")
Back to Journals » Journal of Inflammation Research » Volume 17
Remodeling of Paranasal Sinuses Mucosa Functions in Response to Biofilm-Induced Inflammation
Authors Kaliniak S , Fiedoruk K , Spałek J , Piktel E, Durnaś B, Góźdź S, Bucki R , Okła S
Received 25 October 2023
Accepted for publication 23 January 2024
Published 26 February 2024 Volume 2024:17 Pages 1295—1323
DOI https://doi.org/10.2147/JIR.S443420
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tara Strutt
Szczepan Kaliniak,1 Krzysztof Fiedoruk,2 Jakub Spałek,1,3 Ewelina Piktel,2 Bonita Durnaś,1,3 Stanisław Góźdź,1,3 Robert Bucki,2,3 Sławomir Okła1,3
1Holy-Cross Cancer Center, Kielce, Poland; 2Department of Medical Microbiology and Nanobiomedical Engineering, Medical University of Białystok, Białystok, Poland; 3Institute of Medical Science, Collegium Medicum, Jan Kochanowski University of Kielce, Kielce, 25-317, Poland
Correspondence: Sławomir Okła, Department of Otolaryngology, Head and Neck Surgery, Holy-Cross Cancer Center, Kielce, 25-734, Poland, Tel/Fax +48 41 367 43 36, Email [email protected]
Abstract: Rhinosinusitis (RS) is an acute (ARS) or chronic (CRS) inflammatory disease of the nasal and paranasal sinus mucosa. CRS is a heterogeneous condition characterized by distinct inflammatory patterns (endotypes) and phenotypes associated with the presence (CRSwNP) or absence (CRSsNP) of nasal polyps. Mucosal barrier and mucociliary clearance dysfunction, inflammatory cell infiltration, mucus hypersecretion, and tissue remodeling are the hallmarks of CRS. However, the underlying factors, their priority, and the mechanisms of inflammatory responses remain unclear. Several hypotheses have been proposed that link CRS etiology and pathogenesis with host (eg, “immune barrier”) and exogenous factors (eg, bacterial/fungal pathogens, dysbiotic microbiota/biofilms, or staphylococcal superantigens). The abnormal interplay between these factors is likely central to the pathophysiology of CRS by triggering compensatory immune responses. Here, we discuss the role of the sinonasal microbiota in CRS and its biofilms in the context of mucosal zinc (Zn) deficiency, serving as a possible unifying link between five host and “bacterial” hypotheses of CRS that lead to sinus mucosa remodeling. To date, no clear correlation between sinonasal microbiota and CRS has been established. However, the predominance of Corynebacteria and Staphylococci and their interspecies relationships likely play a vital role in the formation of the CRS-associated microbiota. Zn-mediated “nutritional immunity”, exerted via calprotectin, alongside the dysregulation of Zn-dependent cellular processes, could be a crucial microbiota-shaping factor in CRS. Similar to cystic fibrosis (CF), the role of SPLUNC1-mediated regulation of mucus volume and pH in CRS has been considered. We complement the biofilms’ “mechanistic” and “mucin” hypotheses behind CRS pathogenesis with the “structural” one – associated with bacterial “corncob” structures. Finally, microbiota restoration approaches for CRS prevention and treatment are reviewed, including pre- and probiotics, as well as Nasal Microbiota Transplantation (NMT).
Keywords: rhinosinusitis, chronic rhinosinusitis, nasal polyps, rhinosinusitis pathophysiology, rhinosinusitis pathophysiology, microbiota, nutritional immunity
Introduction
Rhinosinusitis (RS) is an inflammatory disease of the nasal and paranasal sinuses. According to the European Position Paper on Rhinosinusitis and Nasal Polyps 2020 (EPOS 2020), RS is classified into acute (ARS) or chronic (CRS).1 The categorizing factors involve (i) the presence and type of symptoms, such as nasal blockage, facial pain, cough, reduction or loss of smell, and (ii) their duration, ie, ≤12 weeks for ARS and ≥12 weeks for CRS. Both types of RS are among the most common health issues worldwide. It is estimated that 6–15% of the general population suffers from ARS, one of the most common diagnoses for antibiotic prescriptions. Similarly, CRS affects 5–12% of the global population, and differences in risk factors such as smoking or environmental irritants may account for some geographical variation.1–4 The overall prevalence of CRS in the general population in Europe and the United States was estimated to be 10.9% and 11.9%, respectively.1 Comparable percentages were found in the adult general population of other countries, such as South Korea (8.4%), China (8.0%), and Brazil (5.5%).4 CRS significantly reduces the health-related quality of life (HRQoL), mainly due to nasal obstruction, rhinorrhea, and olfactory impairments, as well as substantially burdens the health care system, even more costly than asthma or peptic ulcer disease.1,4–7 The implementation of HRQoL questionnaires demonstrates clinical and predictive treatment efficacy and enhances patient care. The Sino-Nasal Outcome Test-22 (SNOT-22) is the most widely utilized and most suitable questionnaire among those frequently employed in rhinology due to its validity, reliability, and usability.7
Chronic rhinosinusitis (CRS) is a complex condition characterized by numerous variants (phenotypes) with distinct pathophysiologies (endotypes).8 Clinically, the presence or absence of nasal polyps (NPs) has been used to differentiate between the two primary CRS phenotypes – (i) with NPs (CRSwNP) and (ii) without NPs (CRSsNP).9,10 Other CRS phenotypes entail (i) infectious, (ii) fungal, (iii) pediatric, and associated with (iv) cystic fibrosis, (v) aspirin-exacerbated respiratory disease, and (vi) systemic diseases.9
A critical aspect of CRSwNP is the restoration of recovery of olfactory function. The standard of care for CRSwNP, consisting of intranasal corticosteroids (INCS) and short repeated courses of oral corticosteroids (OCS), produced mixed results of olfactory function recovery. In addition, in subjects with CRSwNP, OCS has non-durable effects on olfactory function, often leading to the consumption of high cumulative doses. Therefore, biological medications targeting the IL-4/-13 receptor, such as dupilumab, have been proposed as promising treatments for improving olfactory outcomes in CRSwNP patients with type 2 inflammation.11
In both CRSsNP and CRSwNP, inflammation is highly heterogeneous and manifests as one of three major inflammatory endotypes characterized by various T cell cytokine patterns, including type 1 (T1) with Th1 cytokine IFN-γ, type 2 (T2) with Th2 cytokines IL-4, IL-5, IL-9 and IL-13, and type 3 (T3) with Th17 cytokines IL-17 and IL-22.12 Since type 2 inflammation is distinctive of CRSwNP, CRS is also often divided into type 2 and non-type 2 categories.1,13 Depending on the degree of eosinophilia in sinuses, idiopathic eosinophilic CRS (E-CRS) and non-eosinophilic CRS (NE-CRS) subsets of CRS are also distinguished. Severe tissue eosinophilia is usually associated with more severe symptoms and poorer treatment outcomes.14 Furthermore, CRS is subdivided into primary and secondary, as well as localized and diffused, based on anatomical distribution. Up to 30% of patients with CRS develop NPs, typically as a Th2-dominant eosinophil-driven disease. However, type 1 mediators may also contribute to NPs.15 Consequently, remodeling of the sinus mucosa matter is a common, but not unique, feature of diffused primary CRSwNP. Remodeling of the sinus mucosa may activate several cellular processes such as epithelial-to-mesenchymal transition (EMT) and autophagy.14
Chronic inflammation in CRS generally results in mucosal barrier dysfunction, inflammatory cell infiltration, impaired mucociliary clearance, mucus hypersecretion, and tissue remodeling. However, the stimuli and mechanisms underlying these inflammatory processes remain unclear. CRS etiology has been linked with multiple host and exogenous factors, such as dysfunction of the epithelial barrier, mucociliary clearance and immune responses, environmental exposures, specific bacterial or fungal organisms and their toxins, microbial biofilms, and sinonasal microbiota imbalance (dysbiosis).12,16
Accordingly, several hypotheses underlying CRS development have been proposed, including (i) “immune barrier” hypothesis, (ii) “eicosanoid” hypothesis, (iii) “fungal” hypothesis, (iv) ‘staphylococcal superantigens’ hypothesis, (v) “biofilm” hypothesis, and (vi) “microbiota/microbiome” hypothesis.4,12,17,18 However, the current view of CRS assumes an aberrant interplay between the host and exogenous factors rather than a single cause.17 The hypotheses have been discussed throughout the manuscript. It should be emphasized that single-nucleotide polymorphisms (SNPs) may increase the likelihood of developing CRS.19 Barrier deficiencies, alterations in ion channels, or mutations in genes associated with the TH2 inflammatory response are the major mechanisms that contribute to the development and maintenance of chronic inflammation in CRS. Therefore, understanding the pathophysiology of CRS is improved through genetic analysis of SNPs in patients with CRS.19
The “immune barrier” hypothesis is the most comprehensive and coherent with the other theories’ view on the etiology and pathogenesis of CRS. It is assumed that mechanical or immune-mediated disruption of the mucosal barrier and triggered inflammation enhances exposure to external stimuli and induces dysbiotic changes in biofilms, resulting in excessive compensatory immune responses. Genetic and epigenetic factors are also involved, such as bitter taste receptors (T2Rs) polymorphism, that is receptors that play an essential role in microbiota surveillance by sensing mediators of bacterial quorum sensing (QS) communication.
Recently, a link between CRS and low zinc (Zn) levels in the nasal mucosa of CRS patients, in particular with nasal polyps (CRSwNPs), has been suggested.20 Mucosal zinc (Zn) deficiency may induce several typical CRS pathomechanisms, such as mucosal barrier dysfunction, inflammatory cell infiltration, impaired mucociliary clearance, mucus hypersecretion, and tissue remodeling.20
Like in other chronic diseases, the interplay between sinonasal microbiota and the immune system appears to be an essential but unrevealed trait of CRS-“microbiota” hypothesis.8,21–25 These growths in biofilms microbial communities coexist in a commensal relationship with the human host (eubiosis), and contribute to a nonspecific defense due to their colonization resistance function, ie, the ability of microbiota to limit the introduction of exogenous microorganisms and pathobiont expansion.26,27 If this homeostatic balance is not maintained, certain bacterial species or, more likely, clusters of bacteria (“co-occurrence” or “community” hypothesis) may overgrow and undermine others (dysbiosis) and, in turn, shift from a commensal to parasitic lifestyle. Multiple synergistic and antagonistic interspecies interactions, such as cross-feeding and competitive exclusion, regulate microbiota homeostasis and may contribute to dysbiotic transitions. This interspecies crosstalk is coordinated by quorum sensing (QS) systems. QS is a bacterial cell–cell and population density-dependent communication via signal molecules (QSM), such as N-acyl-homoserine lactone (AHL) and N-3-(oxododecanoyl)-homoserine lactone (C12-HSL), which synchronize metabolic processes and gene expression in a group of microorganisms. In addition, QS systems control several crucial bacterial–host interaction processes such as biofilm formation, expression of virulence factors, stress responses, and metal sequestration. For example, Staphylococcus aureus growth, virulence, and colonization abilities can be impaired by other Staphylococcus species through the accessory gene regulatory (agr) QS system.28 Furthermore, QMS, such as C12-HSL, act as virulence factors and directly or indirectly may affect the epithelial barrier function, inflammation, and even intestinal carcinogenesis (reviewed by Conquant et al, 2021).29 Therefore, QSs maintain a balanced microbiota and, in concert with bitter taste receptors (T2Rs), microbiota–host interactions in homeostasis.30 It has been hypothesized that inflammation and sinus blockage can originate either (i) from immune dysfunction followed by microbiota dysbiosis or, conversely, (ii) that dysbiosis is responsible for immune response dysregulation.31,32 Yet, no clear distinctions between the sinonasal microbiota composition (microbiotypes) of healthy and CRS subjects have been established; hence, the microbiota cannot serve as an independent CRS biomarker.8,22,23
The ineffectiveness of antibiotics in treating CRS supports the “microbiota” hypothesis.33 Nonetheless, like in cystitis fibrosis, antimicrobials with anti-inflammatory properties, such as doxycycline or macrolides, may be advantageous.1,34 For instance, a short course of doxycycline appears beneficial in patients with polyps.1
Microbial biofilms are a ubiquitous form of microbial growth in up to 80% of microorganisms on the Earth, including microbiota in the human body. In medicine, biofilms account for 80% of chronic infections.35,36 These complex and common multispecies communities of microorganisms encased in a self-produced slimy matrix of extracellular polymers (EPS) have an extraordinary capacity for growth and persistence on inanimate and living surfaces, such as mucosal epithelium.
In biofilms growing on the sinus mucosa of patients with CRS, well-recognized pathogens include Staphylococcus aureus, Streptococcus pneumoniae, Pseudomonas aeruginosa, Haemophilus influenzae, and Moraxella catarrhalis. In particular, S. aureus, H. influenzae, and P. aeruginosa are typically identified in CRS-associated biofilms (Figure 1).37–39 In addition, various opportunistic pathogens such as coagulase-negative staphylococci, Corynebacteria, Streptococcus viridans, Enterococcus faecalis, anaerobes, Cutibacterium (Propionibacterium), Porphyromonas, Prevotella, Bacteroides, and Peptoniphilus (Peptostreptococcus). However, comparable biofilm detection rates in patients with CRS and healthy controls raise questions about their role in this disease.40
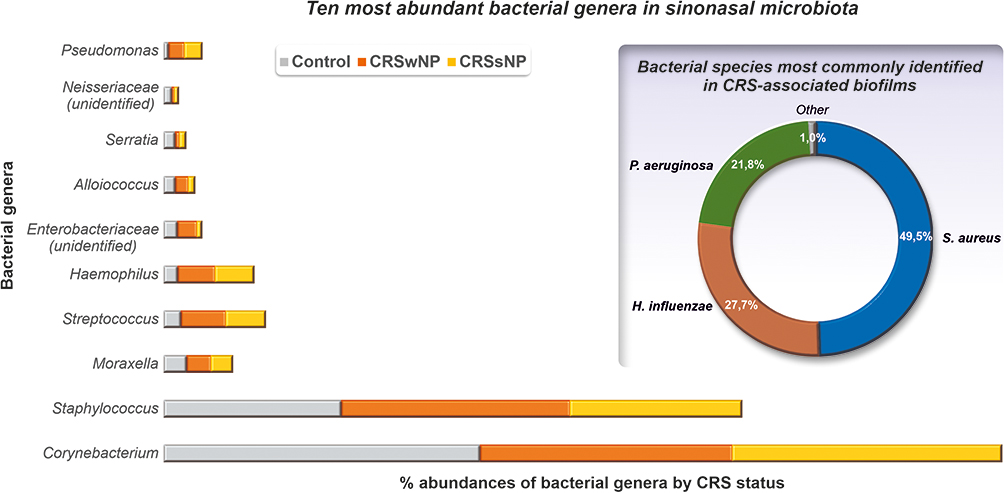 |
Figure 1 Ten most abundant bacterial genera in sinonasal microbiota (Data from Paramasivan S, Bassiouni A, Shiffer A, et al. The international sinonasal microbiome study: a multicentre, multinational characterization of sinonasal bacterial ecology. Allergy. 2020;75(8):2037–2049.23) and bacterial species most commonly identified in CRS-associated biofilms. Data from.37–39 |
However, comparable biofilm detection rates in CRS patients and healthy controls raise questions about the role of these structures in this disease.40 Nonetheless, biofilms in CRS patients are associated with more severe symptoms, poorer outcome, complicated postoperative course, and increased likelihood of revision surgery.41–46 Alterations in the structure and function of the paranasal sinuses associated with epithelial barrier disruption, impaired mucociliary clearance, elevated inflammatory response, fibrosis, hyperplasia, and abnormal collagen deposition, resulting in tissue remodeling, and ultimately, formation of nasal polyps, are also typical manifestations of biofilm-induced sinusitis.47 A “mechanistic” and “mucin” hypotheses have been proposed to explain the potential role of such “pathologic” or “dysbiotic” biofilms in CRS pathogenesis.
In this review, we discuss the potential role of sinonasal microbiota in CRS based on recent metagenomic findings in the context of mucosal zinc deficiency as a possible link between the “host” and “bacterial” hypotheses of CRS. For instance, the potential function of Zn-mediated “nutritional immunity” and calprotectin, a metal-binding antimicrobial peptide (AMP), as microbiota-shaping factors in CRS has been addressed. In addition, another AMP-SPLUNC1, acting as an epithelial Na+ channel (ENaC) pH-dependent inhibitor, may link microbiota via pH with airway surface liquid (ASL) homeostasis and impaired mucociliary clearance (MCC), as shown in cystic fibrosis. In addition, we consider mechanisms behind biofilms’ “mechanistic” and “mucin” hypotheses in the pathogenesis of CRS. Furthermore, alterations in bacterial genetic background as well as inter- and interspecies interactions and processes underlying the transition from “healthy” or eubiotic biofilms toward “pathologic” or “dysbiotic” biofilms are confronted with the host immune responses. We believe that our findings will aid in a better understanding of the biofilm-mediated pathomechanism in CRS and in developing new diagnostic and treatment methods based on microbiota restoration, including nasal microbiota transplantation (NMT).
Role of Sinonasal Microbiota in CRS
Although a metagenomic approach to explore microbial diversity has significantly improved our understanding of the human microbiota, several variables, such as (i) sample type and collection method, (ii) bacterial nucleic acid isolation methodology, and (iii) sequencing technology limitations, may still influence the results and their interpretation. Additional confounders included significant interpersonal, geographical, and racial heterogeneity in the sinonasal microbiota composition. Subsequently, data collected in metagenomic studies on sinonasal microbiota should be carefully interpreted.23–25,40,48 For instance, atypical or unusual microorganisms identified as causative agents of CRS that are not part of the sinonasal microbiome should be thoroughly evaluated.23
Nevertheless, specific sinonasal microbiota patterns and/or interbacterial relationships have been linked to the onset and progression of CRS. Specifically, the predominance of Corynebacterium and Staphylococcus, which likely live in reciprocal or antagonistic relationships, was observed.24,31 Briefly, five genera, Corynebacterium, Staphylococcus, Streptococcus, Haemophilus, and Moraxella, represent the core sinonasal microbiome in both healthy and CRS subjects according to a recent international sinonasal microbiome study involving 461 subjects.23 However, Corynebacterium and Staphylococcus were the far most abundant genera, occurring in samples with a mean relative abundance of 44.02% and 27.34%, respectively (Figure 2). Furthermore, in CRSwNP patients, Corynebacterium abundance was significantly decreased (40.29% vs 50.43%; p = 0.02) and correlated with increased abundance of Streptococcus genus, compared to healthy controls. In contrast, no significant differences were observed between CRSsNP patients and control subjects in the predominance of any of the top ten most abundant bacterial genera (Figure 2). Interestingly, the distributions of the two most frequently isolated pathogens from CRS-biofilms biofilms, Staphylococcus and Pseudomonas, were similar among all the tested groups.23
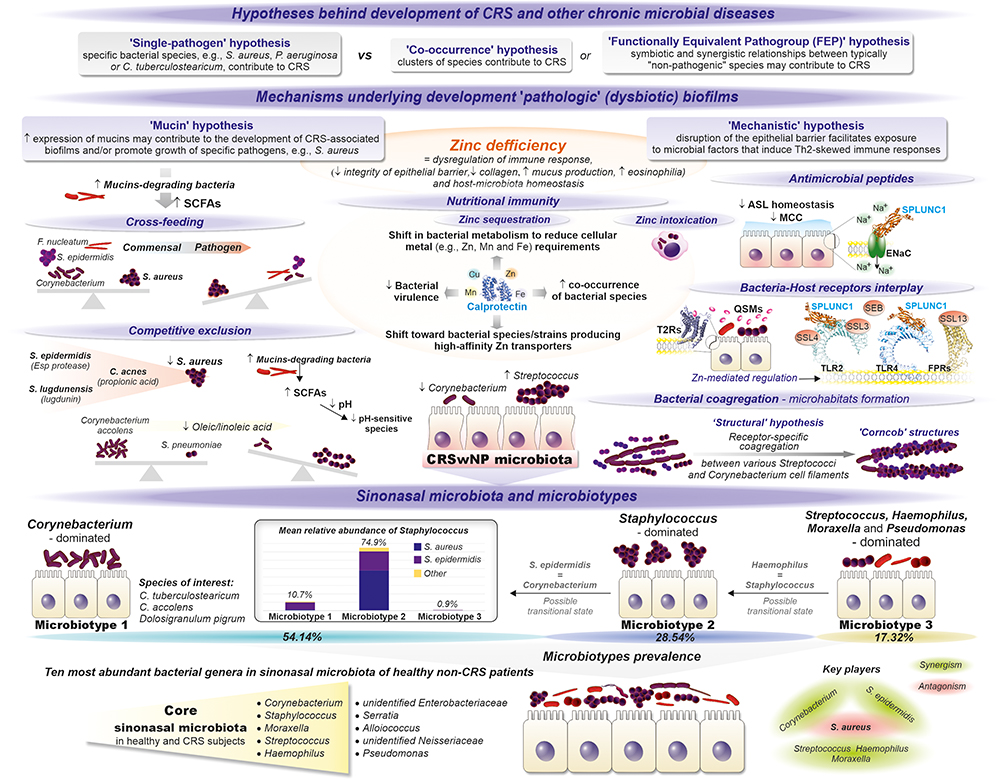 |
Figure 2 The sinonasal microbiota and microbiotypes in patients with CRS and healthy individuals (adapted from Paramasivan et al and Bassiouni et al) and potential hypotheses and mechanisms underlying the development of “pathologic” (dysbiotic) biofilms. Data from 22,23. |
The sinonasal microbiome appears to be most similar to the microbial community of anterior nares, which could explain the abundance of typical respiratory tract pathogens, Streptococcus, Haemophilus, and Moraxella, and implicates sinuses as a potential reservoir for other respiratory tract diseases, such as acute otitis media.49 On the other hand, Streptococcus, Haemophilus, and Moraxella, as a part of the core sinonasal microbiota, likely emphasizing their importance in maintaining sinonasal physiology as commensals.
