")
Back to Journals » International Journal of General Medicine » Volume 17
Prevalence and Patterns of Congenital Heart Defects and Other Major Non-Syndromic Congenital Anomalies Among Down Syndrome Patients: A Retrospective Study
Received 19 January 2024
Accepted for publication 24 March 2024
Published 4 April 2024 Volume 2024:17 Pages 1337—1347
DOI https://doi.org/10.2147/IJGM.S453181
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Woon-Man Kung
Biniam Endale Geleta, Girma Seyoum
Department of Anatomy, Addis Ababa University, Addis Ababa, Ethiopia
Correspondence: Biniam Endale Geleta, Email [email protected]
Background: Children with DS are at higher risk of developing congenital anomalies, particularly cardiac anomalies.
Methods: Medical records of 502 DS patients were reviewed. The logistic regression analyses were performed to determine independent predictors.
Results: Of the total 502 study subjects, 53.4% were males. Only 1.4% of the DS case diagnosis were confirmed by karyotyping. All cases were diagnosed postnatally. The median age at DS diagnosis was 5 months. About 13% were born preterm; 50.2% of the subjects maternal age at conception were thirty-five years and above. Over three-quarters (75.1%) had at least one structural congenital anomaly. Multiple anomalies were diagnosed in 12.8% of the subjects. At least one cardiac congenital anomaly was diagnosed in 67.3% of the study subjects, and 32.8% of them were diagnosed with multiple cardiac anomalies. Patent ductus arteriosus (28.5%), Ventricular septal defect (23.2%), and AVSD (21.9%) were the three common lesions. At least one genitourinary system anomaly was identified in 32 (6.4%) of them. Roughly, 8% of study participants exhibited congenital anomaly of the head, eye, nose, and throat. Anorectal malformation was found as the most common gastrointestinal anomaly. Maternal age at conception was found as independent predictor for presence of structural congenital anomaly (AOR 2.59; 95% CI 1.58– 4.23, p-value < 0.01). Advanced maternal age is also found increasing the risk of developing congenital heart defect (AOR 2.37; 95% CI 1.52– 3.7, p-value < 0.01).
Conclusion: High prevalence of congenital anomalies has been noted in the current study compared to previous studies. Predictive factors increasing risk of congenital anomalies in DS patients have been identified. The current findings may help in developing strategies and more targeted preventive and therapeutic interventions.
Keywords: Down syndrome, congenital anomaly, cardiac anomaly, maternal age, chromosome 21
Introduction
Down syndrome (DS) is a chromosomal abnormality characterized by the presence of an extra copy of human chromosome 21.1 It is the most prevalent chromosomal abnormality that allows for survival beyond the embryonic stage; affecting 1 per 700 to 1000 live births globally.2–4
The incidence of Down syndrome (DS) is decreasing in developed countries due to widespread prenatal testing and elective termination of affected pregnancies.5 In contrast, limited access to antenatal care in Africa and other developing countries poses a significant challenge for prenatal DS diagnosis, leading to late diagnoses after birth.1,6
While karyotyping remains the gold standard for DS diagnosis, its accessibility and affordability are significant barriers in developing countries like Ethiopia. Therefore, clinical diagnosis based on physical features and behavior is commonly employed in these settings, including the one in which this study was conducted.6–8 However, these clinical diagnostic standards were primarily developed in high-income countries and may not be as accurate in LMICs due to potential phenotypic variations across regions and ethnicities.1,8,9 Additionally, studies have shown that the accuracy of clinical diagnosis varies significantly between DS subtypes. One study reported overdiagnosis in up to one-third of cases, while another found accuracy rates of 90% for free trisomy, 100% for translocation, and only 37.5% for mosaicism.9,10
Congenital anomalies are more common among DS patients than the general population. This is attributed to a combination of gene dosage imbalance and epigenetic factors. However, the pattern and prevalence of these anomalies vary from one region to the other.11 Stoll et al reported 64% prevalence of congenital anomalies in children with DS. While the Egyptian study reported 59.9% prevalence among infants.12 Chance of development of these anomalies was found increased by factors like advanced maternal age, paternal age, gestational age, and birth weight.13
Cardiac lesions are the most common congenital anomalies among children with DS. About half of patients with DS have at least one form of cardiac anomaly.12,14 Variations are noted among regions. Accordingly, a study of twenty European countries reported prevalence of 43.6%. Other studies from Urban and rural areas of France and Guatemala reported 44% and 54%, respectively.14,15 While the study from Sub-Saharan Sudan found 91% prevalence among clinically diagnosed children undergone echocardiographic examination.16 Gender variation was noted, with dominating female numbers.16,17
The pattern and clinical phenotypes of cardiac anomalies also vary across geographic regions. Different studies from Asian countries, including China, reported Ventricular septal defect (VSD) (43.6% to 52.9%) and Atrioventricular septal defect (AVSD) (11.8% to 22.0%) as the two most common cardiac anomalies among patients with DS.18,19 While the Korean study indicated Atrial septal defect (ASD) (30.5%), VSD (19.3%), and Patent ductus arteriosus (PDA) (17.5%).13 A study from Saudi Arabia found VSD (33.3%), followed by AVSD (22.8%) and ASD (21.1%). The perimembranous type VSD accounted for 53% of cases, and about 42% were diagnosed as large-sized.20 The Central American Guatemala study identified: PDA (28.6%), VSD (27.5%) and ASD (12.7%) as the three common lesions among children with DS. The diagnosis of perimembranous type dominates with 71% of all VSD cases.14 A literature review by O’Brien & Wong reported about 10% diagnosis of large sized and 30% moderate sized PDA among DS patients.
