")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Recent advances in combinatorial biosynthesis for drug discovery
Authors Sun H, Liu Z, Zhao H, Ang EL
Received 12 October 2014
Accepted for publication 29 November 2014
Published 12 February 2015 Volume 2015:9 Pages 823—833
DOI https://doi.org/10.2147/DDDT.S63023
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Shu-Feng Zhou
Huihua Sun,1,* Zihe Liu,1,* Huimin Zhao,1,2 Ee Lui Ang1
1Metabolic Engineering Research Laboratory, Institute of Chemical and Engineering Sciences, Agency for Science, Technology and Research, Singapore; 2Department of Chemical and Biomolecular Engineering, University of Illinois at Urbana-Champaign, Urbana, IL, USA
*These authors contributed equally to this work
Abstract: Because of extraordinary structural diversity and broad biological activities, natural products have played a significant role in drug discovery. These therapeutically important secondary metabolites are assembled and modified by dedicated biosynthetic pathways in their host living organisms. Traditionally, chemists have attempted to synthesize natural product analogs that are important sources of new drugs. However, the extraordinary structural complexity of natural products sometimes makes it challenging for traditional chemical synthesis, which usually involves multiple steps, harsh conditions, toxic organic solvents, and byproduct wastes. In contrast, combinatorial biosynthesis exploits substrate promiscuity and employs engineered enzymes and pathways to produce novel “unnatural” natural products, substantially expanding the structural diversity of natural products with potential pharmaceutical value. Thus, combinatorial biosynthesis provides an environmentally friendly way to produce natural product analogs. Efficient expression of the combinatorial biosynthetic pathway in genetically tractable heterologous hosts can increase the titer of the compound, eventually resulting in less expensive drugs. In this review, we will discuss three major strategies for combinatorial biosynthesis: 1) precursor-directed biosynthesis; 2) enzyme-level modification, which includes swapping of the entire domains, modules and subunits, site-specific mutagenesis, and directed evolution; 3) pathway-level recombination. Recent examples of combinatorial biosynthesis employing these strategies will also be highlighted in this review.
Keywords: combinatorial biosynthesis, drug discovery, natural products, polyketide synthases, nonribosomal peptide synthetases, biosynthetic pathways
Introduction
The journey of drug discovery and development is long, costly, and risky. On average, it takes about 10–15 years1 and costs approximately US$1.8 billion2 to discover and develop a new drug. Tens of thousands of compounds need to be introduced into the drug discovery pipeline for every successful drug that comes to the market. Therefore, it is critical to improve the diversity and novelty of the compounds to be screened in the process of drug discovery, in order to increase the probability that a compound becomes an approved drug. Analysis of the sources of new drugs from 1981 to 2010 showed that 64% of the 1,073 small molecule drugs are either natural products or natural product derivatives.3 Gifted with extraordinary structural diversity and unparalleled biological activities, natural products are secondary metabolites assembled and modified by dedicated biosynthetic pathways in living organisms.4 The structural complexity of natural products in many cases makes it difficult to produce analogs with improved medicinal activities. In contrast, combinatorial biosynthesis exploits substrate promiscuity and employs engineered enzymes and pathways to produce novel “unnatural” products, which substantially expands the structural diversity of natural products with potential pharmaceutical value.5–8
There are three advantages of combinatorial biosynthesis for drug discovery. Firstly, combinatorial biosynthesis helps to enrich the novelty and diversity of the natural product architectures, which potentially enhances their biological features. Secondly, combinatorial biosynthesis offers an environmentally friendly way to produce natural product analogs, whereas chemical synthesis usually involves multiple protection/deprotection steps, harsh conditions, toxic organic solvents, and byproduct wastes. Thirdly, efficient expression of the combinatorial biosynthetic pathway into genetically tractable heterologous hosts can increase the titer of the compound, eventually resulting in less expensive drugs. This review will focus on combinatorial biosynthesis that generates multiple natural product analogs by the following methods: 1) precursor-directed biosynthesis; 2) enzyme-level modification including swapping of the entire domains, modules and subunits, site-specific mutagenesis, and directed evolution; 3) pathway-level recombination (Figure 1). Recent examples of combinatorial biosynthesis including polyketide, nonribosomal peptide, and saponin biosynthesis are highlighted in this review.
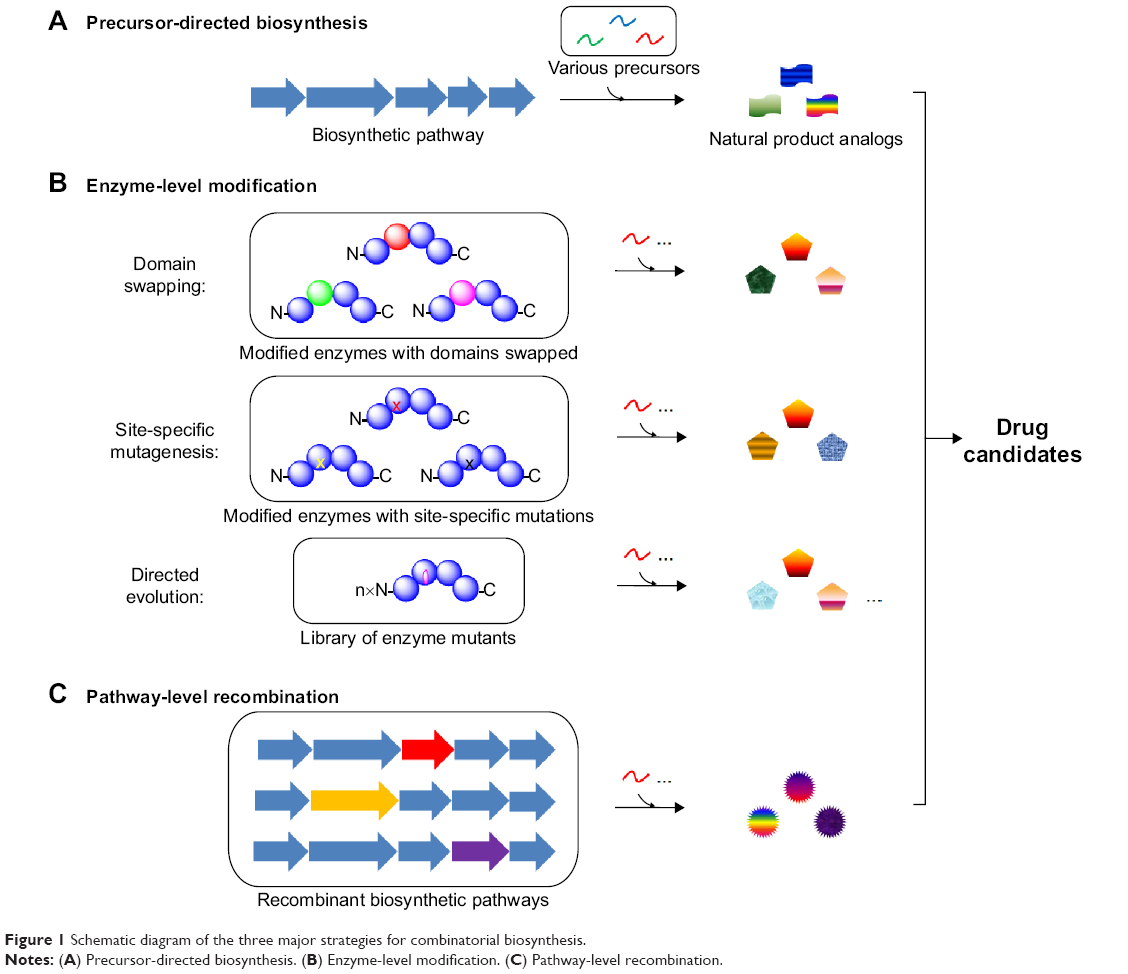 | Figure 1 Schematic diagram of the three major strategies for combinatorial biosynthesis. |
Precursor-directed combinatorial biosynthesis
The structural diversity of natural products comes substantially from diverse building blocks of the natural product assembly lines. Precursor-directed combinatorial biosynthesis takes advantage of the substrate promiscuity of the enzymes in the biosynthetic pathways to incorporate nonnative building blocks, consequently producing various natural product analogs. Modular type I polyketide synthases (mPKSs) are polyketide synthase (PKS) assembly lines that contain sequentially organized modules, each of which harbors a set of catalytic domains required for one cycle of chain extension. The polyether antibiotic monensin is biosynthesized by an mPKS from Streptomyces cinnamonensis. The acyltransferase (AT) domain in the fifth module of the monensin PKS was shown to incorporate nonnatural malonic acid derivatives as building blocks to produce new premonensin analogs.9 Based on the computational model of the AT domain, Bravo-Rodriguez et al9 predicted that the active site should be able to accept the building block with bulky propargyl group. The culture of S. cinnamonensis A495 supplemented with the synthetic propargyl-malonyl-N-acetyl-cysteamine yielded propargyl-premonensin.9 This compound was observed to bind to the human phosphodiesterase δ subunit (PDEδ),9 which could potentially inhibit the PDEδ–KRAS interaction and thus suppress proliferation of KRAS-dependent human pancreatic tumor cells.10 Harvey et al11 developed a chemical genetic strategy that combines precursor-directed biosynthesis and colony bioassay to rapidly discover new macrolide antibiotics. A library of synthetic diketide precursors were fed to cultures of Escherichia coli HYL3 strain evolved for improved polyketide production, with plasmids expressing module 2–6 of the mPKS 6-deoxyerythronolide B synthase (DEBS), and proteins for sugar biosynthesis and glycosyl transfer. Consequently, the precursors were converted to the corresponding glycosylated macrolide and their bioactivities were screened against Bacillus subtilis overlaid lawn. Employing this strategy, the authors identified a new class of alkynyl- and alkenyl-substituted erythromycin analogs, including an equipotent and orthogonally functional analog as a promising lead for antibiotic drug development.11