Furthermore, Bassiouni et al distinguished in this core sinonasal microbiota three major sinonasal “microbiotypes”, ie, Corynebacterium-dominated (microbiotype 1), (ii) Staphylococcus-dominated (microbiotype 2), and (iii) Streptococcus, Haemophilus, Moraxella, and Pseudomonas-dominated (microbiotype 3).22 The prevalence of these microbiotypes was not correlated with healthy and diseased sinuses; however, geographical differences in their distribution were reported.22 The microbiotype 1 was overrepresented in Asia and Australasia, while the prevalence of microbiotype 2 was higher in Europe. However, Europe was represented only by one location (Amsterdam); therefore, it is uncertain whether these results apply to other European areas.
Hoggard et al found a similar sinonasal microbiota profile in controls and idiopathic CRS subjects, with a predominance of Corynebacterium and Staphylococcus genera and lower abundances of Streptococcus, Moraxella, and Haemophilus.40 In contrast, members of the genera Staphylococcus, Streptococcus, Haemophilus, Pseudomonas, Moraxella, and Fusobacterium dominated at various levels in dysbiotic communities, along with a reduced abundance of the dominant Corynebacteria.40
Due to its probiotic potential, Corynebacterium is referred to as a “gatekeeper”, “keystone”, or “cornerstone” member of the upper respiratory tract microbiota that is essential for respiratory health and competitively excludes potential pathogens, such as S. pneumoniae.22,50 Interestingly, Corynebacterium is also the most prevalent and abundant bacterial genus in another chronic condition – diabetic foot ulcers (DFUs).51 In contrast, a high abundance of staphylococci (coagulase-negative commensal species and S. aureus) in the sinonasal microbiota of healthy subjects likely indicates their dual, associated with the ability to shift lifestyle, commensal-pathogen role. The exact factors and mechanisms underlying S. aureus transition from a commensal to pathogenic lifestyle are unknown, but reciprocal or antagonistic interactions via QS systems with Corynebacteria and other Staphylococci are likely involved.22 Indeed, a positive and negative correlation was reported between the co-occurrence of Corynebacterium and S. epidermidis and between this bacterial pair and S. aureus, respectively.22 For example, a serine protease Esp of S. epidermidis disassembles S. aureus biofilms and inhibits its colonization, degrading ~75 S. aureus proteins, including 11 biofilm formation- and colonization-associated proteins as well as several human receptors, such as fibronectin, fibrinogen, and vitronectin, utilized by S. aureus during host colonization or infection.52 Likewise, S. lugdunensis produces lugdunin - a bactericidal peptide that prevents S. aureus nasal colonization.53 Moreover, C. accolens and C. pseudodiphtheriticum are the two most significant determinants of S. aureus nasal carriage, likely due to mutually cooperative - C. accolens and mutually competitive - C. pseudodiphtheriticum interactions. The competitive interactions appear to be microenvironment-dependent, ie, restricted to mucosal sites lined by ciliated pseudostratified columnar epithelium (but not with lined by skin-like squamous epithelium of the anterior naris), possibly as nutrient or other niche-related limitations supporting competitive behavior.54 However, an opposite relationship between C. accolens and S. aureus was reported in other studies, where C. accolens cell-free culture supernatants appear to inhibit the growth of both planktonic and biofilm forms of S. aureus oleic acid.55,56 Subsequently, Tween-80, metabolized by C. accolens to oleic acid, has been proposed as a prebiotic to promote microbiota homeostasis in patients with CRS.57 Also, Dolosigranulum pigrum, as a nasal health-promoting species with immunomodulatory and nasal barrier-enhancing potential as well as antimicrobial activity against S. aureus, was indicated candidate for development into a topical next-generation probiotics or live-biotherapeutic products.58 In addition, Huang et al demonstrated that C. accolens has the potential to mitigate S. aureus-induced mucosal barrier damage.56 Notably, C. accolens antagonizes with S. pneumoniae via oleic and linoleic acid, which likely also explains Streptococcus depletion in Corynebacterium-dominated microbiota in approximately one-fifth of CRS patients.8
Also, antagonism between S. aureus and S. pneumoniae, H. influenzae, or M. catarrhalis is well documented and probably reflects its drastically low abundance (0.53%) in the microbiotype 3 (Figure 2).59–61 Accordingly, Pettigrew et al reported that the elimination from the nasopharyngeal carriage of S. pneumoniae and H. influenzae increases the risk of S. aureus colonization.60 Killing S. aureus by pneumococcal hydrogen peroxide through activation of lytic bacteriophages can partially explain their negative co-occurrence.50 In contrast, positive associations have been identified among S. pneumoniae, H. influenzae and M. catarrhalis.62
The abundance of Corynebacteria or specific Corynebacterium species, such as C tuberculostearicum, was also positively correlated with CRS in certain other studies.8,24,31,63 Accordingly, Cope et al distinguished four central taxonomic states in CRS microbiota, characterized by specifically enriched gene pathways and immune response patterns: (i) Streptococcaceae (DSI) – ansamycin biosynthesis, (ii) Pseudomonadaceae (DSII) – tryptophan metabolism, (iii) Corynebacteriaceae [DSIII(a)] – with enriched the PPAR-γ signaling pathway, and (iv) Staphylococcaceae [DSIII(b)] – two-component response.8 Corynebacteriaceae [DSIII(a)] state was detected in the microbiota of approximately one-fifth of CRS patients and positively correlated with increased expression of IL-5 and IFN-γ and a higher risk for developing polyposis.8 In contrast, Pseudomonadaceae (DSII) and Staphylococcaceae [DSIII(b)] states characterized an increase in a variety of pro-inflammatory Th1 responses.
Similarly, a meta-analysis of sinonasal microbiome studies revealed Staphylococcus, Propionibacterium, Corynebacterium, Streptococcus, and unclassified Actinobacteria were the most abundant bacteria across all subjects. However, only a higher abundance of members of the genus Corynebacterium was consistently linked to CRS; hence, it was proposed as a potential biomarker of CRS-associated sinonasal microbiota.31 Additionally, bacterial communities in this group were characterized by significantly lower diversity and increased dispersion. Among the remaining genera, positive correlations with CRS were observed for Parvimonas, Porphyromonas, Citrobacter, Prevotella, Curtobacterium, and Fusobacterium. Notably, a negative correlation was observed between the abundance of Corynebacterium and Fusobacterium, indicating that these bacteria may compete for the same niche. In addition, the importance of Burkholderia and Propionibacterium (Cutibacterium) in regulating the microbiota communities in healthy sinuses has been suggested. Subsequently, depletion of these “protective” or “gatekeepers” genera significantly increased the susceptibility of the sinonasal microbiota to disturbance.31 This “protection” could be mediated by Cutibacterium acnes (Propionibacterium acnes) via propionic acid-mediated attenuation of macrophage-induced inflammation and inhibition of S. aureus growth.64 Strikingly, Burkholderia and Corynebacterium were found to be negatively correlated. In addition, Abreu et al reported an increase in the relative abundance of a single species, C. tuberculostearicum, concomitant with the depletion of distinct lactic acid bacteria in CRS patients.24 Likewise, Aurora et al reported a statistically significant increase in C. accolens in patients with CRS compared to controls.63
Notably, in dental plaque biofilms, so-called “corncob” or “hedgehog” structures are formed by filaments of Corynebacteria cells (~10–20 μm long) coaggregating with Streptococci, and to a lesser extent with members of Porphyromonas genus and Pasteurellaceae family (Haemophilus and Aggregatibacter) in a specific order.65 Corncobs are well-defined microhabitats within biofilms, where the spatial proximity of their inhabitants facilitates the cross-feeding phenomenon, ie, nutrient exchange between bacteria, where the metabolic products of one species are utilized in the nutrition of another species.65 In this view, Corynebacteria may be essential to the architecture of sinonasal biofilms, and their transition to a dysbiotic state may be associated with structural disturbances affecting corncobs.65 Hence, expanding the mechanistic and mechanistic mucin and mucin hypotheses of biofilms’ role in CRS by the structural hypothesis.
The formation of “corncobs” is a well-described trait of Fusobacterium nucleatum in the context of dental plaque-associated diseases, such as dental caries and periodontitis. In dental plaque, F. nucleatum co-aggregates through receptor-ligand interactions (via RadA and Fap2 fusobacterial receptors) with multiple early (aerobic) and late (anaerobic) colonizers. The receptor–ligand mechanism is likely utilized by Corynebacteria, since co-aggregation with several streptococcal species (S. cristatus, S. mitis, S. oralis, or S. infantis), except S. gordonii, was observed.66 Importantly, a negative correlation between Corynebacterium and Fusobacterium in sinonasal microbiota reported by Mackenzie et al may suggest their distinct roles in sinonasal biofilm formation or participation at various stages of its development.31 For instance, F. nucleatum as an aerotolerant organism may promote the growth of strict anaerobic species. In contrast, a limited number of potential co-aggregations with Corynebacterium species may contribute to the long-term stability of biofilms.65
Role of Zinc in CRS
Zinc is localized in the basal bodies of cilia in airway epithelial cells, and its chelation in human nasal and bronchial airway cells slows ciliary beating by 25%, suggesting that mucociliary function depends on Zn.20 Furthermore, Th2-polarization with elevated production of proinflammatory cytokines (IL-6 and IL-8), tissue eosinophilia, and reduced collagen synthesis were observed under zinc depletion-linking Zn with “immune barrier” hypothesis.67 Suzuki et al hypothesized that efficient Zn sequestration by mucins in the submucosal glands, followed by the release of mucin-Zn complexes in nasal discharge, is responsible for its deficiency. In addition, the authors suggested that this process is continually driven by inflammation in a “vicious cycle” mechanism, where inflammation-induced upregulation of mucins results in hypersecretion of mucus and Zn loss from the mucosa, which, in turn, promotes further Zn depletion and maintains inflammatory process.20,67
Zn is an essential micronutrient required for the activity of >300 enzymes and 1000 transcription factors. For example, intestinal alkaline phosphatase, an enzyme involved in maintaining intestinal barrier integrity and associated with tissue remodeling matrix metalloproteinases (MMPs), are zinc-dependant enzymes.68 Likewise, Zn-dependent proteins account for ~4–8% of the bacterial proteome, ie, ~100–200 S. aureus proteins, including most superantigens. Furthermore, Zn regulates superantigens’ cellular receptor activity, T cell receptors (TCR) signaling activation, and the surface expression of major histocompatibility complex (MHC) class II – linking Zn with “staphylococcal superantigens” hypothesis.69 More importantly, Zn controls the activity of microbial pattern-recognition receptors (PRRs), such as Toll-like receptors (TLRs), receptor for advanced glycation endproducts (RAGEs), and G-protein-coupled receptors (GPRPs), the first line of defense against invading microbial pathogens, recognizing conserved microbial molecules, ie, microbial- or pathogen-associated molecular patterns (MAMPs/PAMPs). For example, Zn modulates LPS-induced and TLR-4-dependent inflammation.68,70
Consequently, a zinc deficiency may serve as an intermediary between exogenous and endogenous factors that contribute to the development and progression of CRS. Indeed, Zn deficiency was connected with hyperpermeability of the airway epithelium in other inflammatory diseases, such as atopic dermatitis and bronchial asthma.20,67 Similarly, low zinc levels impair the stability of intestinal mucosa and can induce gut dysbiosis, whereas adequate Zn levels promote the formation of “healthy” gut microbiota.71 Recently, Yunker et al stated that Zn homeostasis is necessary to regulate host-microbiome interaction via modulation of intestinal barrier permeability and maintenance of Paneth cells.72 Subsequently, Zn homeostatic imbalance was reported in various chronic intestinal diseases, including inflammatory bowel disease (IBD), irritable bowel syndrome (IBS), and colorectal cancer (CRC). Moreover, zinc deficiency in elderly individuals is positively correlated with an increased risk of pneumonia caused by S. aureus, S. pneumoniae, H. influenzae, K. pneumoniae, L. pneumophila.73
It may be assumed that CRS-associated microbiota are under permanent Zn-mediated “nutritional immunity”, that is, the host’s mechanism for inhibiting bacterial growth by restricting the availability of essential metal ions. This process may shape the microbiota composition based on the Zn requirements and tolerances of individual microbiota members and/or promote specific bacterial species or strains adapted to Zn-deprived conditions, for example, equipped with high-affinity Zn transporters such as ZnuABC or less Zn-dependent.73–75
For instance, a high-affinity ZnuABC transporter is crucial for Campylobacter jejuni to survive in chicken intestines colonized with normal microbiota but not with a limited one.74 Likewise, Salmonella enterica can outcompete and survive in the inflamed intestine by overcoming calprotectin-mediated zinc sequestration due to ZnuABC transporters.76 Inactivation of these transporters significantly reduces bacterial virulence. Hence, competition for Zn is part of the host–microbe interplay regulatory network.70,77
Tissue Remodeling
Tissue remodeling is the improper regeneration or restoration of damaged tissues owing to inflammation or mechanical injury. This dynamic process is characterized by both temporary and permanent changes observed in CRS and other inflammatory diseases such as asthma, chronic obstructive pulmonary disease, and bronchiectasis. Tissue remodeling has traditionally been regarded as secondary to persistent inflammation. However, this process has recently been shown to occur concurrently with inflammation after an injury rather than its consequence.78 Therefore, tissue remodeling might be an early stage of CRS, and recognition of its features can improve the identification of patients with a higher risk of recurrent CRS.
However, there are no strict criteria for defining tissue remodeling. In addition, in patients with CRSwNP or CRSsNP the remodeling pattern does not always show a consistent profile. NPs are grape-like, translucent, edematous structures, whereas the ethmoidal mucosa of CRSsNP patients is firm. These differences suggest different types of mucosal remodeling, which are regulated by factors such as the TGF-family and their receptors, matrix metalloproteinases (MMPs; enzymes responsible for extracellular matrix degradation contributing to edema formation), and tissue inhibitors of metalloproteinase (TIMP).10
Epithelial hyperplasia, metaplasia, subepithelial edema, basement membrane thickening, extracellular matrix protein deposition, extracellular matrix degradation, fibrosis, subepithelial collagen deposition, osteitis, and angiogenesis are all commonly involved in this process.79–85 In CRSsNP, tissue remodeling is characterized by transforming growth factor β (TGF-β)-driven fibrotic remodeling of the lamina propria, hyperplasia, excessive collagen production, and thickening of the extracellular matrix.47 Since TGF-β levels are significantly lower in the mucosa of CRSwNP patients, tissue remodeling in this condition is defined by less dense collagen deposition and barrier disruption, as well as by less membrane thickening and interstitial edema, less fibrotic alternations, and glandular hyperplasia.16,47,86
TGF-β is a pleiotropic and multifunctional growth factor that promotes fibroblast proliferation, differentiation, and fibrosis and prevents extracellular matrix (ECM) degradation by inducing tissue inhibitors of metalloproteinase 1 (TIMP-1). Therefore, tissue repair is compromised, which is reflected by the presence of loose connective tissue and edema formation in the severely inflamed tissues in CRSwNP.14 In addition, several other tissue remodeling factors, such as platelet-derived growth factor (PDGF), periostin, and vascular-endothelial growth factor (VEGF), are dysregulated in the CRS mucosa. VEGF can promote nasal epithelial cell growth and resistance to apoptosis, which in turn may play a significant role in nasal polyp formation.47
Tissue remodeling features are also endotype-dependent in CRS. CRSwNP is characterized by a Th1/Th2 cytokine pattern and eosinophil-dominant inflammation, which is typically mediated by S. aureus superantigens, fungi, and allergens. In contrast, neutrophilic infiltration and various Th1, Th2, and Th17 cytokine patterns are the hallmarks of CRSsNP. Clinically, these differences are reflected by a relatively good response to corticosteroid therapy, which effectively reduces Th2-driven eosinophilic inflammatory processes and nasal polyp formation. Conversely, corticosteroids, in general, do not suppress Th17-dependent inflammation.47
Furthermore, the epithelial-to-mesenchymal transition (EMT) and autophagy processes may be activated during sinus mucosa remodeling in CRSwNP and nasal polyp formation.14 EMT is the transformation of adherent epithelial cells into migratory cells that invade the extracellular matrix, resulting in the loss of E-cadherin, a cell-cell adhesion molecule with pivotal roles in epithelial cell behavior. EMT causes neutrophilic inflammation, fibrosis, and tissue remodeling owing to the loss of epithelial mucosal properties.87–91 Factors triggering EMT include microbial infections, hypoxia, allergens, and air pollutions.87–91 EMT is triggered by the disassembly of tight junctions associated with changes in the distribution of occludin, claudin, and zonula occludens 1 (ZO-1), as well as disruption of cell polarity and reorganization of the cytoskeleton. Elevated expression and activity of extracellular proteases, such as matrix metalloproteinase 9 (MMP-9), result in the degradation of ECM proteins.92 Subsequently, epithelial cells acquire mesenchymal characteristics, which are linked with increased levels of N-cadherin, vimentin, α-smooth muscle actin (α-SMA), collagen, fibronectin, and other extracellular matrix proteins.92 EMT is mediated and regulated by several signaling pathways, including TGF-β/Smad, Wnt/β-actin, hypoxia-inducible factor-1α (HIF-1α).92 Autophagy is a process essential for cell survival, differentiation, development, and homeostasis and plays a vital role in some chronic obstructive pulmonary disease (COPD).93
Biofilm Impact on Sinuses Mucosa
The “mechanistic” and “mucin” hypotheses of biofilms’ contribution to CRS have been suggested.94 The former assumes that disruption of the epithelial barrier results in enhanced exposure to microbial factors and overstimulation of the inflammatory response. The “mucin” hypothesis is based on the dysregulated expression of multiple mucins in CRS, which act as attachment ligands for bacterial adhesins initiating bacterial colonization - the first step in biofilm formation.95 Both hypotheses intersect with Zn deficiency due to its pro-inflammatory and barrier-impairing action, mucins’ Zn-binding properties, and their upregulation at low Zn levels.96 In addition, mucins are strictly connected with the “microbiota” hypothesis, being implicated in bacterial cross-feeding and as AMPs binding factors.