The clinical phenotype and patterns of these anomalies vary among different African regions. In Ethiopia, PDA emerged as the most common anomaly (36.5%), followed by VSD (19.9%), ASD (19%), and AVSD (18.6%). Multiple cardiac anomalies were presented in 33.6% of the subjects.7 A study from Egypt identified AVSD (17.7%) and ASD (10.9%).12 However, a different Egyptian study at the Mansoura tertiary hospital reported VSD (39.8%), AVSD (16.4%), and ASD (12.6%). Notably, VSD and PDA were frequently observed together.21 Among Libyan infants with DS, ASD (23%), AVSD (19%), and VSD (14%) were the most prevalent cardiac defects. Multiple lesions were detected in 35% of the infants, with ASD and VSD frequently co-occurring.3 Sudan study reported AVSD (48%), VSD (23%), and PDA (7%).16 While an Algerian study found AVSD (33%), VSD (19%), and ASD (8%). Multiple cardiac lesions were diagnosed in 38% of the subjects, with AVSD and PDA frequently co-occurring lesions.22
Egyptian study found that maternal passive smoking and parental consanguinity were significantly associated with an increased risk of cardiac congenital anomalies.21 Mokhtar and Abdel-Fattah and El-Gilany et al failed to demonstrate a significant association between maternal age, sex, birth order and development of congenital anomaly. Whereas other studies demonstrated a higher chance of developing cardiac congenital anomalies in DS patients with advanced maternal age.13,22,23
Congenital malformations of Gastrointestinal (GI) tract are the second most common birth defects associated with DS. Studies conducted across various continents reported 3 to 7% prevalence. The clinical phenotype of these anomalies also varies across studies, and majority of them exhibited gender disparity with dominating male numbers.12,15,17,24 Duodenal atresia emerged as the most prevalent among French infants with DS, accounting for nearly two-thirds of cases, followed by Hirschsprung disease (HSD) and tracheoesophageal atresia.15 Other studies from Saudi Arabia and Egypt identified duodenal atresia alongside imperforate anus.12,20 Similarly, a fifteen-year population-based study in the United States found duodenal atresia, anal stenosis, and HSD.24 In contrast, a report from the National Institutes of Health indicated anorectal malformation as becoming one of the most prevalent GI anomaly among DS patients.25
Anomalies of the respiratory, urinary, musculoskeletal, and nervous systems are also frequently diagnosed in individuals with DS. The prevalence of these anomalies also varies across different studies. The prevalence of genital anomalies in DS patients was estimated between 0.5% and 1% in previous studies; with hypospadias and micropenis the most prevalent male genital anomalies diagnosed in infants with DS.15,17,20 Regarding congenital anomalies of kidney and urinary tract (CAKUT), Morris et al found 1.9% prevalence among DS infants. Obstructive CAKUT phenotypes accounted for more than half of all CAKUT cases.15
Previous studies reported a 0.3% to 7% prevalence of musculoskeletal anomalies among DS patients.15,23 Other less common congenital anomalies observed in DS patients include eye anomalies (0.1 to 1%), with congenital cataract being the most prevalent;15,17 cleft lip and palate (0.2 to 1.5%), with isolated cleft palate being more common;17,26 central nervous system anomalies (less than 0.5%);26 and respiratory system anomalies (0.3 to 3%), with congenital laryngomalacia constituting the majority.15,25,26
In this study, we investigate the prevalence, patterns, and potential risk factors associated with congenital heart defects and other major non-syndromic congenital anomalies in a population of DS patients in Ethiopia. Previous study from Ethiopia, only focused on congenital cardiovascular anomalies, has offered valuable insights.7 However, it involved patients with established diagnosis and primarily focused on identifying patterns of congenital heart defects and patient survival rather than exploring the overall prevalence and risk factors for this condition.
