")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Relaxin inhibits cardiac fibrosis and endothelial–mesenchymal transition via the Notch pathway
Authors Zhou X, Chen X, Cai JJ, Chen LZ, Gong YS, Wang LX, Gao Z, Zhang HQ, Huang WJ, Zhou H
Received 25 March 2015
Accepted for publication 26 May 2015
Published 11 August 2015 Volume 2015:9 Pages 4599—4611
DOI https://doi.org/10.2147/DDDT.S85399
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Professor Shu-Feng Zhou
X Zhou,1 X Chen,2 JJ Cai,2 LZ Chen,3 YS Gong,4 LX Wang,5 Z Gao,1 HQ Zhang,1 WJ Huang,1 H Zhou1
1Department of Cardiology, The First Affiliated Hospital of Wenzhou Medical University, 2Wenzhou Medical University, 3Department of Clinical Laboratory, Wenzhou Central Hospital, 4Institute of Hypoxia Medicine, Wenzhou Medical University, 5Department of Respiratory Medicine, Wenzhou Medical University, Wenzhou, People’s Republic of China
Background: Relaxin (RLX) can prevent cardiac fibrosis. We aimed to investigate the possible mechanism and signal transduction pathway of RLX inhibiting cardiac fibrosis.
Methods: Isoproterenol (5 mg·kg-1·d-1) was used to establish the cardiac fibrosis model in rats, which were administered RLX. The cardiac function, related targets of cardiac fibrosis, and endothelial–mesenchymal transition (EndMT) were measured. Transforming growth factor β (TGF-β) was used to induce EndMT in human umbilical vein endothelial cells, which were pretreated with RLX, 200 ng·mL-1, then with the inhibitor of Notch. Transwell cell migration was used to evaluate cell migration. CD31 and vimentin content was determined by immunofluorescence staining and Western blot analysis. Notch protein level was examined by Western blot analysis.
Results: RLX improved cardiac function in rats with cardiac fibrosis; it reduced the content of collagen I and III, increased the microvascular density of the myocardium, and suppressed the EndMT in heart tissue. In vitro, RLX decreased the mobility of human umbilical vein endothelial cells induced by TGF-β, increased the expression of endothelial CD31, and decreased vimentin content. Compared to TGF-β and RLX co-culture alone, TGF-β + RLX + Notch inhibitor increased cell mobility and the EndMT, but decreased the levels of Notch-1, HES-1, and Jagged-1 proteins.
Conclusion: RLX may inhibit the cardiac fibrosis via EndMT by Notch-mediated signaling.
Keywords: relaxin, endothelial to mesenchymal transition, transforming growth factor β, myocardial fibrosis, Notch
Corrigendum for this paper has been published.
Introduction
Cardiac fibrosis, associated with a decreased extent of microvasculature and disrupted normal myocardial structures, results from excessive deposition of extracellular cell matrix mediated by recruitment of fibroblasts and endothelial dysfunction, but its contribution to these conditions is unclear.1
Endothelial–mesenchymal transition (EndMT) is a complex biological process whereby endothelial cells lose their specific markers and acquire a mesenchymal or myofibroblastic phenotype and express mesenchymal cell products such as α-smooth muscle actin (α-SMA) and type I collagen.2 EndMT was originally observed during heart development, and recent studies have suggested its role in pathological settings such as cancer and fibrosis. Zeisberg et al first showed that cardiac fibrosis is associated with the emergence of fibroblasts originating from endothelial cells, suggesting an EndMT similar to events that occur during formation of the atrioventricular cushion in the embryonic heart.3 Then it has also been reported that endothelial cell-derived endothelin-1 promoted cardiac fibrosis via the EndMT in a streptozotocin-induced diabetic mouse model.4 In the high-fat-fed apolipoprotein E knockout mice model, it was demonstrated that inflammatory stress exacerbates the progression of cardiac fibrosis via EndMT.5 In acute viral myocarditis, EndMT has also been proved to be involved in cardiac fibrosis.6 According to these findings, we speculated that inhibition of EndMT may be a new target of anti-fibrosis of heart.
The naturally occurring hormone relaxin (RLX) is now regarded as a rapidly occurring antifibrotic that prevents and reduces aberrant collagen deposition and fibrosis progression in various experimental models, regardless of pathogenesis.7–10 Furthermore, it has been reported that RLX may regulate extracellular cell matrix remodeling, such as in the heart,11 liver,12 kidney,13,14 and lungs.15 Male, but not female, RLX-null mice showed increased left ventricular (LV) collagen content and collagen type I expression with aging, which was reversed with exogenous RLX treatment.11 RLX has also shown efficacy in treating experimentally induced cardiac fibrosis. Recent Phase III clinical trials have successfully evaluated the vasodilatory properties of serelaxin (a recombinant form of human gene-2 RLX, which is the major stored and circulating form of RLX in humans) in acute heart failure.16 The antifibrotic efficacy of serelaxin, which is bioactive in mice, has also been demonstrated experimentally.11,15,17,18
Despite the therapeutic potential of RLX as an antifibrotic agent, the mechanism of its antifibrotic effect in the heart was still unclear. So the aim of our study is to investigate whether EndMT maybe the possible mechanism of RLX preventing the cardiac fibrosis.
Notch signaling has been implicated in cardiac morphogenesis19,20 and plays a crucial role in regulating the EndMT in the primary heart tube during septation and formation of the valves.21,22 Notch-1 is the main receptor subtype found in the endocardial layer in the outflow tract of the developing heart.23 Then, the role of Notch in the postnatal heart was revealed. Furthermore, the use of cardiac hypertrophy models revealed that components of the Notch pathway are upregulated in the hypertrophic heart.24 Nowadays, it has been reported that the Notch signaling pathway is involved in the epithelial to mesenchymal transition or EndMT process of the organs or cancers such as corneal cancer;25 atrioventricular canal,26 and foramen ovale closure.27 And what about the relationship between the Notch and RLX? Boccalini et al concluded that Notch was involved in the protection of cardiac muscle cells from hypoxia/reoxygenation injury by RLX28 and that Notch signaling was involved in the cardiac fibroblast to myofibroblast transition induced by transforming growth factor β (TGF-β) and activated by RLX.29 Accordingly, this paper examines whether RLX inhibits the EndMT and exerts its anti-fibrotic effects via Notch-1 signaling.
Materials and methods
Rat model of cardiac fibrosis
In total, 40 male Sprague Dawley rats (200–220 g; approximately 6 weeks old) were supplied by the Wenzhou Medical University Laboratory Animal Centre (Wenzhou, People’s Republic of China). The animals were housed individually in cages under hygienic conditions with 12-hour light/dark cycle at 22°C±3°C and 45%±10% humidity for 7 days before experiments. The animals were allowed free access to a standard commercial diet and tap water.
Rats were randomly divided into five groups (ten per group) for treatment: control; myocardial fibrosis (isoproterenol [Iso]); and low-, middle-, and high-dose RLX (0.2, 2, and 20 μg·kg−1·day−1, respectively. For Iso-model, on days 1–6, Iso (5 mg·kg−1·d−1; Sigma-Aldrich Co., St Louis, MO, USA) was injected subcutaneously in the rats. In the therapeutic groups, Iso administration was the same as in the Iso-model group, and RLX at different concentrations (0.2, 2.0, and 20 μg·kg−1·day−1) were injected at the same time as Iso injection and lasted for 6 days, then RLX injection was continued for another 8 days. The same volume of saline was injected for controls.
Animal welfare and ethics statement
The procedures we used were as humane as possible. The study was evaluated and approved by the Wen Zhou Medical University Animal Care And Use Ethics Committee (permission number wydw2013-0054).