Furthermore, the “mechanistic” and “mucin” hypothesis concepts assume the formation of “pathologic” or dysbiotic biofilms, characterized by specific qualitative and/or quantitative changes in the proportion of bacterial species resulting from dysregulated reciprocal interactions. The latter are strictly associated with niche nutritional resources and are typically coordinated by QS communication systems, allowing unicellular organisms to function as multicellular entities, improving their chances of survival, competition, and cooperation in complex microbial environments.30,36,97 However, QS signal molecules (QMSs), such as acyl-homoserine lactones (AHLs), can be disrupted by several bacterial enzymes, such as lactonases, acylases, or oxidoreductases.98 Additionally, antagonism between QS signal-sensing molecules, such as autoinducers 2 (AI-2) and auto-inducing peptides (AIPs), may affect interspecies QS communication. For instance, four groups of S. aureus species are distinguished based on differences in AIPs, where APIs of one group inhibit QS signaling in the other S. aureus groups.98 In this context, producing antimicrobial substances such as bacteriocins or competition for space, which can be viewed as mechanisms that maximize nutrient absorption in one species while restricting it in another. Therefore, QSMs maintain the balance of the microbial community (eubiosis), which ensures colonization resistance and microbiota–host interactions in homeostasis. Subsequently, QSM-based communication is vital to maintain a balanced microbiota (eubiosis).30 In addition, QMS may act as virulence factors and directly or indirectly influence intestinal epithelial barrier function, inflammation, and intestinal carcinogenesis (reviewed by Conquant et al, 2021).29
Additionally, competitive mechanisms in biofilms can be mediated through so-called public goods, ie, products of one species (siderophores, proteases) beneficial for other community members, eg, siderophore production as competition for iron between S. aureus and P. aeruginosa, supporting kin selection and social cheaters’ interactions99 Furthermore, lifestyle transition, eg, from commensalism to parasitism or dormancy (persister or VBNC state), is a common bacterial strategy of response to changes in environmental conditions.100 For example, a shift of S. aureus from a virulent to a commensal state can be induced by other Staphylococci and bacteria via QS accessory gene regulator (Agr) – a major regulator of virulence controlling over 70 genes, including 23 virulence factors.28,101,102 Accordingly, S. aureus co-culture with C. striatum causes alterations in S. aureus transcriptome involving downregulation of virulence genes, eg, hemolysins, and upregulation of nasal colonization factors, eg, SpA, enhancing S. aureus adhesion to epithelial cells.103 Likewise, S. epidermidis ability to inhibit agr activity of S. aureus via AIP peptide likely explains its domination over S. aureus on the human skin.28 Subsequently, staquorsin (an analog of savarin-triazoloquinazoline derivative), a potent Agr inhibitor, has been proposed as a promising drug candidate for reducing the symptoms of S. aureus infections, including weight loss and the severity of inflammation in the tissues.104 Similarly, in P. aeruginosa, inactivation or loss of QS receptor LasR is a possible indicator of the initial phase of cystic fibrosis (CF) via promoting production of several toxic for other microbes and the host molecules, such as pyocyanin proteases and hydrogen cyanide, resulting in the exaggerated inflammatory response.105–107
From this perspective, positive co-occurrence of Corynebacterium and S. epidermidis in “microbiotype 1” may indicate that both may act as probiotics impeding S. aureus growth (Figure 2). Significantly, bacterial mucin-derived metabolites, eg, short-chain fatty acids (SCFAs), can induce similar changes in bacterial transcriptomes and simultaneously drive cross-feeding interactions, regulate mucosal pH, and upregulate mucin expression (Figure 3).108,109 Therefore, bacterial biofilms should be perceived as a network of reciprocal interactions between coordinated bacterial consortia, actively sensing and responding to changes in the inhabited niche as well as maximizing nutrient acquisition via numerous cooperative and competitive, such as (i) competitive exclusion, (ii) contest and scramble competition, (iii) negative-frequency dependent and Kin selection as well as (i) cross-feeding, commonly associated with lifestyle transitions, rather than passive nutrient exploiters.36,98,110 Also, the spatial structure-dependent coexistence in biofilms, such as bacterial co-aggregation into “corncobs” structures driving interspecies cross-feeding or excessive EPS production elevating bacteria toward oxygen-rich regions at the air-liquid interface, is important mechanisms shaping the composition and structure of bacterial biofilms.65,111
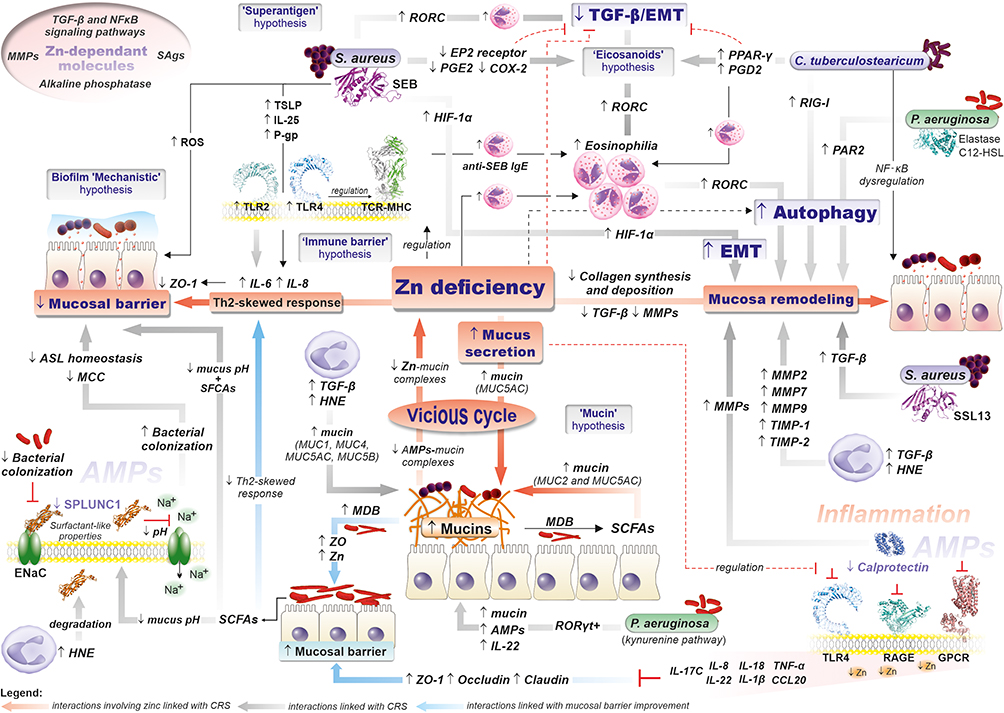 |
Figure 3 Mechanisms of CRS pathogenesis in the light of various hypotheses underlying CRS development, (i) “immune barrier” hypothesis, (ii) “eicosanoid” hypothesis, (iii) “staphylococcal superantigens” hypothesis, (iv) “biofilm” hypothesis, (v) “microbiota” hypothesis, and “Zinc deficiency hypothesis (adapted from Suzuki et al) as well as ‘mechanistic’ and ‘mucin’ hypotheses of biofilms” role in CRS pathogenesis (details are included in the text).20,67. Abbreviations: MDB, Mucin-Degrading Bacterial; SCFAs, Short-chain fatty acids; TLRs, Toll-like receptors; FPRs, N-formyl peptide receptors; T2Rs, Taste Receptor Family 2 receptors; RAGE, Receptor for Advanced Glycation Endproducts; GOCRs, G-protein-coupled receptors; HNE, Human neutrophil elastase; RORC, Retinoic Acid Receptor-Related Orphan Receptor C; HIF-1α, Hypoxia-Inducible Factor 1α Receptor; PAR2, Protease-Activated Receptor 2; PPAR-γ, Peroxisome Proliferator-Activated Receptor Gamma; RIG-1, Retinoic Acid-Inducible Gene I; ENaC, Epithelial Na+ Channel; TGF-β, transforming growth factor β; TIMP-1, tissue inhibitors of metalloproteinase 1; MMPs, matrix metalloproteinases; HNE, Human neutrophil elastase; ASL, airway surface liquid; QSM, quorum sensing molecules; EMT, epithelial-to-mesenchymal transition; SEB, S aureus superantigenic enterotoxin B; SSL, S aureus superantigen-like (SSL) toxins; C12-HSL, N-3-(oxododecanoyl)-homoserine lactone; SPLUNC1, Short-palate lung and nasal epithelial clone 1; ZO-1, zonula occludens; MCC, Mucociliary clearance. |
This viewpoint on bacterial biofilms is adapted to the functionally equivalent pathway (FEP) hypothesis to describe the mechanisms of microbiota-associated chronic infections.51 The FEP postulating that symbiotic and synergistic relationships between typically “non-pathogenic” species (pathobionts) may form communities or consortia (“pathogenic” biofilms), ie, FEPs with pathogenic potential comparable - “functional equivalence” to well-established pathogens, such as S. aureus or P. aeruginosa.51
Role of Epithelial Cell Barrier in CRS
Sinonasal epithelial cells (ciliated, non-ciliated, goblet, and basal cells) are the first line of defense against potentially harmful environmental factors and play a pivotal role in the onset and progression of CRS. Impairment of mucociliary clearance (MCC) and cell–cell contact in the sinonasal epithelium promote bacterial colonization, biofilm formation, dysbiosis, and mucositis. In addition, the sinonasal epithelium, via microbial pattern-recognition receptors (PRRs) and release of cytokines, complements system proteins and exosomes, coordinates the innate and adaptive immune responses and actively participates in the inflammatory process.
In general, the sinonasal mucosa of CRS is characterized by increased ion permeability and decreased tight junctions (TJ) due to decreased expression of TJ proteins occludin-1, claudin-1, and zonula occludens 1 (ZO-1), as well as iE-cadherin and desmosome shortening.12 The epithelial barrier can be compromised by several immune system molecules, eg, IL-4 and IL-13, and IFN-γ released under T2 and T1 conditions, respectively. Granule proteins and eosinophil extracellular traps (EETs) produced by eosinophils and other inflammatory mediators, such as oncostatin M (OSM), are significant epithelial barrier-affecting factors in CRSwNP.12,112
The “vicious cycle” associated with Zn deficiency may be responsible for the enhanced and sustained bacteria-mediated impairment of the epithelial barrier. Low Zn concentrations downregulate ZO-1 expression, likely via significant upregulation of the proinflammatory cytokines IL-6 and IL-8 (Figure 3).20,67
CRS-associated pathogens such as S. aureus, S. pneumoniae, P. aeruginosa, and H. influenzae, produce multiple virulence factors that affect transmembrane proteins (reviewed by Gao et al, 2022).37–39,113 For example, P. aeruginosa elastase downregulates the expression of claudin-1 and −4, occludin, and tricellulin, as well as protease-activated receptor 2 (PAR2), which regulates the expression of TJ proteins and epithelial permeability via autophagy.114,115 P. aeruginosa exoenzymes (Exo) and rhamnolipids also disrupt the actin cytoskeleton and TJ structure.113 Similarly, S. aureus superantigenic enterotoxin B (SEB) and V8 protease downregulate expression of ZO-1, occluding, phospho-occludin, claudin-1,113 whereas S. aureus α-toxin activates the metalloproteinase domain-containing protein 10 (ADAM10), resulting in the cleavage of E-cadherin as well as compromises the actin cytoskeleton.113 Furthermore, several QSMs, eg, P. aeruginosa 3-oxo-C12-HSL, directly or indirectly may disrupt TJ.29 On the other hand, numerous commensal mucin-degrading bacteria, such as Fusobacterium, Prevotella, and Streptococcus, can stabilize the TJ barrier, eg, regulating the expression of ZO-1, occludin, claudin-1, and E-cadherin.116–118
Importantly, the sinonasal epithelium acts not only as a passive barrier but also actively coordinates both innate and adaptive responses through pathogen-recognition receptors (PRRs), such as Toll-like receptors (TLRs), N-formyl peptide receptors (FPRs), and Taste Receptor Family 2 receptors (T2Rs).12 It should be emphasized that the function of PPRs is far beyond pathogen recognition. As part of the cell signaling pathways, PPRs promote the secretion of proinflammatory cytokines, TSLP, IL-25, and IL-33, which recruit multiple immune cells and promote the production of different Th2 cytokines, such as IL-4, IL-5, and IL-13, by activating Th2 cells and ultimately inducing type 2 inflammatory responses.12,119
Moreover, due to recognition by T2Rs bacterial quorum sensing molecules (QSMs), eg, 3-oxo-C12-HSL (C12-HSL), T2Rs provide information about the population density of microorganisms based on the chemical composition of the extracellular environment.120 In addition, T2Rs may mediate tumor-microbiome crosstalk via T2R-driven apoptotic pathways.120 Overall, T2Rs activation led to enhanced bacteria phagocytosis by airway ciliated cells through T2R-eNOS pathway and control AMP secretion. For instance, activation of T2R38 by P. aeruginosa C12-HSL results in a signaling cascade with resultant Ca2+-dependent release of bactericidal nitric oxide and increased ciliary beat frequency.121 Moreover, C12-HSL induces also cell-dependent dysregulation of mucin expression, upregulating of MUC2 and downregulating of MUC3 in the LS174T Caco-2 cell lines, respectively.29 C12-HSL also stimulates MMP-2 and MMP-3 activation via the protease-activated receptor (PAR2) and shows several immunomodulatory properties, eg, inhibition of TNF-α and IL-12 production, promotion of T lymphocytes differentiation toward Th1-like phenotype, and inhibition of the differentiation of Th1 and Th2 T lymphocytes.122 Hence, T2Rs may play a pivotal role in microbial surveillance and host-bacteria interactions that may be augmented by T2Rs gene polymorphisms, eg, altering T2Rs response to sugars and, in turn, T2R-mediated AMP secretion.30,120,123 For example, in CRS patients, glucose concentration in nasal secretions is 3- to 4-fold higher than in healthy individuals, resulting in lower AMP levels.121
In contrast, activation of FPRs (FPR-1, FPR-2, and FPR-) by N-formylated peptides present in bacterial cells or S. aureus superantigen-like (SSL) toxin 13 (SSL13) is associated with increased release of molecules responsible for tissue remodeling by epithelial cells, such as VEGF-A, TGF-β, and MMPs (MMP-2, MMP-7, and MMP-9), parallel to the reduced expression of tissue inhibitors of MMPs (TIMP-1 and TIMP-2), indicating their role in nasal polyp formation (Figure 3).47,124,125
Role of S. Aureus Superantigens in CRS
S. aureus SEB is the single bacterial toxin most frequently linked to CRS, that is, the “superantigen” hypothesis. SEB induces the local production of Th2 cytokines, regulatory T-cell inhibition, and increased eosinophil and mast cell activity, which leads to tissue injury, remodeling, and polyp formation (Figure 3).126
Superantigens (SAgs) are a class of immunostimulatory exotoxins that directly bind to the αβ T-cell receptor (TCR) and major histocompatibility complex (MHC) molecules that bypass the antigen recognition process, resulting in polyclonal T-lymphocyte proliferation and massive cytokine release. At least 80% of S. aureus clinical strains carry at least one SAg gene, and more than 20 different SAgs have been identified in S. aureus.127 Notably, Rha et al reported that among eight studied S. aureus enterotoxins (SEA, SEB, SEC, SED, SEE, SEG, SEH, and SEI), the sei gene was present in all tested patients with nasal polyps (NPs). Subsequently, the fraction of SEI (and SED) SED-responsive TCRVβ+ (TCRVβ1+ and Vβ5.1+) CD4+ T cells was significantly increased in the ethmoidal mucosa of CRSwNP subjects.126 Therefore, SEI was suggested as a leading S. aureus SAgs driving TCRVβ1+ or Vβ5.1+ CD4+ T cell expansion.126 Interestingly, despite the detection of the sei genes in 95% of CRSnNPs and 90% of controls, the skewed distribution of TCRVβ was not observed in these groups, implying the involvement of other factors or mechanisms underlying this process.126 This may be associated with promoting MHC-II surface expression on dendritic cells by a decrease in intracellular zinc, which can be inhibited by artificially increasing intracellular zinc levels.128
SAgs are classified into three groups based on their binding mechanisms with MHC: (i) binding only the MHC α-chain through the low-affinity site (SEB, SEC1-3, SEG, TSST-1, SPEA, SSA); (ii) binding only the MHC β-chain through the zinc-dependent and high-affinity sites (SEH, SEI, SEJ, SEK, SEL, SEM, SPEC, SPEG, SPEH, SPEJ, SMEZ); and (iii) crosslinking two MHC molecules by simultaneously binding the α-chain of one MHC and the β-chain of another MHC via the low- and high-affinity sites, respectively (SEA, SED, SEE).129 Moreover, SAgs show β-chain (Vβ) specificities, for example, SEA and SEB, and preferentially promote the expansion of Vβ11 and Vβ8.1,8.2 T cell subsets, respectively. Importantly, zinc-binding superantigens remain on the cell surface for at least 40 h, whereas single-binding-site superantigens diminish within four hours, which explains the enhanced T cell activation by SEA and other zinc-dependent and cross-linking SAgs.130