Our study expands upon this knowledge base by examining a broader DS population in Ethiopia. We focus on determining the overall prevalence and patterns of congenital heart defects and other major non-syndromic congenital anomalies in DS patients. Additionally, we provide a more detailed analysis of the most prevalent congenital heart defect phenotypes, including information on type and size. We also explore potential risk factors that might contribute to the development of overall congenital anomalies, and particularly congenital heart defects in this group. This comprehensive approach offers a more nuanced understanding of these congenital anomalies landscape within the Ethiopian DS population, which can be crucial for clinicians in risk assessment, early detection, and potentially guiding treatment strategies.
Materials and Methods
Study Design and Setting
This is a retrospective cross-sectional study of patients who attended follow-up at different pediatric units of Tikur Anbessa Specialized Hospital (TASH), between August 2022 and October 2023. All patients diagnosed with DS were included in the study.
Study Subjects and Data Collection
Pediatric DS patients attending their follow-up at pediatric Cardiac, developmental, Neurology, renal, endocrine, orthopedics, high risk, and Psychiatry clinics were included. A list of DS patients who attended follow-up visits during the study period was obtained from administrators of I-CARE, the hospital’s patient database. Then, I-CARE administrators granted the researchers temporary access to review patients’ medical records via a temporary account.
Patient medical records were reviewed by inputting their medical registration number, and relevant data was transferred to an Excel spreadsheet. Data collected included address, telephone number, sex, age at DS diagnosis, time of diagnosis, method of diagnosis, maturity status at birth, plurality status, history of retro viral infection (RVI) exposure in the womb, birth order, maternal age at conception, and the presence, type, and clinical detail of major congenital anomalies. Identified congenital anomalies were categorized into anomalies of the Heart, GI, Genitourinary system (GUS), Head, eye, nose, and throat (HEENT) and Limb. Potential factors contributing to the development of these anomalies, as documented in the medical records, were also collected.
Diagnosis of DS cases in this study was primarily relied on clinical method as documented in patients’ medical records. Karyotyping is prohibitively expensive and geographically limited in Ethiopia, available only in a few private laboratories within the capital city. Consequently, due to accessibility and affordability constraints, karyotyping confirmation was only possible for 1.4% of them in this study. This reliance on clinical diagnosis is an important consideration when interpreting the findings, as it may introduce limitations in diagnostic accuracy.
In cases where medical records lacked complete data, families or childcare providers were contacted via telephone. For major congenital heart anomalies with incomplete size or type information, the diagnosis is classified as “unspecified” type or size.
Statistical Analysis
The data from the Excel spreadsheet was organized, checked for completeness, and then transferred to IBM SPSS software version 24 for statistical analysis. Descriptive statistics were generated using SPSS software. The Shapiro–Wilk test was employed to determine the normality of age at diagnosis of DS. Since the data was found not to be normally distributed, the median was calculated as the measure of central tendency.
Logistic regression analysis was utilized to investigate the potential factors associated with the development of congenital anomalies. Univariate analysis was initially performed to examine the potential factors, and candidate variables with a p-value ≤0.2 were subsequently included in multivariate analysis to identify independent predictors. The Hosmer–Lemeshow test was used to assess the goodness-of-fit of the model. A p-value ≤0.05 was considered statistically significant.
Results
A total of 502 medical records of DS patients attending follow-up care at various pediatric units were reviewed. More than half (53.4%) of the study subjects were males, and nearly a third (29.2%) were the second child in the family. About 13% of them were born preterm, and almost all (99.6%) were born singleton. Only 1.8% of them had RVI exposure in the womb. More than half (50.2%) of the DS patients’ maternal age at conception were thirty-five years and above (Figure 1).
 |
Figure 1 Maternal age at conception of DS patients on follow up at TASH. Explanations: Color Labels: to show distribution of maternal age at conception. |
All DS cases were diagnosed postnatally. The median age at DS diagnosis was calculated as 5 months (birth to 78 months). Clinical diagnosis accounted for nearly all (98.6%) cases. Only seven (1.4%) cases received karyotype confirmation. This low rate of karyotyping confirmation might be attributed to limitations in access to the service and associated cost in the study region, Ethiopia.
The prevalent dysmorphic features considered for clinical diagnosis include up-slanted palpebral fissures, epicanthal folds, low-set ears, hypertelorism, micrognathia, protruding tongue, hypotonia, and a single palmar crease. Various imaging modalities, such as echocardiography, Computed tomography scan and magnetic resonance imaging, confirmed the diagnosis of additional congenital malformations.