Measurement of hemodynamics
Rats were weighed and anesthetized with 1% pentobarbital sodium (40 mg·kg−1) by intraperitoneal injection. Rats were fixed, the right carotid artery was separated, and a catheter filled with 0.05% heparin was inserted into the left ventricle. The other end of the catheter was connected with a pressure-transducer input signal recorder for recording the LV mean systolic pressure, LV end diastolic pressure, and maximum rate of change in LV pressure (+dp/dtmax, −dp/dtmax).
Hematoxylin and eosin staining
Hematoxylin and eosin (HE) staining was performed to evaluate pathological and morphological changes of myocardial tissue. The myocardium sample (approximately 5 mm thick) was placed in 4% paraformaldehyde at 4°C for 72 hours for fixation, then routine paraffin embedding was performed and 4 μm thick slices were taken. Sections were deparaffinized with xylene, hydrated through a graded ethanol series, stained with HE, dehydrated through a graded ethanol series, cleared in xylene, and finally mounted in neutral resin. Pathological and morphological changes were observed under an optical microscope.
Masson’s trichrome staining
Masson’s trichrome staining was performed to assess myocardial fibrosis. Rat myocardial tissue specimens were fixed in 4% paraformaldehyde, routinely paraffin-embedded, and sliced into 4 μm thick sections, then deparaffinized in water, stained with Harris hematoxylin for 3 minutes, and rinsed with running water. Sections were differentiated in 1% hydrochloric acid alcohol solution for 3–5 seconds, rinsed with running water, placed in water for bluing for 1 minute, and washed with running water, then stained in warm Ponceau-acid fuchsin solution for 3 minutes, washed with distilled water, and differentiated in a 1% phosphomolybdic acid solution for 1 minute. After the phosphomolybdic acid residue was wiped from slides, sections were stained in 2% aniline blue solution for 1 minute, rinsed with 95% alcohol, dehydrated in 95% alcohol and anhydrous alcohol, dried with cold air, and mounted in neutral resin. The sections were examined using light microscopy, and photographs were taken at ×200 magnification. Five non-repeating visual fields were randomly selected, myocardial collagen areas were measured using Image-Pro Plus (Media Cybernetics, Inc., Rockville, MD, USA), and the areas were averaged.
ELISA of collagen I and III content, fibrotic protein, in myocardial tissue
A piece of the LV myocardium (100 mg) was cut into smaller pieces, added to 1 mL phosphate buffered saline (PBS) (pH 7.4), and vortexed on ice. After centrifugation at 3,000 rpm for 20 minutes, the supernatant was separated and content of types I and III collagen was measured by use of an ELISA kit (Shanghai Boyun Biotech, Shanghai, People’s Republic of China).
Immunohistochemistry of microvascular density
Von Willebrand factor was used as a marker of vascular endothelial cells to indicate blood vessels. Endogenous peroxidase activity was blocked with 3% methanol–H2O2. Nonspecific sites were blocked with 5%–10% goat serum before incubation with anti-Von Willebrand factor antibody (Abcam, Cambridge, UK; 1:600) in 0.1 M PBS overnight at 4°C. After washing, reagents 1 and 2 of the Polymer HRP Detection System for rabbit primary antibodies (Zhongshan Jinqiao, Beijing, People’s Republic of China) were added, washed sequentially, and developed with horseradish peroxidase and diaminobenzidine chromogen (Zhongshan Jinqiao). Blood vessels were observed at ×100 and ×400 magnifications under a microscope before being imaged and counted at ×200 magnification. Stained single cells or cell clusters clearly separate from surrounding vessels, cardiomyocytes, and other tissues were considered microvessels. The number of microvessels was calculated as the mean microvascular density (MVD) in five randomly selected non-repeating visual fields.
Immunofluorescence staining
For single immunofluorescence staining, tissue sections were incubated with the antibodies for anti-vimentin (1:100), anti-CD31 (1:1,600), and anti-α-SMA (1:1,000) at 4°C overnight, washed with PBS three times, 5 minutes per wash, incubated with DyLight 488/DyLight 594 antibodies (1:300; EarthOX, Millbrae, CA, USA) for 1 hour, then incubated with 1 μg/mL Hoechst 33342 (Sigma-Aldrich Co.) for 20 minutes to stain nuclei. Sections incubated with the appropriate isotype control primary antibodies, and fluorescently labeled secondary antibodies were negative controls. The results were analyzed by fluorescence microscopy (Nikon Corporation, Tokyo, Japan) and processed with Image-Pro Plus (Media Cybernetics, Inc.).
For dual immunofluorescence staining, sections were co-incubated with the antibodies anti-CD31 and anti-vimentin in vitro and anti-CD31 and anti-α-SMA in vivo, then processed as for single immunofluorescence staining.
Cell culture and treatments
Human umbilical vein endothelial cells (HUVECs) were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum, penicillin (100 U·mL−1), and streptomycin (100 U·mL−1) (Sigma, Milan, Italy) at 37°C in a humidified atmosphere of 5% CO2. At the start of the study, cells were randomly divided into five groups for treatment; blank control; negative control which is treated with Notch inhibitor N-[N-(3,5-difluorophenacetyl)-1-alanyl]-S-Phenylglycine t-butyl ester (DAPT); TGF-β induction; TGF-β + RLX (200 ng·mL−1); TGF-β + RLX (200 ng·mL−1) + DAPT.
Transwell assay of cell migration
DMEM/high-glucose medium containing 10% fetal bovine serum was added to the lower chamber of the Transwell system and cell suspension with RLX or negative control was added to the upper chamber for 48 hours. After incubation at 37°C with 5% CO2 for 12–18 hours, the lower chamber was observed by use of an inverted microscope. Incubation was terminated when cells passed into the lower chamber. The inside of the upper chamber was cleaned with a cotton swab, then the chamber was immersed and washed with PBS, fixed with 4% paraformaldehyde, stained with 0.1% crystal violet, washed three times with running water, and photographed, and cell numbers were counted.
Western blot analysis
Cells were resuspended in an appropriate volume of ice-cold cell extraction buffer. After centrifugation at 12,000× g for 10 minutes at 4°C, the supernatant was collected. Total protein content was quantified by use of the Pierce BCA Protein Assay Kit according to its manufacturer (Pierce Biotechnology, Waltham, MA, USA). In total, 400 μg total protein was electrophoresed on 7% gel (Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA; 200 V, 40 minutes) and blotted onto polyvinylidene difluoride membrane (Beyotime Institute of Biotechnology, Haimen, People’s Republic of China; 30 V, 1 hour), which was blocked with blocking solution for 30 minutes at room temperature on a rotary shaker and incubated overnight at 4°C with the antibodies rabbit polyclonal anti-vimentin (1:1,000), mouse polyclonal anti-CD31 (1:1,000), anti-Notch-1 (1:1,000, Cell Signaling Technology, Danvers, MA, USA), anti-Hes-1 (1:1,000, Abcam), and anti-Jagged-1 (1:1,000, Abcam). The housekeeping protein glyceraldehyde 3-phosphate dehydrogenase and β-actin (Bioworld Technology, Nanjing, People’s Republic of China; 1:5,000) were used as loading controls. Immunoreactive bands were detected by use of Chemiluminescent HRP Substrate (Applygen Technologies, Beijing, People’s Republic of China), and scans were obtained by use of the Bio-Rad gel image analysis system (BioRad, Hercules, CA, USA) and processed by use of Image-Pro Plus.
Statistical analysis
Data are expressed as mean ± SEM. Statistical analyses involved use of SPSS v16.0 (SPSS Inc., Chicago, IL, USA) by Student’s t-test for comparing the two groups or one-way ANOVA. P<0.05 was considered statistically significant.