Compared to multimodular PKSs, type III PKSs are structurally simple PKSs, with a single active site catalyzing the priming, extension, and cyclization reactions iteratively. They are widely found in plants, bacteria, and fungi, producing biologically important compounds such as chalcones, pyrones, acridones, phloroglucinols, and stilbenes.12 Due to their remarkable substrate tolerance, type III PKSs offer an excellent platform for precursor-directed combinatorial biosynthesis of novel “unnatural” natural products.13 HsPKS1 from a primitive club moss Huperzia serrata accepted various starter units to produce an array of aromatic tetraketides, including chalcones, benzophenones, phloroglucinols, and acridones. Surprisingly, this type III PKS also accepted bulky starter units such as 4-methoxycinnamoyl-coenzyme A (CoA) and N-methylanthraniloyl-CoA to produce 4-methoxy-2′, 4′, 6′-trihydroxychalcone and 1,3-dihydroxy-N-methylacridone, respectively.14 Furthermore, biologically active and structurally unique polyketide–alkaloid hybrid molecules were synthesized by HsPKS1 which was capable of accepting synthetic nitrogen-containing nonphysiological starter units, including 2-carbamoylbenzoyl-CoA, pyridine-containing 3-carbamoylpicolinoyl-CoA, and naphthalene-containing 3-carbamoyl-2-naphthoyl-CoA.15
With modular organization similar to mPKSs, nonribosomal peptide synthetases (NRPSs) assemble a variety of medicinally important peptides, including antibiotics (actinomycin, daptomycin), immunosuppressants (cyclosporine A), and antitumor drugs (bleomycin).16 Uridyl peptide antibiotics such as pacidamycins and sansanmycins, exhibit good antibacterial activity toward highly refractory pathogens and are assembled by NRPSs with remarkably relaxed substrate specificity.17–19 Grüschow et al17 found that the biosynthetic machinery of pacidamycin exhibited highly relaxed substrate specificity with majority of the tryptophan analogs, which resulted in production of new pacidamycin derivatives. Zhang et al20 identified the nine-enzyme assembly line for the biosynthesis of tetrapeptidyl nucleoside pacidamycin antibiotics. The relaxed substrate specificities of PacI and adenylation (A) domains in the assembly line led to in vitro biosynthesis of nine pacidamycin analogs with varied C-terminal amino acid, central diamino acid, and uridine moieties.20 Additionally, exploration of the NRPSs’ substrate promiscuity in sansanmycin producer strain led to the isolation and structural elucidation of eight new uridyl peptides, sansanmycins H to O.18
Some natural products are biosynthesized by hybrid PKS/NRPS assembly lines. Micacocidin is a thiazoline-containing natural product from the plant pathogenic bacterium Ralstonia solanacearum and used to treat Mycoplasma pneumoniae infections. To guide precursor-directed biosynthesis of micacocidin derivatives, Kreutzer et al21 performed in vitro specificity tests on the starter-unit-activating domain, fatty acid-AMP (adenosine monophosphate) ligase, of the hybrid PKS/NRPS enzyme MicC toward unnatural substrates. Then the most promising nonnative precursors were fed into the micacocidin-producing cell culture, leading to the generation of six unnatural analogs of micacocidin with activity against M. pneumoniae.21
Precursor-directed biosynthesis can also be applied without detailed characterization of the biosynthetic enzymes, as long as the native precursor of certain natural products are known. Given that the pyrrolyl moiety of the fungal metabolite rumbrin originates from pyrrole-2-carboxylic acid, a suit of substituted pyrrole-2-carboxylates were incubated with the rumbrin-producing organism, generating three unnatural rumbrin analogs. The 3-chloro- and 3-bromo-isorumbrin were more potent than rumbrin, and 3-bromo-isorumbrin exhibited improved anticancer activity.22 To yield diazepinomicin analogs with modified ring-A, Ratnayake et al23 fed potential ring-A precursors such as fluorinated tryptophans, halogenated anthranilates, and various substituted indoles to the marine actinomycete culture (Micromonospora sp. strain DPJ15).23 Two fluorinated diazepinomicin analogs were identified and they showed weak to modest antibacterial activity against selected Gram-positive and Gram-negative bacteria.23
Enzyme-level combinatorial biosynthesis
Swapping of the entire domains, modules, and subunits
Swapping of the entire domains, modules, or subunits has been the main classical approach for combinatorial biosynthesis. Modular PKSs24,25 and NRPSs16 are amenable to combinatorial biosynthesis due to their modular organization and stepwise synthetic strategy. One early remarkable example for mPKS is that a large combinatorial library of 61 6-deoxyerythronolide B (6-DEB) analogs was constructed by substituting the AT domains and the β-carbon processing domains of erythromycin mPKS with their respective counterparts from the rapamycin mPKS.26 Domain and module swapping approaches for NRPSs have led to fruitful combinatorial biosynthesis of analogs as demonstrated by the engineering of the daptomycin synthetase.27–30 Subunit and module exchanges between related cyclic lipopeptide A54145 and calcium-dependent antibiotic (CDA), coupled with modifications of glutamine (Gln) at position 12 and variations in lipid side chain, generated a combinatorial library of 72 compounds, many of which were active antibacterial agents.30
Different from mPKSs and NRPSs, iterative type I PKSs (iPKSs) are PKSs that produce polyketides by using a single set of catalytic domains iteratively.31 The iterative synthetic fashion of iPKSs is encoded with smart programming, such as selective reduction, selective methylation, chain-length control, patterned cyclization, and product release, some of which still remain mysterious.32,33 Domain swapping among iPKSs not only enables generation of “unnatural” natural products, but also allows us to decipher the programmed biosynthesis of iPKSs in an effort to generate novel bioactive polyketides through rational reprogramming.34 Liu et al35 reported production of a new “unnatural” polyketide through replacing the starter unit ACP transacylase (SAT) domain from AfoE, the nonreducing iPKS (NR-PKS) for asperfuranone biosynthesis, with the SAT domain from StcA, the NR-PKS for sterigmatocystin biosynthesis. Without any full-length iPKS structure information, the major obstacle of domain swapping is the uncertainty of domain boundaries. They employed the Udwary-Merski algorithm (UMA) to predict the linker region between the SAT and ketosynthase (KS) domains, leading to successful construction of chimeric PKSs.35 Interestingly, the hybrid AfoE accepted the starter unit shorter by two carbon units, but produced a new metabolite sharing the same final chain length with the native AfoE product.35 Recently, SAT domain swapping between AfoE and nine other NR-PKSs showed that the newly constructed hybrid AfoE accepted the starter unit longer by two carbon units and produced a novel aromatic polyketide with the same final chain length with the native AfoE product.36 These findings further support that the final polyketide chain length, instead of the number of extension rounds, could be controlled by the KS domain.