Over three-quarters (75.1%) of the study subjects had at least one structural congenital anomaly. About 11.6% of the subjects had multiple anomalies. Of the total subjects, 67.3% were diagnosed with at least one form of cardiac congenital anomaly (Figure 2). About 28.1% had two different cardiac anomalies, and 4.7% were diagnosed with three or more. Patent ductus arteriosus (28.5%) was the most often diagnosed phenotype of all cardiac anomalies, followed by VSD (23.2%) (Table 1). The two commonly co-occurring congenital cardiac anomalies were PDA and AVSD.
 |
Table 1 Congenital Cardiac Anomaly Phenotypes Among DS Patients on Follow-Up at TASH |
 |
Figure 2 Distribution of congenital anomalies among DS patients on follow-up at TASH. Explanations: X-axis shows types of congenital anomalies diagnosed among DS patients. Y-axis shows percentage of the congenital anomalies diagnosed. Types of congenital anomalies. Cardiac Anomalies (67.3%). HEENT (Head, Eye, Ear, Nose, and throat) Anomalies (8%). GUS (Genito-urinary System) Anomalies (6.4%). GI (Gastro-intestinal) Anomalies (5.2%). RS (Respiratory System) Anomalies (1%). Limb Anomalies (1%). |
Sizes of PDA cases diagnosed include small in 45.1%; medium in 33.1%; and large in 14.3%; while size was not specified in 7.5% of the cases. In the case of VSD, the perimembranous type was the most common, constituting more than half of all VSD cases (56.5%) (Figure 3). Size of VSD lesions diagnosed includes large in 45.4%; moderate in 12.9%; small in 31.5%; and size not specified in 10.2% of the cases.
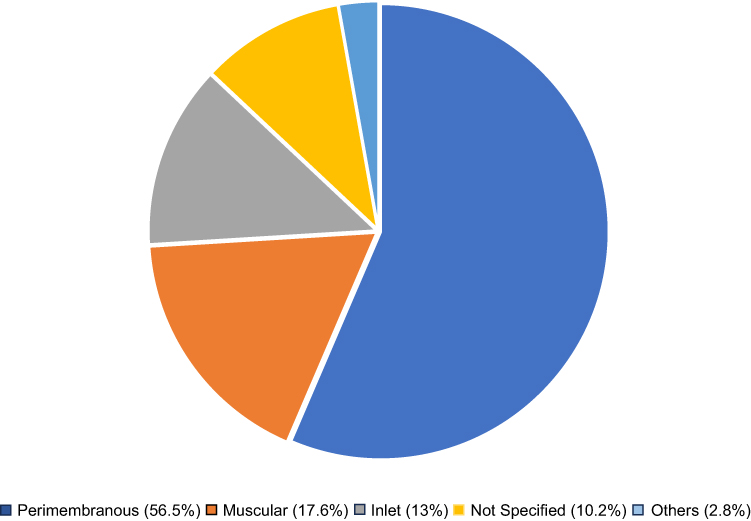 |
Figure 3 Types of VSD cases diagnosed among DS patients on follow-up at TASH. Note: Others include mixed muscular with perimembranous and Swiss cheese type VSD. Explanations: Color Labels: to show different types of ventricular septal defect cases. |
The majority of AVSD cases (79.4%) were complete type; while 8.8% intermediate type, 4.9% incomplete type and type not specified in 6.9%. Of the total ASD cases, about 84% were secundum type ASD, 3.2% premium type, while type not specified in 12.8% of the cases. In terms of their size, 47.9% of ASD cases were small; 18.8% moderate; 21.9% large; and size not specified in 11.5%.
At least one GUS anomaly was identified in 32 (6.4%) of the study subjects, with cryptorchidism accounting for over half of these anomalies (Table 2).