Results
RLX improved the cardiac function of rats with cardiac fibrosis
As compared with controls, Iso-induced cardiac fibrosis affected cardiac function L vs P (108.8±11.6 vs 138.8±11.7 mmHg−1), LV end diastolic pressure (21.59±8.55 vs −7.04±7.37 mmHg−1), +dp/dtmax (3,070.3±1,099.7 vs 9,778.6±2,110.7 mmHg−1·s−1) and −dp/dtmax (−3,095.3±1,249.2 vs −8,524.4±2,678.4 mmHg−1·s−1), P<0.01]. RLX administration could improve cardiac function dose-dependently, for low-, middle-, and high-dose RLX, compared to Iso alone for LV mean systolic pressure, LV-end diastolic pressure, +dp/dtmax, and −dp/dtmax (Figure 1).
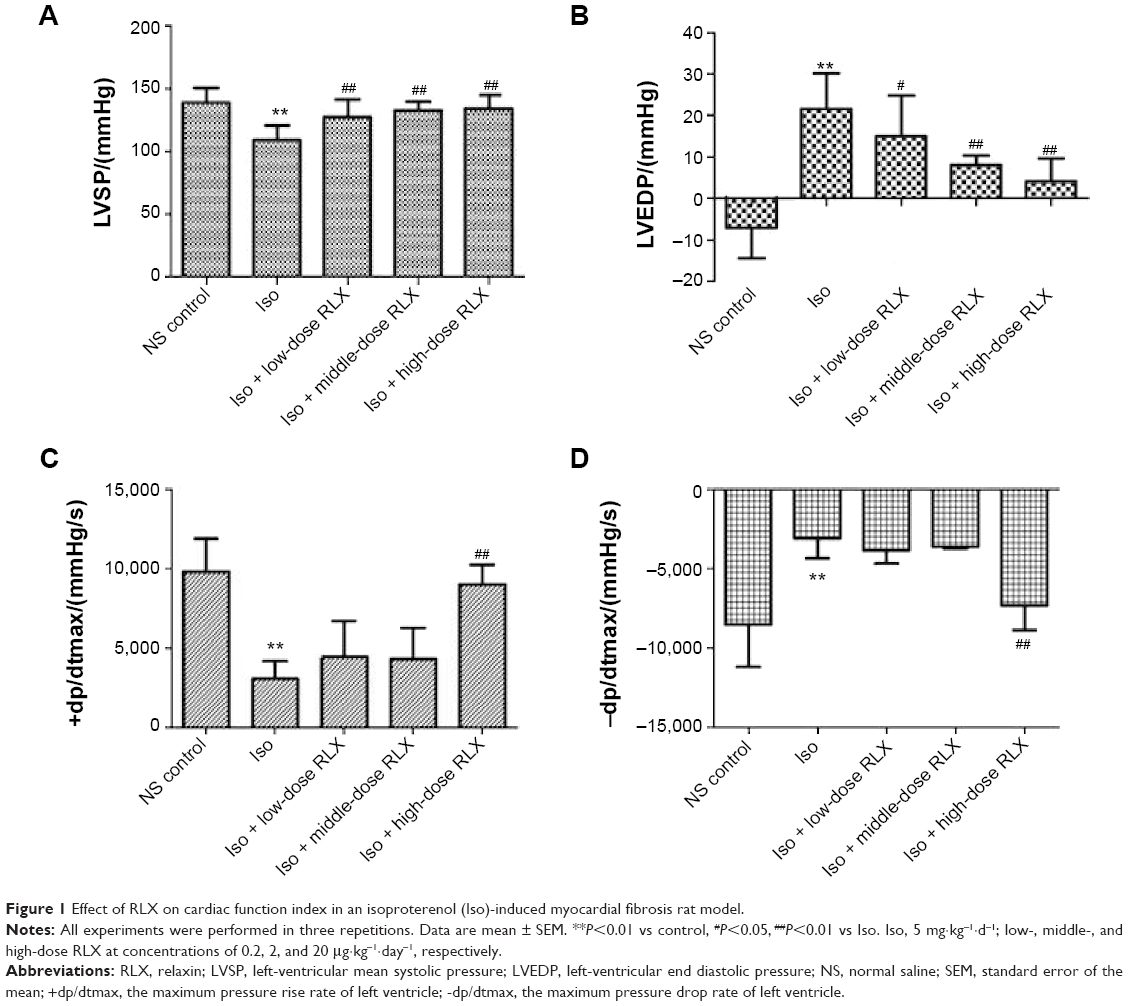 | Figure 1 Effect of RLX on cardiac function index in an isoproterenol (Iso)-induced myocardial fibrosis rat model. |
RLX attenuated fibrosis of the rat heart
From HE staining, myocardial cells from the control group were neatly arranged, with clear cross-striations. However, with Iso, myocardial cells showed an irregular morphology, and were disorganized with considerable fibrous tissue hyperplasia. With RLX treatment, myocardial cells were slightly disorganized and tissue was fibrous as compared with controls. Compared with Iso alone, RLX produced relatively neat myocardial cells, and the degree of fibrous hyperplasia was significantly reduced (Figure 2A). Under normal circumstances, Masson’s trichrome staining produces red myocardium but blue collagen-content fibers. We found that Iso produced a large area of blue-stained collagen fiber between myocardial cells, with fewer blue-stained collagen fibers in controls and with RLX treatment (P<0.01, Figure 2B and C). Therefore, heart failure was accompanied by myocardial fibrosis, and RLX could significantly reduce myocardial fibrosis.
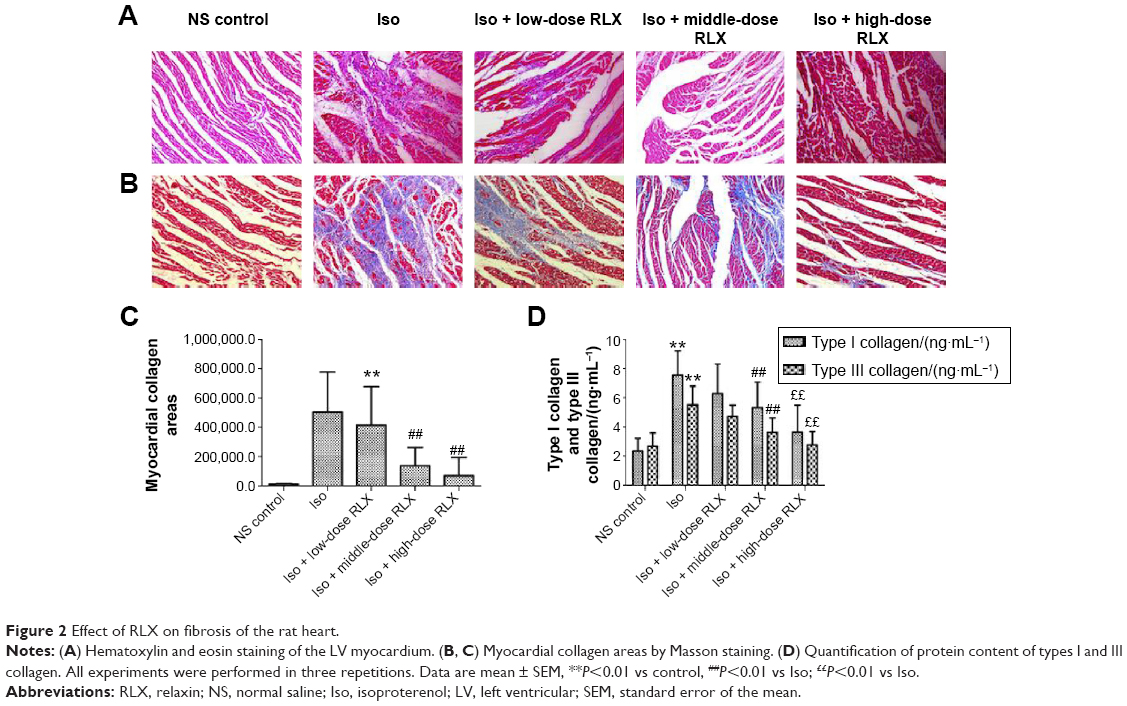 | Figure 2 Effect of RLX on fibrosis of the rat heart. |
As compared with controls, Iso increased the protein levels of type I and III collagen (P<0.01) (Figure 2D). As compared with Iso treatment, RLX reduced type I and III collagen protein content (P<0.01). Therefore, RLX could inhibit the Iso-induced expression of type I and III collagen.