The importance of Zn in the action of SAgs was emphasized by Fraser et al.131 The authors demonstrated that SEA and SEE binding to HLA-DR, but not TSST-1, were entirely abolished by low levels of EDTA and that the addition of Zn2+, over EDTA as low as 2 μM, completely reversed this inhibition.131
Furthermore, TCR function is regulated by intracellular zinc levels.132 Briefly, antigen recognition by TLR increases cytoplasmic uptake of extracellular zinc by Zn transporter – ZIP6, which is crucial for TCRs signaling activation.20 As a result, elevated extracellular Zn bioavailability promotes the induction of T cell proliferative responses to suboptimal antigen concentration through amplifying TCR signaling.69 For example, the frequencies of T cells that responded to the stimulation with mature dendritic cells (DC) and 0.04 ng/mL of TSST-1 increased ~5-fold in medium containing Zn at concentrations of 3 μM, 15 μM, 30 μM, 45 μM, and 75 μM.69 Furthermore, Zn enhances TCR signal antigen reactivity (avidity), ie, lowers TCR activation, eg, TSST-1, which typically activates only Vβ2+ T cells, under Zn-optimized conditions, can also activate a small number of Vβ2- T cells, likely due a low affinity of TSST-1 to Vβ2-.69 On the contrary, a low level of intracellular Zn stimulates the surface expression of MHC-II in TLR4-mediated mechanism involving activation of TRIF and Zn transporters (ZIP and ZNT).128 Notably, SEB upregulates the expression of TLR4 and TLR2, and likely directly interacts with the latter receptor.133 Whereas high Zn levels suppress TLR2 and TLR4.134
Overall, SEB contributes to CRS pathogenesis through several mechanisms, including TLR2-mediated disruption of epithelial barrier integrity and a local Th2/Th17 response, which causes eosinophil degranulation, the release of antimicrobial proteins (AMPs), and localized eosinophilic inflammation. The induction of Th2 cytokines, such as IL-2, IL-4, IL-5, IL-6, and IL-8, is preferred over IL-10 and TGF-β1, and SEB does not appear to cause allergic reactions in the nasal tissue. Additionally, TSLP and IL-25 levels were upregulated by SEB. The former is essential for promoting Th2 cytokines and inhibiting IFN-γ production, whereas IL-25 is associated with the production of multiple inflammatory markers, including Th1/Th2/Th17 inflammation. TSLP upregulation is likely associated with P-glycoprotein (P-gp), an efflux pump that participates in cytokine expression and transport in the sinonasal mucosa.135
Furthermore, SEB-mediated disruption of TJs depends on the activation of TLR2 and triggering the production of pro-inflammatory cytokines IL-6 and IL8 as well as the induction of reactive oxygen species (ROS) and endoplasmic reticulum stress-induced inflammation in CRSwNP.133 Recently, Martens et al revealed in SEB-stimulated polyp epithelial cells from CRSwNP patients a dose-dependent decrease of trans-epithelial electrical resistance (TEER) and an increase of FD4 (Fluorescein isothiocyanate dextran 4 kDa) permeability. Interestingly, SEB did not affect the TEER of primary nasal epithelial cells in the control subjects. Therefore, SEB appears to be a driving factor, rather than a primary cause, of epithelial barrier impairment. The fact that SEB binds to TLR2 and elevates TLR2 expression could explain this observation, because anti-TLR2 antibodies prevent SEB-induced mucosal barrier disruption.135 Subsequently, TLR2-directed treatment may offer a novel therapeutic strategy for preventing the pathophysiological effects of S. aureus in CRSwNP.133 Furthermore, Wang et al reported significantly higher expression of TLR2, TLR4, and TGF-β1 in CRSsNP patients than in CRSwNP patients.136 In addition, in CRSsNP, the expression of TLR2 in patients with severe epithelial damage (score 3) was significantly higher than that in patients with mild epithelial damage (score ≤2). Moreover, the expression of TLR2 correlated negatively with squamous hyperplasia in CRSsNP, and positively correlated with gland hyperplasia in CRSwNP.136 Interestingly, TLR2-induced inflammation is also a property of C. tuberculostearicum, and Abreu et al demonstrated in a mouse model that the pathogen C. tuberculostearicum induces goblet cell hyperplasia and mucin hypersecretion.24,125
However, the significance of these SEB-mediated mechanisms in overall CRS pathogenies.137–144 Moreover, the mode of SEB interaction with TLR2 remains to be elucidated. To predict potential SEB-TLR2 binding sites, we searched the protein 3D model databases for other TLR2 binding proteins sharing structural similarity with SEB (Figure 4).145 For instance, the B subunit of a type II heat-labile enterotoxin of E. coli (LT-IIb-B5) was matched. LT-IIb is a potent immunomodulatory molecule with adjuvant properties and, similar to SEB, induces local production of pro-inflammatory cytokines.144,146–148
It is also reasonable to investigate the role of S. aureus superantigen-like (SSL) toxins such as SSL3 and SSL4-TLR2 antagonists, in CRS pathogenesis. SSLs are an under-examined family of 14 exotoxins that share structural similarity with superantigens, but lack superantigenic activity. Nonetheless, SSL proteins can modulate immune responses by binding to various host receptors.149 For instance, SSL13 is a potent neutrophil activator that interacts with formyl peptide receptor 2 (FPR2).150,151 Accordingly, whole-genome sequencing (WGS) analysis of S. aureus isolates collected from 28 CRSsNP and 30 CRSwNP patients revealed SSL proteins as the only virulence factors correlated with the CRS type. SSL5 was more prevalent in the CRSsNP cohort, whereas SSL14 was more prevalent in the CRSwNP cohort. Notably, SSL5 inhibited neutrophil extravasation by blocking the interaction between PSGL-1 and P-selectin and the enzymatic activities of MMP8 and MMP9. Moreover, SSL5 modulates the host immune defense by interfering with C1Inh in the complement system.152
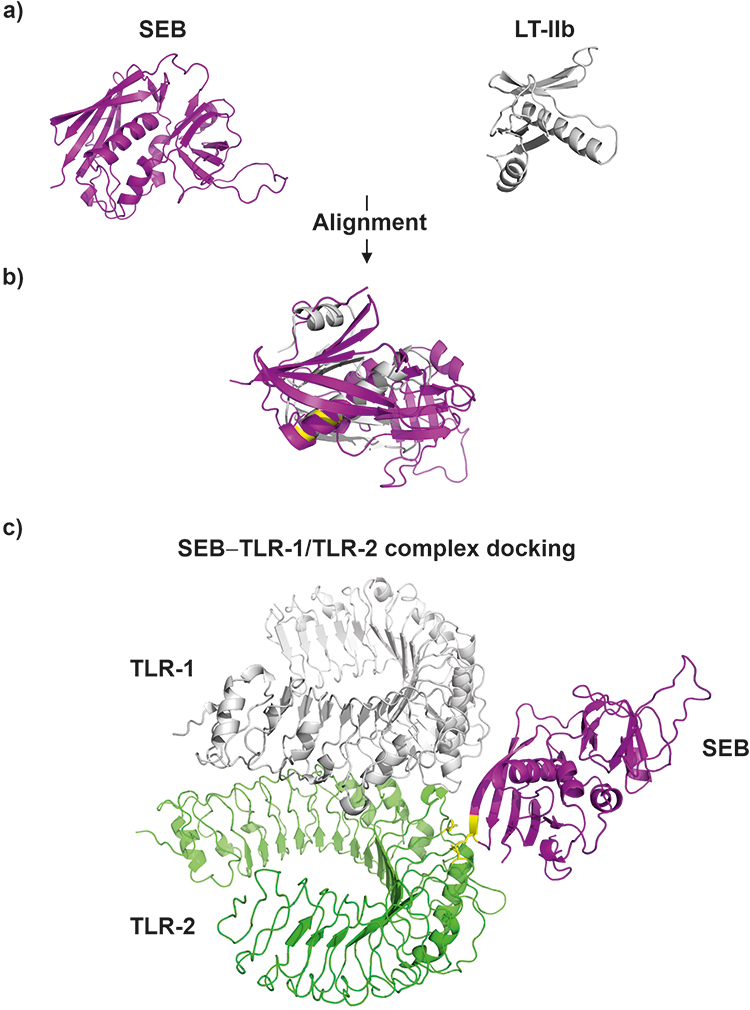 |
Figure 4 3D models S. aureus enterotoxin type B - SEB (3seb.pdb) and B subunit of the type II heat-labile enterotoxin of E. coli monomer (LT-IIb; qb5.pdb) (panel (A); SEB and LT-IIb alignment (TM-score = 0.43; RMSD = 4.44) (panel (B), LT-IIb residues (Met69, Ala70, Leu73 and Ser74) which responsible for binding with TLR-2 (residues Asp235 and Asn290)153 are highlighted on yellow color; SEB and TLR-1 – TLR-2 complex (2z7x.pdb) docking model (panel (C), the predicted SEB residues (Lys141 and Asn142) responsible for interaction with TLR-2 (residues Asp235 and Asn290) are highlighted on yellow color. Protein alignment was performed with TM-align tool (https://zhanggroup.org/TM-align/), Protein-docking was predicted using ZDOCK tool (https://zdock.umassmed.edu/). |
The connection between superantigens and the “eicosanoids” hypothesis is also notable. Eicosanoids, such as prostaglandins (PGs) and leukotrienes (LTs), are lipid-based signaling molecules that play a unique role in innate immune responses through cyclooxygenase (CO)-mediated metabolism of arachidonic acid. Patients with CRSwNP commonly demonstrate increased levels of proinflammatory leukotrienes alongside the downregulation of cyclooxygenase-2 (COX-2) and reduced levels of prostaglandin E2 (PGE2).154 SEB can affect eicosanoid metabolism via downregulation of PGE2, COX-2, and E-prostanoid-2 (EP2) receptor expression resulting in enhanced eosinophilic inflammation.154 This process can be reversed by PGE2 that acts as a dose-dependent inhibitor of SEB-induced eosinophilic inflammation primarily through the EP2-mediated pathway.154 Conversely, this process may downregulate EMT since PGE2 is a potent inhibitor of TGF-β1-induced EMT.155 Hence, PGE derivatives, such as misoprostol, have been indicated as efficient treatment method for CRS and other eosinophilic airway diseases (eg, bronchial asthma or allergic rhinitis).154 On the other hand, this therapy might not be applicable for Corynebacterium tuberculostearicum-induced eosinophilia, which possibly occurs via activation of PPAR-γ receptor and upregulation of PGD2, a potent chemoattractant for eosinophils, and in turn, downregulation of TGF-β production and Th2 cytokines.8,156 Since PGD2, like PGE2, is a potent inhibitor of TGF-β1-induced EMT, an activation of PPAR-γ may affect EMT and tissue remodeling, as reported in a mouse asthma model.155,156 For instance, Asaka et al reported expression PPAR-γ and PGD2 synthase in eosinophils and T cells infiltrating the nasal polyps and administration of PPAR-γ agonist diminishes nasal allergy symptoms and eosinophilic infiltration of the nasal mucosa.157
Furthermore, the biological activity of SEB may be enhanced by other S. aureus virulence factors and may trigger intensive inflammation, resulting in the disruption of epithelial cells and ultimately nasal polyp formation.135 For instance, S. aureus surface protein A (SpA) induces an allergenic response, increasing the release of histamine, LTC4/D4/E4, and PGD2 in nasal tissue, contributing to inflammation in CRS. Additionally, certain S. aureus proteases, such as serine protease-like protein (Spl) D, act as allergens and induce the release of IL-33, production of specific IgE and eosinophils, bronchial hyperreactivity, and airway goblet cell hyperplasia response.158 In addition, delta-toxin and lipoteichoic acid (LTA) have been linked to mast cell degranulation and epithelial colonization, respectively. Finally, S. aureus-induced Th1 and Th17 inflammation via the release of extracellular membrane vesicles (EV) may contribute to the development of CRSsNPs.
Role of Mucins in CRS
Mucus hypersecretion is a hallmark of CRS owing to the dysregulation of mucin expression.95 Neutrophils regulate this process through signaling pathways involving TNF-α, α-convertase, TGF-β, epidermal growth factor receptor (EGFR), and human neutrophil elastase (HNE). For example, HNE and TGF-β1 regulate the expression of several mucins, mainly MUC1, MUC4, MUC5AC, and MUC5B (Figure 3).95 Also, HNE triggers autophagy in nasal epithelial cells - a crucial process for the subsequent HNE-induced mucus hypersecretion through upregulation of MUC5AC via JNK-AP-1 pathway.93
Mucus hypersecretion from the nasal epithelium may contribute to zinc (Zn) depletion due to the efficient Zn-binding properties of mucins and the “vicious cycle” mechanism.67 Accordingly, an inflammation-induced upregulation of mucins results in hypersecretion of mucus and a net loss of Zn (via Zn-mucin complexes) from the mucosa, which promotes further Zn loss, contributing to CRS progression (Figure 3).20,67 In human nasal epithelial cells (HNECs), Zn depletion significantly upregulated the expression of Muc5AC and proinflammatory cytokines IL-6 and IL-8. Notably, both cytokines may be directly responsible for the elevated MuC5AC expression. Neveu et al demonstrated that IL-6 can directly induce the mucin genes Muc5AC and Muc5B in human lung epithelial cell lines.159 Similarly, Bautista et al proposed a novel function for IL-8, that is, maintaining MUC5AC (and MUC5B) gene expression in inflamed airways through a post-transcriptional mechanism based on increasing the abundance of MUC5AC mRNA.160 Furthermore, Zn deficiency can impair mucus stability by interfering with mucin O-glycosylation and their secretion by goblet cells.161
Mucins serve as receptor-binding sites for bacterial adhesion and nutrient sources and are implicated in bacterial cross-feeding interactions. Importantly, the latter influence several virulence-associated processes, such as biofilm formation and motility, as well as may alter the lifestyle of bacteria, hence contributing to maintaining commensal relationships with microbiota.162 Moreover, mucin degradation products, such as SCFAs, are vital for mucus pH homeostasis and “colonization resistance” processes.116,163
Specifically, mucins may contribute to developing CRS-associated biofilms and/or supporting the growth of particular species such as S. aureus.164 Psaltis et al reported a significant enrichment of the CRS-associated microbiome with mucin-degrading genes produced by various mucin-degrading bacteria (MDB), including Prevotella, Fusobacterium, and Streptococcus.108,165 Degradation of mucin degradation generates carbon-source nutrients for pathogens such as SCFAs (propionate, butyrate, and acetate) and other mucin-derived metabolites utilized, for example, by P. aeruginosa, or can shift bacterial lifestyle, for example, from commensalism to parasitism, by interfering with the expression of key metabolism- and virulence-associated pathways.165 Specifically, SCFAs (propionate and butyrate) directly impede S. aureus growth and lead to reduced expression of the accessory gene regulator (agr) quorum sensing system and induction of fatty acid degradation operon (fadXEDBA), likely as an SCFA-mediated stress response.166 In addition, SCFAs are toxic to many bacteria, including antibiotic-resistant Enterobacteriaceae, and can lower the pH. The latter shape microbiota composition and prevent overgrowth by pH-sensitive pathogenic bacteria.116,163 SCFAs also contribute to epithelial barrier stability, promoting ZO-1 and mucin (MUC2 and MUC5AC) expression.116,117 However, a combination of low pH and SCFAs may disrupt TJ and impair barrier function.116,167 Recently, Yuan et al reported that intestinal microbiota-derived SCFAs might attenuate allergic responses by maintaining stable IGF-1 expression. Furthermore, SCFAs-RARα (retinoic acid receptor α) interaction in dendritic cells has been suggested as a crucial immunomodulator of Th2 cell differentiation in a dysbiotic microbiota state.168 Therefore, SCFA deficiency in allergic diseases enhances a Th2-skewed immune response and contributes to allergy development.169
In addition, tryptophan-mediated cross-feeding relationships may link the co-colonization of Pseudomonas and tryptophan-producing bacteria, such as, Achromobacter in Pseudomonadaceae-dominated microbiota, recognized by Cope et al in patients with CRS.8 Products of tryptophan catabolism (anthranilate, kynurenine, and kynurenic acid) possess immunomodulatory properties. For instance, Pseudomonas species catabolize tryptophan to pro-inflammatory kynurenine via the kynurenine pathway, which also provides anthranilate, a precursor of several P. aeruginosa virulence factors.8,170 As a result, the increased capacity for tryptophan production and metabolism by Pseudomonadaceae-dominated microbiota may contribute to enhanced antimicrobial resistance and pathogenicity by upregulating biofilm formation and virulence gene expression.8,170 In the intestinal tract, microbiota via tryptophan-indole derivatives can regulate Th2-driven immunity, inducing Th17 cells and a subset of Treg cells expressing the retinoid-related orphan receptor (RORγt+), and stimulate the production of IL-22. IL-22 aids intestinal homeostasis and promotes epithelial cell regeneration, AMPs, and mucus secretion.47,171
Role of AMPs in CRS
Sinonasal mucus improves the retention of AMPs owing to its direct binding to mucins; for example, MUC5B binds histatin-1, −3, and −5 and statherin.172 Therefore, it might be assumed that mucus hypersecretion in CRS results in a more significant loss of AMPs. AMPs play a pivotal role in several aspects of CRS pathogenesis, including biofilm formation, tissue remodeling, and EMT.