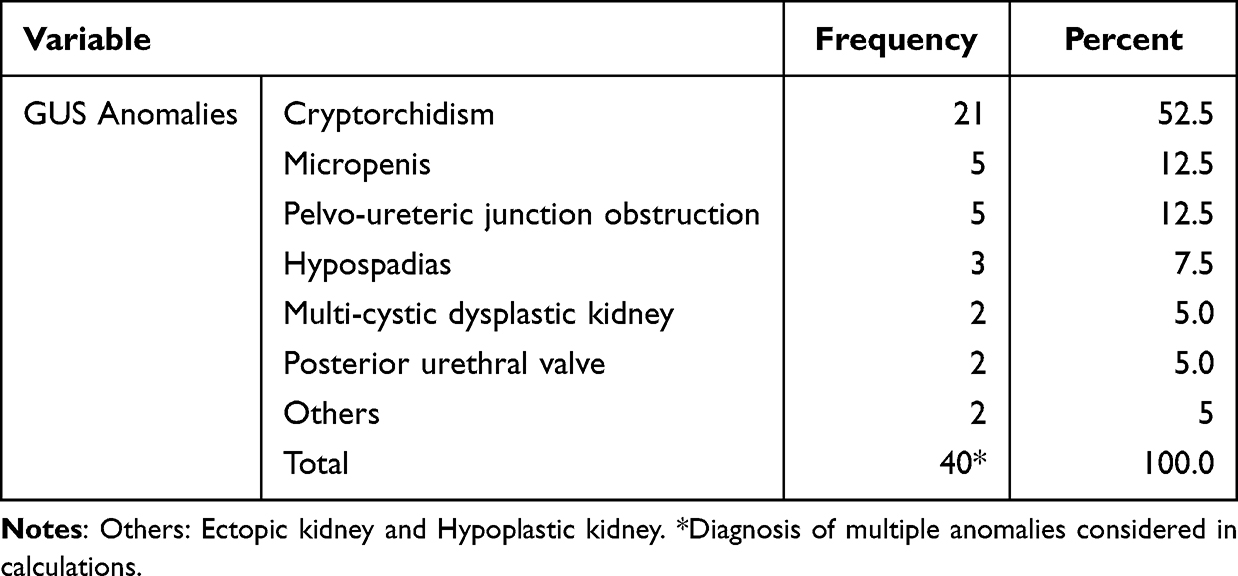 |
Table 2 Congenital GUS Anomalies Among DS Patients on Follow-Up at TASH |
Roughly, 8% of the study participants exhibited at least one congenital anomaly of the HEENT. Among these, congenital eye malformations accounted for the highest proportion at 46.2%, followed by microcephaly (38.5%), cleft palate (5.1%), and craniosynostosis (5.1%).
The three limb anomalies diagnosed in the study subjects were talipes calcaneovalgus (33.3%), talipes equinovarus (33.3%), and radial agenesis (33.3%). From respiratory-related anomalies, only congenital laryngomalacia was diagnosed in five (1%) of the study subjects.
Twenty-six (5.2%) of the study subjects had at least one GI-related congenital anomaly, and 39.3% of these patients were diagnosed with ARM without fistula (Table 3).
 |
Table 3 Congenital GI Anomalies Among DS Patients on Follow-Up at TASH |
Multivariate logistic regression analyses revealed statistically significant association between maternal age at conception and both the presence of at least one structural congenital anomaly (AOR 2.59; 95% CI 1.58–4.23, p-value <0.01) and multiple congenital anomalies (AOR 2.06; 95% CI 1.19–3.58, p-value = 0.01) among DS patients. The odds of developing at least one congenital anomaly among patients with maternal age at conception thirty-five years and older was calculated 2.6.
Statistically significant association was observed between advanced maternal age and development of cardiac congenital anomaly. Gender disparity was also noted among patients with multiple cardiac lesions (Table 4).
 |
Table 4 Logistic Regression Analysis of Predictive Factors for Development of Cardiac Congenital Anomalies Among DS Patients on Follow-Up at TASH |
Discussion
Down syndrome is the most prevalent chromosomal disorder, renowned for its far-reaching socioeconomic implications. Patients with DS exhibit a higher frequency of various congenital anomalies compared to the general population. Notably, the clinical phenotype and patterns of these anomalies vary across geographical regions. Factors like genetics, environmental and difference in healthcare practice are considered responsible for this variation.11,27
Of the total 502 subjects included in this study, more than half (53.4%) were males, which is the global trend of DS.11 No DS case was diagnosed prenatally, as all the diagnoses were made after birth. The median age at diagnosis was 5 months. Controverting to this finding, developed countries have been implementing prenatal testing services particularly for all mothers with advanced age at conception, and prenatal diagnosis account for 54% to 66% of DS cases.11,17 In the case of developing countries, only 6 to 32% of sub-Saharan Africa pregnant mothers attend one-time antenatal care service, and only 10% of them get ultrasonographic examination, and none of them get advanced biochemical and invasive prenatal tests. This inadequate healthcare services in Africa and other developing nations, stemming from the socioeconomic status of these countries, hinder the prenatal and early postnatal diagnosis of DS.6
Clinical diagnosis of DS is known for its less accuracy.5 A previous study by Hindley & Medakkar reported an overdiagnosis rate of about one-third of clinically diagnosed DS patients. In contrast, 98.6% of the DS cases in the present study were diagnosed clinically based on specific dysmorphic features, and karyotyping was only performed in 1.4% of them. Therefore, the high reliance on clinical diagnosis in the current study raises concerns about the potential for overdiagnosis.
Notably, over half (50.2%) of the study subjects in the present study had a maternal age of thirty-five years and older at conception. This reinforces the established link between advanced maternal age and increased risk of DS.2,28
In the current study, over three-quarters (75.1%) of the study subjects had at least one structural congenital anomaly, with 12.8% of the subjects diagnosed with multiple anomalies. This finding is higher compared to other studies from France (64%) and Egypt (59.9%).12,15 These discrepancies could potentially be attributed to a higher likelihood of overdiagnosis in the present study since it relied on clinically diagnosed DS cases. Additionally, the focus on major visceral organ congenital anomalies in the Egyptian study might account for its lower reported prevalence.
