")
Back to Journals » Open Access Rheumatology: Research and Reviews » Volume 14
Predicting the Progression of Very Early Systemic Sclerosis: Current Insights
Authors Bellocchi C , Chung A, Volkmann ER
Received 24 May 2022
Accepted for publication 6 September 2022
Published 15 September 2022 Volume 2022:14 Pages 171—186
DOI https://doi.org/10.2147/OARRR.S285409
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Chuan-Ju Liu
Chiara Bellocchi,1,2 Augustine Chung,3 Elizabeth R Volkmann4
1Scleroderma Unit, Referral Center for Systemic Autoimmune Diseases, Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico di Milano, University of Milan, Milan, Italy; 2Department of Clinical Sciences and Community Health, University of Milan, Milan, Italy; 3Division of Pulmonary and Critical Care, Department of Medicine, University of California, David Geffen School of Medicine, Los Angeles, CA, USA; 4Division of Rheumatology, Department of Medicine, University of California, David Geffen School of Medicine, Los Angeles, CA, USA
Correspondence: Elizabeth R Volkmann, Division of Rheumatology, Department of Medicine, University of California, Los Angeles, David Geffen School of Medicine, 1000 Veteran Avenue, Ste 32-59, Los Angeles, CA, 90095, USA, Tel +1 310-825-5800, Fax +1 310-206-5088, Email [email protected]
Abstract: Systemic sclerosis (SSc) is a complex autoimmune connective tissue disease with distinct pathological hallmarks (ie, inflammation, vasculopathy, fibrosis) that may predominate at different stages in the disease course with varying severity. Initial efforts to classify patients with SSc identified a subset of patients with very early SSc. These patients possessed signs of SSc (eg, Raynaud phenomenon, SSc specific autoantibodies and/or nailfold capillary abnormalities) without fulfilling complete SSc classification criteria. Recognizing the inherent value in early diagnosis and intervention in SSc, researchers have endeavored to identify risk factors for progression from very early SSc to definite SSc. The present review summarizes the clinical phenotype of patients with very early and early SSc. Through a scoping review of recent literature, this review also describes risk factors for progression to definite SSc with a focus on the specific clinical features that arise early in the SSc disease course (eg, diffuse cutaneous sclerosis, interstitial lung disease, esophageal dysfunction, renal crisis, cardiac involvement). In addition to clinical risk factors, this review provides evidence for how biological data (ie, serological, genomic, proteomic profiles, skin bioengineering methods) can be integrated into risk assessment models in the future. Furthering our understanding of biological features of very early SSc will undoubtedly provide novel insights into SSc pathogenesis and may illuminate new therapeutic targets to prevent progression of SSc.
Keywords: systemic sclerosis, scleroderma, early diagnosis, disease progression
Introduction
Systemic sclerosis (SSc) is a complex autoimmune connective tissue disease with significant heterogeneity in disease course and clinical outcomes. Affecting multiple organ systems and to varying degrees, SSc has the highest cause-specific mortality among all connective tissue diseases.1 Marked diversity exists in the evolution of SSc-related disease progression, disease manifestations and treatment response rates between patients.2 For this reason, when a patient receives a diagnosis of SSc, multiple questions immediately emerge, ranging from “How long do I have to live?” to “Will I develop lung disease?” This prognostic uncertainty poses challenges to patients and their providers.
Data from clinical trials and observational cohorts, along with direct clinical observations, have furthered our understanding of the unique clinical phenotypes associated with SSc. Increasingly, specific patient data are gathered to predict disease course and organ involvement early in SSc. These data include demographic characteristics, presenting disease features, laboratory analyses, radiographic studies and functional tests. The seasoned SSc practitioner uses these data to educate the patient on their anticipated prognosis and develop an initial monitoring and treatment plan. However, risk stratification in SSc remains an evolving area of research,3 and valid SSc-prediction tools do not exist.
In addition, patients with very early SSc may exhibit few manifestations of SSc at the time of their presentation (eg, Raynaud phenomenon [RP], positive anti-nuclear antibody [ANA], abnormal nailfold capillaroscopy).4 With these scarce data, the practitioner must then generate a gestalt regarding the likelihood this patient will develop SSc and the time course for disease progression. This review article describes how to predict disease progression in a patient with very early SSc with a focus on practical and readily available clinical and biological assessments. This review also explores novel ways to predict disease progression with a focus on genetic biomarkers and the gastrointestinal (GI) microbiome. The overall goal of this review is to provide a practical guide for monitoring patients with very early SSc through careful history taking, physical examination and evidence-based studies.
Very Early Ssc
In the literature, the terms very early SSc and early SSc have been often used interchangeably to indicate the preclinical phases of SSc with signs of RP, but not skin or internal organ involvement (these terms include very early SSc, early SSc [abbreviated as EaSSc], preclinical systemic sclerosis [PreSSc], undifferentiated connective tissue disease at risk for systemic sclerosis [UCTD-risk-SSc]). However, very early and early SSc have distinct clinical meanings. To avoid confusion, in the present review, we refer to very early SSc as the stages prior to a definite diagnosis of SSc and to early SSc as the first phases of SSc with an already definite, but recent SSc diagnosis (<3 years). Both of these representative disease stages are important to identify, as early recognition of these stages may improve outcomes for patients.
Definitions of Very Early SSc
In 2001, LeRoy and Medsger proposed criteria to individuate and classify patients at a very early stage and who did not completely satisfy The American Rheumatism Association (ARA) preliminary criteria for SSc proposed in the 1980.5 They labeled patients with RP, SSc specific autoantibodies and/or typical abnormalities at nailfold capillaroscopy (Figure 1) as limited SSc (not to be confused with limited cutaneous SSc).5 In 2008, a study from a Canadian group investigated 586 consecutive patients presenting with RP and reported that among patients with capillaroscopy abnormalities and specific autoantibodies, 65.9% and 72.7% of patients progressed to definite SSc in 5 and 10 years, respectively, according to ARA preliminary criteria.6 This study represented a preliminary validation of the LeRoy and Medsger criteria for very early SSc and identified specific risk factors for progression to definite SSc.
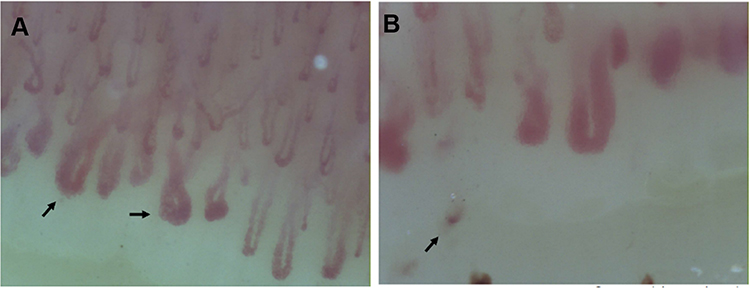 |
Figure 1 (A and B) demonstrate nailfold video-capillaroscopy findings in a patient with very early SSc who presented with RP and anticentromere antibodies. Arrows in (A) identify giant capillaries. (B) demonstrates capillary drop-out; the arrow in this panel identifies a microhemorrhage. |
In 2011, the European League Against Rheumatism (EULAR) Scleroderma Trial and Research (EUSTAR) proposed criteria for a very early diagnosis of SSc (VEDOSS) based on a Delphi consensus study. In this study, physicians from 85 EUSTAR centers identified RP, the presence of puffy fingers and antinuclear antibodies (ANA) positivity as key features and the presence of SSc specific autoantibodies (anti-centromere antibody or anti-topoisomerase I) and SSc pattern on nailfold capillaroscopy as necessary features for a very early diagnosis of SSc.7
In 2013, a EUSTAR study of 469 patients with RP demonstrated that high suspicion of very early SSc was represented by the presence of ANA positivity and puffy fingers.8 In the same year, the 2013 American College of Rheumatology/European League Against Rheumatism (ACR/EULAR) classification criteria for SSc were published, and reported a higher specificity and sensitivity for SSc than the ARA criteria.9 The 2013 ACR/EULAR criteria include clinical features of the LeRoy and Medsger criteria for very early SSc (eg, RP, SSc specific antibodies and/or SSc pattern at capillaroscopy), as well as the VEDOSS criteria (RP, ANA positivity, SSc specific antibodies, SSc pattern at capillaroscopy, puffy swollen digits) (Table 1). While a score of 9 is the minimum score required to classify a patient with definite SSc, patients fulfilling the Le Roy and Medsger criteria for very early SSc and the VEDOSS criteria will have a maximum score of 8 (Figure 2). Identifying preclinical SSc represents an important diagnostic opportunity as preventative treatment strategies (an unmet area of research) could be administered during this time window.10
 |
Table 1 Criteria for Very Early SSc Based on the LeRoy and Medsger Definition5 and the VEDOSS Criteria7 |
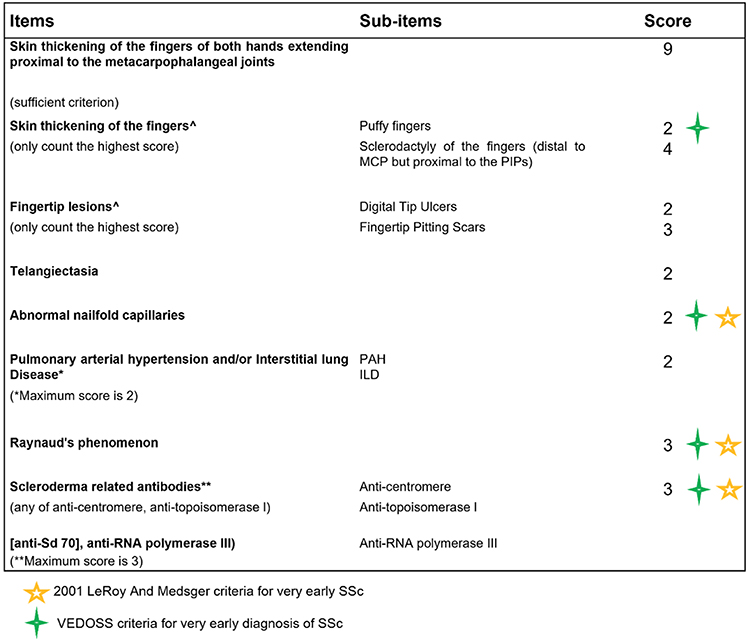 |
Figure 2 The ACR/EULAR 2013 Classification Criteria for SSc, highlighting the criteria for very early SSc. A total score of 9 is sufficient to classify patients with definite SSc. |
Recently, in 2021, VEDOSS criteria were prospectively validated in a EUSTAR multicenter study that investigated the clinical values of VEDOSS criteria in detecting SSc clinical progression. This study followed 1150 subjects with RP, of whom 553 met VEDOSS criteria and had available data at follow-up.11 Disease progression was defined as the fulfillment of ACR/EULAR classification criteria for SSc. Of the 553 patients, 254 experienced progression or reached 5-year follow-up. The overall rate of progression in 5 years was of 52.4%, aligning with the previous data from Koenig et al.6 The study also reported that the absence of the ANA decreased the risk of progression to SSc, while ANA positivity together with the presence of puffy fingers predicted the highest rate of progression to definite SSc. Interestingly, patients with a positive ANA, SSc specific autoantibodies and puffy fingers had the highest progression rate over time (94.1%), followed by patients with ANA, specific autoantibodies and capillaroscopic abnormalities (82.2%).
