")
Back to Journals » Pediatric Health, Medicine and Therapeutics » Volume 15
Potocki-Lupski Syndrome in Ethiopian Child: A Case Report
Authors Deginet E , Abdissa D, Hailu T
Received 12 December 2023
Accepted for publication 22 March 2024
Published 26 March 2024 Volume 2024:15 Pages 129—131
DOI https://doi.org/10.2147/PHMT.S451161
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Roosy Aulakh
Endayen Deginet, Deme Abdissa, Tadele Hailu
Department of Pediatrics and Child Health, School of Medicine, College of Health and Medical Sciences, Saint Paul’s Hospital Millennium Medical College, Addis Ababa, Ethiopia
Correspondence: Endayen Deginet, Email [email protected]
Background: Potocki-Lupski syndrome (PTLS) is a rare developmental disorder resulting from the partial duplication of the short arm of chromosome 17. Affected children may have hypotonia, facial dysmorphism, or neurological abnormalities.
Case Presentation: We present the case of a 5-year-old female patient from Ethiopia diagnosed with Potocki-Lupski syndrome (PTLS)(17p11.2 microduplication) through multiplex ligation-dependent probe amplification (MLPA) testing. This technique identified the duplication of regions of the 17p11.2 chromosome (RAI1, DRC3, USP22, COPS3 and LLGL1). The patient exhibited neurological manifestations including speech delay and mild intellectual disability, along with craniofacial dysmorphism characterized by a triangular face, wide forehead, dental malocclusion, and micrognathia.
Conclusion: A multidisciplinary team approach is imperative for managing patients with PTLS. Parental counseling and genetic advice are crucial for families with children affected by PTLS.
Keywords: Potocki-Lupski syndrome, child, Ethiopia
Background
Potocki-Lupski syndrome (PTLS) is a rare chromosomal anomaly, occurring in approximately 1 in 25,000 individuals. It results from the partial duplication of the short arm of chromosome 17 (17p11.2 micro duplication).1 PTLS is a developmental disorder with patients characterized by hypotonia, developmental and intellectual delay, or congenital anomalies. The spectrum of the clinical manifestations is more important regarding the cognitive level and behavioral disorders.2 Children with PTLS have a characteristic feature with facial dysmorphism and neurological or behavioral abnormalities. Facial dysmorphic characteristics encompass a triangular or oval face, micrognathia, a high-arched palate, downward-slanting palpebral fissures, a broad forehead, a protruding nose, a smooth chin, and dental malocclusion.3 These patients manifest with variable neurological manifestations which determines the prognosis of the disease. Individuals with this condition might exhibit delays in reaching developmental milestones, difficulties in speech and language, intellectual disability, behavioral challenges (such as repetitive behaviors, anxiety, withdrawal, attention deficit hyperactivity disorder - ADHD), as well as motor clumsiness or coordination impairments, and other neuropsychiatric disorders.1,2
Typically, the genetic abnormality arises as a de novo occurrence, with less frequent instances of autosomal dominant transmission.3 Some of the genes implicated in the etiopathogenesis of PTLS include RAI1, SREBF1, DRG2, LLGL1, SHMT1, and ZFP179.4 The confirmation of PTLS diagnosis involves identifying a duplication that encompasses the RAI1 (retinoic acid 1) gene located at chromosome 17p11.2. While this duplicated region may include multiple genes, it is RAI1 that primarily contributes to the key features of PTLS.5
To the best of our knowledge, this is the initial case report documenting the diagnosis of PTLS in Ethiopia through the application of multiplex ligation probe amplification (MLPA).
Case Presentation
We present the case of a 5-year-old female patient residing in Addis Ababa, Ethiopia, diagnosed with Potocki-Lupski syndrome (PTLS). The diagnosis was made by molecular genetic test which showed duplication in the 17p11.2 region.
She was born to non-consanguineous parents presented with delay in developmental mile stones and dimorphic facial features since birth. She has also history of myoclonic type of seizure since the age of four years.
During Physical examination, she displayed unique facial characteristics, including a triangular face, a broad forehead, dental malocclusion, micrognathia and bilateral enlargement of the tonsils. Additionally, hypotonic extremities were observed. (Figure 1)
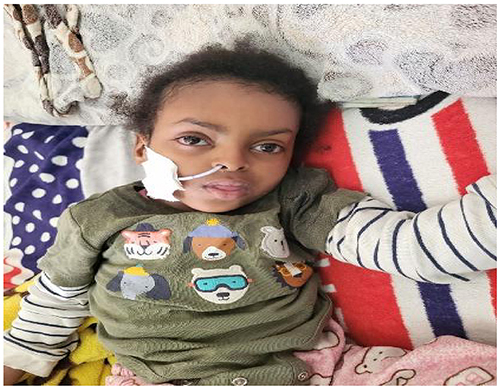 |
Figure 1 The patient with facial dysmorphism including triangular face and wide forehead. |
Hearing and vision showed no clinically apparent deficit. Genetic testing revealed duplication of 17p11.2 region. Following diagnosis, she received treatment with a single antiepileptic medication, effectively controlling her seizures. Additionally, she commenced physiotherapy and speech therapy to address her developmental needs. The parents received counseling and genetic guidance as part of the comprehensive management plan.
