")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Profile of ceftolozane/tazobactam and its potential in the treatment of complicated intra-abdominal infections
Authors Skalweit MJ
Received 3 April 2015
Accepted for publication 2 May 2015
Published 4 June 2015 Volume 2015:9 Pages 2919—2925
DOI https://doi.org/10.2147/DDDT.S61436
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Shu-Feng Zhou
Marion J Skalweit1,2
1Department of Medicine, 2Department of Biochemistry, Louis Stokes Cleveland Department of Veterans Affairs and Case Western Reserve University School of Medicine, Cleveland, OH, USA
Abstract: Drug-resistant pathogens have gained a foothold especially in the most vulnerable patient populations, hospitalized and immunocompromised individuals. Furthermore, extended-spectrum β-lactamase and carbapenemase-producing organisms are finding their way even into the community, with patients presenting to the hospital with established colonization and infection with resistant Enterobacteriaceae in particular. Recently, a novel antipseudomonal cephalosporin in combination with an established Class A β-lactamase inhibitor, ceftolozane/tazobactam has been approved by the FDA for use in the treatment of complicated urinary tract infections and complicated intra-abdominal infections. Ceftolozane is a uniquely potent antipseudomonal cephalosporin because of its high affinity for the penicillin-binding proteins of Pseudomonas aeruginosa, its low affinity for the intrinsic Class C β-lactamases of P. aeruginosa, its ability to enter P. aeruginosa through the outer membrane without the utilization of OprD protein, and the fact that it is not a substrate of the often upregulated MexAB/OprM efflux system of P. aeruginosa. The biological chemistry, pharmacokinetics/pharmacodynamics, microbiologic spectrum, and clinical trials that led to the approval of ceftolozane is reviewed. A discussion regarding its potential role in the treatment of complicated intra-abdominal infections and other infectious disease syndromes associated with drug-resistant pathogens follows.
Keywords: ceftolozane/tazobactam, complicated intra-abdominal infections, Pseudomonas aeruginosa
Introduction
The ongoing emergence of resistance among gram-negative bacteria in community- and hospital-acquired infections is inspiring the development of a variety of new β-lactam/β-lactamase inhibitor combinations for the first time in decades. Among these exciting new therapeutic options is ceftolozane/tazobactam (Zerbaxa™), a combination that contains a novel and robust antipseudomonal cephalosporin with the established Class A β-lactamase inhibitor, tazobactam. Ceftolozane/tazobactam has recently been FDA approved for the treatment of complicated urinary tract infections (cUTI), including pyelonephritis caused by Escherichia coli, Klebsiella pneumoniae, Proteus mirabilis, and Pseudomonas aeruginosa. In combination with metronidazole, ceftolozane/tazobactam is also indicated in the treatment of complicated intra-abdominal infections (cIAI) caused by gram-negative bacteria (Enterobacter cloacae, E. coli, K. pneumoniae, Klebsiella oxytoca, P. mirabilis, P. aeruginosa), gram-positive bacteria (Streptococcus anginosus, Streptococcus constellatus, Streptococcus salivarius) and anaerobic bacteria (Bacteroides fragilis). The microbiology of cIAI, emerging resistance in community-acquired/hospital-acquired cIAI and in cIAI in the immunocompromised host, as well as the potential role and significance of ceftolozane/tazobactam in the clinic as a “carbapenem-sparing” regimen are reviewed.
Microbiology of cIAI: current guidelines for management
The microbiology of cIAI reflects the host’s typical enteric flora, and generally, when a patient presents from the community, current guidelines recommend minimal testing and selection of empiric antibiotics that cover the local resistance patterns eg, CTX-M producing E. coli, potentially carbapenem-resistant Enterobacteriaceae (CREs).1 In hospital-associated cIAI and in cIAI in immunocompromised hosts, additional pathogenic bacteria and yeast must be considered, including P. aeruginosa with resistance patterns that reflect local antibiograms, CREs, extended-spectrum β-lactamase (ESBL) producing gram-negative bacteria, and the so-called “derepressed” AmpC hyperproducing organisms such as E. cloacae.