RLX upregulated the microvascular density and inhibited the EndMT of the fibrotic heart
Iso decreased the MVD in rat hearts as compared with controls (P<0.01). However, RLX increased the MVD of hearts as compared with Iso (P<0.01) (Figure 3). Therefore, the fibrotic heart may show EndMT, and EndMT could be attenuated by RLX. For further study of EndMT in the heart, we examined CD31 content, an endothelial marker and α-SMA, a fibrotic marker, by immunofluorescence. With Iso, green fluorescence was greater than for controls, so the expression of α-SMA was upregulated, whereas red fluorescence was reduced, so the expression of CD31 was downregulated. As compared with Iso, with increasing RLX dose, we found increased expression of CD31 but reduced expression of α-SMA (P<0.01) (Figure 4).
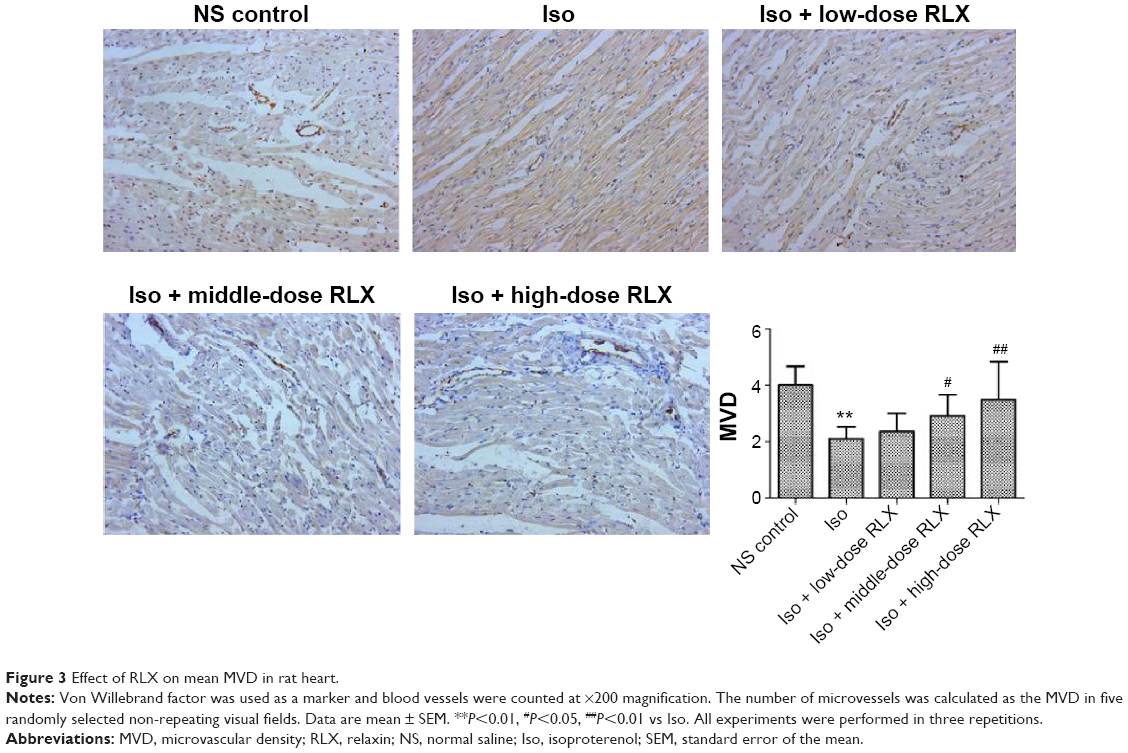 | Figure 3 Effect of RLX on mean MVD in rat heart. |
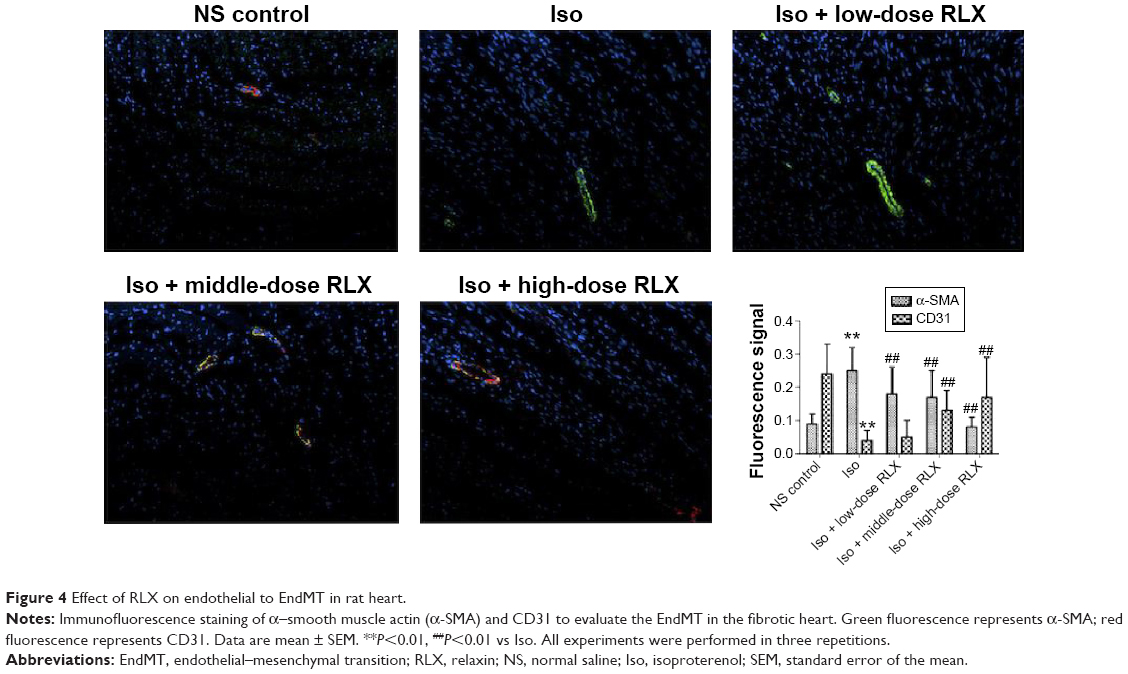 | Figure 4 Effect of RLX on endothelial to EndMT in rat heart. |
RLX inhibits cell migration stimulated by TGF-β in HUVECs
Cell migration ability was analyzed by Transwell assay 48 hours after RLX treatment. The number of cells migrating was lower with TGF-β +200 ng RLX than with TGF-β alone in HUVECs (P<0.05). However, after co-culture with the Notch inhibitor DAPT, the number of migrating cells was increased as compared with TGF-β + RLX (P<0.05). Therefore, RLX could inhibit HUVEC cell migration stimulated by TGF-β, and DAPT could abrogate the effect (Figure 5).