Yeh et al37 reannotated and characterized an NR-PKS DtbA in Aspergillus niger, which belongs to group VI NR-PKS but has a reductase (R) domain instead of a typical thioesterase (TE) domain for product release. They then created chimeric PKSs by replacing the R domain with TE domains from two phylogenetically close NR-PKSs, AusA, and ANID_06448, leading to alternative product release and successful production of new compounds with distinct functional groups.37 However, no products were detected when the R domain was replaced with the TE or the thioesterase/claisen-cyclase (TE/CLC) from three phylogenetically distant NR-PKSs.37 These results suggested that close phylogenetic relationships could guide successful domain swapping probably due to the minimal interruption of domain–domain interaction of the chimeric PKSs. On the other hand, a series of domain swapping between two closely related NR-PKSs, CcRADS2, and AtCURS2, emphasized that the shape and size of the polyketide intermediate is crucial for proper recognition and product release by the TE domain, which in turn determines the success of combinatorial domain swaps.38 Failure of the TE domain to recognize the intermediate structure produced by the upstream domains of chimeric PKSs abolished the product formation. On the other hand, choosing the proper TE domain that can accept altered polyketide intermediates generated by chimeric PKSs successfully led to the production of a novel dihydroxyphenylacetic acid lactone (DAL), radilarin.38 The results indicated the complex and somewhat unpredictable decision gating role of TE domains. Further study is required to establish the rules on choosing TE domains for combinatorial domain swapping of NR-PKSs to create novel polyketides.
Fungal benzenediol lactone (BDL) synthases are quasi-mPKSs, which consist of sequentially acting iPKS subunits.39,40 Co-expressing random pairs of subunits from four BDL synthases in yeast allowed 10 of the possible 12 heterocombinations to produce a library of BDL analogs. Among these novel analogs, radilarin with a novel 14-membered ring exhibited more potent heat shock response-inducing activity than natural dehydrocurvularin.41
Site-specific mutagenesis
The classical domain swapping approach often leads to insoluble protein expression, impaired activities and reduced product yields. This is most probably due to disruption of the protein’s overall structure and thus its function. Moreover, the drastic structural changes of intermediates created by domain swapping may render the intermediates inaccessible by downstream catalytic domains. Modern protein engineering methods, such as site-specific mutagenesis to substitute specific amino acids, are less invasive and offer more effective ways to change the enzyme function.
A minimally invasive mutagenesis scheme was developed to inactivate the targeted reductive domains, such as ketoreductases, dehydratases, and enoylreductases of the chosen modules of monensin PKS, leading to a library of 22 premonensin redox derivatives.42 The derivatives ER20-A, -B, and -C exhibited increased antibacterial activity against Pseudomonas aeruginosa and showed activity against B. subtilis, against which the native premonensin showed negligible activity.42 When a key S2107A mutation was introduced into the AT domain on the sixth module of the mPKS DEBS, the AT substrate specificity was switched from native methylmalonyl-CoA to fluoromalonyl-CoA.43 Taking advantage of the modularity and stepwise synthetic mode of mPKS, mutagenesis of the AT domain on either the module 2 or the module 3 led to site-selective fluorine insertion into the polyketide backbone.43 This study opens the door to generating natural product analogs added with fluorine, which has a great impact in drug optimization owing to its modulation of acidity and basicity, lipophilicity, and metabolic stability of a parent compound.44
Crystal structures of type III PKSs have facilitated structure-based enzyme engineering by mutagenesis of the residues in the active site cavity to significantly expand product diversity.45–47 Based on the crystal structure of the type III PKS HsPKS1 from H. serrata, Morita et al15 performed docking simulations with models of various intermediates and found that active site residue Ser348 was crucial for the cyclization of the polyketide intermediate. A single mutation, S348G, introduced to HsPKS1 successfully extended the product chain length and created a new cyclization pattern to produce a biologically active dibenzoazepine with an expanded 6.7.6-fused ring system.
Multiple sequence alignments of homologous active sites facilitate sequence pattern deduction, which has been useful for altering the NRPS A domain specificity. In light of this, introduction of a single mutation K278Q to the A domain of module 10 of the CDA NRPS changed the A domain specificity from glutamic acid (Glu) to Gln, producing a Gln-containing CDA analog.48 This mutated NRPS was also capable of incorporating synthetic (2S,3R)-3-methyl glutamine (mGln) to produce an mGln-containing CDA analog,48 providing the first example of A domain modification to incorporate nonnatural amino acid into a nonribosomal peptide natural product.
Directed evolution
Directed evolution, a powerful enzyme engineering approach, has not been widely employed on natural product biosynthetic enzymes. However, there are significant advantages of applying directed evolution to combinatorial biosynthesis. Compared to more conservative changes by site-specific mutagenesis, directed evolution approaches can potentially spawn more drastic alterations of substrate specificity of domains, while restoring the impaired activity due to large changes in substrate specificity. In contrast to only one enzyme variant obtained with every successful domain swap, directed evolution methods significantly increase the throughput of enzyme variants beneficial for combinatorial biosynthesis. Last but not least, directed evolution can be accomplished even when the enzyme catalytic mechanism still remains elusive.