Resistance mechanisms in pathogens found in cIAI
A variety of resistance determinants are emerging in bacteria causing cIAI, including in community-acquired infections. These include a variety of common gram-negative bacteria as well as gram-positives and anaerobic organisms. Resistance mechanisms are detailed for β-lactams in Table 1 and below for other agents.
 | Table 1 β-lactam resistance mechanisms in bacteria associated with cIAI |
ESBL-producing organisms in the community and hospital
CTX-M-producing E. coli ST131 are increasing in community-acquired infections including urinary tract infections and cIAI.2,3 The two main enzymes in circulation worldwide are the Type 1 CTX-M-15 β-lactamase and the Type 9 CTX-M-14 β-lactamase. Enterobacteriaceae (mainly E. coli and K. pneumoniae) contain CTX-M ESBLs on plasmids, and the CTX-M resistance genes can be found in association with many other resistance determinants4 including fluoroquinolone resistance (qnr and drug modifying genes).5 The presence of CTX-M ESBLs confers resistance to advanced generation cephalosporins, and in some cases, to β-lactam/β-lactamase inhibitor combinations such as amoxicillin/clavulanate, ampicillin/sulbactam, ticarcillin/clavulanate, and piperacillin/tazobactam in organisms that hyperproduce these enzymes, or in the setting of an abscess (“inoculum effect”), despite the fact that CTX-M β-lactamases are exquisitely sensitive to inhibition by tazobactam.6 Current treatment recommendations for infections caused by ESBLs, including cIAI, include carbapenems.1,7,8 The widespread use of carbapenems in countries where CTX-M ESBLs are especially prevalent, as well as poor sanitation and health infrastructure, and global travel are thought to have led to the spread of CTX-M producers. It is hypothesized that this has led to more carbapenem use and the selection of even more resistant organisms that produce carbapenemases (and references therein).2
Carbapenem-resistant Enterobacteriaceae
CREs are typically organisms that produce a carbapenemase such as the Class A Klebsiella pneumoniae carbapenemase (KPC) β-lactamases, Class D OXA (oxacillinase) β-lactamases especially OXA-48, Class B metallo-β-lactamases eg, NDM, VIM, and IMP types. The hyperproduction of an AmpC β-lactamase in combination with porin loss has also been described in some CREs (reviewed by Livermore and Woodford).9 These β-lactamases cannot be inhibited with traditional Class A β-lactamase inhibitors such as tazobactam. It is estimated that upward of 50% of K. pneumoniae isolates in Italy possess KPC carbapenem resistance determinants,10 and 72% of K. pneumoniae isolates were imipenem resistant in Karachi, Pakistan, where KPC, OXA-48, and NDM metallo-β-lactamases are prevalent.11 Recent CRE hospital outbreaks associated with intensive care units (ICUs) and with endoscopy equipment in the US have garnered more widespread attention12–14 to the problem of CREs and their role in infections. Carbapenemase genes can be found in conjunction with resistance determinants to fluoroquinolones, tetracyclines, sulfonamides, etc. Infections with CREs are associated with high morbidity and mortality,15 and hospital outbreaks with such organisms are growing in frequency.16–18
Nonfermenters, notably P. aeruginosa
Antibiotic resistance among P. aeruginosa strains is due to a variety of resistance mechanisms. β-lactam resistance is typically caused by the presence of upregulated efflux pumps, downregulation of OprD porin, and by the presence of specific β-lactamases (AmpC of P. aeruginosa, OXA, KPC, and metallo-β-lactamases, Class A ESBLs). In one study of 414 ICU patients, 43% had intestinal colonization with P. aeruginosa at admission.19 Resistance to three or more drugs was found in approximately 50% of P. aeruginosa isolates from the intestinal reservoir of these ICU patients who had been hospitalized 10 days or longer and was associated with disease severity and the prior use of antibiotics (fluoroquinolones, meropenem, and ertapenem).
