")
Back to Journals » Hepatic Medicine: Evidence and Research » Volume 14
Post-Marketing Surveillance Study of the Safety and Efficacy of Nalfurafine (Capsules 2.5 μg, Oral Dispersing Tablets 2.5 μg) in 1186 Patients with Chronic Liver Disease and Intractable Pruritus
Authors Yoshitani H, Ito J, Kozono H
Received 6 December 2021
Accepted for publication 8 March 2022
Published 2 May 2022 Volume 2022:14 Pages 37—66
DOI https://doi.org/10.2147/HMER.S352775
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Gerry Lake-Bakaar
Hiroshi Yoshitani, Junko Ito, Hideki Kozono
Pharmaceutical & Medical Device Vigilance Department, Toray Industries, Inc., Tokyo, Japan
Correspondence: Hiroshi Yoshitani, Pharmaceutical & Medical Device Vigilance Department, Toray Industries, Inc., 1-1, Nihonbashi-Muromachi 2-chome, Chuo-ku, Tokyo, 103-8666, Japan, Tel +81 3 3245-8615, Email [email protected]
Background: Nalfurafine (Remitch®, Toray Industries, Inc.) is a selective κ-receptor agonist approved in Japan for the improvement of pruritus in patients with chronic liver diseases (only when existing treatments bring insufficient efficacy) in May 2015.
Methods: A post-marketing Specific Drug Use Survey was conducted in Japan (March 1, 2016 to June 30, 2020) of the safety and efficacy of nalfurafine for the improvement of pruritus in patients with chronic liver disease.
Results: Among 1186 cases analyzed for safety, the incidence of adverse drug reactions was 9.4% (112/1186 cases), lower than 61.4% reported in pre-marketing surveillance (297/484 cases). No specific safety issues were found and no cases of concern for drug dependence identified. Efficacy (itch improvement) was demonstrated in 73.16% (815/1114 cases; 12-week analysis set) and in 85.67% (520/607; general assessment of itch improvement at 1-year analysis set). A significant difference was found in 4 items of itch improvement at 12 weeks and 8 items of itch improvement at 1 year. No noteworthy issues were identified. Mean Visual Analog Scale (VAS) values after 12 weeks and 1 year after the first dose were significantly lower than the baseline (p < 0.0001 for both treatment durations). Mean severity scores (Kawashima’s classification scheme) were significantly lower than the pretreatment score at 12 weeks and 1 year after the first dose (both p < 0.0001). No concerns were identified in the efficacy and safety of nalfurafine in patients with specific background, ie, the elderly (aged ≥ 65 years), those with renal impairment, and those on long-term treatment (≥ 365 days) compared with patients without corresponding background.
Conclusion: No new safety issues of concern or cases of insufficient efficacy were identified in this Specific Drug Use Survey of the safety and efficacy of nalfurafine for the improvement of pruritus in patients with chronic liver diseases.
Keywords: post-marketing surveillance, safety, efficacy, nalfurafine, pruritus, chronic liver disease
Background
With estimated 450,000 people in Japan treated for chronic liver disease1 and as many as 1.7 million yet untreated for hepatitis,2 pruritus is estimated to be highly prevalent in Japan. Itching associated with chronic liver diseases is observed in hepatitis, liver cirrhosis and primary biliary cholangitis (PBC). It is often intractable and can significantly compromise patient quality of life. Patients with PBC in particular suffer from itchiness from an early stage of the disease, which can be so severe as to cause sleep disorders.3–5
Pruritus is reportedly experienced by 2.5% to 69% of patients with chronic liver diseases including PBC, hepatitis B, hepatitis C, and liver cirrhosis, but the incidence varies widely with the underlying diseases or studies. Exact figures are not available due to the difficulty in the objective evaluation of itchiness.6–12 Multiple factors may contribute to itchiness with no association identified between the severity of itchiness and the skin conditions or the level of serum bile acids among patients with intractable pruritus and underlying chronic hepatic disorder.
Common treatments for itchiness include prescription of antihistamines, antiallergic agents, and hypnotics,13 but their efficacy can be insufficient. Therefore, novel treatments for pruritus have recently been developed. Among others, the use of selective κ-opioid receptor agonist has been explored as a possible treatment for pruritus in patients with chronic liver diseases. Studies suggest that the opioid peptide-opioid receptor may control the transmission and amplification of the itching signal in the central nerve system, independent of the action of histamine.14,15
Nalfurafine (Remitch®) is a selective opioid-kappa (κ) receptor (κ-receptor hereafter) agonist developed by the Pharmaceutical Research Laboratories of Toray Industries, Inc. in 1992. Selective binding of nalfurafine to the κ-receptor has been demonstrated in vitro.16 Nonclinical studies have shown its efficacy for itchiness in experimental animal models of pruritus for which antihistamines and other common antipruritics are ineffective.14,17 Unlike morphine and other μ-receptor agonizing agents, nalfurafine caused no dependence,18–20 nor was it associated with aversion, which is often a problem with existing κ-receptor agonists.21,22 Based on these observations, Toray Industries, Inc. launched a program of clinical studies in 1998. The efficacy and safety of nalfurafine were subsequently demonstrated in the treatment of pruritus in patients on hemodialysis who did not respond sufficiently to existing treatments, and the company proceeded to apply for manufacturing and marketing approval. In January 2009, nalfurafine was approved in Japan as the first oral antipruritic anywhere in the world with an indication for the improvement of pruritus in patients on hemodialysis (for use only when existing treatments bring insufficient efficacy). Its efficacy was further demonstrated in Phase III and subsequent long-term follow-up Phase III trials. A multicenter, double-blind controlled Phase III trial23 studied the efficacy of repeated-dose oral nalfurafine (2.5 µg or 5 µg once daily for 12 weeks) in 316 patients with chronic liver diseases using the Visual Analog Scale (VAS). A subsequent open-label extension Phase III trial (approval application data) also used the VAS to evaluate the efficacy of oral nalfurafine (5 µg once daily for 52 weeks) in 122 patients with chronic liver diseases. None of the trials identified either physical or psychological dependence with nalfurafine.
In May 2015, nalfurafine received an approval for the indication of improvement of pruritus in patients with chronic liver diseases based on the results of the studies described above (ie, for use only when existing treatments bring insufficient efficacy).
An open-label clinical trial found the efficacy and safety of nalfurafine in patients on peritoneal dialysis with pruritus refractory to existing treatments, and an approval was obtained in September 2017 for the additional indication of “the improvement of pruritus in patients on peritoneal dialysis (for use only when existing treatments bring insufficient efficacy).” When nalfurafine was approved for this indication, a post-marketing surveillance was required to evaluate the safety and efficacy. More specific requests were made to review the incidence of insomnia and other sleep disorders as well as psychiatric disorders, drug dependence, and effects on serum prolactin and thyroid hormone levels.
Here, we report the results of the Specific Drug Use Survey conducted in Japan between March 1, 2016 and June 30, 2020, examining the safety and efficacy of nalfurafine for “the improvement of pruritus in patients with chronic liver diseases (for use only when existing treatments bring insufficient efficacy).”
Patients and Methods
In accordance with the protocol of the Japanese Ministry of Health, Labor and Welfare, this surveillance was conducted in compliance with the Good Post-marketing Study Practice (GPSP), or the Standard for Conducting Post-marketing Surveillance and Trials of Drugs, which is an ordinance enacted under the Japanese Pharmaceutical Affairs Law. This surveillance did not need informed consent from patients to conduct surveillance under the real-world, excluding bias from informed consent.
Patients
Patients with chronic liver disease with intractable pruritus identified as starting oral nalfurafine at any period between March 1, 2016 and December 25, 2018 were registered. Patients were administered nalfurafine after itch treatment with their existing therapy (ie, itch treatment such as antihistamine/anti-allergic agent, moisturizing agent, topical antihistamine, topical steroid, UV-B light therapy approved in Japan) was thought to be insufficiently effective by the physician.