Another recent study on 217 subjects with RP and at least one manifestation of SSc also demonstrated that the combination of ANA positivity and puffy fingers were the two features most strongly associated with disease progression.12
Clinical Features of Very Early SSc
As described above, based on 2001 LeRoy and Medsger criteria or on VEDOSS criteria, subjects at a very early SSc stage can present with RP plus ANA and/or SSc specific antibodies and/or scleroderma pattern at nailfold capillaroscopy and/or puffy fingers without any other skin or organ involvement.5,7 Nevertheless, some studies have demonstrated subclinical organ involvement in very early SSc stages. For example, one study reported that 42% of 19 patients with very early SSc had signs of early SSc-cardiac involvement (ie, defined as an inverted mitral E/A ratio), diffusion impairment (ie, a diffusing lung capacity for carbon monoxide [DLCO] <80% of the predictive value) and/or esophageal involvement (ie, based on a low esophageal sphincter pressure <15 mmHg).13 In another cohort followed by the same investigators, 48.7% of 39 very early SSc patients presented at baseline at least one of the clinical involvements described above and/or decreased lung function (ie, forced vital capacity [FVC]< 80% predicted).14 Another study found evidence of GI tract involvement in 59 patients meeting VEDOSS criteria compared to 24 healthy controls, reporting alterations of esophageal as well as anorectal manometry in very early SSc patients.15 However, one study investigating cardiac autonomic modulation in SSc patients, healthy controls and very early SSc patients demonstrated that heart rate variability was not impaired in very early SSc subjects who had comparable indexes to the healthy controls.16
Importance of Identifying Very Early SSc
Recognition of the preclinical phase of SSc could potentially help identify early signs of internal organ involvement.17 From a clinical perspective, the early diagnosis may guide screening and risk stratification efforts and facilitate therapeutic interventions at an earlier time point in the disease course. From the research side, understanding the biomolecular and clinical features of the preclinical phases may reveal important insights into the pathogenesis of SSc, as well as therapeutic targets. Research on this preclinical disease stage may help us to understand why some patients progress rapidly into a definite diagnosis, while others exhibit stable very early SSc features for several years before progression to definite SSc. While patients with very early SSc may not present at the same frequency to a rheumatologist as those with obvious SSc, efforts are needed to ensure these patients are seen by rheumatologists and preferably those with an expertise in SSc.
Risk Stratification
As described above, risk stratification is a cornerstone to the initial evaluation of a patient with early SSc. The presence or absence of specific clinical and biological factors can provide insight into the future course of disease.18 A simple way to approach risk stratification is to consider risk by organ system (Table 2; Figure 3). Specifically, the practitioner evaluates the risk of development/progression of specific manifestations, separately by organ system. This organized approach not only allows the practitioner to apply evidence-based medicine to their evaluation process, but it also may improve their ability to effectively communicate their evaluation and findings to the patient. Because this review focuses on progression in very early and early SSc, risk stratification strategies will be described for SSc manifestations that arise earlier in the SSc disease course (cutaneous fibrosis, interstitial lung disease [ILD], renal crisis, cardiac involvement, digital ulcers, and upper GI tract involvement).13,19 While the timing from RP to the first non-RP manifestation of SSc varies, a recent longitudinal study of the EUSTAR cohort (median follow up time of 2.1 years) demonstrated that the median time was 0.9 (IQR 0–4.2) years.19
 |
Table 2 Initial Evaluation of a Patient with Very Early or Early SSc |
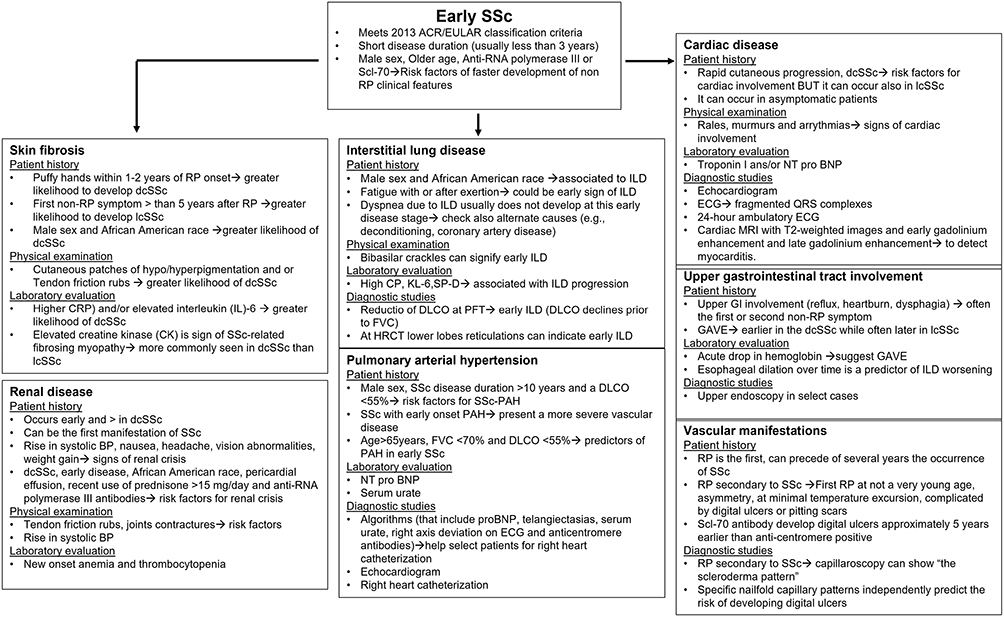 |
Figure 3 Flow chart for the evaluation of patients with early SSc by organ system with description of risk factors and diagnostic testing. |
Skin Fibrosis
The skin is the most common organ system involved in SSc, affecting 95% of patients.19 While a minority of patients may present with scleroderma sine scleroderma,20 most will exhibit skin fibrosis over the course of their disease. The two major subgroups of skin disease in SSc (ie, limited [lcSSc] and diffuse [dcSSc] cutaneous SSc) are determined based on physical examination.21 Patients with lcSSc may have involvement of the hands, forearms, feet, lower legs and face, while patients with dcSSc may have involvement of these areas, as well as the trunk, chest, upper arms and thighs. When evaluating a patient with very early or early SSc, certain historical clues may help the practitioner determine the risk of developing diffuse cutaneous SSc.22 For example, when a given patient develops their first non-Raynaud symptom of SSc (eg, puffy hands) within 1–2 years of the onset of Raynaud phenomenon, this can suggest a greater likelihood that this patient will develop diffuse cutaneous sclerosis.23 In contrast, when a given patient develops their first non-Raynaud symptom of SSc more than 5 years after the onset of Raynaud phenomenon, this can suggest a greater likelihood that this patient will develop limited cutaneous sclerosis.23 Therefore, we recommend that practitioners record the estimated start date for each symptom attributable to SSc and generate a timeline in the patient’s chart. This simple task can provide invaluable insight into the temporal evaluation of SSc.
In addition to obtaining a thorough patient history, documentation and interpretation of demographic characteristics is prudent during risk stratification. Male sex23 and African American race24 are each associated with a greater likelihood of developing diffuse cutaneous sclerosis. This does not mean that male patients will always have diffuse cutaneous disease; however, their probability for having this clinical phenotype is greater.