Ceftolozane/tazobactam
Ceftolozane (Figure 1) (formerly known as CXA-101 and FR264205) is a unique parenteral 3′-aminopyrazolium cephalosporin first discovered by Fujisawa Pharmaceutical Company in 2004.20,21 It has a unique medicinal chemistry reviewed by Zhanel et al.22 Ceftolozane contains the R1 side-chain of ceftazidime, another antipseudomonal cephalosporin as well as a unique R2 side-chain that confers resistance to hydrolysis by the AmpC β-lactamase of P. aeruginosa because of the reduced affinity for these enzymes.20 The compound has a high affinity for the penicillin-binding proteins of P. aeruginosa, accounting for its excellent activity against this organism.23,24 Ceftolozane is not affected by overexpression of MexAB/OprM efflux pumps because it is not a substrate of these pumps, or deletion of OprD porins because it does not enter bacterial cells through OprD.25
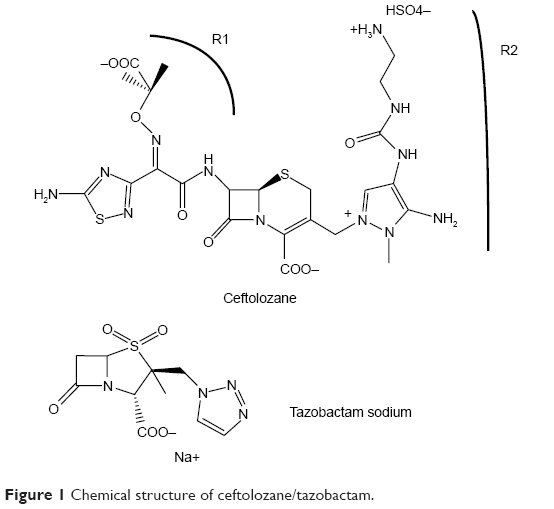 | Figure 1 Chemical structure of ceftolozane/tazobactam. |
When given alone, ceftolozane is active against many clinical strains of P. aeruginosa, as well as Enterobacteriaceae that do not express ESBLs or carbapenemases (Table 2 of typical minimum inhibitory concentrations [MICs]).6,20,25–30 In combination with the Class A β-lactamase inhibitor, tazobactam, the spectrum broadens to include ESBL-producing organisms as well as some anaerobic bacteria, notably B. fragilis.6,20,25–30 The anaerobic spectrum of ceftolozane/tazobactam is further expanded with the addition of metronidazole.29
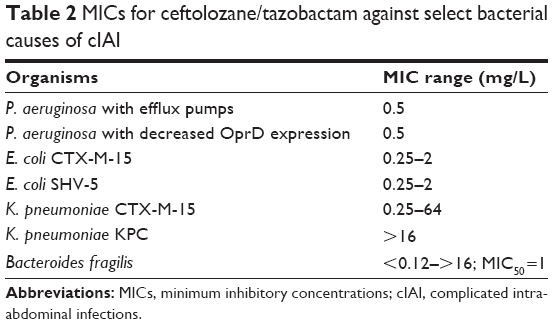 | Table 2 MICs for ceftolozane/tazobactam against select bacterial causes of cIAI |
According to the package insert, ceftolozane/tazobactam is administered parenterally 1.5 g (1 g/0.5 g) every 8 hours with metronidazole 500 mg every 8 hours for the treatment of cIAI. It must be renally adjusted (creatinine clearance [CrCl] 30–50 mL/min 750 mg IV every 8 hours; 15–29 mL/min 375 mg IV every 8 hours; end-stage renal disease or chronic kidney disease V on hemodialysis single loading dose of 750 mg followed by 150 mg IV every 8 hours, with dosing as close as possible to the end of the hemodialysis treatment session) and may have reduced efficacy in patients with CrCl ≤50 mL/min. Adverse reactions include nausea, diarrhea, headache, and fever; ceftolozane/tazobactam is contraindicated in patients with serious β-lactam hypersensitivity reactions. As with all antibiotics that have broad spectrum activity against enteric bacilli, ceftolozane/tazobactam can cause Clostridium difficile-related diarrhea.