Design
Patients were registered at an independent patient registration center for this prospective surveillance with a 1-year observation period. Survey items were baseline patient characteristics, nalfurafine dosage regimen, previous and concomitant treatment for pruritus, concomitant treatment for diseases other than pruritus, improvement in itch severity, symptoms (Child-Pugh grading, encephalopathy, ascites) and laboratory test results associated with hepatic dysfunction, dependence, and adverse events. The target number of cases was set at 1000. The reason being that 750 cases correspond to about 5% of the estimated number of eligible patients in Japan, who will be registered in the study, meaning adverse events at an incidence of 0.4% can be detected with a probability of 95%. In addition, 31 cases of floating dizziness (4.1%), which had the lowest frequency of occurrence among the priority items at the time of the clinical trial, can be predicted to be collected.
Assessment of Itch Improvement
Itch improvement was assessed by three items, namely, general assessment of itch improvement, VAS assessment and Kawashima’s severity classification.
A general assessment of itch improvement was performed by a physician providing an overall assessment of itch improvement and rated by three categories, ie, improved, stable and aggravated. The timing of assessment was decided at 12 ± 2 weeks and 1 year ± 1 month (30 days) after the first dose of nalfurafine. If treatment was terminated before either of these assessment time points, the assessment was to be performed at either ± 2 weeks or ± 1 month (30 days) from termination, respectively. Choices of unevaluable and unknown were additionally provided for rating under consideration for a scenario where the assessment or classification might not be possible for some reasons.
VAS assessment employed a 100-mm vertical linear scale with its left and right ends representing “no itchiness” and “the most severe possible itchiness”, respectively. Patients were asked to rate on the scale for severity of the most intense sensation of itchiness they experienced after breakfast and evening meal, and the distance from the left end on the scale (VAS value [mm]) was recorded. The assessment time points were pre-treatment with nalfurafine (within 1 month [30 days]), 12 ± 2 weeks and 1 year ± 1 month (30 days) after the first dose of nalfurafine. When a patient was terminated before 12 weeks or 1 year from the first dose, the assessment was performed ± 2 weeks or ± 1 month (30 days), respectively, from termination. The test used mean VAS values of baseline and the defined time points after the meal either in the morning or evening. The mean value of VAS was calculated by testing the larger value after breakfast or after dinner before and after the start of treatment in each case. The mean VAS value change was tested by the paired-sample t-test. The significance level was set at 5%.
For Kawashima’s severity classification, patients selected by themselves from the 5-point score (0: none, 1: Mild, 2: Moderate, 3: Severe, 4: Very severe) which best described the intensity of the itchiness at day and night. The assessment was performed before the first dose (within 1 month [30 days]), 12 ± 2 weeks and 1 year ± 1 month (30 days) after the first dose. When a patient terminated after the first dose but before 12 weeks or 1 year after the first dose, the assessment was performed ± 2 weeks or ± 1 month (30 days), respectively, from termination. The mean value of Kawashima’s severity score was using mean score in each patient at the baseline and the defined time points rated either the daytime or nighttime score whichever higher. The difference between the pre- and post-treatment mean scores was tested using the paired-sample t-test. The significance level was set at 5%.
Assessment of Safety
For the safety specifications established for this surveillance based on the drug risk management plan with nalfurafine operated in Japan, the following were investigated: incidence of insomnia, somnolence, (floating) dizziness and aggravated hepatic function (including abnormal laboratory changes related to hepatic functions). These were considered to be the important identified risks because the incidence of endocrine dysfunction such as serum prolactin has been reported to increase. Central adverse effects associated with coadministration of hypnotic, antianxiety, antidepressant, antipsychotic, or anti-epilepsy drugs are also considered to be important potential risks. Furthermore, the incidence of moderate and severe (Child-Pugh grades B and C) adverse drug reactions was also investigated in patients with hepatic impairment associated with nalfurafine as important missing information.
Dependence was evaluated with the Questionnaire of Drug Dependence comprised of questions relating to psychological dependence, physical dependence and tolerance related for the periods between the nalfurafine first dose and 12 weeks later, and between 13 weeks and 1 year after the initial dose. For each period, dependence during nalfurafine treatment was measured by 10 questions in the “on-treatment” (Table 1). Additionally, in case treatment was interrupted during each observation period, dependence during the 4 weeks after the end of treatment was assessed by 6 questions in the “off-treatment”. Each patient was assigned one of 4 options (“remarkable,” “moderate,” “slight,” or “none”). If a patient’s symptoms were described as either “remarkable” or “moderate,” the reasons expressed by the patient and the findings of the physician were also recorded.
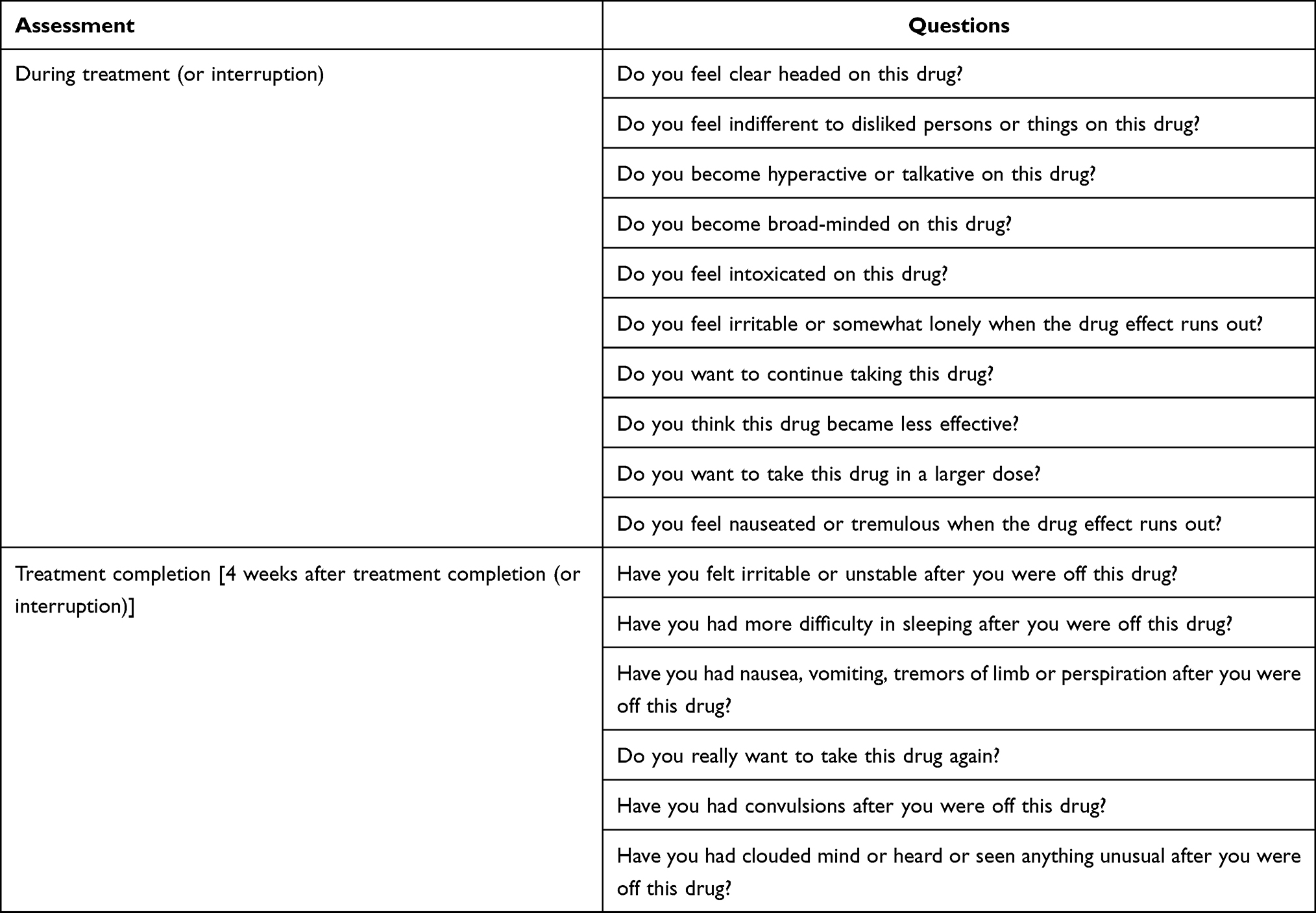 |
Table 1 Dependence Assessment |
Factorial Analysis on the Frequency of Adverse Drug Reactions (ADRs) and Efficacy
Seventeen characteristics were assessed in the factorial analysis, ie sex, age, registered department, Child-Pugh grading before nalfurafine administration, complications (present or absent), complications (each condition-based), medical history, duration of pruritus, allergy, average daily dose, total dose administered, duration of administration (days of administration), previous treatment for pruritus, concomitant medications for pruritus, concomitant medications for other than pruritus and concomitant medications (present or absent), concomitant medications (each medication-based). Additionally, to assess the safety and efficacy of patients with specific characteristics, children (under 15 years), elderly (65 years or above), pregnant women, renal dysfunction and long-term administration were assessed.