The first directed evolution for NRPSs reported that rounds of random mutagenesis followed by in vivo screening successfully restored the activity losses of two chimeric NRPSs due to A domain swapping.49 Then the first directed evolution strategy for combinatorial NRPS biosynthesis was developed. The strategy employed saturation mutagenesis of the active site residues of A domain of AdmK within a hybrid NRPS/PKS pathway for antibiotic andrimid biosynthesis50 (Figure 2). After screening a library of ~14,330 clones via mass spectrometry method, four derivatives of andrimid were generated and three of them showed bioactivity toward E. coli imp ASR in agar overlay bioassays.50 Also there are efforts in directed evolution of isolated A domain to increase the repertoire of A domains for combinatorial biosynthesis. Zhang et al51 dramatically altered the substrate specificity of the DhbE A domain of the bacillibactin NRPS complex toward unnatural aromatic building blocks using a powerful high-throughput screening technique, yeast cell surface display. To display the DhbE library on the yeast cell surface, DhbE mutants were fused to the yeast agglutinin protein Aga2p that is bound via two disulfide bridges to the cell wall component Aga1p. The authors also designed adenosine monosulfamate-biotin (AMS-biotin) probes that were conjugated to nonnative substrates to select mutants based on their affinity with the probes. Through iterative rounds of selection of a library of 5×106 yeast clones, DhbE mutants that can bind to the nonnative substrates 3-hydroxybenzoic acid and 2-aminobenzoic acid were identified.51
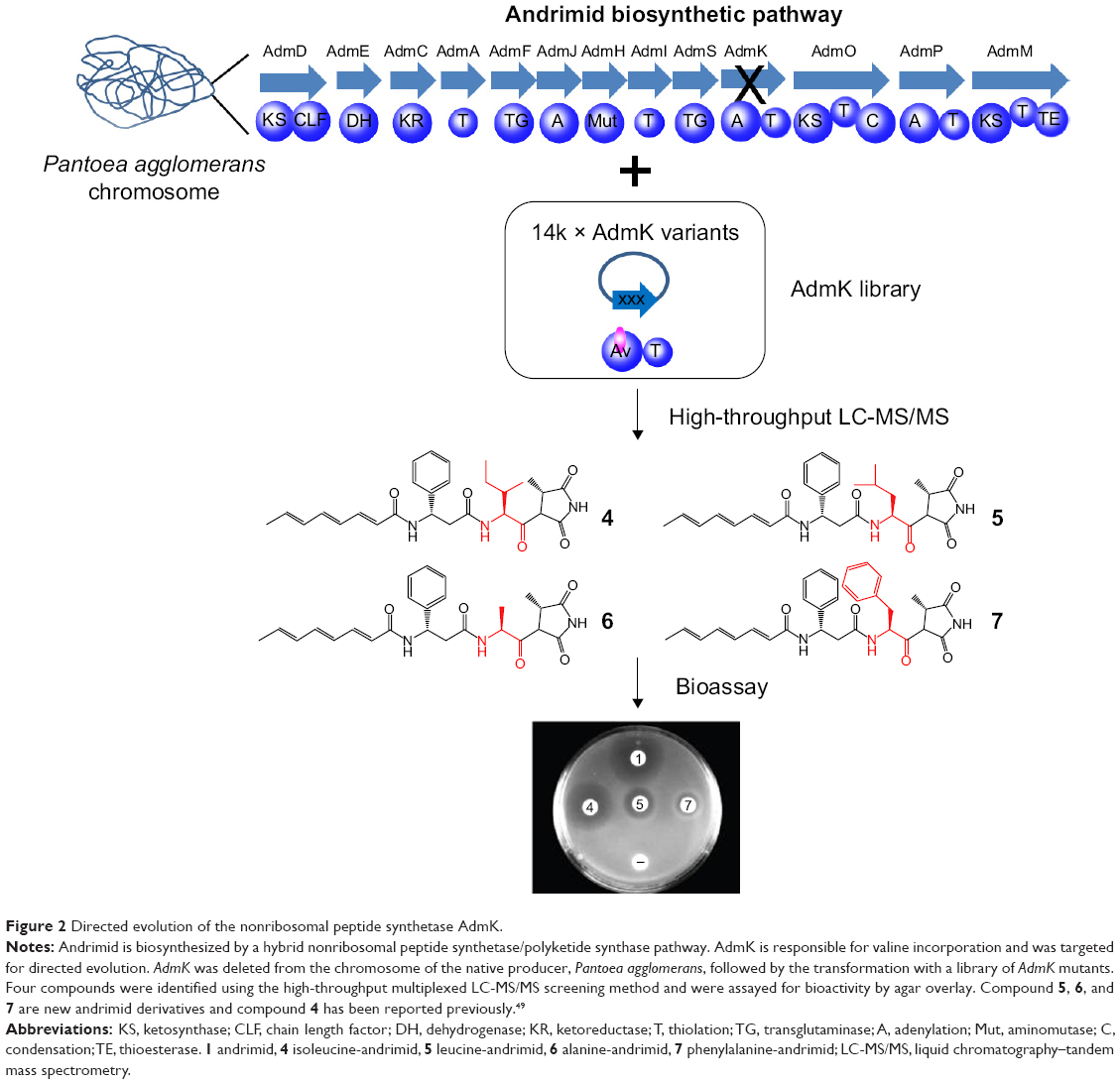 | Figure 2 Directed evolution of the nonribosomal peptide synthetase AdmK. |
Pathway-level combinatorial biosynthesis
The development of molecular and synthetic biology techniques has enabled the heterologous expression of biosynthetic genes from different species in well characterized host organisms.
As early as 1985, Hopwood et al52 explored the feasibility and potential of hybrid antibiotic production and reported the production of a novel antibiotic compound, mederrhodin A, by interchanging and combining genes from multiple species to generate combinatorial pathways. Since then, hybrid pathways have been widely used for production of novel natural products, especially in the field of drug discovery. For example, triterpene saponins, which are secondary metabolites with a wide range of biological activities synthesized by many plant species,53 were heterologously expressed with combinatorial biosynthesis approaches. Recombinant yeast strains expressing the combinations of either the β-amyrin synthase, cytochrome P450 reductase, β-amyrin oxidases CYP93E2 and CYP72A61v2, or the β-amyrin synthase, cytochrome P450 reductase, β-amyrin oxidases CYP716A12 and CYP72A68v2, were able to produce soyasapogenol B and gypsogenic acid, respectively.53 P450s that do not work together in planta were co-expressed in yeast as well. The yeast strains expressing β-amyrin synthase, cytochrome P450 reductase, β-amyrin-30-oxidase CYP72A63,54 and β-amyrin oxidases CYP93E2 or CYP716A12 produced rare triterpenoids that do not occur in M. truncatula.53 Additionally, Saikosaponins are abundant saponins produced by plants with anti-inflammatory effect. Moses et al55 identified a gene, CYP716Y1, encoding a cytochrome P450 monooxygenase from Bupleurum falcatum that was involved in the oxidation of saikosaponins. This enzyme was combined with the oxidosqualene cyclase, P450-dependent monooxygenase, and glycosyltransferase genes available from other plant species in yeast cells to produce nonnatural sapogenins and saponins (Figure 3).