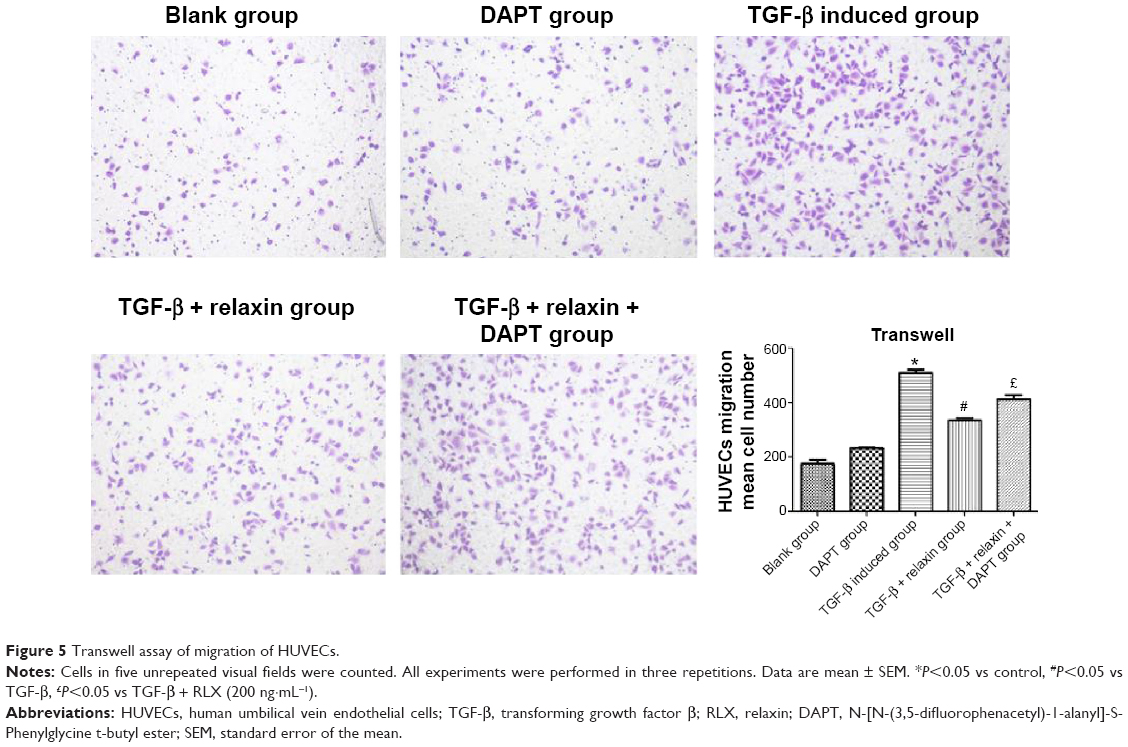 | Figure 5 Transwell assay of migration of HUVECs. |
RLX inhibits the EndMT in vitro with TGF-β stimulation
CD31+ cells were used as a marker of HUVECs and vimentin+ cells as a marker of mesenchymal cells. On immunofluorescence staining, vimentin+ cells were higher with TGF-β than negative induction (P<0.05), but the CD31+ cells were lower (P<0.05). CD31+ cells were higher, but vimentin+ cells were lower with TGF-β + RLX than TGF-β alone. With TGF-β + RLX + DAPT, the fluorescence of CD31 was attenuated and that of vimentin was increased as compared with TGF-β + RLX (P<0.05) (Figure 6). Dual immunofluorescence staining produced the same results, so RLX could inhibit the EndMT of HUVECs induced by TGF-β and, as expected, treatment with DAPT abrogated the inhibitory effect of RLX on TGF-β-induced EndMT (Figure 7).
 | Figure 6 Immunofluorescence staining of CD31 and vimentin to evaluate the EndMT in HUVECs. |
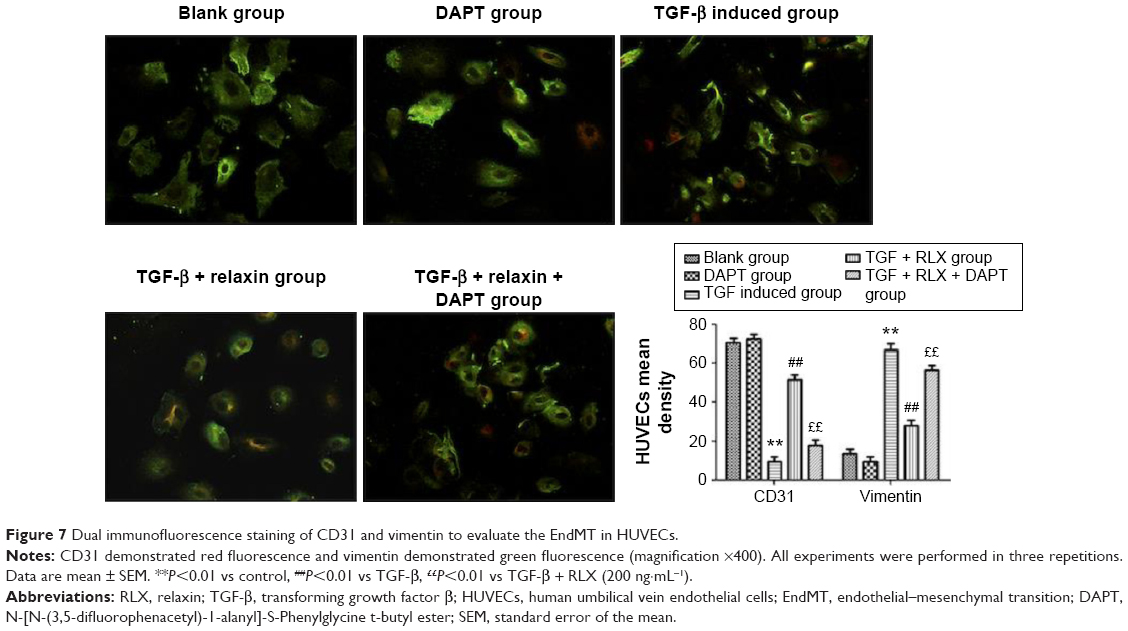 | Figure 7 Dual immunofluorescence staining of CD31 and vimentin to evaluate the EndMT in HUVECs. |
Inhibition of EndMT by Notch-1 with RLX in HUVECs
RLX strongly reduced the phenotypical changes induced by TGF-β. The protein level of CD31 was higher with TGF-β + RLX than TGF-β alone (P<0.05) but that of vimentin, a marker of mesenchymal cells, was lower (P<0.05). Conversely, the addition of the Notch-1 inhibitor DAPT to TGF-β-stimulated cells increased the level of vimentin, decreased that of CD31, and prevented the inhibitory effects of RLX (P<0.05) (Figure 8).