For instance, lower expression of PLUNC family proteins, SPLUNC1 (BPIFA1) and LPLUNC2, has been reported in CRS patients.173 SPLUNC1, in addition to its antibacterial activity, acts as an LPS scavenger to suppress the LPS-induced inflammatory response.174,175 In addition, SPLUNC1 modulates airway surface liquid (ASL) volume, acting as an endogenous inhibitor of the epithelial Na+ channel (ENaC) in a pH-dependent.174 As a result, hyperactivity of ENaC, hence Na+ hyperabsorption and ASL dehydration in cystic fibrosis (CF) airways, is related to low ASL pH, which prevents binding of SPLUNC1 to ENaC. Hence, SPLUNC1 represents a soluble mucus volume sensor, and its low levels may affect MCC.176 On the other hand, SCFAs (butyrate), by increasing ENaC transcription and activity, regulate colonic Na absorption.177 Furthermore, the SPLUNC1-ENaC complex protects normal airway epithelia from Burkholderia cenocepacia invasion, and degradation of SPLUNC1 by HNE and MMPs impairs airway epithelial defense against bacteria (Figure 3).178,179 Subsequently, SPLUNC1 was proposed as a novel marker of disease severity and airway infection in bronchiectasis and CF exacerbations, and ENaC antagonists, for example, SPLUNC1-derived S18 peptide, have been suggested as a therapeutic option for the treatment of CF lung disease.180–182
In addition, significantly lower expression of SLPI and clusterin (CLU) antimicrobial peptides in the nasal tissues of CRSwNP patients was correlated with a low number controlled by Th2 cytokine submucosal glands.183 Th2 inflammatory conditions in CRS reduced SLPI expression, but not CLU, and S100A8/A9 (BPIFA1) were specifically attenuated by IL-13 in eosinophilic CRSwNP.183,184 Moreover, the expression of SLPI and CLU was affected by TGF-β1, an essential factor in the development of EMT and tissue remodeling, and the regulation of various mucins (Figure 3).95,183 Notably, in human airway epithelial cells, CLU also induces MUC5AC expression via the activation of the NF-κB signaling pathway.185
The S100 family proteins, psoriasin (S100A7), calgranulins A (S100A8), B (S100A9), C (S100A12), and calprotectin (S100A8/A9), are examples of other AMPs that are abnormally expressed in CRS. S100 proteins show different expression patterns concerning the location of tissue sampling and tissue subsite, which may explain the disagreement in the literature about whether S100 proteins, such as calprotectin, play a protective or pathological role in patients with CRS.186,187 In contrast, this observation implies that CRS inflammation may manifest differently depending on the sinus location.187 For example, the epithelium of CRS patients expresses significantly less psoriasin and calprotectin, which may contribute to a diminished innate immune response and barrier function. Similarly, Viksne et al reported decreased HBD-2, HBD-3, and LL-37 concentrations in the epithelium of patients with nasal polyps and elevated levels of HBD-3 in subepithelial connective tissue, suggesting its role in the pathogenesis of primary nasal polyps.188 Conversely, elevated calprotectin levels in nasal polyp tissues may reflect neutrophil recruitment as a compensatory mechanism.189
Calprotectin (CP) exerts proinflammatory responses via TLR4, IL-8, and TNF-α, as well as anti-inflammatory and tissue-protective functions, such as inhibition of MMPs by sequestration of zinc and PAR-2-mediated antioxidative effects.190,191 CP is a Mn-, Fe-, Ni-, and Zn-binding antimicrobial protein produced by neutrophils that inhibits the growth of pathogenic microorganisms by sequestering essential metal nutrients and reprogramming bacterial metabolism, for example, toward utilization of Fe- and Zn-dependent cofactors, to overcome their deficiency. For example, in a systemic infection model, inhibition of S. aureus Mn-dependent superoxide dismutase (SOD) by CP increases S. aureus sensitivity to neutrophil-mediated killing and attenuates virulence.192 On the other hand, S. aureus and P. aeruginosa enhance heme utilization via upregulation of heme uptake systems and siderophores to overcome Fe depletion by CP.193 Additionally, CP-mediated repression of P. aeruginosa Zn- and Mn-dependent anti-staphylococcal factors promotes its co-colonization in biofilms with S. aureus, which may account for their frequent co-infections.194 Similarly, Acinetobacter baumannii, in response to CP-mediated Fe and Zn starvation, upregulates flavin biosynthesis pathways, which increases the use of flavodoxins as substitutes for ferredoxins and decreases the cellular metabolic Fe requirement.195 Furthermore, Helicobacter pylori circumvents metal sequestration by CP by modifying the outer membrane and biofilm formation.196 Interestingly, by reducing sensitivity to zinc poisoning, CP may inadvertently benefit bacterial pathogens instead of the host, eg, via sequestration of Zn from high-affinity manganese transporter PsaA of highly sensitive to zinc poisoning pathogen S. pneumoniae.197 Finally, production of high-affinity Zn transporter ZnuABC transporter allows Salmonella enterica to overcome CP-mediated Zn sequestration, and, in turn, to outcompete intestinal commensals and survive in the inflamed intestine.76
Nasal Polyp Development
Nasal polyps (NP) are benign, hyperplastic, inflammatory edema lesions resulting from mucosal linings of the sinuses.84,198–200 Although the formation of NP is still mostly enigmatic, their classification is based on etiology (inflammatory, neoplastic, atopy, Staphylococcus super-antigens) as well as type and area covered (localized, diffused, systemic).199 In general, dysregulated and intense local immune response, triggered by extrinsic factors (fungi, staphylococcal superantigens, biofilms, and imbalanced microbiota) and intrinsic factors like (i) induced by hypoxia EMT, (ii) depletion of Treg lymphocytes, (iii) low local vitamin-D levels, (iv) high levels of leukotrienes, and (v) altered levels of NO have been associated with pathogenesis of nasal polyposis.201 Nevertheless, it appears that the damage of the epithelial barrier, regardless form intrinsic or extrinsic etiology, is a precondition increasing the susceptibility of subepithelial layers to invasion by pathogens that trigger a Th-2-skewed response. Subsequently, Th2 cytokines induce the accumulation of eosinophils and IgE, as well as remodeling of the sub-epithelial stroma, resulting in the formation of nasal polyps.201
Pseudocyst formation, involving edema formation with albumin accumulation and collagen depletion within the extracellular matrix, is a typical remodeling feature of the mucosa in CRSwNP.136 Damage to the epithelial barrier results in an influx of inflammatory cells, tissue swelling, and fibrin deposition. The nasal polyp tissues accumulate dendritic cells (DCs) expressing IL-17Rb, ST2, and TSLPR.14 Exosomes have also been found to be associated with nasal polyp formation via upregulation of coagulation and downregulation of fibrinolysis cascades, causing retention of plasma proteins, intense edema, excessive fibrin deposition in nasal polyps, and subsequent tissue remodeling.14 A nasal polyp forms without submucosal glands through tissue remodeling and intensified inflammation. Therefore, lower expression of specific AMPs, such as SLPI, SPLUNC1, and LPLUNC2, associated with decreased numbers of submucosal glands is likely most frequently linked to nasal polyps in CRS.183 Proinflammatory IL-6 and Oncostatin M (OSM) are highly expressed in CRSwNP tissue and may cause an overreaction to external stimuli, hence contributing to the formation of nasal polyps.14 In addition, OSM can disrupt the TJs of epithelial cells and increase their permeability, leading to epithelial barrier dysfunction.92 Since OSM expression is induced by lipopolysaccharide (LPS), it has been suggested as a therapy target to prevent the progression and exacerbation of severe asthma caused by bacteria.202
Likewise, Zn affects the expression and enzymatic activities of zinc-dependent MMPs that are responsible for collagen removal and are dysregulated in CRS, for example, elevated expression of MMP9 in nasal polyps is well documented. However, the relation between zinc and MMPs is equivocal; according to the literature review by Nosrati et al inhibitory effect of Zn on MMPs production and activity was reported by ~62% (29/47), whereas the remaining papers showed null or opposite action203 The epithelial barrier can also be impaired by decreased expression of tight junction protein - ZO-1 under low Zn concentrations.20
A so-called “staphylococcus superantigen” hypothesis has been suggested, where by S. aureus superantigenic enterotoxins induce local production of Th2 cytokines, regulatory T-cell inhibition, and increased eosinophil and mast cell activity, which led to tissue injury and remodeling and the formation of polyps.126 Indeed, the severity of CRSwNP correlates with the expansion of CD4+ T cells differentiated into the Th2 subset with proliferative features in response to a superantigen that appears to play a vital role in Th2-skewed inflammation.126 In addition, specific anti-superantigen IgE is present in almost half of the nasal tissue homogenates from nasal polyps135,204–206 SEB may play a role in the upregulation of retinoic acid receptor-related orphan receptor C (RORC) and hypoxia-inducible factor 1α (HIF-1α) receptor expression in Tregs and by maintaining inflammation in the sinonasal mucosa, contributing to the pathogenesis of nasal polyposis.133,135 RORC is a multifunctional nuclear receptor on mucosal surfaces that maintains homeostasis with the symbiotic microbiota, synchronizes defenses against extracellular microbes, regulates allergic responses, and induces autophagy. RORC+ Tregs may be associated with eosinophilic nasal polyposis. In addition, it negatively regulates oncogenic TGF-β/EMT and induces autophagy.207–209 HIF-1α induces an imbalance between Treg and Th17 activity and can lead to autoimmune disorders and chronic inflammation.
In CRSwNP, TGF-β1 can induce EMT by modulating various microRNAs, resulting in downregulation of E-cadherin, an epithelial biomarker, and upregulation of mesenchymal biomarkers, such as vimentin and N-cadherin.14 For example, macrophage-derived mucin domain 4 (TIM-4) promotes transforming growth factor-β (TGF-β1)-mediated EMT in nasal epithelial cells, facilitating nasal polyp formation.
Likewise, the abundance of Corynebacteriaceae in CRS-associated microbiota is linked with a higher risk of developing polyposis, possibly via the activation of PPAR-γ and RIG-I receptors.8 PPAR-γ is a lipid-sensing receptor that has recently been shown to regulate eosinophil activation in polyp tissues of CRS patients157 and airway remodeling following allergic inflammation in mice. In contrast, RIG-I is an intracellular sensor of viral DNA that is elevated in nasal polyp tissue [43] and is induced by IFN-γ [53]. In addition, C. tuberculostearicum may be responsible for the NP formation. This lipophilic skin inhibitor shows potent immunomodulatory properties via NF-κB dysregulation.125,210 As NF-κB dysregulation is associated with chronic inflammatory skin diseases, Altonsy et al suggested that this pathogen can initiate and/or sustain a chronic inflammatory process or skin cancers, and its higher occurrence in keratinocyte carcinomas has been reported.125
In other studies, the development of CRSwNP has been described as a complex process, represented mainly by increased epithelial damage and expression of fibronectin.83
Future Perspectives
To date, no clear link has been found between CRS and sinonasal microbiota composition or specific species or toxins, that is, single-species or superantigen hypotheses. Moreover, the lack of validated animal models and the disease’s complexity have hampered research into CRS disease mechanisms.211 Nonetheless, investigating the hypotheses leads to greater understanding of CRS mechanisms.211 Likewise, identifying and characterizing CRS endotypes could ultimately enhance patient care.7,11,211 For instance, the identification of immune factors that regulate the process by which neutrophils influence polyp formation, tissue remodeling, and mucosal barriers has the potential to facilitate diagnostic and therapeutic applications. Recent discoveries suggest that IL-6, IL-8, IL-1β, IL-36γ, TGF-β1, and oncostatin M may be implicated in the pathophysiology of neutrophilic inflammation in CRS. A better understanding of these pathways may guide future research into the use of monoclonal antibodies targeting neutrophilic pathways in CRS.7,11,211
Furthermore, confounding results regarding the importance of certain microorganisms in CRS have been reported in literature. These discrepancies are most likely due to several methodological issues with 16S rRNA sequencing and the significant inter-individual, geographical, and racial heterogeneity of sinonasal microbiota. In addition, it is unclear whether the reported differences in the composition of the sinonasal microbiota, eg, “microbiotypes”, represent specific microbiota community “types” or rather temporary “states”. In addition, transitional or intermediate states between specific microbiotypes are unknown because of the lack of longitudinal data showing their stability over time.22 Also, it cannot be excluded that chronicity of inflammation alone does not predict a dysbiotic microbiome, rather, the presence of a clinically apparent “sinus infection” at the time of sample collection.22 Nevertheless, exploring the hypotheses provides insights into disease mechanisms.
Furthermore, the discriminatory power of the current sequencing technology, based on short 16S rRNA amplicons, ie, generally genus-specific, is too low to resolve mainly species-specific relationships between the microbiota members.22 For instance, S. epidermidis appears to be a mutually cooperative partner for Corynebacteria, whereas mutually competitive for S. aureus.22 Overall, interspecies interactions rely on a delicate balance of competition and cooperation between species and may be substantially affected by environmental physicochemical factors, such as pH or essential micronutrient fluctuations and the genetic background of the interacting species. At the molecular level, these interactions may involve substantial alterations at the transcriptome level, resulting in different metabolic or virulence features, including the transition from a commensal to a pathogenic or dormant lifestyle. For instance, both synergy and antagonism are possible between S. pneumoniae and H. influenzae, depending on the pH and phase of growth. Low pH and stationary growth phases trigger H. influenzae to a viable but nonculturable (VBNC) state.110
In this view, the role of S. aureus (or its superantigens) in CRS should be perceived as a derivative of its genetic and the biofilm “background” as well as physicochemical parameters of the sinonasal mucosa promoting S. aureus lifestyle toward commensalism or parasitism and vice versa - “co-occurrence” hypothesis. Analogically, according to the functionally equivalent pathogroups (FEP) hypothesis, CRS can be triggered by symbiotic/commensal species without the contribution of well-known pathogens such as S. aureus and P. aeruginosa.
This view intersects with “mucin” and “mechanistic” hypotheses of CRS-associated biofilms through mucin-degrading bacteria and mucin-derived metabolites, eg, SCFAs. SCFAs have been implicated in cross-feeding multifunctional molecules that act on both the bacteria and the host. In the intestine, SCFA-mediated lower pH shapes the composition of gut microbiota and prevents the overgrowth of pH-sensitive species. Low mucus pH may interfere with the pH-dependent regulation of ASL homeostasis via inhibition of the Na(+) channel (ENaC) by SPLUNC1, an antimicrobial peptide that is dysregulated in CRS. Hence, as in CF lung disease, therapies based on ENaC antagonists may be beneficial in patients with CRS.
Likewise, because SCFAs commonly modify bacterial transcriptomes, QS-directed treatments interfere with these bacterial communication systems, such as the accessory gene regulator (agr) locus, which is the central S. aureus virulence regulator. The activation of agr results in biofilm dispersion, infection dissemination, and the upregulation of several toxins, including superantigens. Therefore, loss of the agr or its dysfunction enhances S. aureus adhesion and biofilm formation, which likely explains the common association (13–82%) of S. aureus agr-defective strains with hospital settings or hospitalization, where most infections are biofilm-associated, eg, catheter-associated.212–214 Also, the agr is repressed by mildly acidic conditions (pH ~ 5.5) that may facilitate colonization of low pH niches, such as the skin or the vagina.212,215 Thus, the Agr system and its regulation represent promising therapeutic targets, and screening the sinonasal S. aureus population for agr dysfunction or dysregulated expression appears relevant.216
Due to immunomodulatory traits, tryptophan metabolites, such as kynurenine, may contribute to CRS pathogenesis in Pseudomonadaceae-dominated microbiota.8 Notably, tryptophan is the leading mediator in the gut-brain axis (GBA), that is, a bilateral communication network between the gastrointestinal (GI) tract microbiota and the central nervous system.217
In CRS, Zn-mediated nutritional immunity may select specific species and force or drive bacterial adaptation to overcome the Zn limitations. Hence, differences in the genetic background of “healthy” and CRS-microbiota may concern the prevalence and the type of high-affinity Zn transporters, lower number of Zn-dependent proteins, metabolic pathways as adaptations to Zn-depleted mucosa as well as QS and two-component signal transduction system (TCS) patterns along with T2Rs polymorphism. Accordingly, TCS-enriched gene content characterizes the Staphylococcaceae-dominated sinonasal microbiota in CRS patients.8 This assumption is also supported by the observation that the nasal carrier strain of S. aureus shows significantly greater attachment and growth on nasal epithelial cells (NEC) than the non-carrier (laboratory) S. aureus strain.218 Additionally, the susceptibility of the non-carrier S. aureus strain to nasal fluid collected from S. aureus carriers is likely explained by its inability to induce HBD-2 and HBD-3 and its delayed upregulation of TLR2 in NEC.218
Similarly, low Zn levels should be considered in studies on the role of SEB and other superantigens in CRS pathogenesis. Interestingly, upregulation of IL-6 and IL-8, along with downregulation of ZO-1 expression, is a hallmark of the Th2-skewed pattern induced by both Zn depletion and SEB. Furthermore, investigating the role of S. aureus SSL3 or SSL4-TLR2 antagonists or low intracellular Zn concentrations for enhanced MHC-II surface expression may help decipher differences between SEB action in CRS patients and healthy controls as well as create a basis for future TLR2-directed therapies.128,133
Competitive exclusion mediated by anti-streptococcal action of Corynebacterium-produced oleic and linoleic acids was suggested to explain a negative correlation between Corynebacterium and Streptococcus abundance in CRS-associated microbiota.23 However, structural characteristics of CRS-associated biofilms, eg, formation of “corncob” structures via co-aggregation of these bacteria, may also underlay this observation. Therefore, Corynebacteria may be vital compositional and structural members of sinonasal biofilms responsible for their long-term stability, and the “mechanistic” and “mucin” hypotheses of biofilms’ role in CRS pathogenesis may be complemented by the “structural” one.
Importantly, F. nucleatum-mediated co-aggregation is a receptor-ligand-specific interaction that can be inhibited by relatively common compounds. For example, co-aggregation with early colonizers, including Streptococcus and Actinomyces, can be suppressed by L-arginine and L-lysine (via the RadD receptor), whereas D-galactose inhibits (via the Fap2 receptor) the interaction with late colonizers (Actinobacillus and Porphyromonas). In addition, L-arginine- and L-lysine-inhibitable adhesion preferentially affects gram-positive and gram-negative species, respectively. Furthermore, this process is controlled by a two-component signal transduction system CarRS, controlling lysine metabolism and several virulence genes.219 Overall, this system, similarly to the Agr system of S.aureus, may function as a mechanism controlling planktonic – biofilm (co-aggregation) state or spreading – localized infection biofilm, where a lack of lysine promotes F. nucleatum planktonic (spreading) form via downregulation by CarRS of RadD, which acts as a lysine receptor.219 Consequently, understanding the mechanisms underlying Corynebacteria-mediated “corncobs” could be useful for developing CRS therapies, as shown for staquorsin – the Agr inhibitor (Figure 5).
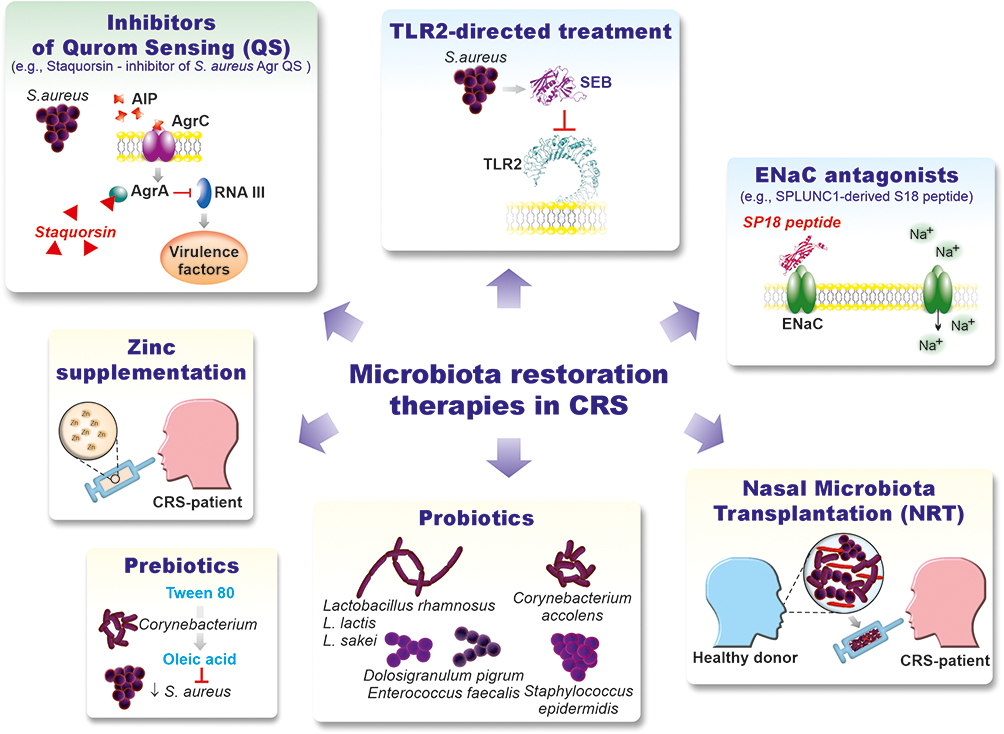 |
Figure 5 Microbiota restoration therapies in CRS. |
Conclusion
In conclusion, biofilm formation depends on various environmental physicochemical parameters such as pH, ion concentration, temperature, and specific nutrients. Fluctuations in the biofilm pH are commonly associated with dysbiotic changes in these bacterial communities. Glycogens drive Lactobacillus-mediated eubiosis associated with low vaginal pH, and an increase in vaginal pH results in dysbiosis and vaginitis. In contrast, sugar-rich saliva and an abundance of streptococci and other lactic acid bacteria result in pH acidification, leading to dental caries. Acidification of the local environment by Dolosigranulum has also been suggested as a factor facilitating the expansion of lipophilic Corynebacteria.220 In comparison, protein-rich gingival fluid promotes the growth of acquiring energy from the metabolism of amino acids periodontal pathogens, such as Porphyromonas gingivalis, and hence a neutral to weakly alkaline pH environment. Identification of the physicochemical characteristics of sinonasal biofilms may be essential for deciphering their compositional and architectural features, which is indispensable for understanding biofilm-associated CRS pathogenesis and developing appropriate therapies to reestablish eubiotic biofilm microbiota restoration therapies, for example, by supporting competitive exclusion, interfering with bacterial QS communication, host receptors, and microbiota transplantation. Successful nasal microbiota transplantation (NMT) using nasal lavages from healthy donors was recently performed in patients with CRSnNP, resulting in significant and long-lasting improvements in symptoms along with a stable increase in the abundance and diversity of resident bacteria.221 These results have now been verified in the Phase II Randomized Control Trial of Nasal Microbiota Transplant Therapy in Chronic Rhinosinusitis Without Nasal Polyps (CRSsNP) (ClinicalTrials.gov Identifier: NCT03122795). The concept of Nasal Microbiota Transplantation (NMT) is analogous to fecal microbiota transplantation (FMT) or Oral Microbiota Transplantation (OMT), that is, transferring bacteria from a healthy donor to a patient.222,223 FMT as a therapy for Clostridioides difficile (CDI) diseases has been used for over two decades, and recently, the US Food and Drug Administration (FDA) approved Vowst capsules, the first oral fecal microbiota formulation (ClinicalTrials.gov Identifier: NCT03183128). OMT treats oral diseases such as dental caries and periodontitis.222,223 Currently, OMT therapy has only been applied to treat periodontitis in dogs.224 Therefore, NMT likely supported by local zinc administration, for example, via nasal irrigation or surgical hydrogels, could be a promising CRS treatment.20
Abbreviations
RS, rhinosinusitis; CRS, chronic rhinosinusitis; NPs, nasal polyps; CRSwNP; chronic rhinosinusitis with nasal polyps; CRSsNP, chronic rhinosinusitis without nasal polyps; E-CRS, idiopathic eosinophilic chronic rhinosinusitis; NE-CRS, and non-eosinophilic chronic rhinosinusitis; EMT, epithelial-to-mesenchymal transition; QS, quorum sensing; QSM, quorum sensing molecules; TCR, T cell receptors; PRRs, pattern-recognition receptors; MAMPs/PAMPs, pathogen/microbe-associated molecular patterns; AHL, N-acyl-homoserine lactone; C12-HSL, N-3-(oxododecanoyl)-homoserine lactone; AMP, antimicrobial peptide; ASL, airway surface liquid; ENaC, epithelial Na+ channel; SPLUNC1, Short-palate lung and nasal epithelial clone 1; NMT, Nasal microbiota Transplantation; DFUs, diabetic foot ulcers; TGF-β, transforming growth factor β; TIMP-1, tissue inhibitors of metalloproteinase 1; VEGF, vascular-endothelial growth factor; PDGF, platelet-derived growth factor, TJ, tight junctions; ZO-1, zonula occludens 1; COPD, chronic obstructive pulmonary disease; CF, cystic fibrosis; AI-2, autoinducers 2; AIPs, auto-inducing peptides; VBNC, viable but non-culturable cells; EPS, extracellular polymeric substances; FEP, Functionally Equivalent Pathogroups (FEP) hypothesis; EETs, eosinophil extracellular traps; OSM, oncostatin M; SEB, S. aureus superantigenic enterotoxin B; MMPs, matrix metalloproteinases; SAgs, superantigens; SSL, S. aureus superantigen-like (SSL) toxins, ROS, reactive oxygen species; LT-IIb, type II heat-labile enterotoxin of E. coli; PGs, prostaglandins; LTs, leukotrienes; PGE2, prostaglandin E2; COX-2, cyclooxygenase-2; EP2, E-prostanoid-2; PGD2, prostaglandin D2; HNE; human neutrophil elastase; EGFR, epidermal growth factor receptor; LPLUNC2, long-palate lung and nasal epithelium clone 2; CLU, clusterin; SLPI, secretory lLeukocyte peptidase inhibitor; HBD, human beta-defensin; CP, calprotectin; NEC, nasal epithelial cells; TCS, two-component signal transduction system; TLRs, Toll-like receptors; FPRs, N-formyl peptide receptors; T2Rs, Taste Receptor Family 2 receptors; RAGE, Receptor for Advanced Glycation Endproducts; GOCRs, G-protein-coupled receptors; RORC, Retinoic Acid Receptor Related Orphan Receptor C; HIF-1α, Hypoxia-Inducible Factor 1α Receptor; PAR2, Protease-Activated Receptor 2; PPAR-γ, Peroxisome Proliferator-Activated Receptor Gamma; RIG-1, Retinoic Acid-Inducible Gene I; ENaC, Epithelial Na+ Channel; TSPL, Thymic Stromal Lymphopoietin; MDB, Mucin-Degrading Bacterial; SCFAs, Short-chain fatty acids; MCC, Mucociliary clearance; agr, the accessory gene regulatory (agr) system; PSGL-1, P-selectin glycoprotein ligand-1.