Pharmacokinetics/pharmacodynamics (PK/PD) studies
A Phase I study was performed initially to evaluate the safety, tolerability, and PK of ceftolozane in healthy adult volunteers.31 This placebo-controlled trial established that ceftolozane was safe and well tolerated in the 64 people studied, up to 2,000 mg in a single dose, and up to 3 g in multiple doses (1,000 mg IV q8h and 1,500 mg q12h), for up to 10 days. This study demonstrated linear dose kinetics with a mean drug plasma half-life of 2.3 hours. More than 90% of the drug was renally eliminated, unchanged. This provided the initial safety and PK data for subsequent trials of efficacy. The PK and safety of the ceftolozane/tazobactam combination was studied in a Phase I trial involving six subjects receiving each component alone, and then the combination with doses as above.32 The half-life (2.48–2.64 hours), total clearance (4.35–6.01 L/h), and volume of distribution (11.0–14.1 L) of ceftolozane were consistent with different dosing schedules and doses, and with the coadministration of tazobactam. No adverse events were noted in this study either. In summary, ceftolozane/tazobactam has linear PK and there is no drug accumulation when dosed every 8 hours. Ceftolozane is renally eliminated mostly via glomerular filtration and clearance is not affected by tazobactam, which is eliminated by renal tubular secretion.
Clinical trials of ceftolozane/tazobactam plus metronidazole for cIAI
Phase II
Ceftolozane/tazobactam (1.5 g IV q8h) plus metronidazole 500 mg IV q8h was compared to meropenem (1 g IV q8h)33 in a prospective, double-blind, randomized, multicenter Phase II clinical trial involving adults with cIAI requiring surgical intervention (ClinicalTrials.gov, No NCT01147640). Patients were randomized in a 2:1 (ceftolozane/tazobactam + metronidazole, 82 patients vs meropenem, 39 patients) and ceftolozane/tazobactam + metronidazole was found to be non-inferior to meropenem with clinically evaluable cure rates of 91% (94% meropenem) and microbiologically evaluable cure rates of 89% (96% meropenem; 87% vs 95% with meropenem for cure of cIAI caused by E. coli). Adverse effect rates were similar between the two groups. This trial provided further support for the development of the drug and was conducted by Cubist Pharmaceuticals.
Phase III
The ASPECT-cIAI trials34 were two identical Phase III prospective, double-blind, randomized, multicenter clinical trials involving adults with cIAI requiring surgical intervention as well (ClinicalTrials.gov, Nos NCT01445665 and NCT01445678). These trials included a total of 993 patients primarily in Europe (76.9%). In these trials, either ceftolozane/tazobactam 1.5 g IV q8h plus metronidazole 500 mg IV q8h or meropenem 1 g IV q8h was administered for 4–14 days to hospitalized patients. Patients with staged repair with delayed fascial closure, poor source control, estimated glomerular filtration rate <30 mL/min, and antimicrobials for cIAI >24 hours prior to the first dose of study drug (unless empiric therapy had failed) were excluded from these studies. Patients were randomized in a 1:1 fashion, and received doses of study or comparator drug that were renally adjusted, using placebo saline infusions to preserve blinding of the patients and clinical staff. Intra-abdominal specimens were collected according to IDSA guidelines.1 Endpoints were prospectively defined as clinical cure rates at a test-of-cure visit taking place 24–32 days from the beginning of therapy. The results were analyzed