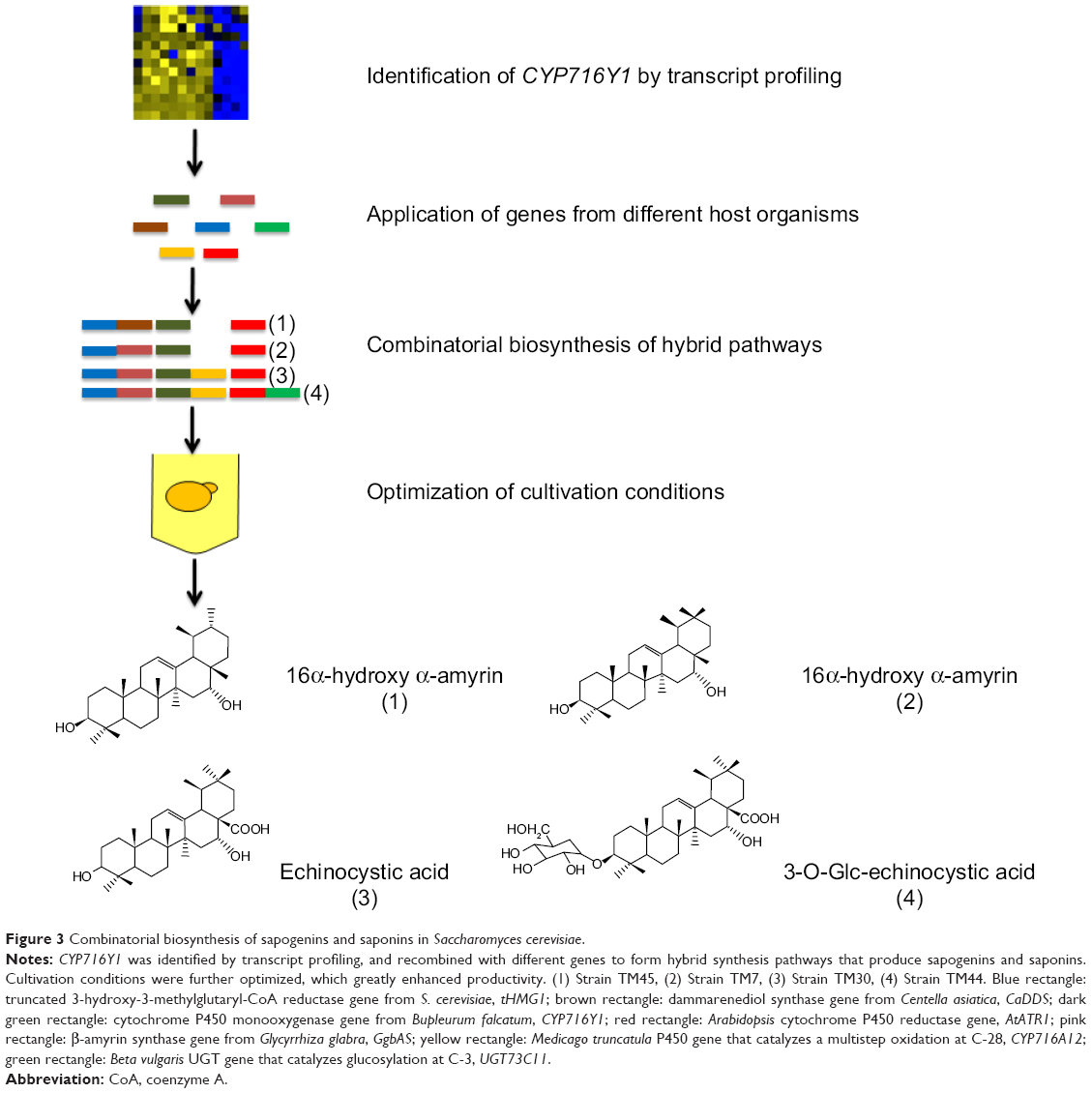 | Figure 3 Combinatorial biosynthesis of sapogenins and saponins in Saccharomyces cerevisiae. |
Sugars are often important to drug-target interactions and in most cases, glycosylation significantly affects the drug solubility and bioactivities.56 A large number of novel glycosylated products can be generated, thus offering greater opportunities for drug discovery. Many important therapeutic compounds, including antiparasitics, antibiotics, antifungals, and anticancer drugs, contain sugars attached to the aglycone core. One well-defined method based on the “sugar biosynthesis plasmids”57 is to obtain the novel sugar-modified drugs, especially for those compounds with good antitumor activities but severe cytotoxic side effects. By glycosylation of a side chain structure, in some cases, the biological activity of the new compounds can be improved.
Mithramycin is a glycosylated polyketide that binds to DNA and inhibits transcription and protein synthesis. It has been used for the treatment of several types of cancers and hypercalcaemia and hypercalciuria. However, its clinical use has been limited because of its life-threatening side effects. Núñez et al58 generated seven novel mithramycin analogs with changes in the sugar profile or in both the sugar profile and the 3-side chain by expressing glycosylated NDP-d-digitoxose synthesis plasmid in the mutant strain S. argillaceus M3W1. One of the constructed compounds, demycarosyl-3D-β-d-digitoxosyl-mithramycin SK, showed high antitumor activity and less toxicity than the parent mithramycin compound during preliminary in vivo evaluation. Another example is the combinatorial biosynthesis of gilvocarcin analogs, the most prominent member of a distinct class of antitumor compounds. Five novel gilvocarcin analogs with varied saccharide moieties were generated through complementation of gilvocarcin mutant Streptomyces lividans TK24 (cosG9B3-U−) with various deoxysugar plasmids.59 Preliminary anticancer assays showed that some analogs exhibit increased antitumor activities against multiple cancer cell lines.59 Additionally, doxorubicin, also known as hydroxydaunorubicin and hydroxydaunomycin, is one of the most commonly used cancer chemotherapy drugs. Han et al60 reported an efficient Streptomyces venezuelae-based system that could convert doxorubicin aglycone ε-rhodomycinone into glycosylated doxorubicin analogs. This was achieved through the use of a S. venezuelae mutant strain bearing a deletion of the whole pikromycin biosynthetic gene cluster (pikAI, pikAII, pikAIII, pikAIV, pikA, desVIII, desVII, desVI, desR, desV, desIV, desIII, desII, and desI).61,62 Different combinations of genes that direct the biosynthesis of various nucleotide activated deoxysugars and their transfer to ε-rhodomycinone, as well as the postglycosylation modifications, were heterologously expressed in the mutant strain and 17 unnatural glycosylated doxorubicin analogs were generated.