Acknowledgments
This study was financially supported by the Medical University of Bialystok (SUB/1/DN/22/002/1122, EP).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Fokkens WJ, Lund VJ, Hopkins C, et al. European position paper on rhinosinusitis and nasal polyps 2020. Rhinology. 2020;58(Suppl S29):1.
2. Fokkens WJ, Lund VJ, Hopkins C, et al. Executive summary of EPOS 2020 including integrated care pathways. Rhinology. 2020;58(2):82–111. doi:10.4193/Rhin20.601
3. Bachert C, Zhang N, van Zele T, Gevaert P. Chronic rhinosinusitis: from one disease to different phenotypes. Pediatr Allergy Immunol. 2012;23(22):2–4. doi:10.1111/j.1399-3038.2012.01318.x
4. Bachert C, Marple B, Schlosser RJ, et al. Adult chronic rhinosinusitis. Nature Reviews Disease Primers. 2020;6(1):86. doi:10.1038/s41572-020-00218-1
5. Rudmik L, Smith TL. Quality of life in patients with chronic rhinosinusitis. Curr Allergy Asthma Rep. 2011;11(3):247–252. doi:10.1007/s11882-010-0175-2
6. Dykewicz MS, Hamilos DL. Rhinitis and sinusitis. J Allergy Clin Immunol. 2010;125(2 Suppl 2):S103–115. doi:10.1016/j.jaci.2009.12.989
7. La Mantia I, Ragusa M, Grigaliute E, et al. Sensibility, specificity, and accuracy of the Sinonasal Outcome Test 8 (SNOT-8) in patients with chronic rhinosinusitis (CRS): a cross-sectional cohort study. Europ Arch Oto Rhino Laryngol. 2023;280(7):3259–3264. doi:10.1007/s00405-023-07855-8
8. Cope EK, Goldberg AN, Pletcher SD, Lynch SV. Compositionally and functionally distinct sinus microbiota in chronic rhinosinusitis patients have immunological and clinically divergent consequences. Microbiome. 2017;5(1):53. doi:10.1186/s40168-017-0266-6
9. Cho SH, Hamilos DL, Han DH, Laidlaw TM. Phenotypes of Chronic Rhinosinusitis. J Aller Clin Immunol Pract. 2020;8(5):1505–1511. doi:10.1016/j.jaip.2019.12.021
10. Akdis CA, Bachert C, Cingi C, et al. Endotypes and phenotypes of chronic rhinosinusitis: APRACTALL document of the European Academy of Allergy and Clinical Immunology and the American Academy of Allergy, Asthma & Immunology. J Aller Clin Immunol. 2013;131(6):1479–1490. doi:10.1016/j.jaci.2013.02.036
11. La Mantia I, Grigaliute E, Ragusa M, et al. Effectiveness and rapidity on olfactory function recovery in CRS patients treated with Dupilumab: a real life prospective controlled study. Europ Arch Oto Rhino Laryngol. 2023;2023:1.
12. Kato A, Schleimer RP, Bleier BS. Mechanisms and pathogenesis of chronic rhinosinusitis. J Aller Clin Immunol. 2022;149(5):1491–1503. doi:10.1016/j.jaci.2022.02.016
13. Tomassen P, Vandeplas G, Van Zele T, et al. Inflammatory endotypes of chronic rhinosinusitis based on cluster analysis of biomarkers. J Allerg Clin Immunol. 2016;137(5):1449–1456.e1444. doi:10.1016/j.jaci.2015.12.1324
14. He Y, Fu Y, Wu Y, Zhu T, Li H. Pathogenesis and treatment of chronic rhinosinusitis from the perspective of sinonasal epithelial dysfunction. Front Med. 2023;2023:10.
15. Stevens WW, Schleimer RP, Kern RC. chronic rhinosinusitis with nasal polyps. J Aller Clin Immunol Pract. 2016;4(4):565–572. doi:10.1016/j.jaip.2016.04.012
16. Schleimer RP. Immunopathogenesis of chronic rhinosinusitis and nasal polyposis. Annu Rev Pathol. 2017;12:331–357. doi:10.1146/annurev-pathol-052016-100401
17. Lam K, Schleimer R, Kern RC. the etiology and pathogenesis of chronic rhinosinusitis: a review of current hypotheses. Curr Aller Asthma Rep. 2015;15(7):41. doi:10.1007/s11882-015-0540-2
18. Cho DY, Hunter RC, Ramakrishnan VR. The Microbiome and chronic rhinosinusitis. Immunol Aller Clin North Am. 2020;40(2):251–263. doi:10.1016/j.iac.2019.12.009
19. Antonino M, Nicolò M, Jerome Renee L, et al. Single-nucleotide polymorphism in chronic rhinosinusitis: a systematic review. Clin Otolaryngol. 2022;47(1):14–23. doi:10.1111/coa.13870
20. Suzuki M, Suzuki T, Watanabe M, et al. Role of intracellular zinc in molecular and cellular function in allergic inflammatory diseases. Allergol Internat. 2021;70(2):190–200. doi:10.1016/j.alit.2020.09.007
21. Aggarwal N, Kitano S, Puah GRY, Kittelmann S, Hwang IY, Chang MW. Microbiome and human health: current understanding, engineering, and enabling technologies. Chem Rev. 2023;123(1):31–72. doi:10.1021/acs.chemrev.2c00431
22. Bassiouni A, Paramasivan S, Shiffer A, et al. Microbiotyping the sinonasal microbiome. Frontiers in Cellular and Infection Microbiology. 2020;10:137. doi:10.3389/fcimb.2020.00137
23. Paramasivan S, Bassiouni A, Shiffer A, et al. The international sinonasal microbiome study: a multicentre, multinational characterization of sinonasal bacterial ecology. Allergy. 2020;75(8):2037–2049. doi:10.1111/all.14276
24. Abreu NA, Nagalingam NA, Song Y, et al. Sinus microbiome diversity depletion and Corynebacterium tuberculostearicum enrichment mediates rhinosinusitis. Sci Transl Med. 2012;4(151):151ra124. doi:10.1126/scitranslmed.3003783
25. Biswas K, Hoggard M, Jain R, Taylor MW, Douglas RG. The nasal microbiota in health and disease: variation within and between subjects. Front Microbiol. 2015;9:134. doi:10.3389/fmicb.2015.00134
26. Radaic A, Kapila YL. The oralome and its dysbiosis: new insights into oral microbiome-host interactions. Computat Struct Biotechnol J. 2021;19:1335–1360. doi:10.1016/j.csbj.2021.02.010
27. Tokajuk J, Deptuła P, Piktel E, et al. Cathelicidin LL-37 in health and diseases of the oral cavity. Biomedicines. 2022;10(5):1086. doi:10.3390/biomedicines10051086
28. Canovas J, Baldry M, Bojer MS, et al. Cross-talk between staphylococcus aureus and other staphylococcal species via the agr quorum sensing system. Front Microbiol. 2016;7:1733. doi:10.3389/fmicb.2016.01733
29. Coquant G, Aguanno D, Pham S, et al. Gossip in the gut: quorum sensing, a new player in the host-microbiota interactions. World J Gastroenterol. 2021;27(42):7247–7270. doi:10.3748/wjg.v27.i42.7247
30. Li K, Ly K, Mehta S, Braithwaite A. Importance of crosstalk between the microbiota and the neuroimmune system for tissue homeostasis. Clin Translat Immunol. 2022;11(5):e1394. doi:10.1002/cti2.1394
31. Wagner mackenzie B, Waite DW, Hoggard M, Douglas RG, Taylor MW, Biswas K. Bacterial community collapse: a meta-analysis of the sinonasal microbiota in chronic rhinosinusitis. Environm Microbiol. 2017;19(1):381–392. doi:10.1111/1462-2920.13632
32. Joss TV, Burke CM, Hudson BJ, et al. Bacterial communities vary between sinuses in chronic rhinosinusitis patients. Front Microbiol. 2016:6. doi:10.3389/fmicb.2016.00006
33. Barshak MB, Durand ML. The role of infection and antibiotics in chronic rhinosinusitis. Laryngos Investigat Otolaryngol. 2017;2(1):36–42. doi:10.1002/lio2.61
34. Chmiel JF, Konstan MW, Elborn JS. Antibiotic and anti-inflammatory therapies for cystic fibrosis. Cold Spring Harbor Perspect Med. 2013;3(10):a009779. doi:10.1101/cshperspect.a009779
35. Jamal M, Ahmad W, Andleeb S, et al. Bacterial biofilm and associated infections. J Chin Med Assoc. 2018;81(1):7–11. doi:10.1016/j.jcma.2017.07.012
36. Penesyan A, Paulsen IT, Kjelleberg S, Gillings MR. Three faces of biofilms: a microbial lifestyle, a nascent multicellular organism, and an incubator for diversity. NPJ Biof Microb. 2021;7(1):80. doi:10.1038/s41522-021-00251-2
37. Huang Y, Qin F, Li S, et al. The mechanisms of biofilm antibiotic resistance in chronic rhinosinusitis: a review. Med. 2022;101(49):e32168. doi:10.1097/MD.0000000000032168
38. Łusiak-szelachowska M, Weber-Dąbrowska B, Żaczek M, Górski A. Anti-biofilm activity of bacteriophages and lysins in chronic rhinosinusitis. Acta Virol. 2021;65(2):127–140. doi:10.4149/av_2021_203
39. Sabino HAC, Valera FCP, Santos DV, et al. Biofilm and planktonic antibiotic resistance in patients with acute exacerbation of chronic rhinosinusitis. Front Cell Infect Microbiol. 2021;11:813076. doi:10.3389/fcimb.2021.813076
40. Hoggard M, Biswas K, Zoing M, Wagner Mackenzie B, Taylor MW, Douglas RG. Evidence of microbiota dysbiosis in chronic rhinosinusitis. Internat Forum Aller Rhinol. 2017;7(3):230–239. doi:10.1002/alr.21871
41. Hochstim CJ, Masood R, Rice DH. Biofilm and persistent inflammation in endoscopic sinus surgery. Otolaryngol Head Neck Surg. 2010;143(5):697–698. doi:10.1016/j.otohns.2010.07.017
42. Foreman A, Wormald PJ. Different biofilms, different disease? A clinical outcomes study. Laryngoscope. 2010;120(8):1701–1706. doi:10.1002/lary.21024
43. Martinez-Paredes JF, Choby G, Marino M, et al. Endoscopic outcomes in patients with AERD treated with topical antibiotics and intranasal corticosteroids. Front Cell Infect Microbiol. 2022;12:812215. doi:10.3389/fcimb.2022.812215
44. Deal RT, Kountakis SE. Significance of nasal polyps in chronic rhinosinusitis: symptoms and surgical outcomes. Laryngoscope. 2004;114(11):1932–1935. doi:10.1097/01.mlg.0000147922.12228.1f
45. Psaltis AJ, Weitzel EK, Ha KR, Wormald PJ. The effect of bacterial biofilms on post-sinus surgical outcomes. Am J Rhinol. 2008;22(1):1–6. doi:10.2500/ajr.2008.22.3119
46. Singhal D, Foreman A, Jervis-Bardy J, Wormald PJ. Staphylococcus aureus biofilms: nemesis of endoscopic sinus surgery. Laryngoscope. 2011;121(7):1578–1583. doi:10.1002/lary.21805
47. Salzano FA, Marino L, Salzano G, et al. Microbiota composition and the integration of exogenous and endogenous signals in reactive nasal inflammation. J Immunol Res. 2018;2018:2724951. doi:10.1155/2018/2724951
48. Kim DY, Cho SH, Takabayashi T, Schleimer RP. Chronic rhinosinusitis and the coagulation system. Allergy Asthma Immunol Res. 2015;7(5):421–430. doi:10.4168/aair.2015.7.5.421
49. De Boeck I, Wittouck S, Martens K, et al. Anterior nares diversity and pathobionts represent sinus microbiome in chronic rhinosinusitis. mSphere. 2019;4(6). doi:10.1128/mSphere.00532-19
50. Man WH, de Steenhuijsen Piters WAA, Bogaert D. The microbiota of the respiratory tract: gatekeeper to respiratory health. Nature Reviews Microbiology. 2017;15(5):259–270. doi:10.1038/nrmicro.2017.14
51. Dowd SE, Wolcott RD, Sun Y, McKeehan T, Smith E, Rhoads D. Polymicrobial nature of chronic diabetic foot ulcer biofilm infections determined using bacterial tag encoded FLX amplicon pyrosequencing (bTEFAP). PLoS One. 2008;3(10):e3326. doi:10.1371/journal.pone.0003326
52. Sugimoto S, Iwamoto T, Takada K, et al. Staphylococcus epidermidis Esp degrades specific proteins associated with Staphylococcus aureus biofilm formation and host-pathogen interaction. Journal of Bacteriology. 2013;195(8):1645–1655. doi:10.1128/JB.01672-12
53. Zipperer A, Konnerth MC, Laux C, et al. Human commensals producing a novel antibiotic impair pathogen colonization. Nature. 2016;535(7613):511–516. doi:10.1038/nature18634
54. Yan M, Pamp SJ, Fukuyama J, et al. Nasal microenvironments and interspecific interactions influence nasal microbiota complexity and S. aureus carriage. Cell Host Microbe. 2013;14(6):631–640. doi:10.1016/j.chom.2013.11.005
55. Menberu MA, Liu S, Cooksley C, et al. Corynebacterium accolens has antimicrobial activity against staphylococcus aureus and methicillin-resistant S. aureus pathogens isolated from the sinonasal niche of chronic rhinosinusitis patients. Pathogens (Basel, Switzerland). 2021;10(2). doi:10.3390/pathogens10020207
56. Huang S, Hon K, Bennett C, et al. Corynebacterium accolens inhibits Staphylococcus aureus induced mucosal barrier disruption. Frontiers in Microbiology. 2022;2022:13.