Statistical Analysis
Fundamental statistics were calculated from the VAS values, Kawashima’s severity classification, and the levels of serum prolactin and thyroid hormones observed in each patient. These were analyzed using the one sample t-test for the difference between the pre- and post-treatment values, with p values less than 0.05 regarded as statistically significant. Factorial analysis was conducted using Fisher’s exact test for 2 × 2 design and χ2 test for others.
Ethics Approval and Consent to Participate
In accordance with the Japanese Ministry of Health, Labor, and Welfare, this surveillance was conducted in compliance with the Good Post-marketing Study Practice and the Standard for Conducting Post-marketing Surveillance and Trials of Drugs, which is an ordinance enacted under Article 14, Section 4, Clause 4 and Article 14, Section 6, Clause 4 of the Japanese Law (ie, Pharmaceuticals and Medical Devices Law) for Ensuring the Quality, Efficacy, and Safety of Drugs and Medical Devices. Separate ethics approval for this surveillance and informed consent to participate in the surveillance were not required under Japanese law. All original data have been completely anonymized such that the privacy of patients or facilities involved was ensured.
Results
Baseline Patient Characteristics
A total of 1195 patients were registered at 210 institutions. Of these, 1186 patients were analyzed for safety and efficacy of nalfurafine (Figure 1). Table 2 shows the major demographic characteristics and the treatment profile of the safety analysis set, respectively. In this surveillance, on the basis of the mean daily dose, 93.51% (1109/1186 patients) were administered nalfurafine at 2.5 μg, 4.38% (52/1186 patients) received over 2.5 μg but <5.0 μg, and 1.94% (23/1186 patients) received 5.0 μg. Most of the registered patients kept taking the regular mean daily dose of 2.5 μg, but at least more than 4% of patients received the elevated dose of 5.0 μg.
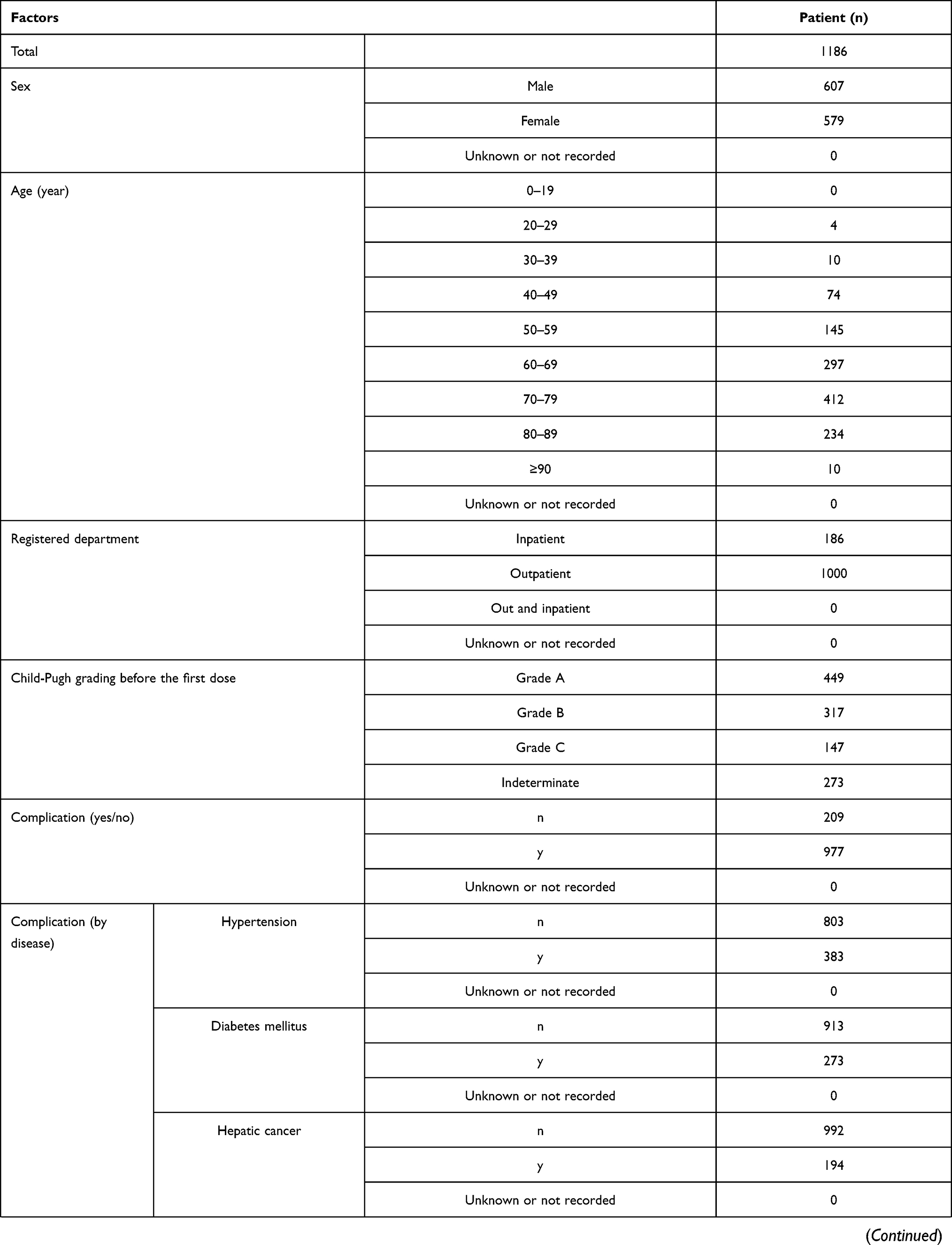 |  |  | 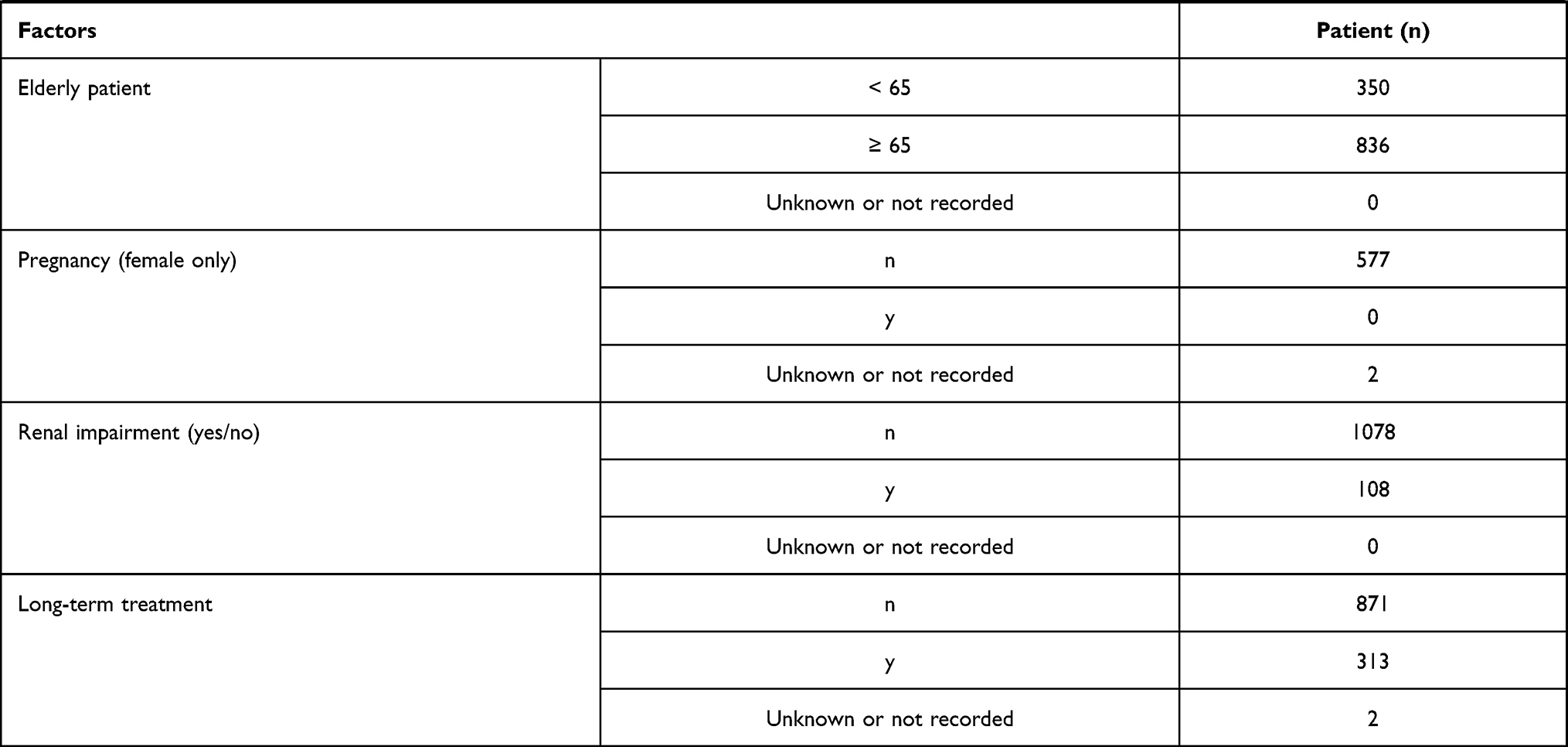 |
Table 2 Baseline Patient Characteristics |
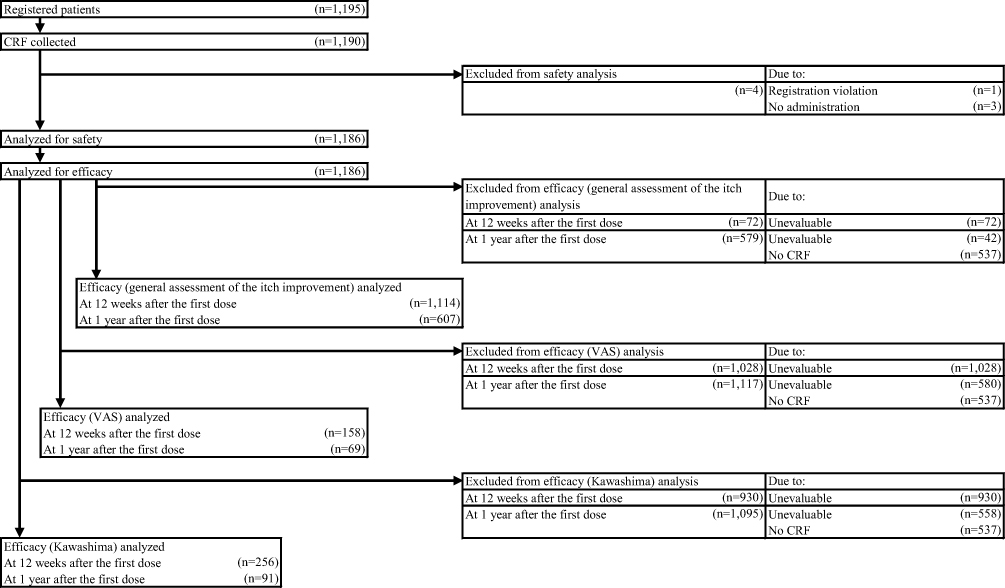 |
Figure 1 Participant flow diagram. |
Safety
Frequency of Adverse Drug Reactions
The frequency of ADRs according to each of the patient characteristics or the treatment characteristics is shown in Table 3. Among the 1186 patients analyzed for safety, 112 (9.44%) patients experienced ADRs. No specific ADR was common in this surveillance. Serious ADRs developed in 10 (0.84%) patients (Table 4). The most common serious ADR was hepatic encephalopathy seen in 4 cases, 3 of whom recovered and one did not. No causality to nalfurafine was confirmed owing to the potential relation to chronic liver disease, complications or concomitant medications. No discrepancy was seen between the ADR profile observed in this surveillance and the precautions for usage on the package insert.