Narbomycin is a 14-membered macrolide antibiotic from the group of ketolides. A diverse set of novel and biologically active analogs of narbomycin with unnatural sugar moieties were generated by heterologous expression of nonnative deoxysugar biosynthetic gene cassettes and the gene encoding a substrate-flexible glycosyltransferase DesVII into a narbonolide-accumulating S. venezuelae mutant strain in which the thymidine-5′-diphospho-d-desosamine biosynthetic gene cluster was deleted.63 Their significant antibacterial activity was demonstrated against clinically isolated erythromycin-resistant pathogens.63 However, because S. venezuelae can also produce the 12-membered ring compounds along with narbonolide, each constructed mutant can only produce small amounts of narbomycin, YC-17.63 Subsequently, the same group successfully made enough YC-17 for in vitro stereochemistry study.64 Additional YC-17 analogs possessing unnatural sugars replacing native d-desosamine were generated through expression of four nonnative sugar biosynthetic gene cassettes in 10-deoxymethynolide-producing mutants. An in vitro study demonstrated that on substitution with l-rhamnose, the l-rhamnosyl-10-deoxymethynolide, displayed outstanding antibacterial activities relative to the parent compound.64
Besides sugar biosynthesis, pathway-level combinatorial biosynthesis was applied in other modifications such as halogenation. Organofluorine compounds play an important role on the market and they are responsible for around 20% of pharmaceuticals and 30%–40% of agrichemicals.65 Eustaquio et al66 reported the biosynthesis of the potent anticancer agent salinosporamide A in the marine bacterium Salinispora tropica from a related pathway involving chlorine assimilation via 5′-chloro-5′-deoxyadenosine in a reaction catalyzed by the chlorinase SalL. Fluorosalinosporamide was produced by replacing the chlorinase gene, salL, with the fluorinase gene, flA. Moreover, halogenation was also applied in the first report of using combinatorial biosynthesis in plants. Chlorination biosynthetic machinery from several soil bacteria was introduced into Catharanthus roseus to produce halogenated tryptophan, which was then used by the endogenous terpenoid indole alkaloids biosynthetic enzymes to yield chlorinated alkaloids.67
Challenges and perspectives
Combinatorial biosynthesis exploits the shuffling of anabolic pathways to produce mutated natural product analogs,68 and has led to a fundamental change in the field of classical synthesis. It will undoubtedly remain very important for drug discovery programs.8,69
Great efforts have been made in the field of drug discovery. However, production of many of the novel compounds are often hampered by low yields, which in turn hinders their commercialization and require substantial further engineering. The low production could be tackled with enzyme engineering, finding appropriate expression hosts, and metabolic engineering.70,71 However, drug discovery always requires large compound libraries and conventional cloning approaches usually limit the throughput of engineered biocatalysts generation and combinatorial biosynthesis. This situation could be circumvented by the application of new and rapid DNA synthesis and assembly techniques.72–74 Longer and cheaper DNA fragments that are synthesized by codons rather than nucleotides would be desirable to reduce the time and cost for large-scale gene applications.75 Moreover, combinatorial biosynthesis usually generates large analog libraries, and screening thousands of candidates consumes time and effort.76 Hence high-throughput screening methods are urgently needed. The issue could be tackled with better designs combining computational approaches with structural and bioactivity analyses to ensure the desired activities are achieved.77,78
A major challenge in domain or module swapping approaches has been the fact that most of the resulting chimeric synthases are insolubly expressed and/or functionally impaired. This is thought to result from the disrupted integrity of interdomain linkers or disruption of protein–protein interactions needed for efficient processing of intermediates. Our understanding of the kinetics of protein folding is incomplete, and the energy landscape model also has trouble accounting for these situations. Computational tools that can accurately predict the protein conformations as well as domain–domain and module–module interactions are needed. Moreover, the molecular mechanism of the bioactivities for many drugs is not well studied, for example, the relationship between the sugar moieties and bioactivity is not clear. To exploit the full potential of combinatorial biosynthesis, better understanding of the dynamics and mechanisms of the key enzymes and the metabolic pathways is essential.75
Recent advances in next-generation sequencing have revolutionized functional genomics.79–81 An unexpectedly large number of cryptic biosynthetic gene clusters in pharmaceutically important strains, as well as largely untapped biosynthetic resources in metabolically diverse unculturable microbes were uncovered,82 which facilitate the generation of a great number of analogs with improved biological properties.83 The discovery of new biocatalysts and biosynthetic pathways will enrich the combinatorial biosynthesis toolbox. However, the enormous amount of data generated worldwide, on the other hand, poses a challenge in identifying relevant information. More organized data deposit databases and more efficient and user-friendly software will alleviate this problem.84
Disclosure
The authors report no conflicts of interest in this work.
References
Pharmaceutical Research and Manufacturers of America. Biopharmaceutical Research Industry Profile. 2014; Available from: http://www.phrma.org/sites/default/files/pdf/2014_PhRMA_PROFILE.pdf. Accessed December 1, 2014. | ||
Paul SM, Mytelka DS, Dunwiddie CT, et al. How to improve R&D productivity: the pharmaceutical industry’s grand challenge. Nat Rev Drug Discov. 2010;9(3):203–214. | ||
Cragg GM, Newman DJ. Natural products: a continuing source of novel drug leads. Biochim Biophys Acta. 2013;1830(6):3670–3695. | ||
Walsh CT, Fischbach MA. Natural products version 2.0: connecting genes to molecules. J Am Chem Soc. 2010;132(8):2469–2493. | ||
Cummings M, Breitling R, Takano E. Steps towards the synthetic biology of polyketide biosynthesis. FEMS Microbiol Lett. 2014;351(2):116–125. | ||
Wong FT, Khosla C. Combinatorial biosynthesis of polyketides – a perspective. Curr Opin Chem Biol. 2012;16(1–2):117–123. | ||
Winter JM, Tang Y. Synthetic biological approaches to natural product biosynthesis. Curr Opin Biotechnol. 2012;23(5):736–743. | ||
Goss RJ, Shankar S, Fayad AA. The generation of “unnatural” products: synthetic biology meets synthetic chemistry. Nat Prod Rep. 2012;29(8):870–889. | ||