During the physical examination, the presence of certain signs may be a harbinger for the development of diffuse cutaneous disease. For example, identification of cutaneous patches of hypo/hyperpigmentation may signify that the patient will develop diffuse cutaneous sclerosis.25 Signs of excoriation from scratching could suggest urticaria, a defining feature of early cutaneous inflammation in diffuse cutaneous SSc. Moreover, the development of forearm inflammation/fibrosis shortly after the onset of puffy hands (ie, within weeks to months) could indicate that diffuse progression of cutaneous sclerosis is imminent.26 Tendon friction rubs are also a feature of early diffuse cutaneous sclerosis.27 In addition, more extensive nailfold capillary changes are often observed in patients with early diffuse versus limited cutaneous SSc.27
Laboratory analysis is an essential component of the initial assessment in a patient with very early or early SSc. Assessment of basic laboratory studies, as well as autoimmune serologies, yields important prognostic data. For example, elevated C-reactive protein (CRP) and/or elevated interleukin (IL)-6 not explained by an alternative cause, such as infection, could also indicate a greater likelihood of diffuse cutaneous sclerosis.28 Similarly, a modestly elevated creatine kinase (CK) is a sign of SSc-related fibrosing myopathy, a condition more commonly seen in patients with dcSSc than lcSSc.29 Certain autoantibodies are associated with cutaneous subtype (Table 3). For example, anti-centromere antibodies are associated with limited cutaneous disease, while anti-Scl-70 antibodies are associated with diffuse cutaneous disease.3 Anti-RNA polymerase III antibodies are also associated with diffuse cutaneous disease; very early and early SSc patients who possess the latter antibodies are at risk for rapidly progressively skin disease.30 Evaluation for these autoantibodies should occur at the time of initial presentation. Moreover, autoantibodies may be present several years before the clinical detection of SSc.31 Care is needed when selecting the autoantibody detection method, as certain methods (eg, multiple-bead, enzyme-linked immunosorbent assay) can lead to a higher likelihood of a false positive result for the anti-Scl-70 antibody test.32
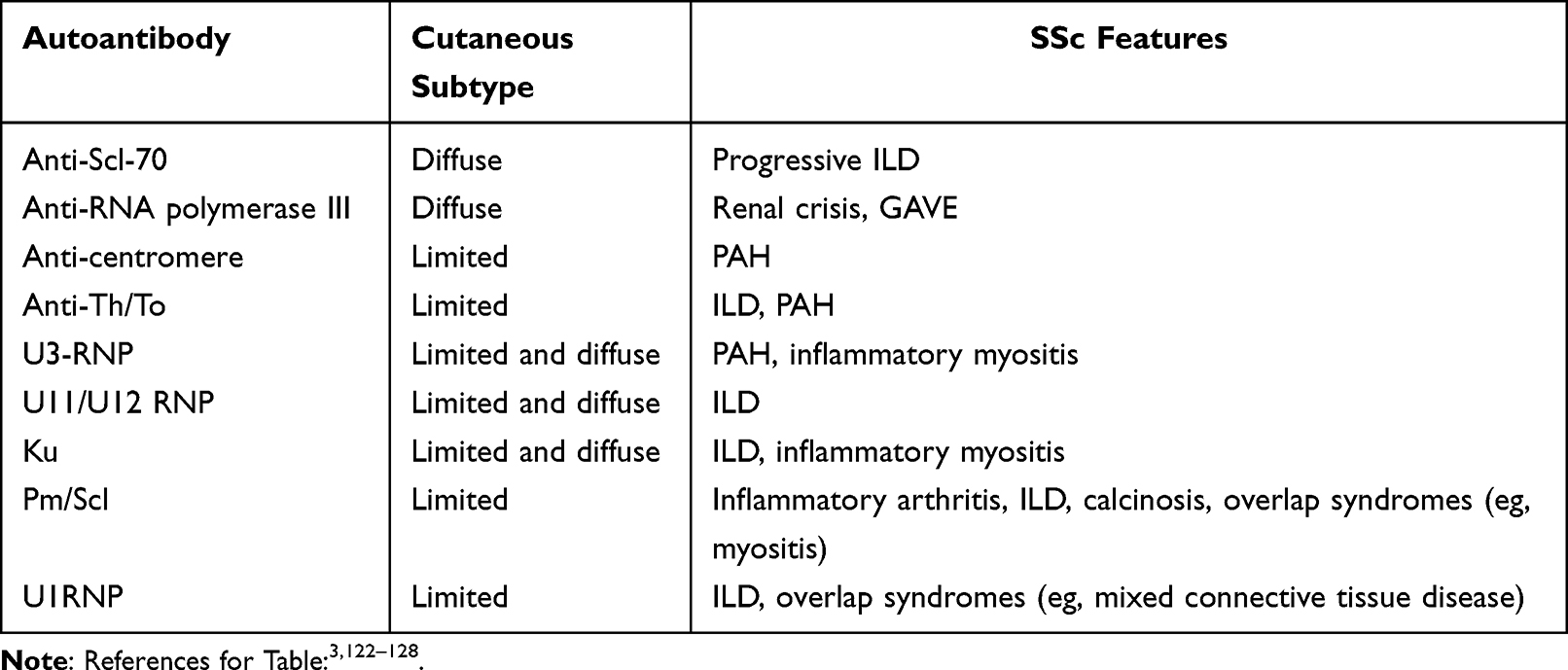 |
Table 3 Clinical Correlates of Autoantibody Profiles in SSc |
Skin biopsies are not commonly used in clinical practice to diagnose SSc; however, emerging research from gene expression studies in the skin of patients with SSc has demonstrated distinct gene expression signatures present at an early stage of disease. For example, in patients with early dcSSc, innate and adaptive immune cell signatures are increased compared with SSc patients with longer disease duration.33
Interstitial Lung Disease
ILD is the leading cause of SSc-related death34 and the complication that many patients fear the most. While the most reliable and sensitive way to detect early ILD is to obtain high-resolution computed tomography of the chest,35 patients with very early SSc may not have access to this type of imaging. Furthermore, the risk of radiation exposure may outweigh the perceivable benefit of obtaining a high-resolution computed tomography (HRCT) for some practitioners. In these scenarios, risk stratification may help guide the decision to obtain an HRCT of the chest.
On history, patients with very early or early SSc may complain of fatigue. While fatigue has multiple etiologies in SSc,36 fatigue that occurs with or after exertion could be an early sign of reduced gas exchange due to parenchymal lung disease. Typically, dyspnea on exertion or at rest due to ILD does not develop at this early disease stage, and alternate causes (eg, deconditioning, coronary artery disease) should be considered if a patient with very early or early SSc presents with dyspnea.
Similar to diffuse cutaneous sclerosis, male sex37 and African American race24 are independently associated with a greater risk of developing ILD. Patients with certain overlap connective tissue diseases also associated with ILD (eg, myositis) may be at higher risk for developing ILD. On physical examination, the presence of bibasilar crackles can signify early interstitial lung abnormality. Clubbing is exceedingly rare in SSc and would not be expected to occur at an early disease stage of SSc. Similarly, resting hypoxemia and desaturation on a six-minute walk (6MWT) are also not expected in very early or early SSc. However, measuring oxygen saturation at rest and during 6MWT in these patients is important for establishing a baseline for future assessments of disease progression. Emerging evidence suggest that specific nailfold capillary patterns may predict the presence of ILD;38 however, larger prospective studies are needed to affirm this hypothesis.
Specific laboratory tests may raise suspicion for the presence of ILD, including an elevated CRP concentration in the circulation.39 Candidate biomarkers currently under investigation, such as Krebs von den Lungen 6 (KL-6) and surfactant protein-D (SP-D), are associated with ILD progression. KL-6 is a pneumoprotein that at baseline predicts the decline of FVC over time and correlates with ILD severity. Moreover, therapeutic intervention for ILD is associated with a decline in KL-6.40–43 SP-D levels correlate with severity of ILD and predict response to rituximab for the treatment of ILD.44–46 Similar to CRP, increased serum levels of interleukin (IL)-6 are associated with progression of ILD and mortality.47 Treatment targeting the IL-6 receptor was found to stabilize lung function compared with placebo in a relatively small study of patients with dcSSc with risk factors for ILD progression at baseline (eg, progressive skin disease, elevated inflammatory markers).48
Serological profiling can also help risk stratify patients. For example, the presence of anti-Scl-70 antibodies, also referred to as anti-topoisomerase antibodies, portend a greater risk of developing progressive ILD, while the presence of anti-centromere and anti-RNA polymerase III antibodies, portend a lower risk of developing progressive ILD (Table 3).3
Rarely, a patient with very early or early SSc will have historical pulmonary function test (PFT) results available for comparison. If available, a reduction in the DLCO can signify early ILD. Typically, the DLCO declines prior to the FVC in early ILD.35 Similar to the 6MWT, PFTs are important to obtain in all patients with very early or early SSc to establish baseline values for monitoring future disease progression. Although PFTs represent a cornerstone for non-invasive monitoring of ILD, HRCT more sensitively detects early ILD and should be performed in all patients with a definite diagnosis of SSc regardless of PFT results.35,49 In early SSc-ILD, radiographic features include ground glass opacity with possible subtle reticulations, in the bilateral lower lobes. The predominant radiographic pattern of SSc-ILD is non-specific interstitial pneumonia (NSIP), characterized by bilateral reticulation, ground glass opacities and traction bronchiectasis. Usual interstitial pneumonia (UIP) occurs in approximately 25% of patients with SSc-ILD.50
In some instances, patients will have undergone an X-ray of the chest for other reasons. Chest X-ray is neither sensitive, nor specific for ILD in SSc;51 however, any signs of bibasilar abnormalities should prompt the practitioner to order an HRCT of the chest. In early SSc-ILD, distinguishing between interstitial abnormality due to SSc and atelectasis can be challenging. Obtaining prone imaging is helpful in these scenarios.52 It is also important to note that ILD may occur as the presenting feature of SSc in some cases. For instance, a recent study of a large US health insurance database demonstrated that 4–6% of patients had claims for ILD more than one year prior to a claim for SSc.53 These patients may have features of SSc at the time of the ILD diagnosis that are more subtle and go unrecognized by their primary physician and/or pulmonologist.