Stoll et al reported about 44% prevalence of cardiac anomalies among DS patients from both rural and urban areas of France. The present study found congenital heart defect in 67.3% of DS patients. A study involved twenty European countries reported a diagnosis rate of 43.6%. Another study conducted in Sudan, a sub-Saharan country, found a 91% diagnosis rate among clinically diagnosed DS patients who were examined by echocardiography.16 The central America Guatemala study reported 54.1% cardiac lesion.14 The observed discrepancies may stem from inherent geographic variations and the likelihood of overdiagnosis in both the Sudanese and present study, both of which relied solely on clinically diagnosed DS cases.
The present study found that PDA, VSD, and AVSD as the three most common cardiac anomalies, with prevalences of 28.5%, 23.2%, and 21.9%, respectively. These findings align with previous research in Ethiopia and Guatemala, which also identified PDA and VSD as the two most prevalent anomalies but followed by ASD.7,14 Previous studies from Algeria,22 Libya,3 and Ethiopia7 reported multiple cardiac anomalies in 38%, 35%, and 33.6% of the study subjects with DS, respectively. Consistent with these findings from Africa, the present study identified multiple congenital cardiac anomalies among 32.8% of DS patients. Furthermore, similar to Abbag and Vida et al, the majority of VSD cases were perimembranous type (56.5%) and large-sized (45.4%). Additionally, consistent with Boussouf et al, we found PDA and AVSD to be commonly co-occurring lesions.
Intriguingly, despite a ten-year gap, our study, conducted at a similar institution as a prior Ethiopian study,7 yielded consistent results regarding the prevalence of multiple cardiac anomalies, and the two most prevalently reported ones, PDA and VSD. This suggests a potential regional consistency in the distribution of these specific cardiac anomalies among DS patients in this area.
The prevalence of GI congenital anomalies also varies across different countries, ranging from 3.0% to 6.9%.12,15,17,24 In this study, 5.2% of patients with DS had GI congenital anomalies; with ARM (39.3%) and HSD (21.4%) being the two most prevalent anomalies. Most studies have reported duodenal atresia as the most frequently diagnosed GI anomaly, followed by HSD.15,17,20 However, according to the National Institutes of Health, ARM is becoming one of the most common GI anomalies among DS patients.25 Consistent with previous studies of Freeman et al and Morris et al, a gender disparity was noted in this study, with males exhibiting a higher prevalence. However, this difference was not found statistically significant.
The prevalence of genital anomalies in DS patients was estimated between 0.5% and 1%.17 While the current study found a higher prevalence of 4.4%. Majority of these anomalies are undescended testes (71.4%), micropenis (17.9%) and hypospadias (10.7%). Regarding CAKUT, like the finding of Morris et al, 1.9% of study participants in the present study had at least one form of CAKUT. The Pelviureteric junction obstruction (PUJO) phenotype (41.7%) was the most common, followed by posterior urethral valve (PUV) and Multi-cystic dysplastic kidney (MCDK), both at 16.7%. Consistent with Stoll et al, obstructive CAKUT phenotypes account for more than half of all CAKUT cases in the current study.
Previous studies reported a 0.3% to 7% prevalence of musculoskeletal anomalies among DS patients.15,26 Consistent with these findings, the present study report shows 0.6% prevalence. Other less common congenital anomalies observed in DS patients include eye anomalies (0.1 to 1%), with congenital cataract being the most prevalent.15,17 Cleft lip and palate (0.2 to 1.5%), with isolated cleft palate being more common.17,26 Central nervous system anomalies (less than 0.5%);26 and respiratory system anomalies (0.3 to 3%), with congenital laryngomalacia constituting the majority.15,26,29 Similarly, the current study identified congenital cataract as the most prevalent eye anomaly; 0.4% prevalence of facial cleft with dominating isolated cleft palate; and 1% prevalence of respiratory anomaly.
In line with a previous study by Kim et al, the present study demonstrated a significant association between maternal age and the development of congenital anomalies among DS patients. This association is clearly demonstrated in this study, where maternal age at conception emerged as an independent predictor for both the presence of at least one congenital anomaly (OR 2.59; 95% CI 1.58–4.23, p-value < 0.01), and multiple anomalies (OR 2.06; 95% CI 1.19–3.58, p-value = 0.01). Moreover, the odd of developing a congenital anomaly is 2.6 times higher in DS patients with maternal age at conception greater than or equal to thirty-five compared to younger mothers, further solidifying the link between advanced maternal age and an increased risk of congenital anomalies in DS patients. These findings emphasize the critical role of maternal age in assessing the risk of congenital anomalies in DS patients.