Bravo-Rodriguez K, Ismail-Ali AF, Klopries S, et al. Predicted Incorporation of non-native substrates by a polyketide synthase yields bioactive natural product derivatives. Chembiochem. 2014;15(13):1991–1997. | ||
Zimmermann G, Papke B, Ismail S, et al. Small molecule inhibition of the KRAS-PDEdelta interaction impairs oncogenic KRAS signalling. Nature. 2013;497(7451):638–642. | ||
Harvey CJ, Puglisi JD, Pande VS, Cane DE, Khosla C. Precursor directed biosynthesis of an orthogonally functional erythromycin analogue: selectivity in the ribosome macrolide binding pocket. J Am Chem Soc. 2012;134(29):12259–12265. | ||
Yu D, Xu F, Zeng J, Zhan J. Type III polyketide synthases in natural product biosynthesis. IUBMB Life. 2012;64(4):285–295. | ||
Wakimoto T, Mori T, Morita H, Abe I. Cytotoxic tetramic acid derivative produced by a plant type-III polyketide synthase. J Am Chem Soc. 2011;133(13):4746–4749. | ||
Wanibuchi K, Zhang P, Abe T, et al. An acridone-producing novel multifunctional type III polyketide synthase from Huperzia serrata. FEBS J. 2007;274(4):1073–1082. | ||
Morita H, Yamashita M, Shi SP, et al. Synthesis of unnatural alkaloid scaffolds by exploiting plant polyketide synthase. Proc Natl Acad Sci U S A. 2011;108(33):13504–13509. | ||
Condurso HL, Bruner SD. Structure and noncanonical chemistry of nonribosomal peptide biosynthetic machinery. Nat Prod Rep. 2012;29(10):1099–1110. | ||
Grüschow S, Rackham EJ, Elkins B, Newill PLA, Hill LM, Goss RJM. New pacidamycin antibiotics through precursor-directed biosynthesis. Chembiochem. 2009;10(2):355–360. | ||
Xie Y, Cai Q, Ren H, et al. NRPS substrate promiscuity leads to more potent antitubercular sansanmycin analogues. J Nat Prod. 2014;77(7):1744–1748. | ||
Ragab AE, Gruschow S, Rackham EJ, Goss RJ. New pacidamycins biosynthetically: probing N- and C-terminal substrate specificity. Org Biomol Chem. 2010;8(14):3128–3129. | ||
Zhang W, Ntai I, Bolla ML, et al. Nine enzymes are required for assembly of the pacidamycin group of peptidyl nucleoside antibiotics. J Am Chem Soc. 2011;133(14):5240–5243. | ||
Kreutzer MF, Kage H, Herrmann J, et al. Precursor-directed biosynthesis of micacocidin derivatives with activity against Mycoplasma pneumoniae. Org Biomol Chem. 2014;12(1):113–118. | ||
Clark BR, O’Connor S, Fox D, Leroy J, Murphy CD. Production of anticancer polyenes through precursor-directed biosynthesis. Org Biomol Chem. 2011;9(18):6306–6311. | ||
Ratnayake AS, Janso JE, Feng X, Schlingmann G, Goljer I, Carter GT. Evaluating indole-related derivatives as precursors in the directed biosynthesis of diazepinomicin analogues. J Nat Prod. 2009;72(3):496–499. | ||
Dutta S, Whicher JR, Hansen DA, et al. Structure of a modular polyketide synthase. Nature. 2014;510(7506):512–517. | ||
Whicher JR, Dutta S, Hansen DA, et al. Structural rearrangements of a polyketide synthase module during its catalytic cycle. Nature. 2014;510(7506):560–564. | ||
McDaniel R, Thamchaipenet A, Gustafsson C, Fu H, Betlach M, Ashley G. Multiple genetic modifications of the erythromycin polyketide synthase to produce a library of novel “unnatural” natural products. Proc Natl Acad Sci U S A. 1999;96(5):1846–1851. | ||
Baltz RH, Miao V, Wrigley SK. Natural products to drugs: daptomycin and related lipopeptide antibiotics. Nat Prod Rep. 2005;22(6):717–741. | ||
Doekel S, Coeffet-Le Gal MF, Gu JQ, Chu M, Baltz RH, Brian P. Non-ribosomal peptide synthetase module fusions to produce derivatives of daptomycin in Streptomyces roseosporus. Microbiology. 2008;154(Pt 9):2872–2880. | ||
Nguyen KT, He X, Alexander DC, et al. Genetically engineered lipopeptide antibiotics related to A54145 and daptomycin with improved properties. Antimicrob Agents Chemother. 2010;54(4):1404–1413. | ||
Baltz RH, Brian P, Miao V, Wrigley SK. Combinatorial biosynthesis of lipopeptide antibiotics in Streptomyces roseosporus. J Ind Microbiol Biotechnol. 2006;33(2):66–74. | ||
Staunton J, Weissman KJ. Polyketide biosynthesis: a millennium review. Nat Prod Rep. 2001;18(4):380–416. | ||
Crawford JM, Townsend CA. New insights into the formation of fungal aromatic polyketides. Nat Rev Microbiol. 2010;8(12):879–889. | ||
Sun H, Ho CL, Ding F, Soehano I, Liu XW, Liang ZX. Synthesis of (R)-mellein by a partially reducing iterative polyketide synthase. J Am Chem Soc. 2012;134(29):11924–11927. | ||
Fisch KM, Bakeer W, Yakasai AA, et al. Rational domain swaps decipher programming in fungal highly reducing polyketide synthases and resurrect an extinct metabolite. J Am Chem Soc. 2011;133(41):16635–16641. | ||
Liu T, Chiang YM, Somoza AD, Oakley BR, Wang CC. Engineering of an “unnatural” natural product by swapping polyketide synthase domains in Aspergillus nidulans. J Am Chem Soc. 2011;133(34):13314–13316. | ||
Liu T, Sanchez JF, Chiang YM, Oakley BR, Wang CC. Rational domain swaps reveal insights about chain length control by ketosynthase domains in fungal nonreducing polyketide synthases. Org Lett. 2014;16(6):1676–1679. | ||
Yeh HH, Chang SL, Chiang YM, et al. Engineering fungal nonreducing polyketide synthase by heterologous expression and domain swapping. Org Lett. 2013;15(4):756–759. | ||
Xu Y, Zhou T, Zhang S, Xuan LJ, Zhan J, Molnar I. Thioesterase domains of fungal nonreducing polyketide synthases act as decision gates during combinatorial biosynthesis. J Am Chem Soc. 2013;135(29):10783–10791. | ||
Chooi YH, Tang Y. Navigating the fungal polyketide chemical space: from genes to molecules. J Org Chem. 2012;77(22):9933–9953. | ||
Xu Y, Espinosa-Artiles P, Schubert V, et al. Characterization of the biosynthetic genes for 10,11-dehydrocurvularin, a heat shock response-modulating anticancer fungal polyketide from Aspergillus terreus. Appl Environ Microbiol. 2013;79(6):2038–2047. | ||
Xu Y, Zhou T, Zhang S, et al. Diversity-oriented combinatorial biosynthesis of benzenediol lactone scaffolds by subunit shuffling of fungal polyketide synthases. Proc Natl Acad Sci U S A. 2014;111(34):12354–12359. | ||
Kushnir S, Sundermann U, Yahiaoui S, Brockmeyer A, Janning P, Schulz F. Minimally invasive mutagenesis gives rise to a biosynthetic polyketide library. Angew Chem Int Ed Engl. 2012;51(42):10664–10669. | ||
Walker MC, Thuronyi BW, Charkoudian LK, Lowry B, Khosla C, Chang MC. Expanding the fluorine chemistry of living systems using engineered polyketide synthase pathways. Science. 2013;341(6150):1089–1094. | ||
Wang J, Sanchez-Rosello M, Acena JL, et al. Fluorine in pharmaceutical industry: fluorine-containing drugs introduced to the market in the last decade (2001–2011). Chem Rev. 2014;114(4):2432–2506. | ||
Shi SP, Wanibuchi K, Morita H, Endo K, Noguchi H, Abe I. Enzymatic formation of unnatural novel chalcone, stilbene, and benzophenone scaffolds by plant type III polyketide synthase. Org Lett. 2009;11(3):551–554. | ||