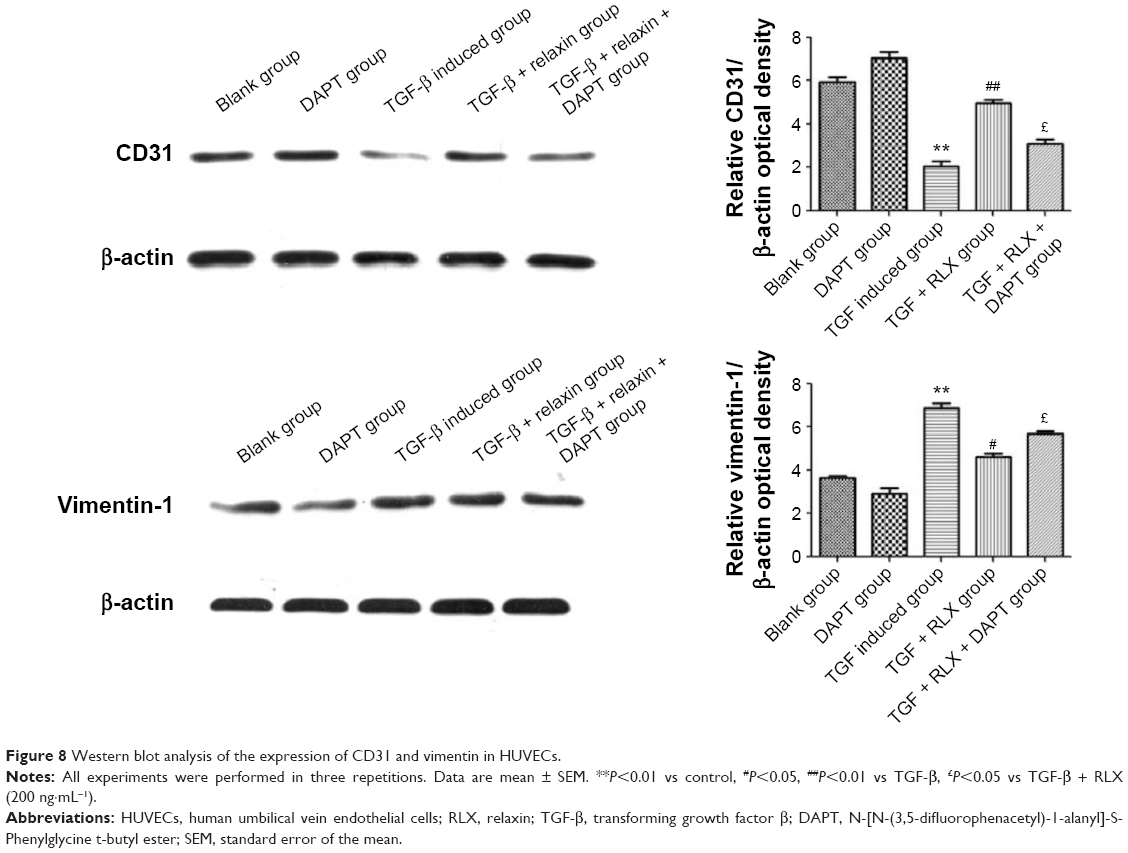 | Figure 8 Western blot analysis of the expression of CD31 and vimentin in HUVECs. |
Notch-1 pathway is involved in RLX-mediated inhibition of EndMT
The Notch-1 and its downstream proteins HES-1 and Jagged-1 were detected in HUVECs. In vitro, TGF-β produced lower protein expression of Notch-1 than control treatment (0.27±0.011 vs 0.61±0.018, P<0.01), but the protein level of Notch-1 was higher with TGF-β + RLX than TGF-β alone (0.43±0.033 vs 0.27±0.011, P<0.01). With Notch-1 inhibitor added to HUVECs, the addition of RLX downregulated the expression of Notch-1 as compared with TGF-β + RLX treatment (0.24±0.031 vs 0.43±0.033, P<0.01). Furthermore, TGF-β decreased the protein expression of HES-1 than control treatment (0.25±0.020 vs 0.45±0.023, P<0.01), but the protein level was higher with TGF-β + RLX than TGF-β alone (0.41±0.010 vs 0.25±0.020, P<0.01). When the Notch-1 inhibitor was employed into HUVECs, the addition of RLX downregulated the expression of HES-1 as compared with TGF-β + RLX treatment (0.22±0.021 vs 0.41±0.010, P<0.01). For detecting the expression of Jagged-1, it showed that DAPT could also decrease the expression of Jagged-1. TGF-β group had lower expression level of Jagged-1 than the control (0.27±0.005 vs 0.42±0.016, P<0.01) while TGF-β + RLX could increase the Jagged-1 compared to the TGF-β group (0.27±0.005 vs 0.43±0.021, P<0.01). Added the DAPT to the TGF-β + RLX group, it demonstrated that Jagged-1 was inhibited compared to the TGF-β + RLX group (0.25±0.020 vs 0.43±0.021, P<0.01) (Figure 9).
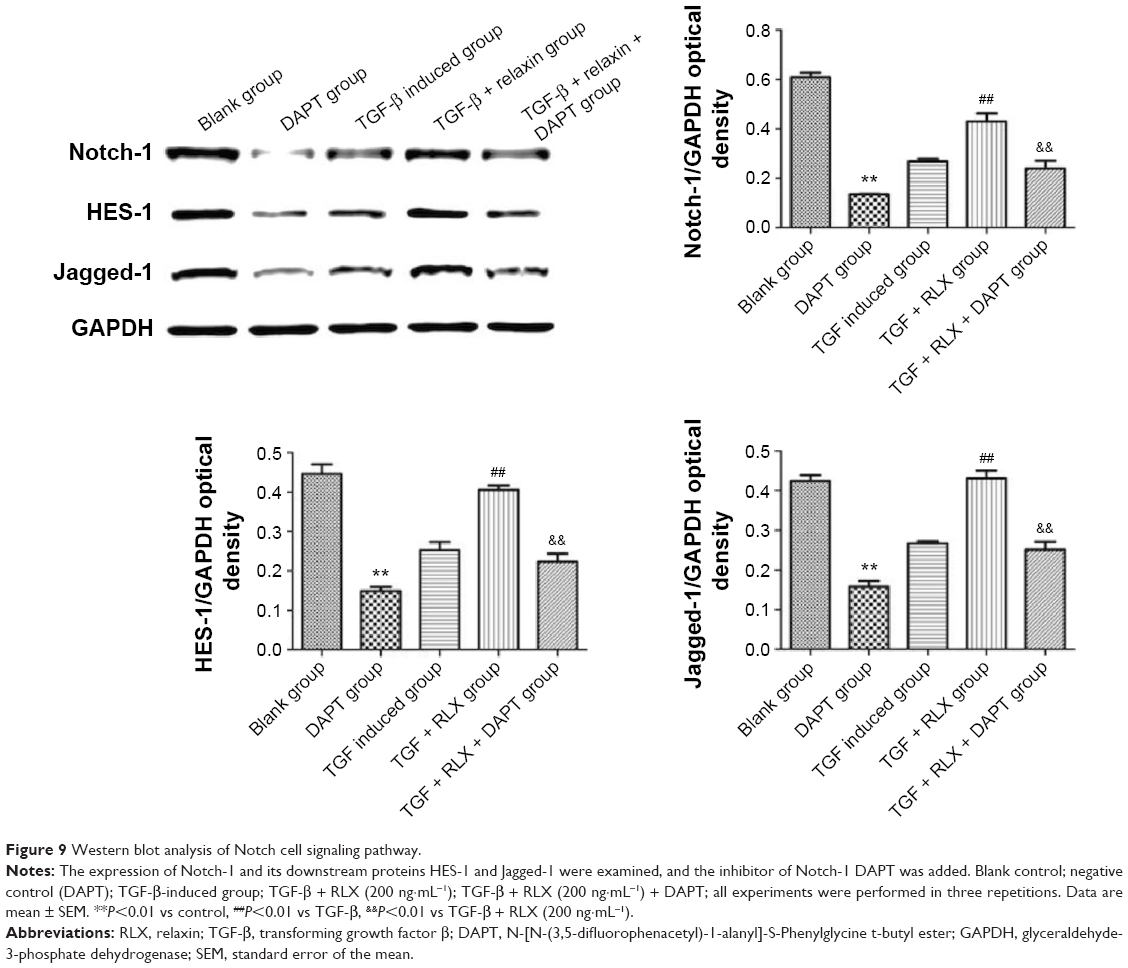 | Figure 9 Western blot analysis of Notch cell signaling pathway. |
Discussion
In our study, we confirmed that RLX can improve the cardiac function of rats; it attenuated the degree of fibrous hyperplasia and downregulated the expression of collagen I and III, which defined the fibrosis as previously research showed. But furthermore, we found that RLX improved the cardiac function and inhibited the fibrosis of heart dose-dependently. Also, in this paper we indicated that RLX could increase the microvascular density which has not been reported before. It implied that the microvascular density of the heart may be involved in the mechanism of the anti-fibrotic effect of RLX.
Furthermore, this research is aimed at investigating the possible mechanism of inhibition of cardiac fibrosis of RLX. As we mentioned earlier, Zeisberg et al first showed that EndMT contributed to the pressure overload-induced progression of cardiac fibrosis.3 While in our Iso-induced fibrotic rat hearts, the protein expression of α-SMA was upregulated and that of CD31 was downregulated which were not reported before. Although several papers have convincingly documented that EndMT is not the predominant source of new fibroblasts in cardiac fibrosis,30 the model of cardiac fibrosis they used was the pressure overload-induced cardiac fibrosis. However, our research used injection of isoproterenol to set up the cardiac fibrosis model. As we know, the mechanism of isoproterenol-induced cardiac fibrosis is ischemia related. According to our results, we supposed that EndMT may be the predominant mechanism of ischemia-induced fibrosis.