57. Menberu MA, Hayes AJ, Liu S, Psaltis AJ, Wormald PJ, Vreugde S. Tween 80 and its derivative oleic acid promote the growth of Corynebacterium accolens and inhibit Staphylococcus aureus clinical isolates. Internat Foru Aller Rhinol. 2021;11(4):810–813. doi:10.1002/alr.22730
58. De Boeck I, Wittouck S, Martens K, et al. The nasal mutualist Dolosigranulum pigrum AMBR11 supports homeostasis via multiple mechanisms. iScience. 2021;24(9):102978. doi:10.1016/j.isci.2021.102978
59. Dunne EM, Murad C, Sudigdoadi S, et al. Carriage of Streptococcus pneumoniae, Haemophilus influenzae, Moraxella catarrhalis, and Staphylococcus aureus in Indonesian children: a cross-sectional study. PLoS One. 2018;13(4):e0195098. doi:10.1371/journal.pone.0195098
60. Pettigrew MM, Gent JF, Revai K, Patel JA, Chonmaitree T. Microbial interactions during upper respiratory tract infections. Emerging Infectious Diseases. 2008;14(10):1584–1591. doi:10.3201/eid1410.080119
61. Regev-Yochay G, Dagan R, Raz M, et al. Association between carriage of streptococcus pneumoniae and staphylococcus aureus in children. JAMA. 2004;292(6):716–720. doi:10.1001/jama.292.6.716
62. Dunne EM, Smith-Vaughan HC, Robins-Browne RM, Mulholland EK, Satzke C. Nasopharyngeal microbial interactions in the era of pneumococcal conjugate vaccination. Vaccine. 2013;31(19):2333–2342. doi:10.1016/j.vaccine.2013.03.024
63. Aurora R, Chatterjee D, Hentzleman J, Prasad G, Sindwani R, Sanford T. Contrasting the microbiomes from healthy volunteers and patients with chronic rhinosinusitis. JAMA Otolaryngology-- Head & Neck Surgery. 2013;139(12):1328–1338. doi:10.1001/jamaoto.2013.5465
64. Huntley KS, Raber J, Fine L, Bernstein JA. Influence of the microbiome on chronic rhinosinusitis with and without polyps: an evolving discussion. Frontiers in Allergy. 2021;2:737086. doi:10.3389/falgy.2021.737086
65. Diaz PI, Valm AM. Microbial interactions in oral communities mediate emergent biofilm properties. J Dent Res. 2020;99(1):18–25. doi:10.1177/0022034519880157
66. Morillo-Lopez V, Sjaarda A, Islam I, Borisy GG, Mark Welch JL. Corncob structures in dental plaque reveal microhabitat taxon specificity. Microbiome. 2022;10(1):145. doi:10.1186/s40168-022-01323-x
67. Suzuki M, Ramezanpour M, Cooksley C, et al. Zinc-depletion associates with tissue eosinophilia and collagen depletion in chronic rhinosinusitis. Rhinology. 2020;58(5):451–459. doi:10.4193/Rhin19.383
68. Islam T, Albracht-Schulte K, Ramalingam L, et al. Anti-inflammatory mechanisms of polyphenols in adipose tissue: role of gut microbiota, intestinal barrier integrity and zinc homeostasis. The Journal of Nutritional Biochemistry. 2023;115:109242. doi:10.1016/j.jnutbio.2022.109242
69. Yu M, Lee -W-W, Tomar D, et al. Regulation of T cell receptor signaling by activation-induced zinc influx. Journal of Experimental Medicine. 2011;208(4):775–785. doi:10.1084/jem.20100031
70. Cerasi M, Ammendola S, Battistoni A. Competition for zinc binding in the host-pathogen interaction. Frontiers in Cellular and Infection Microbiology. 2013;3:3. doi:10.3389/fcimb.2013.00003
71. Wan Y, Zhang B. The impact of zinc and zinc homeostasis on the intestinal mucosal barrier and intestinal diseases. Biomolecules. 2022;12(7):900. doi:10.3390/biom12070900
72. Yunker R, Han GG, Luong H, Vaishnava S. Intestinal epithelial cell intrinsic zinc homeostasis is critical for host-microbiome symbiosis. J Immunol. 2023;210(1_Supplement):18–82. doi:10.4049/jimmunol.210.Supp.82.18
73. Lopez CA, Skaar EP. The impact of dietary transition metals on host-bacterial interactions. Cell Host Microbe. 2018;23(6):737–748. doi:10.1016/j.chom.2018.05.008
74. Gielda LM, DiRita VJ. Zinc competition among the intestinal microbiota. MBio. 2012;3(4):10.1128/mbio.00171–00112. doi:10.1128/mBio.00171-12
75. Djoko KY. Control of nutrient metal availability during host-microbe interactions: beyond nutritional immunity. J Biol Inorg Chem. 2023;28(5):451–456. doi:10.1007/s00775-023-02007-z
76. Liu JZ, Jellbauer S, Poe AJ, et al. Zinc sequestration by the neutrophil protein calprotectin enhances Salmonella growth in the inflamed gut. Cell Host Microbe. 2012;11(3):227–239. doi:10.1016/j.chom.2012.01.017
77. Xia P, Lian S, Wu Y, Yan L, Quan G, Zhu G. Zinc is an important inter-kingdom signal between the host and microbe. Veterin Res. 2021;52(1):39. doi:10.1186/s13567-021-00913-1
78. Amirapu S, Biswas K, Radcliff FJ, Wagner Mackenzie B, Ball S, Douglas RG. Sinonasal tissue remodelling during chronic rhinosinusitis. Int J Otolaryngol. 2021;2021:7428955. doi:10.1155/2021/7428955
79. Xiang R, Zhang QP, Zhang W, et al. Different effects of allergic rhinitis on nasal mucosa remodeling in chronic rhinosinusitis with and without nasal polyps. Eur Arch Otorhinolaryngol. 2019;276(1):115–130. doi:10.1007/s00405-018-5195-x
80. Lee HY, Pyo JS, Kim SJ. Distinct patterns of tissue remodeling and their prognostic role in chronic rhinosinusitis. ORL J Otorhinolaryngol Relat Spec. 2021;83(6):457–463. doi:10.1159/000515005
81. Kuhar HN, Tajudeen BA, Mahdavinia M, Gattuso P, Ghai R, Batra PS. Inflammatory infiltrate and mucosal remodeling in chronic rhinosinusitis with and without polyps: structured histopathologic analysis. Int Forum Allergy Rhinol. 2017;7(7):679–689. doi:10.1002/alr.21943
82. Do TQ, Barham HP, Earls P, et al. Clinical implications of mucosal remodeling from chronic rhinosinusitis. Int Forum Allergy Rhinol. 2016;6(8):835–840. doi:10.1002/alr.21754
83. Meng J, Zhou P, Liu Y, et al. The development of nasal polyp disease involves early nasal mucosal inflammation and remodelling. PLoS One. 2013;8(12):e82373. doi:10.1371/journal.pone.0082373
84. Radajewski K, Kalińczak-Górna P, Zdrenka M, et al. Short term pre-operative oral corticosteroids-tissue remodeling in chronic rhinosinusitis with nasal polyps. J Clin Med. 2021;10(15). doi:10.3390/jcm10153346
85. Watelet JB, Eloy PH, van Cauwenberge PB. Drug management in chronic rhinosinusitis: identification of the needs. Ther Clin Risk Manag. 2007;3(1):47–57. doi:10.2147/tcrm.2007.3.1.47
86. Lee K, Tai J, Lee SH, Kim TH. Advances in the knowledge of the underlying airway remodeling mechanisms in chronic rhinosinusitis based on the endotypes: a review. Int J Mol Sci. 2021;22:2.
87. Ryu G, Mo JH, Shin HW. Epithelial-to-mesenchymal transition in neutrophilic chronic rhinosinusitis. Curr Opin Allergy Clin Immunol. 2021;21(1):30–37. doi:10.1097/ACI.0000000000000701
88. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15(3):178–196. doi:10.1038/nrm3758
89. Yan B, Wang Y, Li Y, Wang C, Zhang L. Inhibition of arachidonate 15-lipoxygenase reduces the epithelial-mesenchymal transition in eosinophilic chronic rhinosinusitis with nasal polyps. Int Forum Allergy Rhinol. 2019;9(3):270–280. doi:10.1002/alr.22243
90. Zhong B, Seah JJ, Liu F, Ba L, Du J, Wang Y. The role of hypoxia in the pathophysiology of chronic rhinosinusitis. Allergy. 2022;77(11):3217–3232. doi:10.1111/all.15384
91. Cheng J, Chen J, Zhao Y, Yang J, Xue K, Wang Z. MicroRNA-761 suppresses remodeling of nasal mucosa and epithelial-mesenchymal transition in mice with chronic rhinosinusitis through LCN2. Stem Cell Res Ther. 2020;11(1):151. doi:10.1186/s13287-020-01598-7
92. Xia Y, Wang H, Yin J. The role of epithelial-mesenchymal transition in chronic rhinosinusitis. Internat Arch Aller Immunol. 2022;183(10):1029–1039. doi:10.1159/000524950
93. Ye Y, Zhao J, Ye J, et al. The role of autophagy in the overexpression of MUC5AC in patients with chronic rhinosinusitis. Internat Immunopharmacol. 2019;71:169–180. doi:10.1016/j.intimp.2019.03.028
94. Hoggard M, Wagner Mackenzie B, Jain R, Taylor MW, Biswas K, Douglas RG. Chronic rhinosinusitis and the evolving understanding of microbial ecology in chronic inflammatory mucosal disease. Clin Microbiol Rev. 2017;30(1):321–348.
95. Tong J, Gu Q. Expression and clinical significance of mucin gene in chronic rhinosinusitis. Curr All Asthma Rep. 2020;20(11):63. doi:10.1007/s11882-020-00958-w
96. Suzuki M, Ramezanpour M, Cooksley C, et al. Metallothionein-3 is a clinical biomarker for tissue zinc levels in nasal mucosa. Auris Nasus Larynx. 2021;48(5):890–897. doi:10.1016/j.anl.2021.01.019
97. Hooshangi S, Bentley WE. From unicellular properties to multicellular behavior: bacteria quorum sensing circuitry and applications. Curr Opin Biotechnol. 2008;19(6):550–555. doi:10.1016/j.copbio.2008.10.007
98. Hibbing ME, Fuqua C, Parsek MR, Peterson SB. Bacterial competition: surviving and thriving in the microbial jungle. Nature Reviews Microbiology. 2010;8(1):15–25. doi:10.1038/nrmicro2259
99. Harrison F, Paul J, Massey RC, Buckling A. Interspecific competition and siderophore-mediated cooperation in Pseudomonas aeruginosa. ISME J. 2008;2(1):49–55. doi:10.1038/ismej.2007.96
100. Desai SK, Kenney LJ. Switching lifestyles is an in vivo adaptive strategy of bacterial pathogens. FrontCellular Infec Microb. 2019;9. doi:10.3389/fcimb.2019.00421
101. Yarwood JM, Schlievert PM. Quorum sensing in Staphylococcus infections. J Clin Invest. 2003;112(11):1620–1625. doi:10.1172/JCI200320442
102. George EA, Muir TW. Molecular mechanisms of agr quorum sensing in virulent staphylococci. Chembiochem. 2007;8(8):847–855. doi:10.1002/cbic.200700023
103. Ramsey MM, Freire MO, Gabrilska RA, Rumbaugh KP, Lemon KP. Staphylococcus aureus shifts toward commensalism in response to Corynebacterium species. Front Microbiol. 2016;7:1230. doi:10.3389/fmicb.2016.01230
104. Mahdally NH, George RF, Kashef MT, Al-Ghobashy M, Murad FE, Attia AS. Staquorsin: a novel staphylococcus aureus agr-mediated quorum sensing inhibitor impairing virulence in vivo without notable resistance development. Front Microbiol. 2021;12:700494. doi:10.3389/fmicb.2021.700494
105. D’Argenio DA, Wu M, Hoffman LR, et al. Growth phenotypes of Pseudomonas aeruginosa lasR mutants adapted to the airways of cystic fibrosis patients. Mol Microbiol. 2007;64(2):512–533. doi:10.1111/j.1365-2958.2007.05678.x
106. Mateu-Borrás M, González-Alsina A, Doménech-Sánchez A, et al. Pseudomonas aeruginosa adaptation in cystic fibrosis patients increases C5a levels and promotes neutrophil recruitment. Virulence. 2022;13(1):215–224. doi:10.1080/21505594.2022.2028484
107. Hennemann LC, LaFayette SL, Malet JK, et al. LasR-deficient Pseudomonas aeruginosa variants increase airway epithelial mICAM-1 expression and enhance neutrophilic lung inflammation. PLoS Pathog. 2021;17(3):e1009375. doi:10.1371/journal.ppat.1009375
108. Cho DY, Skinner D, Hunter RC, et al. Contribution of Short Chain Fatty Acids to the Growth of Pseudomonas aeruginosa in Rhinosinusitis. Front Cell Infect Microbiol. 2020;10:412. doi:10.3389/fcimb.2020.00412
109. Flynn JM, Niccum D, Dunitz JM, Hunter RC. Evidence and Role for Bacterial Mucin Degradation in Cystic Fibrosis Airway Disease. PLoS Pathog. 2016;12(8):e1005846. doi:10.1371/journal.ppat.1005846
110. Tikhomirova A, Trappetti C, Paton JC, Kidd SP. The outcome of H. influenzae and S. pneumoniae inter-species interactions depends on pH, nutrient availability and growth phase. Int J Med Microbiol. 2015;305(8):881–892. doi:10.1016/j.ijmm.2015.09.003
111. Xavier JB, Foster KR. Cooperation and conflict in microbial biofilms. Proc Natl Acad Sci U S A. 2007;104(3):876–881. doi:10.1073/pnas.0607651104
112. Shin S-H, M-K Y, Park J, Geum S-Y. Immunopathologic Role of Eosinophils in Eosinophilic Chronic Rhinosinusitis. Int J Mol Sci. 2022;23(21):13313. doi:10.3390/ijms232113313
113. Gao N, Rezaee F. Airway Epithelial Cell Junctions as Targets for Pathogens and Antimicrobial Therapy. Pharmaceutics. 2022;14(12):2619. doi:10.3390/pharmaceutics14122619
114. Nomura K, Obata K, Keira T, et al. Pseudomonas aeruginosa elastase causes transient disruption of tight junctions and downregulation of PAR-2 in human nasal epithelial cells. Respir Res. 2014;15(1):21. doi:10.1186/1465-9921-15-21
115. Kim Y, Lee Y, Heo G, et al. Modulation of intestinal epithelial permeability via protease-activated receptor-2-induced autophagy. Cells. 2022;11:5.
116. Horrocks V, King OG, Yip AYG, Marques IM, McDonald JAK. Role of the gut microbiota in nutrient competition and protection against intestinal pathogen colonization. Microbiology. 2023;169(8). doi:10.1099/mic.0.001377
117. Giromini C, Baldi A, Rebucci R, et al. Role of short chain fatty acids to counteract inflammatory stress and mucus production in human intestinal HT29-MTX-E12 cells. Foods. 2022;11(13):1983. doi:10.3390/foods11131983
118. Pan M, Barua N, Ip M. Mucin-degrading gut commensals isolated from healthy faecal donor suppress intestinal epithelial inflammation and regulate tight junction barrier function. Front Immunol. 2022;2022;13.
119. Hong H, Liao S, Chen F, Yang Q, Wang DY. Role of IL-25, IL-33, and TSLP in triggering united airway diseases toward type 2 inflammation. Allergy. 2020;75(11):2794–2804. doi:10.1111/all.14526
120. Kouakou YI, Lee RJ. Interkingdom detection of bacterial quorum-sensing molecules by mammalian taste receptors. Microorganisms. 2023;11(5):1295. doi:10.3390/microorganisms11051295
121. Martens K, Steelant B, Bullens DMA. Taste receptors: the gatekeepers of the airway epithelium. Cells. 2021;10(11):2889. doi:10.3390/cells10112889
122. Eum SY, Jaraki D, Bertrand L, András IE, Toborek M. Disruption of epithelial barrier by quorum-sensing N-3-(oxododecanoyl)-homoserine lactone is mediated by matrix metalloproteinases. Am J Physiol Gastrointest Liver Physiol. 2014;306(11):G992–g1001. doi:10.1152/ajpgi.00016.2014
123. Nagi M, Chapple ILC, Sharma P, Kuehne SA, Hirschfeld J. Quorum sensing in oral biofilms: influence on host cells. Preprints. 2023;11(7):1688.
124. Prevete N, Salzano FA, Rossi FW, et al. Role(s) of formyl-peptide receptors expressed in nasal epithelial cells. J Biol Regul Homeost Agents. 2011;25(4):553–564.
125. Altonsy MO, Kurwa HA, Lauzon GJ, et al. Corynebacterium tuberculostearicum, a human skin colonizer, induces the canonical nuclear factor-κB inflammatory signaling pathway in human skin cells. Immun Inflamm Dis. 2020;8(1):62–79. doi:10.1002/iid3.284
126. Rha MS, Kim SW, Chang DY, et al. Superantigen-related T(H)2 CD4(+) T cells in nonasthmatic chronic rhinosinusitis with nasal polyps. J Allergy Clin Immunol. 2020;145(5):1378–1388.e1310. doi:10.1016/j.jaci.2019.12.915
127. Xu SX, McCormick JK. Staphylococcal superantigens in colonization and disease. Front Cell Infect Microbiol. 2012;2:52. doi:10.3389/fcimb.2012.00052
128. John E, Laskow TC, Buchser WJ, et al. Zinc in innate and adaptive tumor immunity. J Transl Med. 2010;8(1):118. doi:10.1186/1479-5876-8-118
129. Fernández MM, Guan R, Swaminathan CP, Malchiodi EL, Mariuzza RA. Crystal structure of staphylococcal enterotoxin I (SEI) in complex with a human major histocompatibility complex class II molecule. J Biol Chem. 2006;281(35):25356–25364. doi:10.1074/jbc.M603969200
130. Pless DD, Ruthel G, Reinke EK, Ulrich RG, Bavari S. Persistence of zinc-binding bacterial superantigens at the surface of antigen-presenting cells contributes to the extreme potency of these superantigens as T-cell activators. Infect Immun. 2005;73(9):5358–5366. doi:10.1128/IAI.73.9.5358-5366.2005
131. Fraser JD, Urban RG, Strominger JL, Robinson H. Zinc regulates the function of two superantigens. Proc Natl Acad Sci U S A. 1992;89(12):5507–5511. doi:10.1073/pnas.89.12.5507
132. Suzuki M, Cooksley C, Suzuki T, et al. TLR signals in epithelial cells in the nasal cavity and paranasal sinuses. Front Allergy. 2021;2021:2.
133. Martens K, Seys SF, Alpizar YA, et al. Staphylococcus aureus enterotoxin B disrupts nasal epithelial barrier integrity. Clin Exp Allergy. 2021;51(1):87–98. doi:10.1111/cea.13760
134. Suvanprakorn P, Tongyen T, Prakhongcheep O, Laoratthaphong P, Chanvorachote P. Establishment of an anti-acne vulgaris evaluation method based on TLR2 and TLR4-mediated interleukin-8 production. Vivo. 2019;33(6):1929–1934.
135. Chegini Z, Didehdar M, Khoshbayan A, Karami J, Yousefimashouf M, Shariati A. The role of Staphylococcus aureus enterotoxin B in chronic rhinosinusitis with nasal polyposis. Cell Commun. 2022;20(1):29. doi:10.1186/s12964-022-00839-x
136. Wang X, Zhao C, Ji W, Xu Y, Guo H. Relationship of TLR2, TLR4 and tissue remodeling in chronic rhinosinusitis. Int J Clin Exp Pathol. 2015;8(2):1199–1212.