 |  | 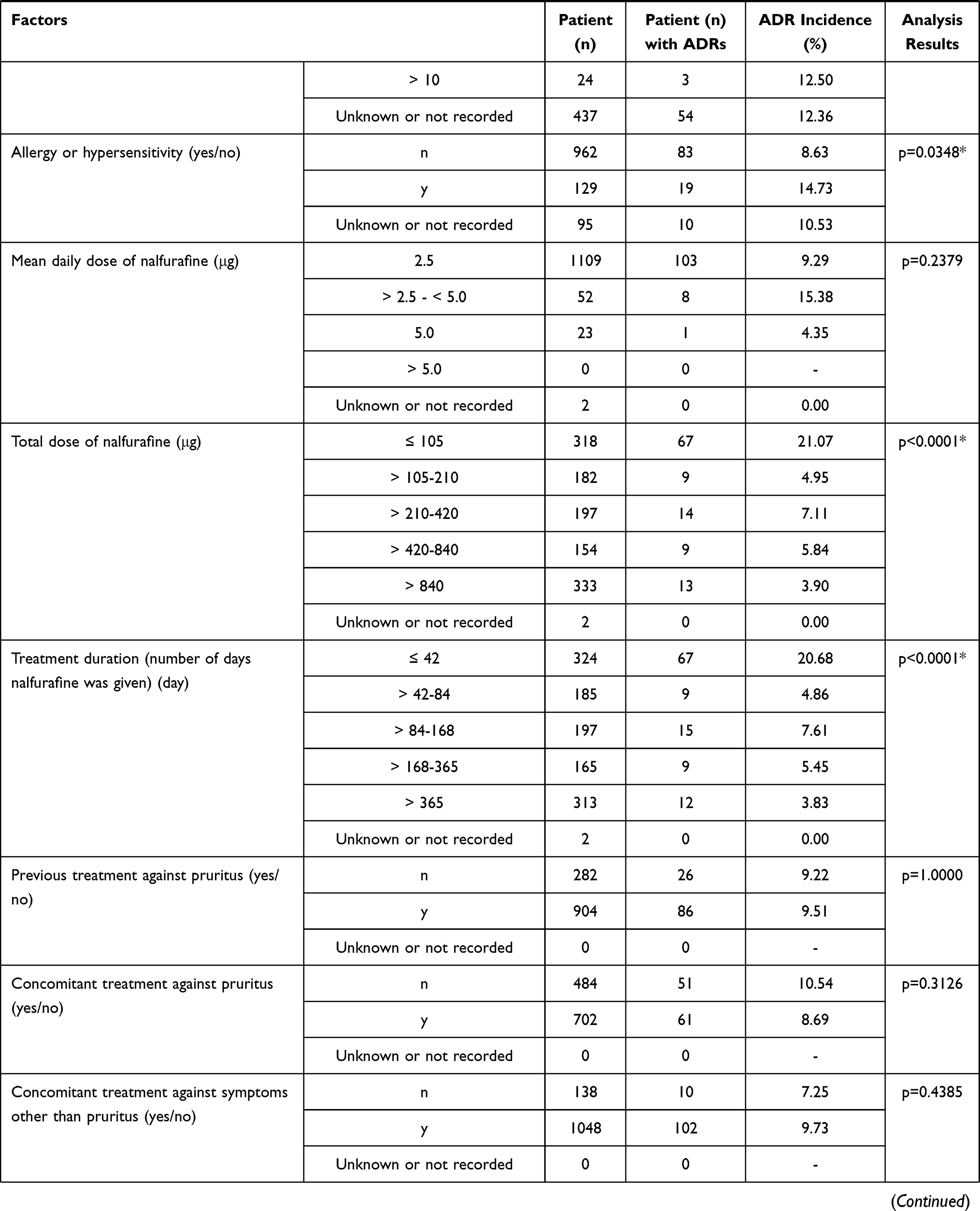 | 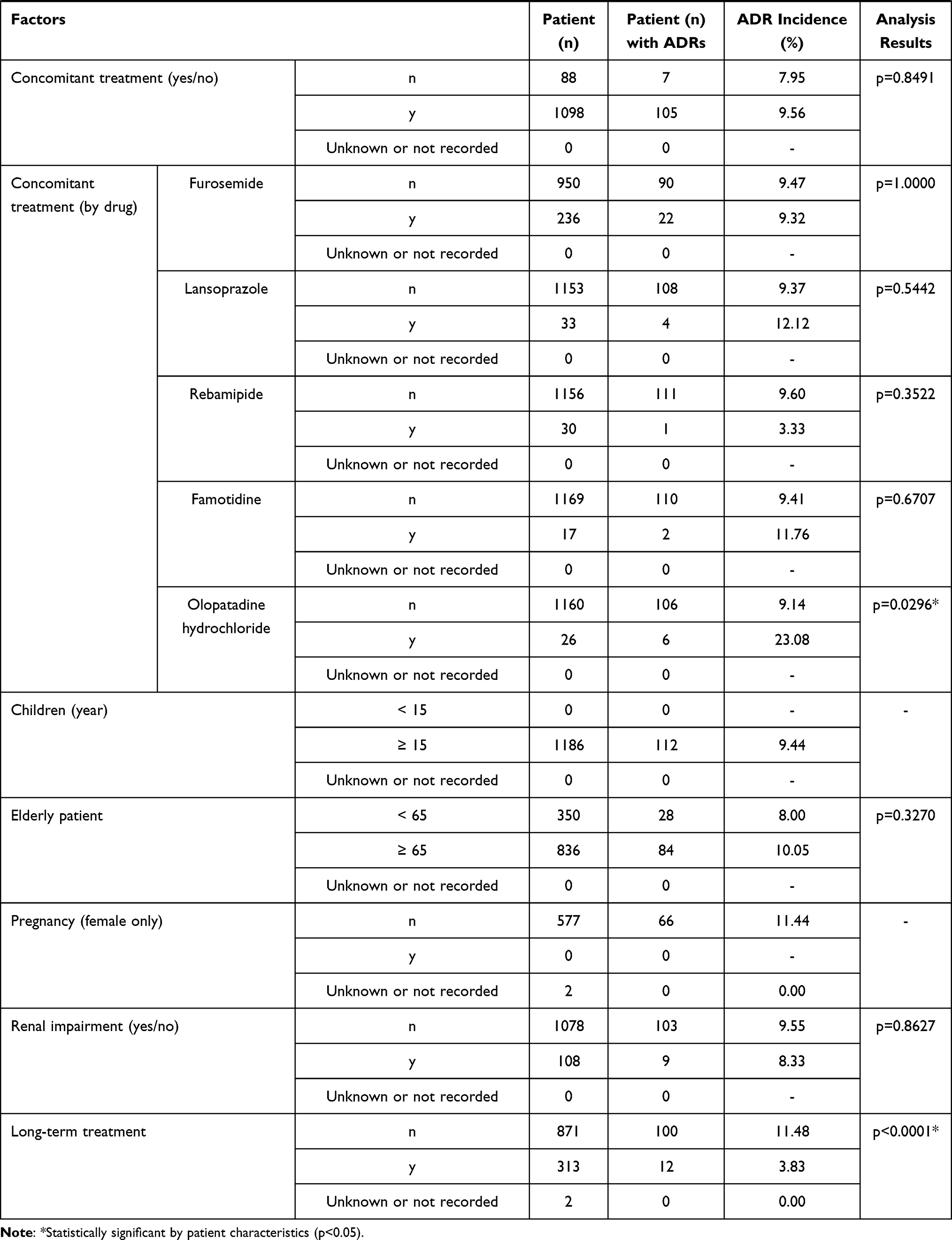 |
Table 3 Baseline Patient Characteristics and Frequency of ADRs |
 |
Table 4 Number and Incidence of Serious ADRs by Surveillance |
Factorial Analysis of ADR Frequency
Sex, complication of ascites, allergy, total dose administered, duration of administration (days of administration), concomitant use of olopatadine and long-term administration were found to be significantly higher in frequency for all ADRs.
The frequency of all ADRs was significantly higher in female patients (p = 0.0286). Significant differences were seen with “nervous system disorders” (p = 0.0408) and “kidney and urinary tract disorders” (p = 0.0433), but no common symptoms were identified. The reason for the difference was uncertain but no sex-specific trend in ADRs was seen, therefore no issue was raised based on this result.
The frequency of all ADRs was significantly higher in patients without ascites (p = 0.0473). The reason for this is uncertain and having no ascites should not be a risk for ADRs. There was no significant difference in the frequencies of each ADR, therefore no issue was raised based on this result.
The frequency of all ADRs was significantly higher in patients with allergy (p = 0.0348), with the frequency of “psychiatric disease” significantly higher in these patients (p = 0.0004). The most common ADR in the category of “psychiatric disease” in the patients with allergy was insomnia but as all 6 cases were not serious and all recovered this was considered not to be clinically relevant. The allergens for the 6 patients varied including drugs, foods and pollen without any tendency.
The incidence of ADRs was significantly higher in the group that received ≤ 105 µg by total dose (p < 0.0001) and in the group with ≤ 42 day by treatment duration (number of days nalfurafine was given) (p < 0.0001). The significantly higher incidence in these two groups can be explained by the larger number of cases in which the treatment was stopped or interrupted as a result of the ADRs that had occurred in an early stage after the first dose. The total administration dose was positively correlated (contribution rate: 0.9147, correlation coefficient: 0.9564) with the treatment duration (number of days nalfurafine was given) (Figure 2), and an excessive number of ADRs was observed in the group of ≤ 105 µg by total dose and the group of treatment for ≤ 42 days by treatment duration (number of days nalfurafine was given) populations.
 |
Figure 2 Association of total dose to dosing duration (number of days nalfurafine was given). |
The frequency of ADRs for each concomitant treatment drug was examined. The incidence of ADRs showed no significant difference in those patients who co-administered furosemide (p = 1.0000), lansoprazole (p = 0.5442), rebamipide (p=0.3522), or famotidine (p = 0.6707). In contrast, the frequency of ADRs was significantly higher in those who received concomitant olopatadine hydrochloride (p = 0.0296). A comparison of the frequency of each ADR in populations with and without olopatadine coadministration identified a significant difference in standard of care of “skin and subcutaneous tissue disorders” (2/26 cases, p = 0.0407) and “renal and urinary disorders” (2/26 cases, p = 0.0459). Specifically, these were generalized systemic dermatitis exfoliative, eczema, renal disorder, and renal impairment reported in one patient each. The higher incidence appears to be attributed to the small size of the olopatadine coadministration population (26 patients). Although the reason remains unknown for the higher incidence of ADRs only in the group with concomitant olopatadine hydrochloride, the surveillance found no trend of ADRs specifically associated with this concomitant drug.