Renal Disease
Renal crisis typically occurs early in the course of SSc, particularly in patients with dcSSc.18 Early signs of renal crisis include a rise in systolic blood pressure, as well as nausea with or without vomiting. Also, non-specific symptoms such as headache, vision abnormalities and weight gain can occur along with new onset anemia and thrombocytopenia. These signs and symptoms are due to new onset of hypertension, progressive renal failure, congestive heart failure, hypertensive encephalopathy and microangiopathic hemolytic anemia.54 Many patients are asymptomatic in the early phases of renal crisis and careful identification of risk factors for renal crisis is vital at this disease stage. Known risk factors for renal crisis include diffuse cutaneous skin disease, early disease, African American race, pericardial effusion, recent use of prednisone >15 mg/day and the presence of anti-RNA polymerase III antibodies.55 In a retrospective study performed on military medical records, the presence of three or more of the following features at the time of SSc diagnosis predicted future renal crisis: proteinuria, anemia, hypertension, chronic kidney disease, elevated erythrocyte sedimentation rate, thrombocytopenia, hypothyroidism and serum anti-Ro antibody positivity.56 Other risk factors that are associated with SRC are the presence of tendon friction rubs, joints contractures and pericardial effusion.57 Acute kidney injury is typically not present in the early stages of renal crisis; however, measuring the creatinine and estimating the glomerular filtration rate in the initial evaluation of a patient with early or very early SSc is important for establishing a baseline level of kidney function. Also, measuring blood pressure is an easy tool for the early detection of renal crisis in SSc and can be performed at home by the patient between clinic visits. The differential diagnosis for scleroderma renal crisis includes pre-renal causes such as volume depletion, toxicity from medications, such as calcineurin inhibitors, membranous nephropathy, pre-eclampsia and ANCA-associated vasculitis.57
Cardiac Complications
Cardiac involvement is an uncommon presenting feature of very early or early SSc. Diverse manifestations of cardiac involvement exist, ranging from arrhythmias to impaired ventricular relaxation.58 Patients with SSc may exhibit signs of pericardial disease (eg, acute or chronic pericarditis, pericardial fibrosis, pericardial effusion, tamponade), myocardial disease (eg, fibrosis, ventricular diastolic or systolic dysfunction, myocarditis), conduction system disease (eg, heart block, supraventricular arrhythmias, ventricular arrhythmias, autonomic dysfunction) and vascular disease (eg, mural fibrosis, intimal proliferation, platelet-fibrin clotting).59
Predicting the likelihood of developing cardiac disease in a patient with early or very early SSc is challenging. Due to the heterogeneity of cardiac manifestations and the lack of clinical trials in this area, our understanding of risk factors for specific cardiac features is limited. The skin-thickness-progression rate may help identify patients at risk for cardiac disease. A single-center study demonstrated that patients with rapid cutaneous progression were more likely to have severe cardiac disease compared with patients with slow cutaneous progression; however, the difference was relatively small (3% versus 1%, respectively).22 While the presence of diffuse cutaneous disease is a risk factor for cardiac involvement in SSc, patients with limited cutaneous sclerosis are not immune to this feature of SSc.60 Cardiac involvement may occur in both cutaneous subtypes.61
On physical examination, signs of early SSc-cardiac involvement may include presence of rales, murmurs and arrhythmias. Jugular venous distension, hepatomegaly and lower extremity are less likely to occur early in the SSc disease course. Various diagnostic modalities for SSc-cardiac involvement exist. In a patient with early or very early SSc, obtaining an ECG, chest X-ray and echocardiogram may help screen for early cardiac dysfunction. If there is strong clinical suspicion for an arrhythmia (eg, palpitations, syncope), a 24-hour ambulatory ECG detects a higher prevalence of conduction abnormalities compared with resting ECG in patients with SSc.62
Also, recent evidences show that cardiac involvement can occur in asymptomatic patients with SSc.63 Certain biomarkers and studies can suggest the presence of cardiac involvement; these include troponin I, N-terminal pro-hormone BNP (NT-proBNP), echocardiography and ECG.63 For example, the presence of fragmented QRS complexes on ECG (defined as an additional R wave, or notching of the R or S wave, or the presence of fragmentation in two contiguous leads) could suggest cardiac fibrosis.64–66 In addition, cardiac MRI with T2-weighted images and early gadolinium enhancement and late gadolinium enhancement are used to evaluate for myocardial involvement. A recent study demonstrated that the measurement of T1 signal (the T1 mapping) can detect early cardiac fibrosis.67 One of the most common findings in asymptomatic cardiac involvement is the presence of a diastolic cardiac dysfunction but, of note, some early alterations detected in asymptomatic SSc patients can be reversible while symptomatic patients are more prone to show a progression of the cardiac impairment.68,69 While this is an evolving area of clinical research, an accurate evaluation of the cardiac function should also be performed in early SSc.
Pulmonary Hypertension
Pulmonary hypertension (PH) often develops later in the course of SSc.70 However, sometimes patients with pulmonary arterial hypertension (PAH) are misdiagnosed with idiopathic PAH if they have more subtle signs of SSc (mild Raynaud phenomenon, telangiectasias) at their time of initial presentation. These patients usual experience a delayed referral to rheumatology contingent on their development of more overt clinical signs of SSc and/or their insufficient response to PAH therapies. Patients with SSc-PAH often have worse outcomes than patients with idiopathic PAH.71
Observational studies have demonstrated that male sex, SSc disease duration >10 years and a DLCO <55% are risk factors for SSc-PAH.72 Studies have also reported early onset (within 5 years of SSc diagnosis since first non-Raynaud symptoms) of PAH in dcSSc patients. Moreover, early onset of SSc-PAH is associated with more severe vascular disease than late onset SSc-PAH.73,74
Valid algorithms exist to help select patients for evaluation for PAH using a right heart catheterization.75 These algorithms include the measurement of proBNP, presence of telangiectasias on physical examination, measurement of serum urate, presence of right axis deviation on ECG and the detection of anticentromere antibodies. While it may be premature to obtain a proBNP on all patients with early or very early SSc, the other algorithm components are feasible to obtain and may help identify early pulmonary vascular disease. However, even in the absence of the aforementioned risk factors, routine annual screening for PAH via echocardiogram is recommended by the American Heart Association and American College of Cardiology Foundation.76 Patients who have suspected PAH from echocardiogram should be referred for right heart catheterization to obtain direct measurements of pulmonary arterial pressures. Patients whose diagnosis of PAH resulted from routine screening had improved survival compared with patients who were not screened.77 Regular screening is thus of fundamental importance to detect PAH in its earliest phases. Discovering additional biomarkers to identify patients at risk for PAH is an ongoing area of research. A recent study measured 313 proteins from the serum of patients with SSc both with and without PAH in the DETECT study cohort. After performing validation in an external cohort, eight proteins (among them NT-ProBNP) were able to distinguish PAH from non-PAH.78
Upper Gastrointestinal Tract Involvement
The upper GI tract is typically affected early in the course of SSc.79 After Raynaud phenomenon, symptoms of upper GI tract involvement (eg, reflux, heartburn, dysphagia) often occur as the first or second non-Raynaud symptom in SSc.80 Any patient with a history of new onset reflux disease (in the absence of a change in dietary patterns, lifestyle or body habitus or pregnancy), who also reports a relatively recent history of Raynaud phenomenon should be evaluated for SSc. Predicting progression of upper GI involvement in very early or early SSc is difficult as some patients lack symptoms, even when there is objective evidence of esophageal disease (eg, esophagitis).
In a patient with newly diagnosed early SSc, an acute drop in hemoglobin could suggest the presence of gastric antral vascular ectasias (GAVE), a serious complication that can result in rapid blood loss.81 GAVE occurs earlier in the disease course of patients with dcSS and often later in the disease course of patients with lcSSc.82
Given the association between esophageal dysfunction and ILD,83,84 patients who exhibit signs of ILD progression may also have progressive involvement of their esophagus (ie, absent contractility). Esophageal dilation on HRCT of the chest is a common finding in SSc patients and its clinical significance is unknown. While some studies have demonstrated correlations between esophageal dilation and severity of ILD, a recent study found no relationship between two quantitative radiographic measures of esophageal dilation and ILD severity or progression.85,86 In many patients, esophageal-related symptoms may be silent or may be less apparent to the patient than their respiratory symptoms. These patients should undergo thorough evaluation for upper GI involvement. This may include manometry with pH monitoring and upper endoscopy.
Vascular Manifestations
RP is often the first sign of SSc and can occur several years before the onset of the first non-RP sign of SSc in some patients. While primary RP is relatively common in women, certain characteristics can suggest the presence of secondary RP, such as first appearance of RP in later adulthood, asymmetry of the RP, presence of RP with a minimal temperature changes, and the presence of digital ulcers or pitting scars.87 In SSc, RP is associated with structural vascular modifications with imbalance of vasoconstrictor and vasodilator factors.88 Capillaroscopy and autoantibody profiling are recommended to help identify very early stage SSc. Capillaroscopy can show a “scleroderma pattern”, characterized by giant capillaries, hemorrhages, ramified capillary loops and/or avascular areas.89 Currently the classification includes three distinct phases, early, active and late, that are associated with SSc disease duration.90
Certain patients with very early or early SSc are at risk for the development of digital ulcer disease.91 Early identification of these at-risk patients is central to therapeutic decision making for the management of RP. Studies have demonstrated that specific nailfold capillary patterns independently predict the risk of developing digital ulcers in patients without a history of digital ulcers.92 For example, the presence of avascular areas on capillaroscopy is associated with a higher risk of digital ulcers.93 In addition, multi-center observational studies have demonstrated that SSc patients with an Scl-70 antibody develop digital ulcers approximately 5 years earlier than those who were anti-centromere positive.94
Novel Methods to Identify Risk of Disease Progression
Novel methods are under investigation to help identify patients with very early or early SSc most at risk for disease progression. Below is a summary of some of the most promising methods and their clinical applicability.