137. Conley DB, Tripathi A, Seiberling KA, et al. Superantigens and chronic rhinosinusitis: skewing of T-cell receptor V beta-distributions in polyp-derived CD4+ and CD8+ T cells. Am J Rhinol. 2006;20(5):534–539. doi:10.2500/ajr.2006.20.2941
138. Van Bruaene N, Bachert C. Tissue remodeling in chronic rhinosinusitis. Curr Opin Allergy Clin Immunol. 2011;11(1):8–11. doi:10.1097/ACI.0b013e32834233ef
139. Bassiouni A, Chen PG, Wormald PJ. Mucosal remodeling and reversibility in chronic rhinosinusitis. Curr Opin Allergy Clin Immunol. 2013;13(1):4–12. doi:10.1097/ACI.0b013e32835ad09e
140. Sanclement JA, Webster P, Thomas J, Ramadan HH. Bacterial biofilms in surgical specimens of patients with chronic rhinosinusitis. Laryngoscope. 2005;115(4):578–582. doi:10.1097/01.mlg.0000161346.30752.18
141. Hsu J, Peters AT. Pathophysiology of chronic rhinosinusitis with nasal polyp. Am J Rhinol Allergy. 2011;25(5):285–290. doi:10.2500/ajra.2011.25.3680
142. Bachert C, Zhang N, Patou J, van Zele T, Gevaert P. Role of staphylococcal superantigens in upper airway disease. Curr Opin Allergy Clin Immunol. 2008;8(1):34–38. doi:10.1097/ACI.0b013e3282f4178f
143. Ou J, Wang J, Xu Y, et al. Staphylococcus aureus superantigens are associated with chronic rhinosinusitis with nasal polyps: a meta-analysis. Eur Arch Otorhinolaryngol. 2014;271(10):2729–2736. doi:10.1007/s00405-014-2955-0
144. Ferreira-Duarte AP, Pinheiro-Torres AS, Takeshita WM, et al. Airway exposure to Staphylococcal enterotoxin type B (SEB) enhances the number and activity of bone marrow neutrophils via the release of multiple cytokines. Int Immunopharmacol. 2020;78:106009. doi:10.1016/j.intimp.2019.106009
145. Holm L. Dali server: structural unification of protein families. Nucleic Acids Res. 2022;50(W1):W210–W215. doi:10.1093/nar/gkac387
146. Greene CJ, Hu JC, Vance DJ, et al. Enhancement of humoral immunity by the type II heat-labile enterotoxin LT-IIb is dependent upon IL-6 and neutrophils. J Leukoc Biol. 2016;100(2):361–369. doi:10.1189/jlb.3A0415-153RR
147. Hajishengallis G, Tapping RI, Martin MH, et al. Toll-like receptor 2 mediates cellular activation by the B subunits of type II heat-labile enterotoxins. Infect Immun. 2005;73(3):1343–1349. doi:10.1128/IAI.73.3.1343-1349.2005
148. Odumosu O, Nicholas D, Yano H, Langridge W. AB Toxins: a Paradigm Switch from Deadly to Desirable. Toxins. 2010;2(7):1612–1645. doi:10.3390/toxins2071612
149. Yokoyama R, Itoh S, Kamoshida G, et al. Staphylococcal superantigen-like protein 3 binds to the Toll-like receptor 2 extracellular domain and inhibits cytokine production induced by Staphylococcus aureus, cell wall component, or lipopeptides in murine macrophages. Infect Immun. 2012;80(8):2816–2825. doi:10.1128/IAI.00399-12
150. Zhao Y, van Kessel KPM, de Haas CJC, Rogers MRC, van Strijp JAG, Haas PA. Staphylococcal superantigen-like protein 13 activates neutrophils via formyl peptide receptor 2. Cell Microbiol. 2018;20(11):e12941. doi:10.1111/cmi.12941
151. Koymans KJ, Goldmann O, Karlsson CAQ, et al. The TLR2 antagonist staphylococcal superantigen-like protein 3 acts as a virulence factor to promote bacterial pathogenicity in vivo. J Innate Immun. 2017;9(6):561–573. doi:10.1159/000479100
152. Oku T, Kurisaka C, Ando Y, Tsuji T. Identification of human plasma C1 inhibitor as a target protein for staphylococcal superantigen-like protein 5 (SSL5). Biochem Biophys Res Commun. 2019;508(4):1162–1167. doi:10.1016/j.bbrc.2018.12.026
153. Cody V, Pace J, Nawar HF, et al. Structure-activity correlations of variant forms of the B pentamer of Escherichia coli type II heat-labile enterotoxin LT-IIb with Toll-like receptor 2 binding. Acta Crystallogr D Biol Crystallogr. 2012;68(Pt 12):1604–1612. doi:10.1107/S0907444912038917
154. Okano M, Fujiwara T, Haruna T, et al. Prostaglandin E(2) suppresses staphylococcal enterotoxin-induced eosinophilia-associated cellular responses dominantly through an E-prostanoid 2-mediated pathway in nasal polyps. J Allergy Clin Immunol. 2009;123(4):868–874.e813. doi:10.1016/j.jaci.2009.01.047
155. Zhang A, Dong Z, Yang T. Prostaglandin D2 inhibits TGF-beta1-induced epithelial-to-mesenchymal transition in MDCK cells. Am J Physiol Renal Physiol. 2006;291(6):F1332–1342. doi:10.1152/ajprenal.00131.2006
156. Honda K, Marquillies P, Capron M, Dombrowicz D. Peroxisome proliferator-activated receptor gamma is expressed in airways and inhibits features of airway remodeling in a mouse asthma model. J Allergy Clin Immunol. 2004;113(5):882–888. doi:10.1016/j.jaci.2004.02.036
157. Asaka C, Honda K, Ito E, Fukui N, Chihara J, Ishikawa K. Peroxisome proliferator-activated receptor-γ is expressed in eosinophils in nasal polyps. Int Arch Allergy Immunol. 2011;155 Suppl 1:57–63. doi:10.1159/000327294
158. Xu X, Reitsma S, Wang Y, Fokkens WJ. Highlights in the advances of chronic rhinosinusitis. Allergy. 2021;76(11):3349–3358. doi:10.1111/all.14892
159. Neveu WA, Allard JB, Dienz O, et al. IL-6 is required for airway mucus production induced by inhaled fungal allergens. J Immunol. 2009;183(3):1732–1738. doi:10.4049/jimmunol.0802923
160. Bautista MV, Chen Y, Ivanova VS, Rahimi MK, Watson AM, Rose MC. IL-8 regulates mucin gene expression at the posttranscriptional level in lung epithelial cells12. J Immunol. 2009;183(3):2159–2166. doi:10.4049/jimmunol.0803022
161. Maares M, Keil C, Straubing S, Robbe-Masselot C, Haase H. Zinc Deficiency Disturbs Mucin Expression, O-Glycosylation and Secretion by Intestinal Goblet Cells. Int J Mol Sci. 2020;21(17):6149. doi:10.3390/ijms21176149
162. Valle Arevalo A, Nobile CJ. Interactions of microorganisms with host mucins: a focus on Candida albicans. FEMS Microbiol Rev. 2020;44(5):645–654. doi:10.1093/femsre/fuaa027
163. den Besten G, van Eunen K, Groen AK, Venema K, Reijngoud DJ, Bakker BM. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J Lipid Res. 2013;54(9):2325–2340. doi:10.1194/jlr.R036012
164. Mao YJ, Chen HH, Wang B, Liu X, Xiong GY. Increased expression of MUC5AC and MUC5B promoting bacterial biofilm formation in chronic rhinosinusitis patients. Auris Nasus Larynx. 2015;42(4):294–298. doi:10.1016/j.anl.2014.12.004
165. Lucas SK, Villarreal AR, Ahmad MM, et al. Anaerobic microbiota derived from the upper airways impact staphylococcus aureus physiology. Infect Immun. 2021;89(9):e0015321. doi:10.1128/IAI.00153-21
166. Fletcher JR, Villareal AR, Penningroth MR, Hunter RC. Staphylococcus aureus Overcomes Anaerobe-Derived Short-Chain Fatty Acid Stress via FadX and the CodY Regulon. J Bacteriol. 2022;204(5):e00064–00022. doi:10.1128/jb.00064-22
167. Meissner S, Hagen F, Deiner C, et al. Key role of short-chain fatty acids in epithelial barrier failure during ruminal acidosis. J Dairy Sci. 2017;100(8):6662–6675. doi:10.3168/jds.2016-12262
168. Yuan X, Tang H, Wu R, et al. Short-Chain fatty acids calibrate RARα activity regulating food sensitization. Front Immunol. 2021;2021:12.
169. Cait A, Hughes MR, Antignano F, et al. Microbiome-driven allergic lung inflammation is ameliorated by short-chain fatty acids. Mucosal Immunol. 2018;11(3):785–795. doi:10.1038/mi.2017.75
170. Bortolotti P, Hennart B, Thieffry C, et al. Tryptophan catabolism in Pseudomonas aeruginosa and potential for inter-kingdom relationship. BMC Microbiol. 2016;16(1):137. doi:10.1186/s12866-016-0756-x
171. Patnaude L, Mayo M, Mario R, et al. Mechanisms and regulation of IL-22-mediated intestinal epithelial homeostasis and repair. Life Sci. 2021;271:119195. doi:10.1016/j.lfs.2021.119195
172. Linden SK, Sutton P, Karlsson NG, Korolik V, McGuckin MA. Mucins in the mucosal barrier to infection. Mucosal Immunol. 2008;1(3):183–197. doi:10.1038/mi.2008.5
173. Seshadri S, Lin DC, Rosati M, et al. Reduced expression of antimicrobial PLUNC proteins in nasal polyp tissues of patients with chronic rhinosinusitis. Allergy. 2012;67(7):920–928. doi:10.1111/j.1398-9995.2012.02848.x
174. Tarran R, Redinbo MR. Mammalian short palate lung and nasal epithelial clone 1 (SPLUNC1) in pH-dependent airway hydration. Int J Biochem Cell Biol. 2014;52:130–135. doi:10.1016/j.biocel.2014.03.002
175. Di YP. Functional roles of SPLUNC1 in the innate immune response against Gram-negative bacteria. Biochem Soc Trans. 2011;39(4):1051–1055. doi:10.1042/BST0391051
176. Garcia-Caballero A, Rasmussen JE, Gaillard E, et al. SPLUNC1 regulates airway surface liquid volume by protecting ENaC from proteolytic cleavage. Proc Natl Acad Sci. 2009;106(27):11412–11417. doi:10.1073/pnas.0903609106
177. Zeissig S, Fromm A, Mankertz J, et al. Butyrate induces intestinal sodium absorption via Sp3-mediated transcriptional up-regulation of epithelial sodium channels. Gastroenterology. 2007;132(1):236–248. doi:10.1053/j.gastro.2006.10.033
178. Ahmad S, Kim CSK, Tarran R. The SPLUNC1-βENaC complex prevents Burkholderia cenocepacia invasion in normal airway epithelia. Respir Res. 2020;21(1):190. doi:10.1186/s12931-020-01454-5
179. Jiang D, Wenzel SE, Wu Q, Bowler RP, Schnell C, Chu HW. Human neutrophil elastase degrades SPLUNC1 and impairs airway epithelial defense against bacteria. PLoS One. 2013;8:5.
180. Keir HR, Shoemark A, Huang JTJ, Chalmers JD. SPLUNC1 is a novel marker of disease severity and airway infection in bronchiectasis. Eur Respir J. 2021;58(5). doi:10.1183/13993003.01840-2021
181. Khanal S, Webster M, Niu N, et al. SPLUNC1: a novel marker of cystic fibrosis exacerbations. Eur Respir J. 2021;58(5). doi:10.1183/13993003.00507-2020
182. Terryah ST, Fellner RC, Ahmad S, et al. Evaluation of a SPLUNC1-derived peptide for the treatment of cystic fibrosis lung disease. Am J Physiol Lung Cell Mol Physiol. 2018;314(1):L192–l205. doi:10.1152/ajplung.00546.2016
183. Huang Y, Wang M, Hong Y, et al. Reduced expression of antimicrobial protein secretory leukoprotease inhibitor and clusterin in chronic rhinosinusitis with nasal polyps. J Immunol Res. 2021;2021:1057186. doi:10.1155/2021/1057186
184. Tsou Y-A, Lin C-D, Chen H-C, et al. Interleukin-13 inhibits lipopolysaccharide-induced BPIFA1 expression in nasal epithelial cells. PLoS One. 2015;10:e0143484.
185. Bae CH, Na HG, Choi YS, Song SY, Kim YD. Clusterin induces MUC5AC expression via activation of NF-κB in human airway epithelial cells. Clin Exp Otorhinolaryngol. 2018;11(2):124–132. doi:10.21053/ceo.2017.00493
186. De Corso E, Baroni S, Onori ME, et al. Calprotectin in nasal secretion: a new biomarker of non-type 2 inflammation in CRSwNP. Acta Otorhinolaryngol Ital. 2022;42(4):355–363. doi:10.14639/0392-100X-N1800
187. Sumsion JS, Pulsipher A, Alt JA. Differential expression and role of S100 proteins in chronic rhinosinusitis. Curr Opin Allergy Clin Immunol. 2020;20(1):14–22. doi:10.1097/ACI.0000000000000595
188. Viksne RJ, Sumeraga G, Pilmane M. Antimicrobial and defense proteins in chronic rhinosinusitis with nasal polyps. Medicina. 2023;59(7):1259. doi:10.3390/medicina59071259
189. Tieu DD, Peters AT, Carter RG, et al. Evidence for diminished levels of epithelial psoriasin and calprotectin in chronic rhinosinusitis. J Allergy Clin Immunol. 2010;125(3):667–675. doi:10.1016/j.jaci.2009.11.045
190. Isaksen B, Fagerhol MK. Calprotectin inhibits matrix metalloproteinases by sequestration of zinc. Mol Pathol. 2001;54(5):289–292. doi:10.1136/mp.54.5.289
191. Sroussi HY, Lu Y, Villines D, Sun Y. The down regulation of neutrophil oxidative metabolism by S100A8 and S100A9: implication of the protease-activated receptor-2. Mol Immunol. 2012;50(1–2):42–48. doi:10.1016/j.molimm.2011.12.001
192. Kehl-Fie Thomas E, Chitayat S, Hood MI, et al. Nutrient metal sequestration by calprotectin inhibits bacterial superoxide defense, enhancing neutrophil killing of staphylococcus aureus. Cell Host Microbe. 2011;10(2):158–164. doi:10.1016/j.chom.2011.07.004
193. Obisesan AO, Zygiel EM, Nolan EM. Bacterial responses to iron withholding by calprotectin. Biochemistry. 2021;60(45):3337–3346. doi:10.1021/acs.biochem.1c00572
194. Wakeman CA, Moore JL, Noto MJ, et al. The innate immune protein calprotectin promotes Pseudomonas aeruginosa and Staphylococcus aureus interaction. Nat Commun. 2016;7:11951. doi:10.1038/ncomms11951
195. Wang J, Lonergan ZR, Gonzalez-Gutierrez G, et al. Multi-metal restriction by calprotectin impacts de novo flavin biosynthesis in Acinetobacter baumannii. Cell Chem Biol. 2019;26(5):745–755.e747. doi:10.1016/j.chembiol.2019.02.011
196. Gaddy JA, Radin JN, Cullen TW, et al. Helicobacter pylori resists the antimicrobial activity of calprotectin via lipid a modification and associated biofilm formation. MBio. 2015;6(6):10.1128/mbio.01349–01315. doi:10.1128/mBio.01349-15
197. Rosen T, Hadley RC, Bozzi AT, Ocampo D, Shearer J, Nolan EM. Zinc sequestration by human calprotectin facilitates manganese binding to the bacterial solute-binding proteins PsaA and MntC. Metallomics. 2022;14(2). doi:10.1093/mtomcs/mfac001
198. Kwah JH, Peters AT. Nasal polyps and rhinosinusitis. Allergy Asthma Proc. 2019;40(6):380–384. doi:10.2500/aap.2019.40.4252
199. Ta NH. Will we ever cure nasal polyps? Ann R Coll Surg Engl. 2019;101(1):35–39. doi:10.1308/rcsann.2018.0149
200. Eloy P, Poirrier AL, De Dorlodot C, Van Zele T, Watelet JB, Bertrand B. Actual concepts in rhinosinusitis: a review of clinical presentations, inflammatory pathways, cytokine profiles, remodeling, and management. Curr Allergy Asthma Rep. 2011;11(2):146–162. doi:10.1007/s11882-011-0180-0
201. Goulioumis AK, Kourelis K, Gkorpa M, Danielides V. Pathogenesis of Nasal Polyposis: current Trends. Indian J Otolaryngol Head Neck Surg. 2023;75(Suppl S1):733–741. doi:10.1007/s12070-022-03247-2
202. Headland SE, Dengler HS, Xu D, et al. Oncostatin M expression induced by bacterial triggers drives airway inflammatory and mucus secretion in severe asthma. Sci Transl Med. 2022;14(627):eabf8188. doi:10.1126/scitranslmed.abf8188
203. Nosrati R, Kheirouri S, Ghodsi R, Ojaghi H. The effects of zinc treatment on matrix metalloproteinases: a systematic review. J Trace Elem Med Biol. 2019;56:107–115. doi:10.1016/j.jtemb.2019.08.001
204. Van Zele T, Gevaert P, Watelet JB, et al. Staphylococcus aureus colonization and IgE antibody formation to enterotoxins is increased in nasal polyposis. J Allergy Clin Immunol. 2004;114(4):981–983. doi:10.1016/j.jaci.2004.07.013
205. Seiberling KA, Conley DB, Tripathi A, et al. Superantigens and chronic rhinosinusitis: detection of staphylococcal exotoxins in nasal polyps. Laryngoscope. 2005;115(9):1580–1585. doi:10.1097/01.mlg.0000168111.11802.9c
206. Chen JB, James LK, Davies AM, et al. Antibodies and superantibodies in patients with chronic rhinosinusitis with nasal polyps. J Allergy Clin Immunol. 2017;139(4):1195–1204.e1111. doi:10.1016/j.jaci.2016.06.066
207. Yan G, Lei H, He M, et al. Melatonin triggers autophagic cell death by regulating RORC in Hodgkin lymphoma. Biomed Pharmacother. 2020;123:109811. doi:10.1016/j.biopha.2020.109811
208. Eberl G. RORγt, a multitask nuclear receptor at mucosal surfaces. Mucosal Immunol. 2017;10(1):27–34. doi:10.1038/mi.2016.86
209. Oh TG, Wang SM, Acharya BR, et al. The nuclear receptor, RORγ, regulates pathways necessary for breast cancer metastasis. EBioMedicine. 2016;6:59–72. doi:10.1016/j.ebiom.2016.02.028
210. Hinic V, Lang C, Weisser M, Straub C, Frei R, Goldenberger D. Corynebacterium tuberculostearicum: a potentially misidentified and multiresistant Corynebacterium species isolated from clinical specimens. J Clin Microbiol. 2012;50(8):2561–2567. doi:10.1128/JCM.00386-12
211. Succar EF, Turner JH. Recent advances in understanding chronic rhinosinusitis endotypes. F1000Res. 2018;7. doi:10.12688/f1000research.16222.1
212. Weinrick B, Dunman PM, McAleese F, et al. Effect of mild acid on gene expression in staphylococcus aureus. J Bacteriol. 2004;186(24):8407–8423. doi:10.1128/JB.186.24.8407-8423.2004
213. Yang X, Dong F, Qian S, et al. Accessory gene regulator (agr) dysfunction was unusual in Staphylococcus aureus isolated from Chinese children. BMC Microbiol. 2019;19(1):95. doi:10.1186/s12866-019-1465-z
214. Shopsin B, Drlica-Wagner A, Mathema B, Adhikari RP, Kreiswirth BN, Novick RP. Prevalence of agr dysfunction among colonizing Staphylococcus aureus strains. J Infect Dis. 2008;198(8):1171–1174. doi:10.1086/592051
215. Boles BR, Horswill AR. Agr-mediated dispersal of Staphylococcus aureus biofilms. PLoS Pathogens. 2008;4(4):e1000052. doi:10.1371/journal.ppat.1000052
216. Tan L, Li SR, Jiang B, Hu XM, Li S. Therapeutic targeting of the staphylococcus aureus accessory gene regulator (agr) System. Front Microbiol. 2018;9:55. doi:10.3389/fmicb.2018.00055
217. Gao K, Mu CL, Farzi A, Zhu WY. Tryptophan metabolism: a link between the gut microbiota and brain. Adv Nutr. 2020;11(3):709–723. doi:10.1093/advances/nmz127
218. Quinn GA, Cole AM. Suppression of innate immunity by a nasal carriage strain of Staphylococcus aureus increases its colonization on nasal epithelium. Immunology. 2007;122(1):80–89. doi:10.1111/j.1365-2567.2007.02615.x
219. Wu C, Chen YW, Scheible M, et al. Genetic and molecular determinants of polymicrobial interactions in Fusobacterium nucleatum. Proc Natl Acad Sci U S A. 2021;118:23.
220. de Steenhuijsen Piters WA, Sanders EA, Bogaert D. The role of the local microbial ecosystem in respiratory health and disease. Philos Trans R Soc London Series B Biol Sci. 2015;370:1675.
221. Mårtensson A, Cervin-Hoberg C, Huygens F, et al. Upper airway microbiome transplantation for patients with chronic rhinosinusitis. Int Forum Allergy Rhinol. 2023;13(6):979–988. doi:10.1002/alr.23122
222. Nath S, Zilm P, Jamieson L, et al. Development and characterization of an oral microbiome transplant among Australians for the treatment of dental caries and periodontal disease: a study protocol. PLoS One. 2021;16(11):e0260433. doi:10.1371/journal.pone.0260433
223. Nascimento MM. Oral microbiota transplant: a potential new therapy for oral diseases. J Calif Dent Assoc. 2017;45(10):565–568. doi:10.1080/19424396.2017.12222506
224. Beikler T, Bunte K, Chan Y, et al. Oral microbiota transplant in dogs with naturally occurring periodontitis. J Dent Res. 2021;100(7):764–770. doi:10.1177/0022034521995423
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.