The proportion of specific patient subgroups in the safety analysis set was 0% for children (< 15 years old; data not collected), 70.49% (836/1186 cases) for elderly patients (≥ 65 years), 0% for pregnant or postpartum women (data not collected), 9.11% (108/1186) for patients with renal impairment, and 26.39% (313/1186) for patients with long-term treatment (≥ 365 days). The frequency of ADRs in specific patient subgroups was not significantly different to the overall population in elderly (≥ 65 years old or older, p = 0.3270) or patients with renal impairment (p = 0.8627) but was significantly higher than the overall population in patients that received long-term treatment (p < 0.0001). The significantly higher frequency of ADRs in the population with extended treatment can be at least partially attributed to a larger number of cases in which the onset of ADRs at an earlier stage of the treatment required termination or interruption of nalfurafine, as seen with the assessments by total dose administered and duration of administration (days of administration).
Important Identified Risks
The incidence rates of insomnia, somnolence, and dizziness were 1.6% (19/1186 patients), 1.1% (13/1186), and 1.0% (12/1186), respectively. The frequency of aggravated hepatic function was 0.4% (5/1186), which were hepatic encephalopathy in 4 patients and hepatic function abnormal in 1 patient. Other relevant laboratory test results that suggested possible aggravation of hepatic function were increased aspartate aminotransferase, increased blood lactate dehydrogenase, hypoalbuminemia, and decreased total protein in one patient each (0.1%, 1/1186). Included in these events, a total of 9 events were reported in 7 patients, and the ADR incidence was 0.6% (7/1186), suggesting no specific issues without relatively common ADRs including those of laboratory test values.
Important Potential Risks
One sample t-tests (5% significance level) of the blood prolactin, thyroid stimulating hormone and free thyroxine levels showed no significant difference between the laboratory test results before and after the first dose of nalfurafine. Reported ADRs of endocrine disorders such as increased blood prolactin were hyperprolactinemia in 1 patient (0.1%, 1/1186) and increased blood prolactin in 2 patients (0.2%, 2/1186).
Some relatively common ADRs of the central nervous system were found in cases where hypnotics, etc., was co-administered with nalfurafine: insomnia was experienced by 3.70% (6/162 cases) of patients with concomitant hypnotic or antianxiety drugs, and hepatic encephalopathy by 8.33% (1/12) of patients with concomitant anti-epilepsy drugs. All cases of insomnia reported in patients with concomitant hypnotic or antianxiety drugs were non-serious. Hepatic encephalopathy occurred in 1 patient with concomitant anti-epilepsy drug and this high incidence was attributed to the small size of the population (12 cases).
Patients with Moderate to Severe Hepatic Impairment (Child-Pugh Grade B or C)
As shown in Table 3, the frequency of ADRs did not differ according to the Child-Pugh grades assessed prior to the first dose of nalfurafine.
Dependence
Responses to a questionnaire on dependence were collected and evaluated from the 1186 patients included in the safety analysis set. For the questions that allowed multiple responses, the lowest score was adopted for counting. For each question, patients who rated “remarkable” or “moderate” were examined for suspected dependence based on reasons for rating of the question and findings of the physicians.
A “remarkable or moderate” response was collected from 1/1051 cases for the question (psychological dependence-related), “Do you feel clearheaded on this drug?” The explanations and findings for this case are read as “Explained by the patient, who passed away before actual evaluation of dependence was commenced.” Since the patient died of hepatocellular carcinoma one week after the first dose of nalfurafine, it was impossible to evaluate dependence in this patient. No ADRs of psychological or nervous disorders were identified in this patient. Therefore, it appeared that the rating on the question might have been influenced by improvement in pruritus as described in the paragraph for the question, “Do you want to continue taking this drug?”.
In 3/1051 cases, “remarkable or moderate” was given in response to the question “Do you feel indifferent to disliked persons or things on this drug?”. Two cases were explained the basis for the evaluation as “Because (this drug) reduces the itchiness” and “Because of the casual, carefree personality even prior to the drug administration”, which suggests no dependence. The other patient was assigned “remarkable” on the question at 3 weeks after the first dose of nalfurafine and the physician stated, “No explanation can be provided.” The drug administration was continued, and the patient was ultimately assigned “none” for the answer to the question at 61 weeks after the first dose of nalfurafine. Hence, the last patient was not suspected of dependence.
In 1/1051 cases, a “remarkable or moderate” grading was given in response to the question “Do you become hyperactive or talkative on this drug?” This case was interpreted as “an expression of increased daily activity due to remarkably alleviated itchiness.” Again, the response seemed simply to reflect improvements in pruritus.
In 4/1051 cases, “remarkable or moderate” grading was given in response to the question “Do you feel intoxicated on this drug?”. In one case, the reason was explained as, “The itchiness does not immediately go away. Feels drowsy during daytime,” and somnolence was reported as an ADR. In another case, the physician found “vertigo on Days 1 and 2 after drug administration,” and vertigo and delirium were reported as ADRs. Still, another case came with the physician’s finding, “the hallucination had improved before the patient came to my office. The patient had already stopped taking the drug. Considering the visit was on May 2nd, entry is as it is,” and hallucination was reported as an ADR. In the other case, the physician’s findings said, “Details unknown,” and the event was not considered as an ADR; no other findings suggesting dependence were provided.
In 1/1051 cases, “remarkable or moderate” was given in response to the question “Have you felt irritable or unstable after the effect of this drug you are taking wears off?” The explanations and findings said, “According to the patient’s description of the symptoms.” We interpreted the entry was not associated with constant cravings for the drug and the case was not suspected of dependence because the physician had noted, “the patient neither asked nor wished for treatment” and the response to the same question was “none” for the dependence assessment 5 months after the first dose of nalfurafine.
Of the 44/1051 cases where “remarkable or moderate” was given in response to the question “Do you want to continue taking this drug?”, 43 provided explanations expressing hopes to benefit from the efficacy of nalfurafine such as, “Want to continue to alleviate the itch.” Another example of the explanations and findings entry was, “Orally explained by the patient, who passed away before actual evaluation of dependence was commenced.” As the patient died of hepatocellular carcinoma one week after the first dose of nalfurafine, it is impossible to evaluate the dependence in this case. However, the efficacy evaluation entries (general assessment of the itch improvement “improved,” and Kawashima’s severity classification downgraded from 4 to 1) suggested the entry may reflect the patient’s hopeful expectation for drug efficacy.
All of the 5/1051 cases in which “remarkable or moderate” was given in response to the question, “Do you want to take this drug in larger doses?” expressed their expectation for the drug efficacy, saying, “I’m not bothered by the itch when I’m on this drug,” or “for the aggravated scratch-induced lesions in the legs,” for instance.
The 1/568 case of “remarkable or moderate” given in response to the question “Have you had more difficulty in sleeping after stop using this drug you take?” and the explanations and findings were provided as follows: “The symptom was described by the patient.” We interpret the explanation as an expression of the hope for the drug efficacy, as is stated in the next section on the question, “Do you really want to take this drug again?”
The explanations and findings given by the 2/568 cases of “remarkable or moderate” given in response to the question “Do you really want to take this drug again?” were interpreted as the hope for the drug efficacy saying, “The itch has decreased after started to take the drug.” and “The wish to relieve the itch symptom.”