Gene and Gene Expression Studies
Multiple genetic variations of human leukocyte antigen (HLA) region associated genes are associated with SSc.95,96 In addition, polymorphisms among non-HLA genes may increase the risk of developing SSc, including polymorphisms of type I interferon regulatory factors, as well as of genes expressed by T lymphocytes (STAT4, CD247, IL12) or by B lymphocytes.97,98
Biomolecular profiling via gene expression studies may help to identify patients at greater risk of disease progression based on studies performed in cohorts of patients with established SSc.99 Prediction studies on early SSc often focus on early dcSSc. For example, recent studies reported that both innate and adaptive immunity signatures are present in early dcSSc,33 and that these skin signatures can normalize over time in dcSSc.100
Biomolecular alterations are likely present in the preclinical stages of SSc. A study analyzed the whole blood of 19 patients with very early SSc, 30 healthy controls, 12 patients with primary RP and 7 patients with early SSc and evaluated the expression of a limited panel of IFN type I inducible genes. This study detected a type I IFN signature in very early and early SSc patients and found a correlation between monocyte IFN scores and monocyte BAFF mRNA levels.101
Protein Studies
Studies investigating circulating proteins in patients with very early SSc have largely examined a limited number of serum proteins involved in pro-inflammatory and pro-fibrotic pathways (Figure 4). In a study of 24 patients with very early SSc, 48 patients with early SSc and 24 control patients with osteoarthritis/fibromyalgia, higher levels of sICAM-1, CCL2, CXCL8, IL-13 and IL-33 were present in very early SSc patients compared with controls.102 Another study evaluated cytokine production from CD56+ NK/NKT cells in 24 patients with very early SSc (labeled as EaSSc), 10 patients with primary RP, 12 patients with early SSc and 9 healthy controls. This study demonstrated an increase of IL-6, TNF-α and MIP-1α/CCL3 following TLR1/2 stimulation in very early SSc and especially in early SSc compared with controls.103 Significantly elevated serum levels of CXCL10, CXCL11, TNFR2 and CHI3L1 were observed in two different cohorts of very early SSc and early SSc compared with healthy controls.104 Also, a study on 34 patients fulfilling VEDOSS criteria and 29 patients with definite SSc demonstrated higher serum levels of CXCL10 and CXCL11 in patients fulfilling the VEDOSS criteria.105 Studies have also evaluated the role of angiogenic molecules in the earliest phases of SSc. In one study of 43 patients with SSc, 9 patients with very early SSc (labeled as PreSSc) and healthy controls, VEGF serum levels were increased in very early SSc compared with controls.106 Moreover, a subsequent study found that endostatin serum levels were higher not only in established SSc but also in a subgroup of patients with very early SSc suggesting that vascular modifications are already ongoing in the preclinical stages of SSc.107 Consistent with these findings, other studies have demonstrated increased VEGF, reduced capillarogenesis, increased level of the antiangiogenic chemokine CXCL4 and activation of antiangiogenic mechanisms in patients meeting VEDOSS criteria.108–110 While the sample sizes of these studies are small, increasing the likelihood of type 1 error, the findings seem to suggest a pro-inflammatory, anti-angiogenic immune signature in patients with very early SSc.
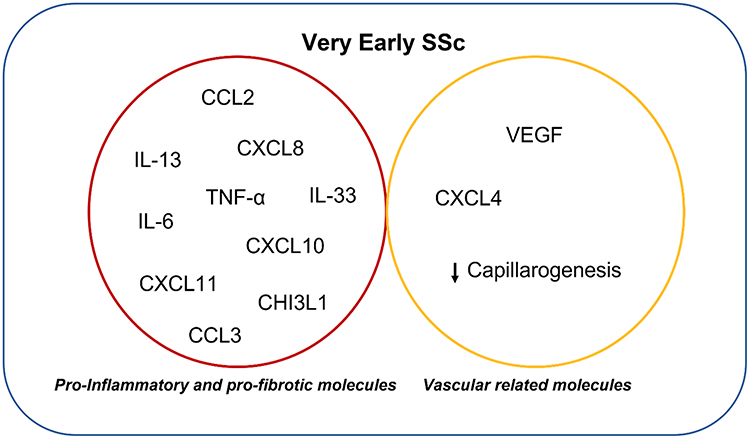 |
Figure 4 Proteins related to very early SSc disease stages. |
Microbiome Studies
Investigation of the GI microbiota provides insights into the complex interaction between the immune system with the bacterial flora.111 Several studies were conducted in inflammatory bowel diseases as well as in systemic autoimmune diseases (SADs) finding specific alterations of GI microbiota in patients compared with healthy controls or within different groups of SADs.112–114 In SSc, recent studies demonstrated a reduction of pro-tolerogenic bacteria species in SSc versus healthy controls, as well a different composition of gut microbiota in SSc with or without GI involvement.115–117 An important question arises about whether these microbiota alterations, toward a decrease of commensal bacteria and increase of pro-inflammatory strains, are more a cause or a consequence of the autoimmune diseases especially when a GI disease involvement is present.118 To answer to this question, studies on very early SSc are needed as well as longitudinal studies to assess the modification of gut microbiota composition over time. Interestingly, a study to evaluate gut microbiota in two geographically different cohorts (one from Sweden and the UCLA cohort from the USA), evaluated SSc patients with long disease duration as well as early SSc with recent diagnosis (median disease duration of 2.0 years).85 This study demonstrated a reduction of commensal bacteria and increase of pathobiont genera in the early SSc group when compared to healthy controls.119 Interestingly, in both of the cohorts, the presence of interstitial lung disease, small intestinal bacterial overgrowth, immunosuppression and disease duration was associated with beta diversity (ie, the microbial composition similarity between-subject, which enables the identification of differences between samples within a group).119 Future, longitudinal studies of patients with very early and early SSc are needed to explore the role of microbiome in disease progression.
Skin Bioengineering Methods
Evaluation of skin mechanical properties could be a way to detect skin progression at a very early stage of disease; ie, the analysis of viscoelasticity of skin in association with data from capillaroscopy could intercept SSc skin involvement and progression.120 Changes in viscoelasticity have been observed in skin of patients with secondary RP distinguishing them from subjects with a primary RP.121
Conclusion
In summary, very early SSc and early SSc are clinically important disease stages of SSc and represent an evolving area of research. While these patients often comprise a minority of SSc patients followed at expert SSc centers, and while the clinical burden of disease is low at this stage, these patients should nonetheless be followed closely with comprehensive clinical evaluation. In addition to a careful history and physical examination, complete autoantibody profile and capillaroscopy, PFT with DLCO assessment, HRCT chest, serological studies, as well as echocardiography are needed. When clinical risk factors for disease progression or for disease worsening are present, further diagnostic testing is needed to evaluate for cutaneous and internal organ involvement and progression.
While our ability to predict the overall risk of progression from very early SSc to definite SSc is still somewhat limited, recent advancements in research (eg, the longitudinal validation of VEDOSS criteria) have propelled this field forward paving the way for future studies examining clinical and biomolecular profiles of the preclinical phase. These future studies have the potential to provide novel insights on SSc pathogenesis and reveal new therapeutic targets aimed at curtailing or even reversing the progression of this chronic and disabling disease.
Acknowledgments
NIH/NHLBI (K23 HL150237) provided support to ERV. They had no role in the writing of the report; and in the decision to submit the paper for publication.
Disclosure
Dr Elizabeth R Volkmann reports grants, personal fees from Boehringer Ingelheim, grants from Horizon and grants from Kadmon, outside the submitted work; also awarded grants from Prometheus. The authors report no other conflicts of interest in this work.
References
1. Denton CP, Khanna D. Systemic sclerosis. Lancet. 2017;390(10103):1685–1699. doi:10.1016/S0140-6736(17)30933-9
2. Wielosz E, Majdan M. Clinical and serological parameters of progression and prognosis in patients with systemic sclerosis - a state of the art review. Wiad Lek. 2020;73(7):1528–1532.
3. Yang C, Tang S, Zhu D, Ding Y, Qiao J. Classical disease-specific autoantibodies in systemic sclerosis: clinical features, gene susceptibility, and disease stratification. Front Med. 2020;7:587773. doi:10.3389/fmed.2020.587773
4. Bellando-Randone S, Matucci-Cerinic M. Very early systemic sclerosis. Best Pract Res Clin Rheumatol. 2019;33(4):101428. doi:10.1016/j.berh.2019.101428
5. LeRoy EC, Medsger TA. Criteria for the classification of early systemic sclerosis. J Rheumatol. 2001;28(7):1573–1576.
6. Koenig M, Joyal F, Fritzler MJ, et al. Autoantibodies and microvascular damage are independent predictive factors for the progression of Raynaud’s phenomenon to systemic sclerosis: a twenty-year prospective study of 586 patients, with validation of proposed criteria for early systemic sclerosis. Arthritis Rheum. 2008;58(12):3902–3912. doi:10.1002/art.24038
7. Avouac J, Fransen J, Walker UA, et al. Preliminary criteria for the very early diagnosis of systemic sclerosis: results of a Delphi Consensus Study from EULAR Scleroderma Trials and Research Group. Ann Rheum Dis. 2011;70(3):476–481. doi:10.1136/ard.2010.136929
8. Minier T, Guiducci S, Bellando-Randone S, et al. Preliminary analysis of the very early diagnosis of systemic sclerosis (VEDOSS) EUSTAR multicentre study: evidence for puffy fingers as a pivotal sign for suspicion of systemic sclerosis. Ann Rheum Dis. 2014;73(12):2087–2093. doi:10.1136/annrheumdis-2013-203716
9. van den Hoogen F, Khanna D, Fransen J, et al. 2013 classification criteria for systemic sclerosis: an American college of rheumatology/European league against rheumatism collaborative initiative. Ann Rheum Dis. 2013;72(11):1747–1755. doi:10.1136/annrheumdis-2013-204424
10. Lepri G, Bellando Randone S, Matucci Cerinic M, Guiducci S. Early diagnosis of systemic sclerosis, where do we stand today? Expert Rev Clin Immunol. 2022;18(1):1–3. doi:10.1080/1744666X.2022.2015327
11. Bellando-Randone S, Galdo PFD, Lepri G, et al. Progression of patients with Raynaud’s phenomenon to systemic sclerosis: a five-year analysis of the European Scleroderma Trial and Research group multicentre, longitudinal registry study for Very Early Diagnosis of Systemic Sclerosis (VEDOSS). Lancet Rheumatol. 2021;3(12):E834–843.