using intention to treat (ITT), microbiological intention to treat (MITT), per protocol clinically evaluable, and per protocol microbiologically evaluable groups. Patients with missing data were considered failures in the ITT analyses, and were excluded from the per protocol analyses. The non-inferiority margin was set at −10%. Adverse effects were also evaluated between the two treatment arms. The ceftolozane/tazobactam plus metronidazole was non-inferior to meropenem in the ITT group (83% vs 87.3% meropenem group, weighted difference −4.2%, 95% CI −8.91% to 0.54%); and in the secondary outcome, in the MITT group (94.2% vs 94.7% meropenem group, difference −1%, 95% CI −4.52 to 2.59). Among the 806 patients in the MITT evaluation, 525 samples (65.1%) grew E. coli, 76 (9.4%) K. pneumoniae, and 72 (8.9%) P. aeruginosa. The majority of infections in each group were polymicrobial. Resistant bacteria were found in equal numbers in each arm including 29 ESBL + Enterobacteriaceae. Among the 52 P. aeruginosa isolates found in the whole study, only three were found to be resistant to three or more antipseudomonal antibiotics. Notably, cure rates were higher in patients with infections caused by ESBL+ Enterobacteriaceae treated with ceftolozane/tazobactam plus metronidazole (95.8% [23/24] vs 88.5% meropenem [23/26]). When CTX-M-producing organisms were considered (the major ESBL family in community acquired Enterobacteriaceae, especially E. coli), 100% (13/13) of the microbiologically evaluable patients were cured with ceftolozane/tazobactam plus metronidazole compared to 72.7% (8/11) of the patients who received meropenem. Similar to the Phase II trial, adverse events, mainly nausea and diarrhea, were similar in the two arms.
Ceftolozane/tazobactam has been studied in Phase II and III clinical trials of cUTI (NCT 01345929) and is approved for use in this infectious disease syndrome; it is currently being investigated for use in ventilated nosocomial pneumonia (ASPECT-NP NCT 02070757).
Ceftolozane/tazobactam affords clinicians with an additional agent that has good activity against P. aeruginosa and some ESBL-producing gram-negative bacteria. As empiric therapy for community-acquired cIAI, ceftolozane/tazobactam in combination with metronidazole offers a carbapenem-sparing alternative. Unfortunately, there is no evidence to show that limiting the use of carbapenems prevents the emergence of CREs and other multi-drug resistant organisms, as resistance determinants such as carbapenemases are typically found in genetic constructs that can be selected by a variety of broad spectrum antibiotics in frequent use among patients with cIAI and other serious infections.35 In the setting of hospital-associated cIAI and potentially in immunocompromised hosts (febrile neutropenia with abdominal infection), ceftolozane/tazobactam may provide additional antipseudomonal coverage over combinations such as piperacillin/tazobactam. At this time, studies demonstrating the efficacy and safety of ceftolozane/tazobactam plus metronidazole to treat cIAI in immunocompromised hosts have not been conducted. Ceftolozane/tazobactam combined with metronidazole is unlikely to replace piperacillin/tazobactam or carbapenems as first-line therapy for cIAI, but more likely will be used in settings where local resistance trends suggest an advantage.
Future concerns/future combinations: what will this β-lactam/β-lactamase inhibitor combination offer in the future compared to others waiting to enter the market?