No patients were rated as “remarkable or moderate” for the following questions: “Do you become broad-minded on this drug?”, “When this drug wears off, do you experience nausea or shaking of the arms and legs?”, “After stopping to use this drug, do you feel restless or irritated?”, “Have you had nausea, vomiting, tremors of limb after you stopping to use this drug?”, “Have you had convulsions after you stopping to use this drug?” and “Have you had clouded mind or heard or seen anything unusual after you stopping to use this drug?”.
To the tolerance-related question, “Do you think this drug became less effective?”, a response of “remarkable or moderate” was given in 8/1051 cases, of which 3 expressed nalfurafine as being less effective than expected. Further 3 cases provided the explanations and findings, “The itchy skin has exacerbated”, “Improvement is noticed in comparison with the baseline, but systemic itchy sensation has intensified” and “for the aggravated scratch-induced lesions in the legs.” Further two were suspected to have developed tolerance to nalfurafine on the basis of the explanations and findings, noting that “The itch is not alleviated as effectively as before,” and “Nalfurafine was resumed to address recurrent itchiness. The patient experienced less effective improvement of the subjective symptom than during the previous administration”.
However, we did not find any other entries in the explanations and findings for other questions that suggested dependence.
In conclusion, dependence was not suspected in any patient. Nalfurafine was administered in a single case among those excluded from the safety analysis, but neither “remarkable” nor “moderate” was assigned to any of the question items in this case.
Efficacy
General Assessment of Itch Improvement
At 12 weeks, 1114 patients were analyzed, excluding 72 whose data were unevaluable (Figure 1). At 1-year, 607 patients were analyzed, while 579 whose data were unevaluable or missing. Nalfurafine was determined to be effective (ie, as evidenced by improvement in itch severity) in 73.16% (815/1114 patients) at 12 weeks and 85.67% (520/607 patients) at 1 year.
Factorial Analysis of Efficacy