Discussion
We introduced a child diagnosed with PTLS (17p11.2 microduplication) who exhibited the primary clinical manifestations of the disorder, including craniofacial dysmorphism and neurological impairment. Fortunately, she did not show any signs of heart damage, kidney issues, osteoarticular anomalies, or ADHD, which are often reported in other patients with the condition. The syndrome characterized by duplication in the 17p11.2 region is often linked to failure to thrive, a condition observed in our patients. Malnutrition in these cases may result from gastroesophageal reflux or difficulties in swallowing (dysphagia), which can manifest as feeding difficulty during infancy.5
The suspicion of PTLS may arise from the child’s physical appearance, although the phenotype is not always highly indicative of the disease. Genetic testing becomes essential when dealing with a patient exhibiting intellectual disability or other psychiatric or neurological signs in conjunction with facial dysmorphism, congenital anomalies, and/or failure to thrive.5 The specific diagnostic method utilized for this patient was multiplex ligation-dependent probe amplification (MLPA). MLPA is a molecular technique used to detect copy number variations (CNVs) in specific genomic regions. It is particularly useful for identifying microduplications or microdeletions, such as the 17p11.2 micro duplication associated with Potocki-Lupski syndrome (PTLS). The case of the first Romanian family, comprising a mother and her five children, diagnosed with PTLS implies that PTLS can be familial despite majority of literatures reaffirm it arises as a de novo occurrence.5 Despite a less severe form of the disease, the children exhibited neurological manifestations such as speech delay and mild intellectual disability, along with craniofacial dysmorphism. The diagnosis was confirmed through genetic testing, specifically multiplex ligation-dependent probe amplification (MLPA), which detected the duplication of three regions of chromosome 17p11.2. Children diagnosed with PTLS do not have a specific treatment protocol. Each child will receive a tailored treatment plan that caters to their specific needs, incorporating elements such as speech therapy, physical therapy, behavioral and communication therapies, or specialized education services. Handling children with severe forms (such as congenital heart defects, renal, or gastrointestinal disorders) necessitates a collaborative, multi-disciplinary team approach.6,7 Consistent with our patient, a case report of a 4-year-old female from Sri Lanka who presented with severe expressive speech impairment with distinctive facial features typical of PTLS was diagnosed with duplication of 17p11.2 using MLPA.1 In summary, children with PTLS face the risk of neurodevelopmental disorders, including learning and language disabilities, or ASD. Early diagnosis and an approach involving a multi-disciplinary team are crucial for effective management. Parents should receive counseling and assistance in addressing medical, psychological, and neuropsychiatric challenges. Genetic advice is also of paramount importance.
Conclusion
This case report outlines the clinical characteristics and genetic results of a five-year-old girl diagnosed with Potocki–Lupski syndrome (PTLS). Her symptoms and signs align with documented clinical presentations of PTLS. Utilizing multiplex ligation probe amplification (MLPA), genetic analysis identified duplications in the 17p11.2 region, encompassing crucial genes such as RAI1, DRC3, USP22, COPS3 and LLGL1, known to be associated with PTLS.
Consent for Publication
The patient’s mother provided written informed consent for the publication of this case report. No institutional approval is required to publish the case details.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Sumathipala DS, Mandawala EN, Sumanasena SP, et al. 17p112 and Xq28 duplication detected in a girl diagnosed with Potocki-Lupski syndrome. BMC Res Notes. 2015;8(1):506. doi:10.1186/s13104-015-1439-7
2. Ciaccio C, Pantaleoni C, Milani D, et al. Neurological phenotype of Potocki–Lupski syndrome. Am J Med Genet A. 2020;182(10):2317–2324. doi:10.1002/ajmg.a.61789
3. Praticò AD, Falsaperla R, Rizzo R, et al. New patient with Potocki-Lupski syndrome: a literature review. J Pediatr Genet. 2018;7(01):29–34. doi:10.1055/s-0037-1604479
4. Shuib S, Saaid NN, Zakaria Z, Ismail J, Abdul Latiff Z. Duplication 17p11. 2 (Potocki-Lupski Syndrome) in a child with developmental delay Malays. J Pathol. 2017;39:77–81.
5. Grama A, Sîrbe C, Miclea D, et al. Case report: Potocki-Lupski syndrome in five siblings. Front Pediatr. 2021;9:698629. doi:10.3389/fped.2021.698629
6. Potocki L, James R, Stankiewicz P. Genomic disorders: molecular mechanisms for rearrangements and conveyed phenotypes. PLoS Genet. 2007;1:e49.
7. Molina J, Carmona-Mora P, Chrast J, et al. Abnormal social behaviors and altered gene expression rates in a mouse model for Potocki-Lupski syndrome (PDF). Hum Mol Genet. 2008;17(16):2486–2495. doi:10.1093/hmg/ddn148
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.