Since ceftolozane/tazobactam was approved for clinical use in December 2014, an additional β-lactam/β-lactamase inhibitor combination has been FDA approved for similar clinical indications: ceftazidime/avibactam (AVYTAZ™; AstraZeneca). In this combination, a relatively less effective antipseudomonal cephalosporin is combined with a novel non-β-lactam/β-lactamase diazabicyclooctane (DBO) inhibitor, avibactam (formerly NXL-104; Novexel Pharma) to afford coverage against P. aeruginosa including KPC, OXA, AmpC, and ESBL-producing organisms, as well as KPC-producing CREs. DBO inhibitor combinations are not effective against organisms such as P. aeruginosa that harbor efflux and porin deletion resistance mechanisms. Additional combinations of avibactam with ceftaroline, an anti-MRSA (methicillin resistant Staphylococcus aureus) cephalosporin and with aztreonam, an older monobactam agent with antipseudomonal activity as well as relative resistance to hydrolysis by metallo beta-lactamase, will be forthcoming. Merck Pharmaceuticals, which recently acquired Cubist Pharmaceuticals, the manufacturer of ceftolozane/tazobactam, has a similar inhibitor, relebactam in combination with imipenem–cilastatin. This combination consists of an antipseudomonal carbapenem that is not subject to known efflux resistance mechanisms in P. aeruginosa, together with a DBO β-lactamase inhibitor that will be effective in the setting of KPCs, OXA carbapenemases, and ESBLs. Imipenem–cilastatin/relebactam will not be effective against metallo-β-lactamase producing or porin-deficient P. aeruginosa strains. It would seem that perhaps the best of all possible combinations is yet to come: ceftolozane/DBO inhibitor, perhaps combined with aztreonam to combat metallo-β-lactamase producing organisms and metronidazole to provide additional anaerobic coverage. Treatment of infections with resistant Acinetobacter baumannii remains an additional challenge. For some CREs, combinations of colistin and tigecycline may represent an effective treatment regimen; for extensively drug resistant P. aeruginosa, colistin alone, or in some settings, IV fosfomycin may represent the only treatment option. For extensively drug resistant A. baumannii, sulbactam containing combinations, as well as colistin, tigecycline, and fosfomycin may provide effective therapy together with source control.36 Other options may include the combination of carbapenems and colistin, tigecycline, or an aminoglycoside with appropriate dosing determined through PK37 but these approaches need to be studied to yield best practice recommendations. Because of the complex co-expression of resistance determinants, the use of “carbapenem-sparing” agents like ceftolozane/tazobactam will likely have limited impact on the spread of CREs in the hospital.
Summary
Ceftolozane/tazobactam combined with metronidazole is a safe and effective new antimicrobial combination approved for the treatment of complicated intra-abdominal and urinary tract infections. Its precise role as empiric and specific treatment for cIAI in the setting of the ever-evolving enteric microbiome remains to be defined.
Disclosure
The author is an employee of the United States Department of Veterans Affairs (USDVA). Views expressed in this review are solely those of the author, and do not reflect the opinions of the USDVA. The author has no other conflicts of interest to disclose.
References
Solomkin JS, Mazuski JE, Bradley JS, et al. Diagnosis and management of complicated intra-abdominal infection in adults and children: guidelines by the Surgical Infection Society and the Infectious Diseases Society of America. Clin Infect Dis. 2010;50(2):133–164. | ||
Hawkey PM. Multidrug-resistant Gram-negative bacteria: a product of globalization. J Hosp Infect. 2015;89(4):241–247. | ||