Anti-fibrotic effects of RLX of the heart to inhibit cardiac remodeling and improve cardiac function has been definitely documented.11,16,18,31 Indeed, RLX treatment of primary rat atrial and ventricular fibroblasts caused alterations in fibrotic markers, including decreased content of collagen types I and III, the fibroblast to myofibroblast transition, and cell proliferation.11 In the Iso-induced cardiac ischemia model, RLX reduced interstitial collagen accumulation and cardiac hypertrophy.32 However, in our paper, after administration of RLX, it was first found that the level of α-SMA protein decreased and that of the endothelial cell marker, CD31, increased in the fibrotic heart of mice. The results partly indicated that RLX could prevent cardiac fibrosis via EndMT in ischemic-associated cardiac fibrosis.
TGF-β has long been shown to induce EndMT in many normal or malignant cells. Some studies demonstrated that TGF-β treatment of endothelial cells induced the expression of mesenchymal cell markers, including α-SMA, vimentin, transgelin, and calponin, and reduced that of the endothelial cell marker claudin 5, CD31.33 TGF-β induced EndMT in primary cultures of murine pulmonary endothelial cells.34 Along these lines, we used TGF-β to induce EndMT in HUVECs. The results of this paper showed that RLX decreased the expression of vimentin but increased that of CD31, showing endothelial cells, as compared with TGF-β alone. It represented that RLX may inhibit the EndMT of HUVECs which has not been demonstrated before. Sassoli et al have examined that RLX inhibited TGF-β-induced fibroblast–myofibroblast transition in neonatal cardiac stromal cells. The conclusion of their paper may provide another possibility of the role of RLX in the cardiac fibrosis.29 However, it will be better if they had an in vivo model to prove their conclusion. And the cell line they used in the research was stromal cells which presented the mesenchyme cells only. Compared to the study mentioned earlier, our data showed that RLX may inhibit the cardiac fibrosis via EndMT in vivo and the cell line that we used was endothelial cell line. The results in vitro definitely showed that RLX could inhibit the TGF-β-induced EndMT.
TGF-β signals through trans-membrane receptors, which activate Smad2/3 phosphorylation; in turn, pSmad2/3 complex translocates into the nucleus, where it induces the expression of pro-fibrotic target genes.35,36 Several distinct mechanisms have been postulated or demonstrated to be involved in RLX-mediated inhibition of the TGF-β–Smad axis including the PI3K-Akt and NOS-NO-cGMP cascades.37–39 Notch protein is a part of a highly conserved signaling pathway that mediates key cell fate decisions during development and in differentiated tissues. Notch signaling has been recently shown to be involved in tissue fibrosis. TGF-β limits Notch-1 activation in mouse fibroblasts by inducing the expression of ADAM12,40 a member of the metalloprotease-disintegrin family, which can cleave Notch ectodomain after ligand–receptor interaction. In transgenic mice overexpressing the Notch ligand Jagged-1, Notch inhibited the development of cardiomyocyte hypertrophy and TGF-β/connective TGF-mediated cardiac fibrosis.41 Moreover, in vitro study revealed that Notch signaling influenced the differentiation of fibroblasts into myofibroblasts.42,43 In contrast, RLX prevented cardiac fibroblast–myofibroblast transition via Notch-mediated inhibition of TGF-β signaling.29 Our results expand the list of possible mechanisms underlying the anti-fibrotic action of RLX, demonstrating that this hormone also acts through Notch-1 and its downstream proteins HES-1, Jagged-1 upregulation. We inferred that RLX treatment of TGF-β-induced EndMT downregulated multiple processes involved in inhibiting the EndMT but upregulating the expression of Notch protein. Furthermore, DAPT, a γ-secretase-specific inhibitor, as well as silencing of Notch-1 ligand, showed the RLX–Notch–EndMT signaling pathway. Indeed, DAPT prevented the reduced migration of HUEVCs induced by RLX, and the pharmacological inhibition of endogenous Notch-1 signaling by DAPT blocked the effect of RLX on inhibition of the EndMT, the reduced CD31 levels, and the upregulation of vimentin in response to TGF-β stimulation.
In a word, one of the probable mechanisms of RLX inhibiting fibrosis of the heart may be its inhibition of EndMT in the fibrotic heart. The Notch signal pathway may mediate the process. However, our study has limitations. First, it should be noted that our study in vivo has only examined the expression of CD31 and α-SMA, which do not adequately represent EndMT. The increased expression of CD31 and decreased expression of α-SMA can only indicate that RLX can inhibit the fibrosis while improve the endothelial expression. Unfortunately, we cannot determine EndMT definitely from this data. However, these problems could be solved if we consider more specific marker in the fibrotic heart in future. Second, the anti-fibrotic effect of RLX was dose-dependent, but high-dose RLX is expensive. As a result, drug combinations are needed. Further studies assessing how Notch pathways are interlinked and cooperate to mediate the effects of RLX on the EndMT and whether another pathway mediates the effects will be required to further clarify the mechanism of the anti-fibrotic action of RLX. Our findings may provide promising prospects for the treatment of myocardial fibrosis with RLX.
Acknowledgments
The authors thank Professor Chaoshu Tang for the valuable discussion. This study was supported by the Zhejiang Provincial Nature Science Foundation of China (LY12H02004) and in part by the Science and Technology Department of Zhejiang Province of China (2014C33166), Provincial Health Department of Zhejiang Province of China (2014KYA136), and Wenzhou Science and Technology program (Y20140074).