Studies from Korea, Algeria, and Lebanon had consistently demonstrated an increased risk of cardiac congenital anomalies in DS patients with advanced maternal age.13,23,29 Aligned with these findings, this study revealed a statistically significant association between advanced maternal age at conception and development of cardiac congenital anomalies among DS patients. The odd of developing cardiac anomaly is 2.4 times higher in DS patients with maternal age thirty-five and older compared to younger mothers (95% CI 1.52–3.7, p-value < 0.01). In accordance with Morris et al and Ali, risk of developing congenital heart defects in the current study is higher in females compared to males. However, this association was not found statistically significant, like prior observation of Ali.
Additionally, a significant association is observed between maternal age (OR 1.73; 95% CI 1.12–2.66, p-value = 0.01), sex (OR 1.72; 95% CI 1.12–2.64, p-value = 0.01), and development of multiple cardiac anomalies among DS patients. Notably, more females were found to develop multiple cardiac defects with an adjusted odds ratio of 1.7.
Conclusion
The present study has successfully identified a range of congenital anomalies associated with DS and explored the potential risk factors contributing to their development. Notably, it has found a high prevalence of congenital anomalies including cardiac lesions compared to similar studies. The study also clearly elucidates the pattern and phenotypes of cardiac anomalies diagnosed. Furthermore, a significant association between advanced maternal age and the occurrence of congenital anomalies in DS patients is also demonstrated.
The high prevalence of these congenital anomalies compared to previous studies necessitates the implementation of effective screening and diagnostic strategies, particularly in developing regions with limited healthcare resources like Ethiopia.
Abbreviations
ASD, Atrial Septal Defect; AVSD, Atrioventricular Septal Defect; CAKUT, Congenital Anomalies of the Kidney and Urinary tract; GI, Gastrointestinal; GUS, Genitourinary system; HSD, Hirschsprung Disease; PDA, Patent Ductus Arteriosus; PUJO, Pelvo-Ureteric Junction Obstruction; PUV, Posterior Urethral Valve; TASH, Tikur Anbessa Specialized Hospital; VSD, Ventricular Septal Defect.
Data Sharing Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Ethics Approval and Consent to Participate
The study was conducted according to the 1964 Helsinki declaration and ethical standards of the College of Health Science of Addis Ababa University. The study protocol was approved by Addis Ababa University College of Health science institutional review board. All important steps were followed including applying to the clinical service directorate to get a supporting letter for I-CARE, the hospital database center. After explaining the objectives of the study, written informed consent was obtained from each family or legal guardians of the study subjects.
Acknowledgments
The authors of this study acknowledge the Department of Anatomy, staff at Clinical service directorate; Pediatric units; I-CARE, and ICT department, as well as the librarians, for their support in making this work easier. Patients whose charts were reviewed for this study are also acknowledged.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors declare that they have no competing interests in this work.
References
1. Korlimarla A, Hart SJ, Spiridigliozzi GA, Kishnani PS. Down syndrome. In: Carey JC, Battaglia A, Viskochil D, Cassidy SB, editors. Cassidy and Allanson’s Management of Genetic Syndromes. 2021. doi:10.1002/9781119432692.ch24
2. Chen L, Wang L, Wang Y, et al. Global, regional, and national burden and trends of Down syndrome from 1990 to 2019. Front Genetics. 2002;13:908482. doi:10.3389/fgene.2022.908482
3. Elmagrpy Z, Rayani A, Shah A, et al. Down syndrome and congenital heart disease: why the regional difference as observed in the Libyan experience? Cardiovasc J Afr. 2011;22(6):306–309. doi:10.5830/CVJA-2010-072
4. Sherman SL, Allen EG, Bean LH, et al. Epidemiology of Down syndrome. Ment Retard Dev Disabil Res Rev. 2007;13(3):221–227. doi:10.1002/mrdd.20157
5. Kuppermann M, Learman LA, Gates E, et al. Beyond race or ethnicity and socioeconomic status: predictors of prenatal testing for Down syndrome. Obstetrics Gynecol. 2006;107(5):1087–1097. doi:10.1097/01.aog.0000214953.90248.db
6. Aliyu LD. Fetal Congenital Anomalies in Africa: Diagnostic and Management Challenges. IntechOpen; 2021. doi:10.5772/intechopen.91994
7. Muntha A, Moges T. Congenital cardiovascular anomalies among cases of Down syndrome: a hospital based review of cases in Tikur Anbessa Specialized Hospital, Ethiopia. Ethiop J Health Sci. 2019;29(2):165–174. doi:10.4314/ejhs.v29i2.3
8. Carey JC, Cassidy SB, Battaglia A, Viskochil D, editors.. Cassidy and Allanson’s Management of Genetic Syndromes. John Wiley & Sons; 2020.