Petty NK, Ben Zakour NL, Stanton-Cook M, et al. Global dissemination of a multidrug resistant Escherichia coli clone. Proc Natl Acad Sci U S A. 2014;111(15):5694–5699. | ||
Li JJ, Spychala CN, Hu FP, Sheng JF, Doi Y. Complete nucleotide sequences of blaCTX-M-harboring IncF plasmids from community-associated Escherichia coli strains in the United States. Antimicrob Agents Chemother. 2015;59(6):3002–3007. | ||
Rogers BA, Sidjabat HE, Paterson DL. Escherichia coli O25b-ST131: a pandemic, multiresistant, community-associated strain. J Antimicrob Chemother. 2011;66(1):1–14. | ||
Estabrook M, Bussell B, Clugston SL, Bush K. In vitro activity of ceftolozane-tazobactam as determined by broth dilution and agar diffusion assays against recent U.S. Escherichia coli isolates from 2010 to 2011 carrying CTX-M-type extended-spectrum β-lactamases. J Clin Microbiol. 2014;52(11):4049–4052. | ||
Harris PN. Clinical management of infections caused by Enterobacteriaceae that express extended-spectrum β-lactamase and AmpC enzymes. Semin Respir Crit Care Med. 2015;36(1):56–73. | ||
Paterson DL. Recommendation for treatment of severe infections caused by Enterobacteriaceae producing extended-spectrum β-lactamases (ESBLs). Clin Microbiol Infect. 2000;6(9):460–463. | ||
Livermore DM, Woodford N. The β-lactamase threat in Enterobacteriaceae, Pseudomonas and Acinetobacter. Trends Microbiol. 2006;14(9):413–420. | ||
Munoz-Price LS, Poirel L, Bonomo RA, et al. Clinical epidemiology of the global expansion of Klebsiella pneumoniae carbapenemases. Lancet Infect Dis. 2013;13(9):785–796. | ||
Saleem AF, Qamar FN, Shahzad H, Qadir M, Zaidi AK. Trends in antibiotic susceptibility and incidence of late-onset Klebsiella pneumoniae neonatal sepsis over a six-year period in a neonatal intensive care unit in Karachi, Pakistan. Int J Infect Dis. 2013;17(11): e961–e965. | ||
Mathers AJ, Cox HL, Kitchel B, et al. Molecular dissection of an outbreak of carbapenem-resistant Enterobacteriaceae reveals intergenus KPC carbapenemase transmission through a promiscuous plasmid. MBio. 2011;2(6):e204–e211. | ||
Mathers AJ, Stoesser N, Sheppard AE, et al. Klebsiella pneumoniae carbapenemase (KPC)-producing K. pneumoniae at a single institution: insights into endemicity from whole-genome sequencing. Antimicrob Agents Chemother. 2015;59(3):1656–1663. | ||
Muscarella LF. Risk of transmission of carbapenem-resistant Enterobacteriaceae and related “superbugs” during gastrointestinal endoscopy. World J Gastrointest Endosc. 2014;6(10):457–474. | ||
Bilavsky E, Schwaber MJ, Carmeli Y. How to stem the tide of carbapenemase-producing Enterobacteriaceae? Proactive versus reactive strategies. Curr Opin Infect Dis. 2010;23(4):327–331. | ||
Gupta N, Limbago BM, Patel JB, Kallen AJ. Carbapenem-resistant Enterobacteriaceae: epidemiology and prevention. Clin Infect Dis. 2011;53(1):60–67. | ||
Logan LK. Carbapenem-resistant Enterobacteriaceae: an emerging problem in children. Clin Infect Dis. 2012;55(6):852–859. | ||
Temkin E, Adler A, Lerner A, Carmeli Y. Carbapenem-resistant Enterobacteriaceae: biology, epidemiology, and management. Ann N Y Acad Sci. 2014;1323:22–42. | ||
Gómez-Zorrilla S, Camoez M, Tubau F, et al. Antibiotic pressure is a major risk factor for rectal colonization by multidrug-resistant Pseudomonas aeruginosa in critically ill patients. Antimicrob Agents Chemother. 2014;58(10):5863–5870. | ||
Takeda S, Nakai T, Wakai Y, Ikeda F, Hatano K. In vitro and in vivo activities of a new cephalosporin, FR264205, against Pseudomonas aeruginosa. Antimicrob Agents Chemother. 2007;51(3):826–830. | ||
Toda A, Ohki H, Yamanaka T, et al. Synthesis and SAR of novel parenteral anti-pseudomonal cephalosporins: discovery of FR264205. Bioorg Med Chem Lett. 2008;18(17):4849–4852. | ||
