A research team demonstrated that a reduction of GluN2B expression was associated with the appearance of dyskinesia.26,27 The discrepancy may be related to different L-DOPA dosages for preparing LID rats and different methods for detecting surface receptors. Although the result was inconsistent, this suggested that inability to maintain proper GluN1, GluN2A, and GluN2B distribution is involved in the development of LID. Future studies will elucidate whether similar changes would occur in subcellular expression of NMDA receptors at synaptic sites and whether these biochemical changes can translate to corresponding modifications of receptor function.
Another important finding of our study was the role of CaMKII in processing the L-DOPA-induced NMDA response. It was interesting to find that T286 autophosphorylation of CaMKII was increased in LID rats. The levels of CaMKII autophosphorylation elevated in both lesioned side and un-lesioned side of striatum may be caused by systemic L-DOPA delivery. Chronic L-DOPA administration might activate CaMKII. However, this activation was more obvious in the lesioned side of 6-OHDA rats. In our study, we focused on the ipsilateral side to 6-OHDA administration. Increased T286 phosphorylation indicates an enhanced state of CaMKII activity and potential of the kinase in processing the effect of L-DOPA on NMDA receptor expression. Indeed, it is known that various interacting proteins can regulate NMDA receptors.28 Central among these interacting partners is CaMKII. As a kinase mostly abundant within the postsynaptic density and directly binding to GluN2B,15,29 CaMKII is actively involved in the regulation of NMDA receptors.12 It phosphorylates GluN2B at a specific serine residue (S1303) and generally enhances NMDA receptor activity.16,17 In this study, CaMKII was found involved at least in part in mediating the L-DOPA-induced changes in subcellular expression of NMDA receptor subunits, suggested by the finding that the inhibition of CaMKII with an inhibitor KN-93 reversed the effect of L-DOPA. It is currently unclear how L-DOPA would activate CaMKII. We may assume that activation of Ca2+ permeable NMDA receptors triggers a cytoplasmic Ca2+ rise which activates CaMKII further and recruits active CaMKII to GluN2B to regulate trafficking and expression of the receptors. Future studies will clarify, in response to L-DOPA, how this feedback mechanism linking CaMKII to NMDA receptors works and the role of this feedback in the pathophysiology of LID.
Acknowledgments
The study was supported by the Projects of National Science Foundation of China (No 81171203, 81471148, 81200871 and 81400925), and Projects of the Shanghai Committee of Science and Technology, People’s Republic of China (12XD1403800).
Disclosure
The authors declare that there are no potential conflicts of interest.
References
Gottwald MD, Aminoff MJ. Therapies for dopaminergic-induced dyskinesias in Parkinson disease. Ann Neurol. 2011;69(6):919–927. | ||
Morin N, Morissette M, Gregoire L, Gomez-Mancilla B, Gasparini F, Di Paolo T. Chronic treatment with MPEP, an mGlu5 receptor antagonist, normalize basal ganglia glutamate neurotransmission in L-DOPA-treated parkinsonian monkeys. Neuropharmacology. 2013;73:216–231. | ||
Calon F, Rajout AH, Hornykiewicz O, Bedard PJ, Di Paolo T. Levodopa-induced motor complications are associated with alterations of glutamate receptors in Parkinson’s disease. Neurobiol Dis. 2003;14(3):404–416. | ||
Ahmed I, Bose SK, Pavese N, et al. Glutamate NMDA receptor dysregulation in Parkinson’s disease with dyskinesias. Brain. 2011;134(Pt 4):979–986. | ||
Maranis S, Stamatis D, Tsironis C, Konitsiotis S. Investigation of the antidyskinetic site of action of metabotropic and ionotropic glutamate receptor antagonists. Intracerebral infusions in 6-hydroxydopamine-lesioned rats with levodopa-induced dyskinesia. Eur J Pharmacol. 2012;683(1–3):71–77. | ||
Standaert DG, Testa CM, Young AB, Penny JB Jr. Organization of N-methyl-D-aspartate glutamate receptor gene expression in the basal ganglia of the rat. J Comp Neurol. 1994;343(1):1–16. | ||
Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacol Rev. 1999;51(1):7–61. | ||