Author contributions
H Zhou and X Zhou contributed to the conception and study design. H Zhou, X Chen, JJ Cai, LX Wang, LZ Chen, and Z Gao contributed to data acquisition. X Chen, JJ Cai, LX Wang, LZ Chen, and Z Gao contributed to data analysis. H Zhou, X Chen, JJ Cai, LZ Chen, and X Zhou drafted the manuscript. YS Gong, LX Wang, HQ Zhang, WJ Huang, and X Zhou critically revised the manuscript. H Zhou, YS Gong, LX Wang, HQ Zhang, WJ Huang, and X Zhou gave final approval. All authors contributed toward data analysis, drafting and critically revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Zeisberg EM, Kalluri R. Origins of cardiac fibroblasts. Circ Res. 2010;107:1304–1312. | ||
Piera-Velazquez S, Li Z, Jimenez SA. Role of endothelial-mesenchymal transition (EndoMT) in the pathogenesis of fibrotic disorders. Am J Pathol. 2011;179:1074–1080. | ||
Zeisberg EM, Tarnavski O. Endothelial to mesenchymal transition contributes to cardiac fibrosis. Nat Med. 2007;13:952–961. | ||
Widyantoro B, Emoto N, Nakayama K, et al. Endothelial cell-derived endothelin-1 promotes cardiac fibrosis in diabetic hearts through stimulation of endothelial-to-mesenchymal transition. Circulation. 2010;121(22):2407–2418. | ||
Kun Ling Ma, Jing Liu, Jie Ni, et al. Inflammatory stress exacerbates the progression of cardiac fibrosis in high-fat-fed apolipoprotein e knockout mice via endothelial-mesenchymal transition. Int J Med Sci. 2013;10(4):420–426. | ||
Hua JY, Zhang ZC, Jiang XH, et al. Relationship between endothelial-to-mesenchymal transition and cardiac fibrosis in acute viral myocarditis. Zhejiang Da Xue Xue Bao Yi Xue Ban. 2012;41(3):298–304. | ||
Dschietzig T, Bartsch C, Baumann G, Stangl K. Relaxin-a pleiotropic hormone and its emerging role for experimental and clinical therapeutics. Pharmacol Ther. 2006;112:38–56. | ||
Samuel CS, Hewitson TD, Unemori EN, Tang ML. Drugs of the future: the hormone relaxin. Cell Mol Life Sci. 2007;64:1539–1557. | ||
Bani D, Yue SK, Bigazzi M. Clinical profile of relaxin, a possible new drug for human use. Curr Drug Saf. 2009;4:238–249. | ||
Bennett RG. Relaxin and its role in the development and treatment of fibrosis. Transl Res. 2009;154:1–6. | ||
Samuel CS, Unemori EN, Mookerjee I, et al. Relaxin modulates cardiac fibroblast proliferation, differentiation, and collagen production and reverses cardiac fibrosis in vivo. Endocrinology. 2004;145:4125–4133. | ||
Bani D, Nistri S, Quattrone S, Bigazzi M, Bani Sacchi T. The vaso relaxant hormone relaxin induces changes in liver sinusoid microcirculation: a morphologic study in the rat. J Endocrinol. 2001;71:541–549. | ||
Heeg MH, Koziolek MJ, Vasko R, et al. The antifibrotic effects of relaxin in human renal fibroblasts are mediated in part by inhibition of the Smad2 pathway. Kidney Int. 2005;68:96–109. | ||
Masterson R, Hewitson TD, Kelynack K, et al. Relaxin down-regulates renal fibroblast function and promotes matrix remodelling in vitro. Nephrol Dial Transplant. 2004;19:544–552. | ||
Unemori EN, Pickford LB, Salles AL, et al. Relaxin induces an extracellular matrix-degrading phenotype in human lung fibroblasts in vitro and inhibits lung fibrosis in a murine model in vivo. J Clin Invest. 1996;98:2739–2745. | ||
Teerlink JR, Cotter G, Davison BA, et al; RELAXIN in Acute Heart Failure (RELAX-AHF) Investigators. Serelaxin, recombinant human relaxin-2, for treatment of acute heart failure (RELAX-AHF): a randomised, placebo-controlled trial. Lancet. 2013;381:29–39. | ||
Hewitson TD, Ho WY, Samuel CS. Antifibrotic properties of relaxin: in vivo mechanism of action in experimental renal tubulointerstitial fibrosis. Endocrinology. 2010;151:4938–4948. | ||
Samuel CS, Cendrawan S, Gao XM, et al. Relaxin remodels fibrotic healing following myocardial infarction. Lab Invest. 2011;91:675–690. | ||
Pedrazzini T. Control of cardiogenesis by the notch pathway. Trends Cardiovasc Med. 2007;17:83–90. | ||
High FA, Epstein JA. The multifaceted role of Notch in cardiac development and disease. Nat Rev Genet. 2008;9:49–61. | ||
Timmerman LA, Grego-Bessa J, Raya A, et al. Notch promotes epithelial-mesenchymal transition during cardiac development and oncogenic transformation. Genes Dev. 2004;18:99–115. | ||
Grego-Bessa J, Díez J, Timmerman L, et al. Notch and epithelial-mesenchyme transition in development and tumor progression: another turn of the screw. Cell Cycle. 2004;3:718–721. | ||
Croquelois A, Domenighetti AA, Nemir M, et al. Control of the adaptive response of the heart to stress via the Notch1 receptor pathway. J Exp Med. 2008;205:3173–3185. | ||
Li C, Dong F, Jia Y, et al. Notch signal regulates corneal endothelial-to-mesenchymal transition. Am J Pathol. 2013;183(3):786–795. | ||
Loomes KM, Taichman DB, Glover CL, et al. Characterization of Notch receptor expression in the developing mammalian heart and liver. Am J Med Genet. 2002;112:181–189. | ||
Chang AC, Fu Y, Garside VC, et al. Notch initiates the endothelial-to-mesenchymal transition in the atrioventricular canal through autocrine activation of soluble guanylyl cyclase. Dev Cell. 2011;21(2):288–300. | ||
Elliott GC, Gurtu R, McCollum C, Newman WG, Wang T. Foramen ovale closure is a process of endothelial-to-mesenchymal transition leading to fibrosis. PLoS One. 2014;9(9):e107175. | ||
Boccalini G, Sassoli C, Formigli L, Bani D, Nistri S. Relaxin protects cardiac muscle cells from hypoxia/reoxygenation injury: involvement of the Notch-1 pathway. FASEB J. 2015;29(1):239–249. | ||
Sassoli C, Chellini F, Pini A, et al. Relaxin prevents cardiac fibroblast-myofibroblast transition via notch-1-mediated inhibition of TGF-β/Smad3 signaling. PLoS One. 2013;8(5):e63896. | ||
Moore-Morris T, Guimarães-Camboa N, Banerjee I, et al. Resident fibroblast lineages mediate pressure overload-induced cardiac fibrosis. J Clin Invest. 2014;124(7):2921–2934. | ||
Bani D, Nistri S, Formigli L, Meacci E, Francini F, Zecchi-Orlandini S. Prominent role of relaxin in improving postinfarction heart remodeling. Ann N Y Acad Sci. 2009;1160:269–277. | ||
Zhang J, Qi Y-F, Geng B, et al. Effect of relaxin on myocardial ischemia injury induced by isoproterenol. Peptides. 2005;26:1632–1639. | ||
Kokudo T, Suzuki Y, Yoshimatsu Y, Yamazaki T, Watabe T, Miyazono K. Snail is required for TGF beta-induced endothelial-mesenchymal transition of embryonic stem cell-derived endothelial cells. J Cell Sci. 2008;121:3317–3324. | ||
Li Z, Jimenez SA. Protein kinase C delta and the c-abl kinase are required for transforming growth factor-beta induction of endothelial- mesenchymal transition in vitro. Arthritis Rheum. Epub 2011 Mar 18. | ||
Visse R, Nagase H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function, and biochemistry. Circ Res. 2003;92:827–839. | ||
Samarakoon R, Overstreet JM, Higgins PJ. TGF-β signaling in tissue fibrosis: redox controls, target genes and therapeutic opportunities. Cell Signal. 2013;25:264–268. | ||
Samuel CS, Mookerjee I, Halls ML, et al. Investigations into the inhibitory effects of relaxin on renal myofibroblast differentiation. Ann N Y Acad Sci. 2009;1160:294–299. | ||
Ahmad N, Wang W, Nair R, Kapila S. Relaxin induces matrix-metalloproteinases-9 and -13 via RXFP1: induction of MMP-9 involves the PI3K, ERK, Akt and PKC-f pathways. Mol Cell Endocrinol. 2012;363:46–61. | ||
Chow BS, Chew EG, Zhao C, Bathgate RA, Hewitson TD, Samuel CS. Relaxin signals through a RXFP1-pERK-nNOS-NO-cGMP-dependent pathway to up-regulate matrix metalloproteinases: the additional involvement of iNOS. PLoS One. 2012;7(8):e42714. | ||
Solomon E, Li H, Duhachek Muggy S, Syta E, Zolkiewska A. The role of SnoN in transforming growth factor beta1-induced expression of metallopro- tease-disintegrin ADAM12. J Biol Chem. 2010;285:21969–21977. | ||
Nemir M, Metrich M, Plaisance I, et al. The Notch pathway controls fibrotic and regenerative repair in the adult heart. Eur Heart J. 2014;35(32):2174–2185. | ||
Pan Z, Feng T, Shan L, et al. Scutellarin induced endothelium-independent relaxation in rat aorta. Phytother Res. 2008;22(11):1428–1433. doi: 10.1002/ptr.2364. | ||
Luo P, Tan ZH, Zhang ZF, Zhang H, Liu XF, Mo ZJ. Scutellarin isolated from Erigeron multiradiatus inhibits high glucose-mediated vascular inflammation. Yakugaku. 2008;128(9):1293–1299. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.