Abe I. Engineered biosynthesis of plant polyketides: structure-based and precursor-directed approach. Top Curr Chem. 2010;297:45–66. | ||
Morita H, Wanibuchi K, Nii H, Kato R, Sugio S, Abe I. Structural basis for the one-pot formation of the diarylheptanoid scaffold by curcuminoid synthase from Oryza sativa. Proc Natl Acad Sci U S A. 2010;107(46):19778–19783. | ||
Thirlway J, Lewis R, Nunns L, et al. Introduction of a non-natural amino acid into a nonribosomal peptide antibiotic by modification of adenylation domain specificity. Angew Chem Int Ed. 2012;51(29):7181–7184. | ||
Fischbach MA, Lai JR, Roche ED, Walsh CT, Liu DR. Directed evolution can rapidly improve the activity of chimeric assembly-line enzymes. Proc Natl Acad Sci U S A. 2007;104(29):11951–11956. | ||
Evans BS, Chen Y, Metcalf WW, Zhao H, Kelleher NL. Directed evolution of the nonribosomal peptide synthetase AdmK generates new andrimid derivatives in vivo. Chem Biol. 2011;18(5):601–607. | ||
Zhang K, Nelson KM, Bhuripanyo K, et al. Engineering the substrate specificity of the DhbE adenylation domain by yeast cell surface display. Chem Biol. 2013;20(1):92–101. | ||
Hopwood DA, Malpartida F, Kieser H, et al. Production of ‘hybrid’antibiotics by genetic engineering. Nature. 1985;314:642–644. | ||
Seki H, Sawai S, Ohyama K, et al. Combinatorial biosynthesis of legume natural and rare triterpenoids in engineered yeast. Plant Cell Physiol. 2013;54(5):740–749. | ||
Seki H, Sawai S, Ohyama K, et al. Triterpene functional genomics in licorice for identification of CYP72A154 involved in the biosynthesis of glycyrrhizin. Plant Cell. 2011;23(11):4112–4123. | ||
Moses T, Pollier J, Almagro L, et al. Combinatorial biosynthesis of sapogenins and saponins in Saccharomyces cerevisiae using a C-16α hydroxylase from Bupleurum falcatum. Proc Natl Acad Sci U S A. 2014;111(4):1634–1639. | ||
Unsin CE-M, Rajski SR, Shen B. The role of genetic engineering in natural product-based anticancer drug discovery. In: Koehn FE, editor. Natural Products and Cancer Drug Discovery. New York, NY: SpringerScience+BusinessMedia; 2013:175–191. | ||
Olano C, Méndez C, Salas JA. Harnessing sugar biosynthesis and glycosylation to redesign natural products and to increase structural diversity. In: Osbourn A Goss RJ, Carter GT, editors. Natural Products: Discourse, Diversity, and Design. Hoboken, NJ: John Wiley & Sons; 2014:317–340. | ||
Núñez LE, Nybo SE, González-Sabín J, et al. A novel mithramycin analogue with high antitumor activity and less toxicity generated by combinatorial biosynthesis. J Med Chem. 2012;55(12):5813–5825. | ||
Shepherd MD, Liu T, Méndez C, Salas JA, Rohr J. Engineered biosynthesis of gilvocarcin analogues with altered deoxyhexopyranose moieties. Appl Environ Microbiol. 2011;77(2):435–441. | ||
Han AR, Park JW, Lee MK, et al. Development of a Streptomyces venezuelae-based combinatorial biosynthetic system for the production of glycosylated derivatives of doxorubicin and its biosynthetic intermediates. Appl Environ Microbiol. 2011;77(14):4912–4923. | ||
Jung WS, Han AR, Hong JSJ, et al. Bioconversion of 12-, 14-, and 16-membered ring aglycones to glycosylated macrolides in an engineered strain of Streptomyces venezuelae. Appl Microbiol Biotechnol. 2007;76(6):1373–1381. | ||
Jung WS, Lee SK, Hong JSJ, et al. Heterologous expression of tylosin polyketide synthase and production of a hybrid bioactive macrolide in Streptomyces venezuelae. Appl Microbiol Biotechnol. 2006;72(4):763–769. | ||
Han AR, Shinde PB, Park JW, et al. Engineered biosynthesis of glycosylated derivatives of narbomycin and evaluation of their antibacterial activities. Appl Microbiol Biotechnol. 2012;93(3):1147–1156. | ||
Shinde PB, Han AR, Cho J, et al. Combinatorial biosynthesis and antibacterial evaluation of glycosylated derivatives of 12-membered macrolide antibiotic YC-17. J Biotechnol. 2013;168(2):142–148. | ||
Thayer AM. Fabulous fluorine. Chem Eng News. 2006;84(23):15–24. | ||
Eustáquio AS, O’Hagan D, Moore BS. Engineering fluorometabolite production: fluorinase expression in Salinispora tropica yields fluorosalinosporamide. J Nat Prod. 2010;73(3):378–382. | ||
Runguphan W, Qu X, O’Connor SE. Integrating carbon-halogen bond formation into medicinal plant metabolism. Nature. 2010;468(7322):461–464. | ||
Xu Y, Zhou T, Zhang S, et al. Diversity-oriented combinatorial biosynthesis of benzenediol lactone scaffolds by subunit shuffling of fungal polyketide synthases. Proc Natl Acad Sci U S A. 2014;111(34):12354–12359. | ||
Mignani S, Patek M. Chirality and combinatorial libraries for drug discovery, an overview. In: Carreira EM, Yamamoto H, editors. Comprehensive Chirality. Amsterdam, Netherlands: Elsevier; 2012:217–245. | ||
Pickens LB, Tang Y, Chooi YH. Metabolic engineering for the production of natural products. Annu Rev Chem Biomol Eng. 2011;2:211–236. | ||
Liu L, Martínez JL, Liu Z, Petranovic D, Nielsen J. Balanced globin protein expression and heme biosynthesis improve production of human hemoglobin in Saccharomyces cerevisiae. Met Eng. 2014;21:9–16. | ||
Shao Z, Luo Y, Zhao H. Rapid characterization and engineering of natural product biosynthetic pathways via DNA assembler. Mol BioSyst. 2011;7(4):1056–1059. | ||
Cobb RE, Ning JC, Zhao H. DNA assembly techniques for next-generation combinatorial biosynthesis of natural products. J Ind Microbiol Biotechnol. 2014;41(2):469–477. | ||
Chao R, Yuan Y, Zhao H. Recent advances in DNA assembly technologies. FEMS Yeast Res. 2014. DOI: 10.1111/1567–1364.12171. | ||
Bornscheuer U, Huisman G, Kazlauskas R, Lutz S, Moore J, Robins K. Engineering the third wave of biocatalysis. Nature. 2012;485(7397):185–194. | ||
Sjostrom SL, Bai Y, Huang M, et al. High-throughput screening for industrial enzyme production hosts by droplet microfluidics. Lab Chip. 2014;14(4):806–813. | ||
Damborsky J, Brezovsky J. Computational tools for designing and engineering enzymes. Curr Opin Chem Biol. 2014;19(0):8–16. | ||
Winter JM, Behnken S, Hertweck C. Genomics-inspired discovery of natural products. Curr Opin Chem Biol. 2011;15(1):22–31. | ||
Eid J, Fehr A, Gray J, et al. Real-time DNA sequencing from single polymerase molecules. Science. 2009;323(5910):133–138. | ||
Metzker ML. Sequencing technologies – the next generation. Nat Rev Genet. 2009;11(1):31–46. | ||
Liu Z, Liu L, Österlund T, et al. Improved production of a heterologous amylase in Saccharomyces cerevisiae by inverse metabolic engineering. Appl Environ Microbiol. 2014;80(17):5542–5550. | ||
Singh BK, Macdonald CA. Drug discovery from uncultivable microorganisms. Drug Discov Today. 2010;15(17–18):792–799. | ||
Olano C, Méndez C, Salas JA. Molecular insights on the biosynthesis of antitumour compounds by actinomycetes. Microb Biotechnol. 2011;4(2):144–164. | ||
Pauwels L, Inzé D, Goossens A. Jasmonate-inducible gene: what does it mean? Trends Plant Sci. 2009;14(2):87–91. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.