Kew JN, Kemp JA. Ionotropic and metabotropic glutamate receptor structure and pharmacology. Psychopharmacology (Berl). 2005;179(1):4–29. | ||
Blandini F, Armentero MT. New pharmacological avenues for the treatment of L-DOPA-induced dyskinesias in Parkinson’s disease: targeting glutamate and adenosine receptors. Expert Opin Investig Drugs. 2012;21(2):153–168. | ||
Hallett PJ, Dunah AW, Ravenscroft P, et al. Alterations of striatal NMDA receptor subunits associated with the development of dyskinesia in the MPTP-lesioned primate model of Parkinson’s disease. Neuropharmacology. 2005;48(4):503–516. | ||
Nutt JG, Gunzler SA, Kirchhoff T, et al. Effects of a NR2B selective NMDA glutamate antagonist, CP-101, 606, on dyskinesia and Parkinsonism. Mov Disord. 2008;23(13):1860–1866. | ||
Simola N, Morelli M, Carta AR. The 6-hydroxydopamine model of Parkinson’s disease. Neurotoxicity Res. 2007;11(3–4):151–167. | ||
Picconi B, Gardoni F, Centonze D, et al. Abnormal Ca2+–Calmodulin-dependent Protein Kinase II Function Mediates Synaptic and Motor Deficits in Experimental Parkinsonism. J Neurosci. 2004;24(23):5283–5291. | ||
Griffith LC, Lu CS, Sun XX. CaMKII, an enzyme on the move: regulation of temporospatial localization. Mol Interv. 2003;3(7):386–403. | ||
Gardoni F, Caputi A, Cimino M, Pastorino L, Cattabeni F, Di Luca M. Calcium/calmodulin-dependent protein kinase II is associated with NR2A/B subunits of NMDA receptor in postsynaptic densities. J Neurochem. 1998;71(4):1733–1741. | ||
Omkumar RV, Kiely MJ, Rosenstein AJ, Min KT, Kennedy MB. Identification of a phosphorylation site for calcium/calmodulindependent protein kinase II in the NR2B subunit of the N-methyl-D-aspartate receptor. J Biol Chem. 1996;271(49):31670–31678. | ||
Liao D, Scannevin RH, Huganir R. Activation of silent synapses by rapid activity-dependent synaptic recruitment of AMPA receptors. J Neurosci. 2001;21(16):6008–6017. | ||
Oh JD, Vaughan CL, Chase TN. Effect of dopamine denervation and dopamine agonist administration on serine phosphorylation of striatal nmda receptor subunits. Brain Res. 1999;821(2):433–442. | ||
Yang XX, Wu N, Song L, Liu Z. Intrastriatal injections of KN-93 ameliorates levodopa-induced dyskinesia in a rat model of Parkinson’s disease. Neuropsychiatr Dis Treat. 2013;9:1213–1220. | ||
Gan J, Qi C, Mao LM, Liu ZG. Changes in surface expression of N-methyl-D-aspartate receptors in the striatum in a rat model of Parkinson’s disease. Drug Des Devel Ther. 2014;8:165–173. | ||
Paxinos G, Watson C. The rat brain in stereotaxic coordinates. 6th ed. Academic Press; 2006. | ||
Cenci MA, Lundblad M. Rating of L-DOPA-induced dyskinesia in the unilateral 6-OHDA lesion model of Parkinson’s disease in rats and mice. Curr Protoc Neurosci. 2007;Chapter 9:Unit 9.25. | ||
Rylander D, Recchia A, Mela F, Dekundy A, Danysz W, Cenci MA. Pharmacological modulation of Glutamate transmission in a rat model of L-Dopa- induced dyskinesia: Effects on motor behavior and striatal nuclear signaling. J Pharmacol Exp Ther. 2009;330(1):227–235. | ||
Mao LM, Wang W, Chu XP, et al. Stability of surface NMDA receptors controls synaptic and behavioral adaptations to amphetamine. Nat Neurosci. 2009;12(5):602–610. | ||
Boudreau AC, Milovanovic M, Conrad KL, Nelson C, Ferrario CR, Wolf ME. A protein cross-linking assay for measuring cell surface expression of glutamate receptor subunits in the rodent brain after in vivo treatments. Curr Protoc Neurosci. 2012;Chapter 5:Unit 5.30.1–19. | ||
Tronci E, Fidalgo C, Zianni E, et al. Effect of memantine on L-DOPA-induced dyskinesia in the 6-OHDA-lesioned rat model of Parkinson’s disease. Neuroscience. 2014;265:245–252. | ||
Gardoni F, Sgobio C, Pendolino V, et al. Targeting GluN2A-containing NMDA receptors reduces L-Dopa-induced dyskinesias. Neurobiol Aging. 2012;33(9):2138–2144. | ||
Picconi B, Ghiglieri V, Bagetta V, Barone I, Sgobio C, Calabresi P. Striatal synaptic changes in experimental parkinsonismL role of NMDA receptor trafficking in PSD. Parkinsonism Relat Disord. 2008;14 Suppl 2:S145–S149. | ||
Strack S, McNeill RB, Colbran RJ. Mechanism and regulation of calcium/-calmodulin-dependent protein kinase II targeting to the NR2B subunit of the N-methyl-D-aspartate receptor. J Biol Chem. 2000;275(31):23798–23806. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.