9. Hindley D, Medakkar S. Diagnosis of Down’s syndrome in neonates. Arch Dis childhood. 2002;87(3):F220–F221. doi:10.1136/fn.87.3.f220
10. Devlin L, Morrison PJ. Accuracy of the clinical diagnosis of Down syndrome. Ulster Med J. 2004;73(1):4–12. PMID: 15244118; PMCID: PMC2475449.
11. Bull MJ, Ropper AH, Ropper AH. Down syndrome. New Engl J Med. 2020;382(24):2344–2352. doi:10.1056/NEJMra1706537
12. Mokhtar MM, Abdel-Fattah M. Major birth defects among infants with Down syndrome in Alexandria, Egypt (1995–2000): trends and risk factors. East Mediterr Health J. 2001;7(3):441–451. PMID: 12690765. doi:10.26719/2001.7.3.441
13. Kim MA, Lee YS, Yee NH, et al. Prevalence of congenital heart defects associated with Down syndrome in Korea. J Korean Med Sci. 2014;29(11):1544–1549. doi:10.3346/jkms.2014.29.11.1544
14. Vida VL, Barnoya J, Larrazabal LA, et al. Congenital cardiac disease in children with Down’s syndrome in Guatemala. Cardiol Young. 2005;15(3):286–290. doi:10.1017/S1047951105000582
15. Stoll C, Dott B, Alembik Y, et al. Associated congenital anomalies among cases with Down syndrome. Eur J Med Genet. 2015;58(12):674–680. doi:10.1016/j.ejmg.2015.11.003
16. Ali SK. Cardiac abnormalities of Sudanese patients with Down’s syndrome and their short-term outcome. Cardiovasc J Afr. 2009;20(2):112–115.
17. Morris JK, Garne E, Wellesley D, et al. Major congenital anomalies in babies born with Down Syndrome: a EUROCAT Population-Based Registry Study. Am J Med Genet B Neuropsychiatr Genet. 2014;164A(12):2979–2986. doi:10.1002/ajmg.a.36780
18. Hijii T, Fukushige J, Igarashi H, et al. Life expectancy and social adaptation in individuals with Down syndrome with and without surgery for congenital heart disease. Clin Pediatr. 1997;36(6):327–332. doi:10.1177/000992289703600603
19. Lo NS, Leung PM, Lau KC, et al. Congenital cardiovascular malformations in Chinese children with Down’s syndrome. Chinese Med J. 1989;102(5):382–386. PMID: 2530065.
20. Abbag FI. Congenital heart diseases and other major anomalies in patients with Down syndrome. Saudi Med J. 2006;27(2):219–222. PMID: 16501680.
21. El-Gilany AH, Yahia S, Wahba Y. Prevalence of congenital heart diseases in children with Down syndrome in Mansoura, Egypt: a retrospective descriptive study. Ann Saudi Med. 2017;37(5):386–392. doi:10.5144/0256-4947.2017.386
22. Boussouf K, Zaidi Z, Amrane M, et al. Study of congenital heart diseases in patients with Down syndrome in Algeria. East Mediterr Health J. 2017;23(9):632–636. doi:10.26719/2017.23.9.632
23. Chéhab G, Fakhoury H, Saliba Z, et al. congenital heart disease associated with gastrointestinal malformations. Lebanese Med J. 2007;55(2):70–74.
24. Freeman SB, Torfs CP, Romitti PA, et al. Congenital gastrointestinal defects in Down syndrome: a report from the Atlanta and National Down Syndrome Projects. Clin Genet. 2009;75(2):180–184. doi:10.1111/j.1399-0004.2008.01110.x
25. National Institutes of Health (NIH). Gastrointestinal (GI) anomalies in Down syndrome (DS) patients [Internet]. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4017552/.
26. Cleves MA, Hobbs CA, Cleves PA, et al. Congenital defects among liveborn infants with Down syndrome. Birth Defects Res Part a Clin Mol Teratol. 2007;79(9):657–663. doi:10.1002/bdra.20393
27. De la Cruz FM, Freijo CM. Down syndrome: current perspectives. Revista Española de Cardiología. 2008;61(10):914–923.
28. Leon-Luis J D, Gamez F, Bravo C, et al. Second-trimester fetal aberrant right subclavian artery: original study, systematic review and meta-analysis of performance in Detection of Down Syndrome. Int Soc Ultrasound Obstetr Gynecol. 2014;44(2):147–153. doi:10.1002/uog.13336
29. Bertrand P, Navarro H, Caussade S, et al. Airway anomalies in children with Down syndrome: endoscopic findings. Pediatr Pulmonol. 2003;36(2):137–141. doi:10.1002/ppul.10332
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.