12. Siqueira VS, Helbingen MFS, Medeiros-Ribeiro AC, et al. Predictors of progression to systemic sclerosis: analysis of Very Early Disease of Systemic Sclerosis (VEDOSS) in a large single cohort. Rheumatology. 2022. doi:10.1093/rheumatology/keac006
13. Valentini G, Vettori S, Cuomo G, et al. Early systemic sclerosis: short-term disease evolution and factors predicting the development of new manifestations of organ involvement. Arthritis Res Ther. 2012;14(4):R188. doi:10.1186/ar4019
14. Valentini G, Cuomo G, Abignano G, et al. Early systemic sclerosis: assessment of clinical and pre-clinical organ involvement in patients with different disease features. Rheumatology. 2011;50(2):317–323. doi:10.1093/rheumatology/keq176
15. Lepri G, Guiducci S, Bellando-Randone S, et al. Evidence for oesophageal and anorectal involvement in very early systemic sclerosis (VEDOSS): report from a single VEDOSS/EUSTAR centre. Ann Rheum Dis. 2015;74(1):124–128. doi:10.1136/annrheumdis-2013-203889
16. Rodrigues GD, Tobaldini E, Bellocchi C, et al. Cardiac autonomic modulation at rest and during orthostatic stress among different systemic sclerosis subsets. Eur J Intern Med. 2019;66:75–80. doi:10.1016/j.ejim.2019.06.003
17. Matucci-Cerinic M, Bellando-Randone S, Lepri G, Bruni C, Guiducci S. Very early versus early disease: the evolving definition of the ‘many faces’ of systemic sclerosis. Ann Rheum Dis. 2013;72(3):319–321. doi:10.1136/annrheumdis-2012-202295
18. Walker UA, Tyndall A, Czirjak L, et al. Clinical risk assessment of organ manifestations in systemic sclerosis: a report from the EULAR scleroderma trials and research group database. Ann Rheum Dis. 2007;66(6):754–763. doi:10.1136/ard.2006.062901
19. Jaeger VK, Wirz EG, Allanore Y, et al. Incidences and risk factors of organ manifestations in the early course of systemic sclerosis: a Longitudinal EUSTAR Study. PLoS One. 2016;11(10):e0163894. doi:10.1371/journal.pone.0163894
20. Diab S, Dostrovsky N, Hudson M, et al. Systemic sclerosis sine scleroderma: a multicenter study of 1417 subjects. J Rheumatol. 2014;41(11):2179–2185. doi:10.3899/jrheum.140236
21. Krieg T, Takehara K. Skin disease: a cardinal feature of systemic sclerosis. Rheumatology. 2009;48(Suppl 3):iii14–iii18. doi:10.1093/rheumatology/kep108
22. Domsic RT, Rodriguez-Reyna T, Lucas M, Fertig N, Medsger TA. Skin thickness progression rate: a predictor of mortality and early internal organ involvement in diffuse scleroderma. Ann Rheum Dis. 2011;70(1):104–109. doi:10.1136/ard.2009.127621
23. Medsger TA. Natural history of systemic sclerosis and the assessment of disease activity, severity, functional status, and psychologic well-being. Rheum Dis Clin North Am. 2003;29(2):255–73, vi. doi:10.1016/s0889-857x(03)00023-1
24. Steen V, Domsic RT, Lucas M, Fertig N, Medsger TA. A clinical and serologic comparison of African American and Caucasian patients with systemic sclerosis. Arthritis Rheum. 2012;64(9):2986–2994. doi:10.1002/art.34482
25. Volkmann ER, Furst DE. Management of systemic sclerosis-related skin disease: a review of existing and experimental therapeutic approaches. Rheum Dis Clin North Am. 2015;41(3):399–417. doi:10.1016/j.rdc.2015.04.004
26. Razykov I, Levis B, Hudson M, Baron M, Thombs BD; Canadian Scleroderma Research G. Prevalence and clinical correlates of pruritus in patients with systemic sclerosis: an updated analysis of 959 patients. Rheumatology. 2013;52(11):2056–2061. doi:10.1093/rheumatology/ket275
27. Ostojic P, Damjanov N, Pavlov-Dolijanovic S, Radunovic G. Peripheral vasculopathy in patients with systemic sclerosis: difference in limited and diffuse subset of disease. Clin Hemorheol Microcirc. 2004;31(4):281–285.
28. Muangchant C, Pope JE. The significance of interleukin-6 and C-reactive protein in systemic sclerosis: a systematic literature review. Clin Exp Rheumatol. 2013;31(2 Suppl 76):122–134.
29. Paik JJ, Wigley FM, Shah AA, et al. Association of fibrosing myopathy in systemic sclerosis and higher mortality. Arthritis Care Res. 2017;69(11):1764–1770. doi:10.1002/acr.23291
30. Terras S, Hartenstein H, Hoxtermann S, Gambichler T, Kreuter A. RNA polymerase III autoantibodies may indicate renal and more severe skin involvement in systemic sclerosis. Int J Dermatol. 2016;55(8):882–885. doi:10.1111/ijd.13032
31. Burbelo PD, Gordon SM, Waldman M, et al. Autoantibodies are present before the clinical diagnosis of systemic sclerosis. PLoS One. 2019;14(3):e0214202. doi:10.1371/journal.pone.0214202
32. Homer KL, Warren J, Karayev D, et al. Performance of anti-topoisomerase I antibody testing by multiple-bead, enzyme-linked immunosorbent assay and immunodiffusion in a university setting. J Clin Rheumatol. 2020;26(3):115–118. doi:10.1097/RHU.0000000000000971
33. Skaug B, Khanna D, Swindell WR, et al. Global skin gene expression analysis of early diffuse cutaneous systemic sclerosis shows a prominent innate and adaptive inflammatory profile. Ann Rheum Dis. 2020;79(3):379–386. doi:10.1136/annrheumdis-2019-215894
34. Tyndall AJ, Bannert B, Vonk M, et al. Causes and risk factors for death in systemic sclerosis: a study from the EULAR Scleroderma Trials and Research (EUSTAR) database. Ann Rheum Dis. 2010;69(10):1809–1815. doi:10.1136/ard.2009.114264
35. Bernstein EJ, Jaafar S, Assassi S, et al. Performance characteristics of pulmonary function tests for the detection of interstitial lung disease in adults with early diffuse cutaneous systemic sclerosis. Arthritis Rheumatol. 2020;72(11):1892–1896. doi:10.1002/art.41415
36. Basta F, Afeltra A, Margiotta DPE. Fatigue in systemic sclerosis: a systematic review. Clin Exp Rheumatol. 2018;113(4):150–160.
37. Peoples C, Medsger TA, Lucas M, Rosario BL, Feghali-Bostwick CA. Gender differences in systemic sclerosis: relationship to clinical features, serologic status and outcomes. J Scleroderma Relat Disord. 2016;1(2):177–240. doi:10.5301/jsrd.5000209
38. Smith V, Vanhaecke A, Guerra MG, et al. May capillaroscopy be a candidate tool in future algorithms for SSC-ILD: are we looking for the holy grail? A systematic review. Autoimmun Rev. 2020;19(9):102619. doi:10.1016/j.autrev.2020.102619
39. Muangchan C, Harding S, Khimdas S, et al. Association of C-reactive protein with high disease activity in systemic sclerosis: results from the Canadian Scleroderma Research Group. Arthritis Care Res. 2012;64(9):1405–1414. doi:10.1002/acr.21716
40. Assassi S, Denton C, Cutolo M, et al. Effect of Nintedanib on KL-6 in patients with systemic sclerosis-associated interstitial lung disease [abstract]. Arthritis Rheumatol. 2021;6(suppl 10):248.
41. Kuwana M, Shirai Y, Takeuchi T. Elevated serum krebs von den lungen-6 in early disease predicts subsequent deterioration of pulmonary function in patients with systemic sclerosis and interstitial lung disease. J Rheumatol. 2016;43(10):1825–1831. doi:10.3899/jrheum.160339
42. Volkmann ER, Tashkin DP, Kuwana M, et al. Progression of interstitial lung disease in systemic sclerosis: the importance of pneumoproteins Krebs von den Lungen 6 and CCL18. Arthritis Rheumatol. 2019;71(12):2059–2067. doi:10.1002/art.41020
43. Salazar GA, Kuwana M, Wu M, et al. KL-6 but not CCL-18 is a predictor of early progression in systemic sclerosis-related interstitial lung disease. J Rheumatol. 2018;45(8):1153–1158. doi:10.3899/jrheum.170518
44. Bonella F, Volpe A, Caramaschi P, et al. Surfactant protein D and KL-6 serum levels in systemic sclerosis: correlation with lung and systemic involvement. Sarcoidosis Vasc Diffuse Lung Dis. 2011;28(1):27–33.