Zhanel GG, Chung P, Adam H, et al. Ceftolozane/tazobactam: a novel cephalosporin/β-lactamase inhibitor combination with activity against multidrug-resistant Gram-negative bacilli. Drugs. 2014;74(1):31–51. | ||
Moyá B, Beceiro A, Cabot G, et al. Pan-β-lactam resistance development in Pseudomonas aeruginosa clinical strains: molecular mechanisms, penicillin-binding protein profiles, and binding affinities. Antimicrob Agents Chemother. 2012;56(9):4771–4778. | ||
Moya B, Zamorano L, Juan C, Ge Y, Oliver A. Affinity of the new cephalosporin CXA-101 to penicillin-binding proteins of Pseudomonas aeruginosa. Antimicrob Agents Chemother. 2010;54(9):3933–3937. | ||
Castanheira M, Mills JC, Farrell DJ, Jones RN. Mutation-driven β-lactam resistance mechanisms among contemporary ceftazidime-nonsusceptible Pseudomonas aeruginosa isolates from U.S. hospitals. Antimicrob Agents Chemother. 2014;58(11):6844–6850. | ||
Takeda S, Ishii Y, Hatano K, Tateda K, Yamaguchi K. Stability of FR264205 against AmpC β-lactamase of Pseudomonas aeruginosa. Int J Antimicrob Agents. 2007;30(5):443–445. | ||
Sader HS, Farrell DJ, Flamm RK, Jones RN. Ceftolozane/tazobactam activity tested against aerobic Gram-negative organisms isolated from intra-abdominal and urinary tract infections in European and United States hospitals (2012). J Infect. 2014;69(3):266–277. | ||
Sader HS, Rhomberg PR, Farrell DJ, Jones RN. Antimicrobial activity of CXA-101, a novel cephalosporin tested in combination with tazobactam against Enterobacteriaceae, Pseudomonas aeruginosa, and Bacteroides fragilis strains having various resistance phenotypes. Antimicrob Agents Chemother. 2011;55(5):2390–2394. | ||
Snydman DR, McDermott LA, Jacobus NV. Activity of ceftolozane-tazobactam against a broad spectrum of recent clinical anaerobic isolates. Antimicrob Agents Chemother. 2014;58(2):1218–1223. | ||
Titelman E, Karlsson IM, Ge Y, Giske CG. In vitro activity of CXA-101 plus tazobactam (CXA-201) against CTX-M-14- and CTX-M-15-producing Escherichia coli and Klebsiella pneumoniae. Diagn Microbiol Infect Dis. 2011;70(1):137–141. | ||
Ge Y, Whitehouse MJ, Friedland I, Talbot GH. Pharmacokinetics and safety of CXA-101, a new anti-pseudomonal cephalosporin, in healthy adult male and female subjects receiving single- and multiple-dose intravenous infusions. Antimicrob Agents Chemother. 2010;54(8):3427–3431. | ||
Miller B, Hershberger E, Benziger D, Trinh M, Friedland I. Pharmacokinetics and safety of intravenous ceftolozane-tazobactam in healthy adult subjects following single and multiple ascending doses. Antimicrob Agents Chemother. 2012;56(6):3086–3091. | ||
Lucasti C, Hershberger E, Miller B, et al. Multicenter, double-blind, randomized, phase II trial to assess the safety and efficacy of ceftolozane-tazobactam plus metronidazole compared with meropenem in adult patients with complicated intra-abdominal infections. Antimicrob Agents Chemother. 2014;58(9):5350–5357. | ||
Solomkin J, Hershberger E, Miller B, et al. Ceftolozane/tazobactam plus metronidazole for complicated intra-abdominal infections in an era of multidrug resistance: results from a randomized, double-blind, phase 3 trial (ASPECT-cIAI). Clin Infect Dis. 2015;60(10):1462–1471. | ||
Amit S, Mishali H, Kotlovsky T, Schwaber MJ, Carmeli Y. Bloodstream infections among carriers of carbapenem-resistant Klebsiella pneumoniae: etiology, incidence and predictors. Clin Microbiol Infect. 2015;21(1):30–34. | ||
Karaiskos I, Giamarellou H. Multidrug-resistant and extensively drug-resistant Gram-negative pathogens: current and emerging therapeutic approaches. Expert Opin Pharmacother. 2014;15(10):1351–1370. | ||
Doi Y, Paterson DL. Carbapenemase-producing Enterobacteriaceae. Semin Respir Crit Care Med. 2015;36(1):74–84. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.