45. Arron JR. Biomarkers in systemic sclerosis: mechanistic insights into pathogenesis and treatment. Curr Opin Rheumatol. 2021;33(6):480–485. doi:10.1097/BOR.0000000000000827
46. Ebata S, Yoshizaki A, Fukasawa T, Asano Y, Oba K, Sato S. Rapid decrease of serum surfactant protein-D levels predicts the reactivity of rituximab therapy in systemic sclerosis-associated interstitial lung disease. J Dermatol. 2020;47(7):796–800. doi:10.1111/1346-8138.15379
47. De Lauretis A, Sestini P, Pantelidis P, et al. Serum interleukin 6 is predictive of early functional decline and mortality in interstitial lung disease associated with systemic sclerosis. J Rheumatol. 2013;40(4):435–446. doi:10.3899/jrheum.120725
48. Khanna D, Lin CJF, Furst DE, et al. Long-term safety and efficacy of tocilizumab in early systemic sclerosis-interstitial lung disease: open-label extension of a Phase 3 randomized controlled trial. Am J Respir Crit Care Med. 2022;205(6):674–684. doi:10.1164/rccm.202103-0714OC
49. Khanna SA, Nance JW, Suliman SA. Detection and monitoring of interstitial lung disease in patients with systemic sclerosis. Curr Rheumatol Rep. 2022;24(5):166–173. doi:10.1007/s11926-022-01067-5
50. Desai SR, Veeraraghavan S, Hansell DM, et al. CT features of lung disease in patients with systemic sclerosis: comparison with idiopathic pulmonary fibrosis and nonspecific interstitial pneumonia. Radiology. 2004;232(2):560–567. doi:10.1148/radiol.2322031223
51. Strollo D, Goldin J. Imaging lung disease in systemic sclerosis. Curr Rheumatol Rep. 2010;12(2):156–161. doi:10.1007/s11926-010-0095-0
52. Chung JH, Walker CM, Hobbs S. Imaging features of systemic sclerosis-associated interstitial lung disease. J Vis Exp. 2020;(160). doi:10.3791/60300
53. Assassi S, Shao N, Yin Z, Volkmann ER, Zoz DF, Leonard TB. Understanding diagnostic pathways in systemic sclerosis and systemic sclerosis-associated interstitial lung disease: a retrospective cohort study. Medicine. 2022;101(32):e29993. doi:10.1097/MD.0000000000029993
54. Forman CJ, Olson SW, Gordon SM, et al. Association of race and risk of future scleroderma renal crisis at systemic sclerosis diagnosis. Arthritis Care Res. 2021. doi:10.1002/acr.24807
55. Woodworth TG, Suliman YA, Li W, Furst DE, Clements P. Scleroderma renal crisis and renal involvement in systemic sclerosis. Nat Rev Nephrol. 2016;12(11):678–691. doi:10.1038/nrneph.2016.124
56. Gordon SM, Stitt RS, Nee R, et al. Risk factors for future scleroderma renal crisis at systemic sclerosis diagnosis. J Rheumatol. 2019;46(1):85–92. doi:10.3899/jrheum.171186
57. Hudson M, Ghossein C, Steen V. Scleroderma renal crisis. Presse Med. 2021;50(1):104063. doi:10.1016/j.lpm.2021.104063
58. Butt SA, Jeppesen JL, Torp-Pedersen C, et al. Cardiovascular manifestations of systemic sclerosis: a Danish Nationwide Cohort Study. J Am Heart Assoc. 2019;8(17):e013405. doi:10.1161/JAHA.119.013405
59. Lambova S. Cardiac manifestations in systemic sclerosis. World J Cardiol. 2014;6(9):993–1005. doi:10.4330/wjc.v6.i9.993
60. Hachulla AL, Launay D, Gaxotte V, et al. Cardiac magnetic resonance imaging in systemic sclerosis: a cross-sectional observational study of 52 patients. Ann Rheum Dis. 2009;68(12):1878–1884. doi:10.1136/ard.2008.095836
61. de Groote P, Gressin V, Hachulla E, et al. Evaluation of cardiac abnormalities by Doppler echocardiography in a large nationwide multicentric cohort of patients with systemic sclerosis. Ann Rheum Dis. 2008;67(1):31–36. doi:10.1136/ard.2006.057760
62. Vacca A, Meune C, Gordon J, et al. Cardiac arrhythmias and conduction defects in systemic sclerosis. Rheumatology. 2014;53(7):1172–1177. doi:10.1093/rheumatology/ket377
63. Bruni C, Ross L. Cardiac involvement in systemic sclerosis: getting to the heart of the matter. Best Pract Res Clin Rheumatol. 2021;35(3):101668. doi:10.1016/j.berh.2021.101668
64. Bayar N, Cay HF, Erkal Z, et al. The importance of fragmented QRS in the early detection of cardiac involvement in patients with systemic sclerosis. Anatol J Cardiol. 2015;15(3):209–212. doi:10.5152/akd.2014.5191
65. Kadi H, Inanir A, Habiboglu A, et al. Frequency of fragmented QRS on ECG is increased in patients with rheumatoid arthritis without cardiovascular disease: a pilot study. Mod Rheumatol. 2012;22(2):238–242. doi:10.1007/s10165-011-0493-9
66. Basaran Y, Tigen K, Karaahmet T, et al. Fragmented QRS complexes are associated with cardiac fibrosis and significant intraventricular systolic dyssynchrony in nonischemic dilated cardiomyopathy patients with a narrow QRS interval. Echocardiography. 2011;28(1):62–68. doi:10.1111/j.1540-8175.2010.01242.x
67. Mavrogeni SI, Bratis K, Karabela G, et al. Cardiovascular magnetic resonance imaging clarifies cardiac pathophysiology in early, asymptomatic diffuse systemic sclerosis. Inflamm Allergy Drug Targets. 2015;14(1):29–36. doi:10.2174/1871528114666150916112551
68. Foocharoen C, Pussadhamma B, Mahakkanukrauh A, Suwannaroj S, Nanagara R. Asymptomatic cardiac involvement in Thai systemic sclerosis: prevalence and clinical correlations with non-cardiac manifestations (preliminary report). Rheumatology. 2015;54(9):1616–1621. doi:10.1093/rheumatology/kev096
69. Pussadhamma B, Mahakkanukrauh A, Suwannaroj S, Nanagara R, Foocharoen C. Clinical outcomes of asymptomatic cardiac involvement in systemic sclerosis patients after a 2-year follow-up (extended study). Am J Med Sci. 2021;362(6):570–577. doi:10.1016/j.amjms.2021.05.027
70. Lefevre G, Dauchet L, Hachulla E, et al. Survival and prognostic factors in systemic sclerosis-associated pulmonary hypertension: a systematic review and meta-analysis. Arthritis Rheum. 2013;65(9):2412–2423. doi:10.1002/art.38029
71. Ramjug S, Hussain N, Hurdman J, et al. Idiopathic and systemic sclerosis-associated pulmonary arterial hypertension: a comparison of demographic, hemodynamic, and MRI characteristics and outcomes. Chest. 2017;152(1):92–102. doi:10.1016/j.chest.2017.02.010
72. Chaisson NF, Hassoun PM. Systemic sclerosis-associated pulmonary arterial hypertension. Chest. 2013;144(4):1346–1356. doi:10.1378/chest.12-2396
73. Hachulla E, Launay D, Mouthon L, et al. Is pulmonary arterial hypertension really a late complication of systemic sclerosis? Chest. 2009;136(5):1211–1219. doi:10.1378/chest.08-3042
74. Krikeerati T, Pussadhamma B, Mahakkanukrauh A, Suwannaroj S, Nanagara R, Foocharoen C. Associated factors of early-onset pulmonary hypertension and clinical difference between early- and late-onset pulmonary hypertension in Thai systemic sclerosis. Mod Rheumatol. 2021;31(3):649–656. doi:10.1080/14397595.2020.1823067
75. Coirier V, Chabanne C, Jouneau S, et al. Impact of three different algorithms for the screening of SSc-PAH and comparison with the decisions of a multidisciplinary team. Diagnostics. 2021;11(10). doi:10.3390/diagnostics11101738
76. McLaughlin VV, Archer SL, Badesch DB, et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension: a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association: developed in collaboration with the American College of Chest Physicians, American Thoracic Society, Inc., and the Pulmonary Hypertension Association. Circulation. 2009;119(16):2250–2294. doi:10.1161/CIRCULATIONAHA.109.192230
77. Brown Z, Proudman S, Morrisroe K, Stevens W, Hansen D, Nikpour M. Screening for the early detection of pulmonary arterial hypertension in patients with systemic sclerosis: a systematic review and meta-analysis of long-term outcomes. Semin Arthritis Rheum. 2021;51(3):495–512. doi:10.1016/j.semarthrit.2021.03.011
78. Bauer Y, de Bernard S, Hickey P, et al. Identifying early pulmonary arterial hypertension biomarkers in systemic sclerosis: machine learning on proteomics from the DETECT cohort. Eur Respir J. 2021;57(6). doi:10.1183/13993003.02591-2020
79. McMahan ZH. Gastrointestinal involvement in systemic sclerosis: an update. Curr Opin Rheumatol. 2019;31(6):561–568. doi:10.1097/BOR.0000000000000645
80. Petcu A, Ghib LJ, Grad SM, et al. Upper gastrointestinal involvement in systemic sclerosis: findings in a real-life setting. Exp Ther Med. 2019;18(6):5095–5100. doi:10.3892/etm.2019.8125
81. El-Gendy H, Shohdy KS, Maghraby GG, Abadeer K, Mahmoud M. Gastric antral vascular ectasia in systemic sclerosis: where do we stand? Int J Rheum Dis. 2017;20(12):2133–2139. doi:10.1111/1756-185X.13047
82. Hung EW, Mayes MD, Sharif R, et al. Gastric antral vascular ectasia and its clinical correlates in patients with early diffuse systemic sclerosis in the Scot trial. J Rheumatol. 2013;40(4):455–460. doi:10.3899/jrheum.121087
83. Christmann RB, Wells AU, Capelozzi VL, Silver RM. Gastroesophageal reflux incites interstitial lung disease in systemic sclerosis: clinical, radiologic, histopathologic, and treatment evidence. Semin Arthritis Rheum. 2010;40(3):241–249. doi:10.1016/j.semarthrit.2010.03.002
84. Savarino E, Bazzica M, Zentilin P, et al. Gastroesophageal reflux and pulmonary fibrosis in scleroderma: a study using pH-impedance monitoring. Am J Respir Crit Care Med. 2009;179(5):408–413. doi:10.1164/rccm.200808-1359OC
85. Volkmann E, Tashkin D, Leng M, Kim G, Goldin J, Roth M. Symptoms of Gastroesophageal reflux are a better predictor of systemic sclerosis-related interstitial lung disease progression than quantitative radiographic assessment of esophageal paramters [Abstract]. Ann Rheum Dis. 2022;81:718–719.
86. Wangkaew S, Losuriya P, Euathrongchit J. Worsening of esophageal dilatation is associated with increase in a high-resolution computed tomography (HRCT) score in early systemic sclerosis-associated interstitial lung disease (SSc-ILD). Clin Rheumatol. 2021;40(3):955–963. doi:10.1007/s10067-020-05346-3
87. Wigley FM, Flavahan NA. Raynaud’s Phenomenon. N Engl J Med. 2016;375(6):556–565. doi:10.1056/NEJMra1507638
88. Herrick AL, Wigley FM. Raynaud’s phenomenon. Best Pract Res Clin Rheumatol. 2020;34(1):101474. doi:10.1016/j.berh.2019.101474
89. Maricq HR, LeRoy EC. Patterns of finger capillary abnormalities in connective tissue disease by “wide-field” microscopy. Arthritis Rheum. 1973;16(5):619–628. doi:10.1002/art.1780160506
90. Cutolo M, Sulli A, Pizzorni C, Accardo S. Nailfold videocapillaroscopy assessment of microvascular damage in systemic sclerosis. J Rheumatol. 2000;27(1):155–160.
91. Hachulla E, Clerson P, Launay D, et al. Natural history of ischemic digital ulcers in systemic sclerosis: single-center retrospective longitudinal study. J Rheumatol. 2007;34(12):2423–2430.
92. Silva I, Teixeira A, Oliveira J, et al. Endothelial dysfunction and nailfold videocapillaroscopy pattern as predictors of digital ulcers in systemic sclerosis: a cohort study and review of the literature. Clin Rev Allergy Immunol. 2015;49(2):240–252. doi:10.1007/s12016-015-8500-0
93. Lambova SN, Muller-Ladner U. Nailfold capillaroscopy in systemic sclerosis - state of the art: the evolving knowledge about capillaroscopic abnormalities in systemic sclerosis. J Scleroderma Relat Disord. 2019;4(3):200–211. doi:10.1177/2397198319833486
94. Denton CP, Krieg T, Guillevin L, et al. Demographic, clinical and antibody characteristics of patients with digital ulcers in systemic sclerosis: data from the DUO Registry. Ann Rheum Dis. 2012;71(5):718–721. doi:10.1136/annrheumdis-2011-200631
95. Acosta-Herrera M, Kerick M, Lopez-Isac E, et al. Comprehensive analysis of the major histocompatibility complex in systemic sclerosis identifies differential HLA associations by clinical and serological subtypes. Ann Rheum Dis. 2021. doi:10.1136/annrheumdis-2021-219884
96. Angiolilli C, Marut W, van der Kroef M, Chouri E, Reedquist KA, Radstake T. New insights into the genetics and epigenetics of systemic sclerosis. Nat Rev Rheumatol. 2018;14(11):657–673. doi:10.1038/s41584-018-0099-0
97. Xu Y, Wang W, Tian Y, Liu J, Yang R. Polymorphisms in STAT4 and IRF5 increase the risk of systemic sclerosis: a meta-analysis. Int J Dermatol. 2016;55(4):408–416. doi:10.1111/ijd.12839
98. Lopez-Isac E, Martin JE, Assassi S, et al. Brief report: IRF4 newly identified as a common susceptibility locus for systemic sclerosis and rheumatoid arthritis in a cross-disease meta-analysis of genome-wide association studies. Arthritis Rheumatol. 2016;68(9):2338–2344. doi:10.1002/art.39730
99. Assassi S, Volkmann ER, Zheng WJ, et al. Peripheral blood gene expression profiling shows predictive significance for response to mycophenolate in systemic sclerosis-related interstitial lung disease. Ann Rheum Dis. 2022;81(6):854–860. doi:10.1136/annrheumdis-2021-221313
100. Skaug B, Lyons MA, Swindell WR, et al. Large-scale analysis of longitudinal skin gene expression in systemic sclerosis reveals relationships of immune cell and fibroblast activity with skin thickness and a trend towards normalisation over time. Ann Rheum Dis. 2022;81(4):516–523. doi:10.1136/annrheumdis-2021-221352
101. Brkic Z, van Bon L, Cossu M, et al. The interferon type I signature is present in systemic sclerosis before overt fibrosis and might contribute to its pathogenesis through high BAFF gene expression and high collagen synthesis. Ann Rheum Dis. 2016;75(8):1567–1573. doi:10.1136/annrheumdis-2015-207392
102. Vettori S, Cuomo G, Iudici M, et al. Early systemic sclerosis: serum profiling of factors involved in endothelial, T-cell, and fibroblast interplay is marked by elevated interleukin-33 levels. J Clin Immunol. 2014;34(6):663–668. doi:10.1007/s10875-014-0037-0
103. Cossu M, van Bon L, Nierkens S, et al. The magnitude of cytokine production by stimulated CD56(+) cells is associated with early stages of systemic sclerosis. Clin Immunol. 2016;173:76–80. doi:10.1016/j.clim.2016.09.004
104. Cossu M, van Bon L, Preti C, Rossato M, Beretta L, Radstake T. Earliest Phase of systemic sclerosis typified by increased levels of inflammatory proteins in the serum. Arthritis Rheumatol. 2017;69(12):2359–2369. doi:10.1002/art.40243
105. Crescioli C, Corinaldesi C, Riccieri V, et al. Association of circulating CXCL10 and CXCL11 with systemic sclerosis. Ann Rheum Dis. 2018;77(12):1845–1846. doi:10.1136/annrheumdis-2018-213257
106. Distler O, Del Rosso A, Giacomelli R, et al. Angiogenic and angiostatic factors in systemic sclerosis: increased levels of vascular endothelial growth factor are a feature of the earliest disease stages and are associated with the absence of fingertip ulcers. Arthritis Res. 2002;4(6):R11. doi:10.1186/ar596
107. Almeida I, Oliveira Gomes A, Lima M, Silva I, Vasconcelos C. Different contributions of angiostatin and endostatin in angiogenesis impairment in systemic sclerosis: a cohort study. Clin Exp Rheumatol. 2016;100(5):37–42.
108. Chora I, Romano E, Manetti M, et al. Evidence for a derangement of the microvascular system in patients with a very early diagnosis of systemic sclerosis. J Rheumatol. 2017;44(8):1190–1197. doi:10.3899/jrheum.160791
109. Romano E, Manetti M, Rosa I, et al. Slit2/Robo4 axis may contribute to endothelial cell dysfunction and angiogenesis disturbance in systemic sclerosis. Ann Rheum Dis. 2018;77(11):1665–1674. doi:10.1136/annrheumdis-2018-213239
110. Jiang Z, Chen C, Yang S, He H, Zhu X, Liang M. Contribution to the peripheral vasculopathy and endothelial cell dysfunction by CXCL4 in Systemic Sclerosis. J Dermatol Sci. 2021;104(1):63–73. doi:10.1016/j.jdermsci.2021.07.006
111. Bellocchi C, Volkmann ER. Update on the gastrointestinal microbiome in systemic sclerosis. Curr Rheumatol Rep. 2018;20(8):49. doi:10.1007/s11926-018-0758-9
112. Seksik P, Rigottier-Gois L, Gramet G, et al. Alterations of the dominant faecal bacterial groups in patients with Crohn’s disease of the colon. Gut. 2003;52(2):237–242. doi:10.1136/gut.52.2.237
113. Chen BD, Jia XM, Xu JY, et al. An autoimmunogenic and proinflammatory profile defined by the gut microbiota of patients with untreated systemic lupus erythematosus. Arthritis Rheumatol. 2021;73(2):232–243. doi:10.1002/art.41511
114. Bellocchi C, Fernandez-Ochoa A, Montanelli G, et al. Identification of a shared microbiomic and metabolomic profile in systemic autoimmune diseases. J Clin Med. 2019;8(9). doi:10.3390/jcm8091291
115. Bellocchi C, Fernandez-Ochoa A, Montanelli G, et al. Microbial and metabolic multi-omic correlations in systemic sclerosis patients. Ann N Y Acad Sci. 2018;1421(1):97–109. doi:10.1111/nyas.13736
116. Andreasson K, Alrawi Z, Persson A, Jonsson G, Marsal J. Intestinal dysbiosis is common in systemic sclerosis and associated with gastrointestinal and extraintestinal features of disease. Arthritis Res Ther. 2016;18(1):278. doi:10.1186/s13075-016-1182-z
117. Patrone V, Puglisi E, Cardinali M, et al. Gut microbiota profile in systemic sclerosis patients with and without clinical evidence of gastrointestinal involvement. Sci Rep. 2017;7(1):14874. doi:10.1038/s41598-017-14889-6
118. Levin D, De Palma G, Zou H, et al. Fecal microbiome differs between patients with systemic sclerosis with and without small intestinal bacterial overgrowth. J Scleroderma Relat Disord. 2021;6(3):290–298. doi:10.1177/23971983211032808
119. Andreasson K, Lee SM, Lagishetty V, et al. Disease features and gastrointestinal microbial composition in patients with systemic sclerosis from two independent cohorts. ACR Open Rheumatol. 2022;4(5):417–425. doi:10.1002/acr2.11387
120. Dobrev H. Novel ideas: the increased skin viscoelasticity - a possible new fifth sign for the very early diagnosis of systemic sclerosis. Curr Rheumatol Rev. 2013;9(4):261–267. doi:10.2174/157339710904140417125455
121. Dobrev H. In vivo study of skin mechanical properties in Raynaud’s phenomenon. Skin Res Technol. 2007;13(1):91–94. doi:10.1111/j.1600-0846.2007.00197.x
122. Aggarwal R, Lucas M, Fertig N, Oddis CV, Medsger TA. Anti-U3 RNP autoantibodies in systemic sclerosis. Arthritis Rheum. 2009;60(4):1112–1118. doi:10.1002/art.24409
123. Fertig N, Domsic RT, Rodriguez-Reyna T, et al. Anti-U11/U12 RNP antibodies in systemic sclerosis: a new serologic marker associated with pulmonary fibrosis. Arthritis Rheum. 2009;61(7):958–965. doi:10.1002/art.24586
124. Hoa S, Hudson M, Troyanov Y, et al. Single-specificity anti-Ku antibodies in an international cohort of 2140 systemic sclerosis subjects: clinical associations. Medicine. 2016;95(35):e4713. doi:10.1097/MD.0000000000004713
125. Koschik RW, Fertig N, Lucas MR, Domsic RT, Medsger TA. Anti-PM-Scl antibody in patients with systemic sclerosis. Clin Exp Rheumatol. 2012;30(2 Suppl 71):S12–6.
126. Wielosz E, Dryglewska M, Majdan M. The prevalence and significance of anti-PM/Scl antibodies in systemic sclerosis. Ann Agric Environ Med. 2021;28(1):189–192. doi:10.26444/aaem/127801
127. Asano Y, Ihn H, Yamane K, Kubo M, Tamaki K. The prevalence and clinical significance of anti-U1 RNA antibodies in patients with systemic sclerosis. J Invest Dermatol. 2003;120(2):204–210. doi:10.1046/j.1523-1747.2003.12028.x
128. Ihn H, Yamane K, Yazawa N, et al. Distribution and antigen specificity of anti-U1RNP antibodies in patients with systemic sclerosis. Clin Exp Immunol. 1999;117(2):383–387. doi:10.1046/j.1365-2249.1999.00961.x
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.