A significant difference was observed in the stratified response rate by the general assessment of itch improvement at 12 weeks in the following 4 items: a) total dose (p < 0.0001), b) treatment duration (number of days nalfurafine was given) (p < 0.0001), c) concomitant treatment (present or absent) (p = 0.0344), and d) long-term treatment (p < 0.0001) (Table 5). And at 1 year, in the following 8 specifications. a) The Child-Pugh grade before the first dose (p = 0.0254), b) complication (hepatic cancer) (p = 0.0128), c) mean daily dose (p = 0.0070), d) total dose (p = 0.0001), e) treatment duration (number of days nalfurafine was given) (p < 0.0001), f) concomitant treatment against symptoms other than pruritus (p = 0.0318), g) renal impairment (p = 0.0215), and h) long-term treatment (p < 0.0001) (Table 6).
 | 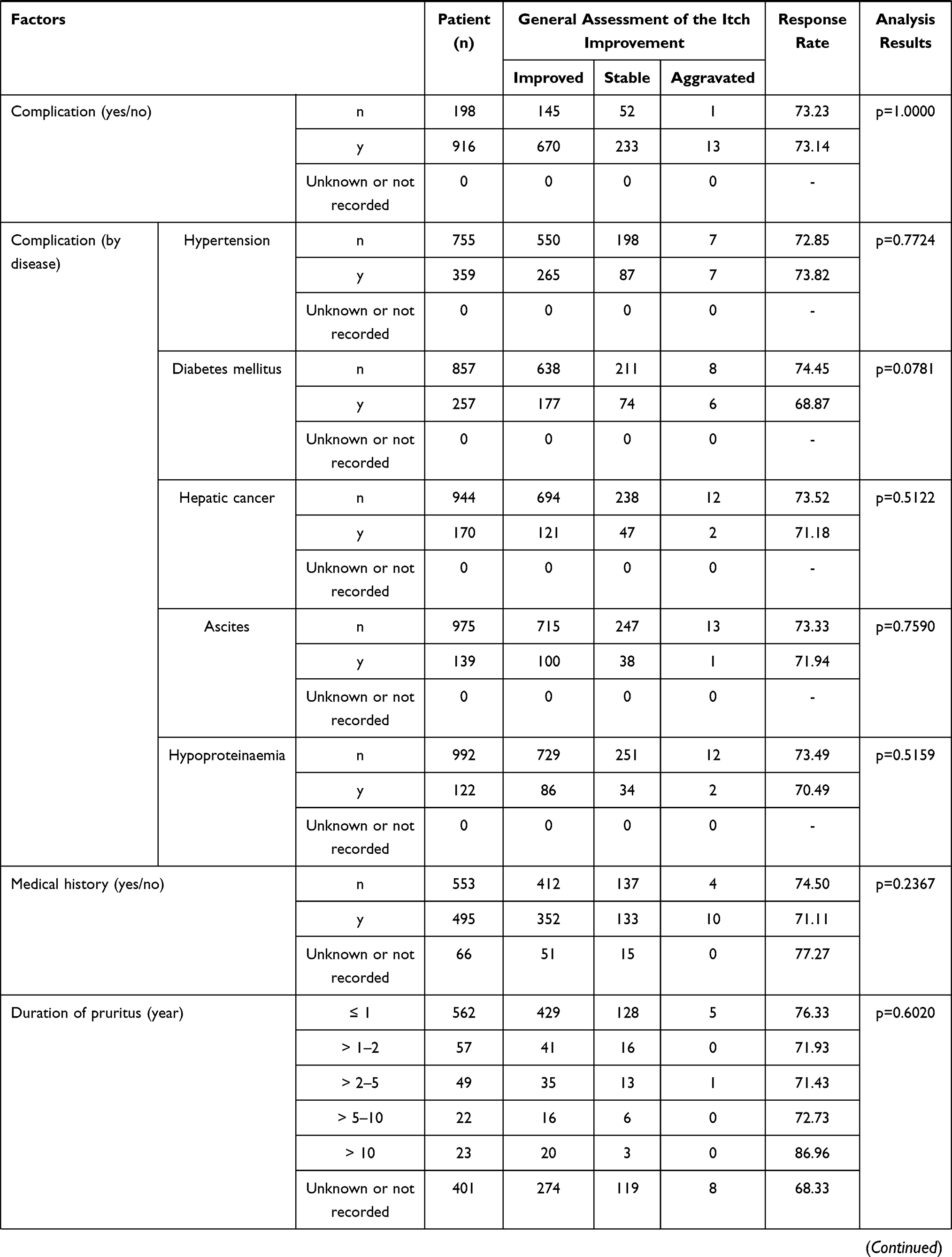 | 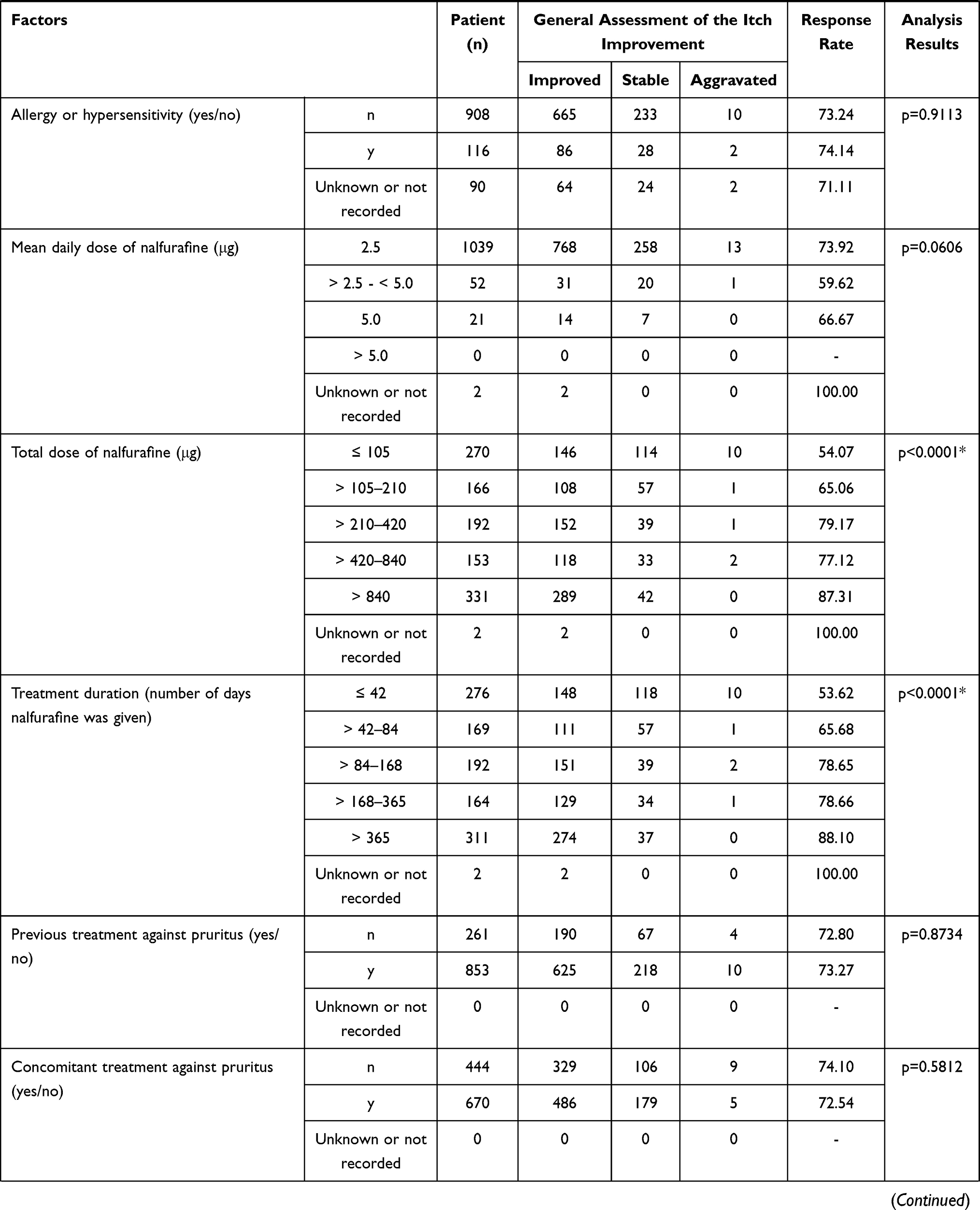 |  |
Table 5 Response Rate (General Assessment of the Itch Improvement at 12 Weeks) by Baseline Patient Characteristics |
 | 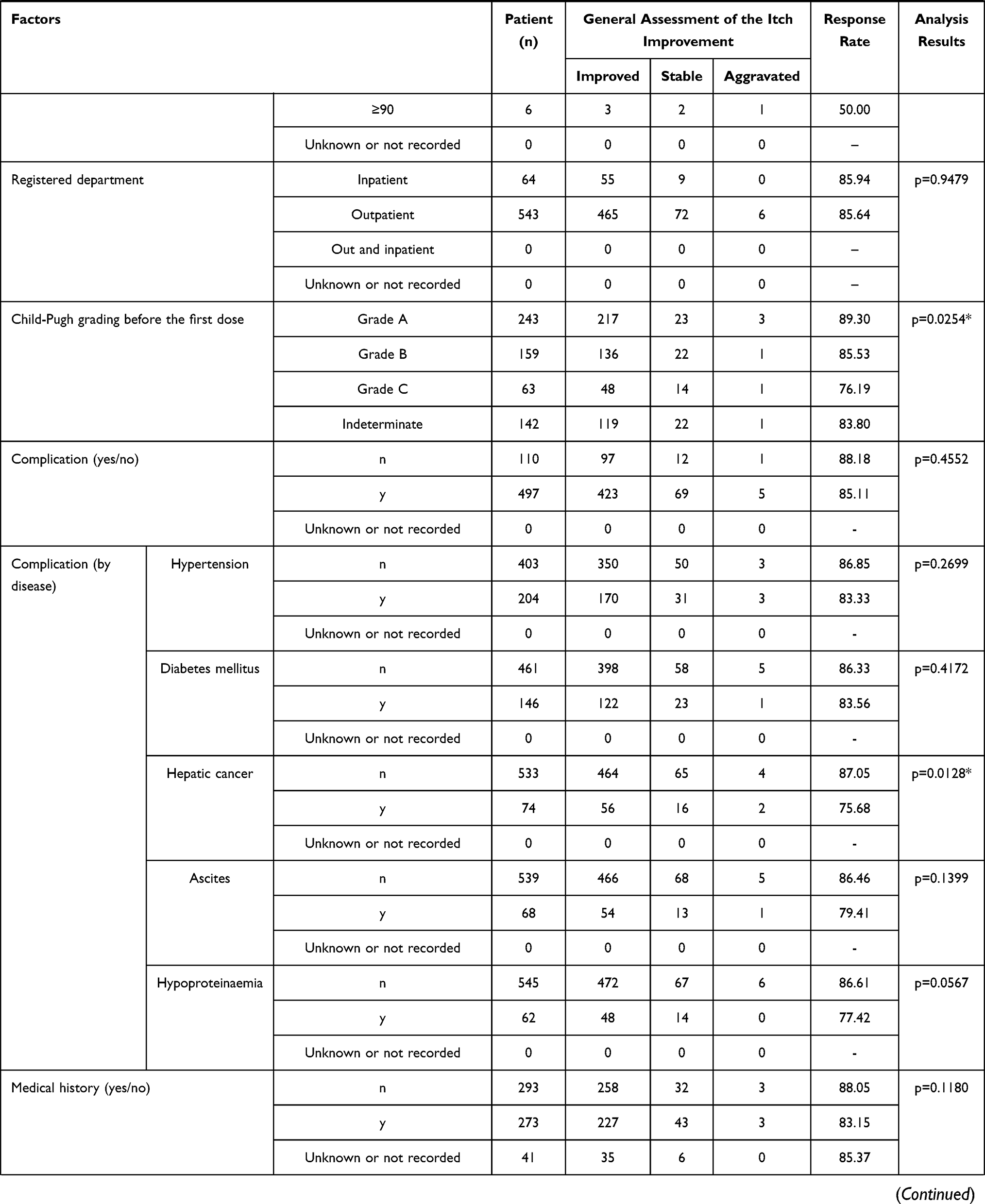 |  |  |
Table 6 Response Rate (General Assessment of the Itch Improvement at 1 Year) by to Patient Characteristics |
Pre-Administration Child-Pugh Grading
The general assessment of itch improvement at 1 year showed a lower response rate in the grade C population at 76.19% (48/63) compared with grade A (89.3%, 217/243) and grade B (85.53%, 136/159; overall p = 0.0254). Importantly, the response rate in the grade C patients cannot be considered extremely low, and therefore should not be interpreted as poor efficacy.
Complication (Hepatic Cancer)
The general assessment of itch improvement at 1 year found a lower response rate at 75.68% (56/74 cases) in the population with a complication (hepatic cancer), compared with the response rate of 87.05% (464/533) in those without hepatic cancer (p = 0.0128). An analysis of variance of the complication (hepatic cancer) and mean daily doses found that the population with complication (hepatic cancer) comprised a larger proportion of patients treated with the mean daily dose of > 2.5 µg to < 5.0 µg (Table 7). The response rate in the general assessment of itch improvement at 1 year was lower in that population treated with the mean daily dose of > 2.5 µg to < 5.0 µg. The statistically significant difference by complication (hepatic cancer) could have been confounded by the different composition of each population with different ratio of mean daily-dose groups, with a larger proportion of patients that received the mean daily dose of > 2.5 µg to < 5.0 µg in the population with complication (hepatic cancer).
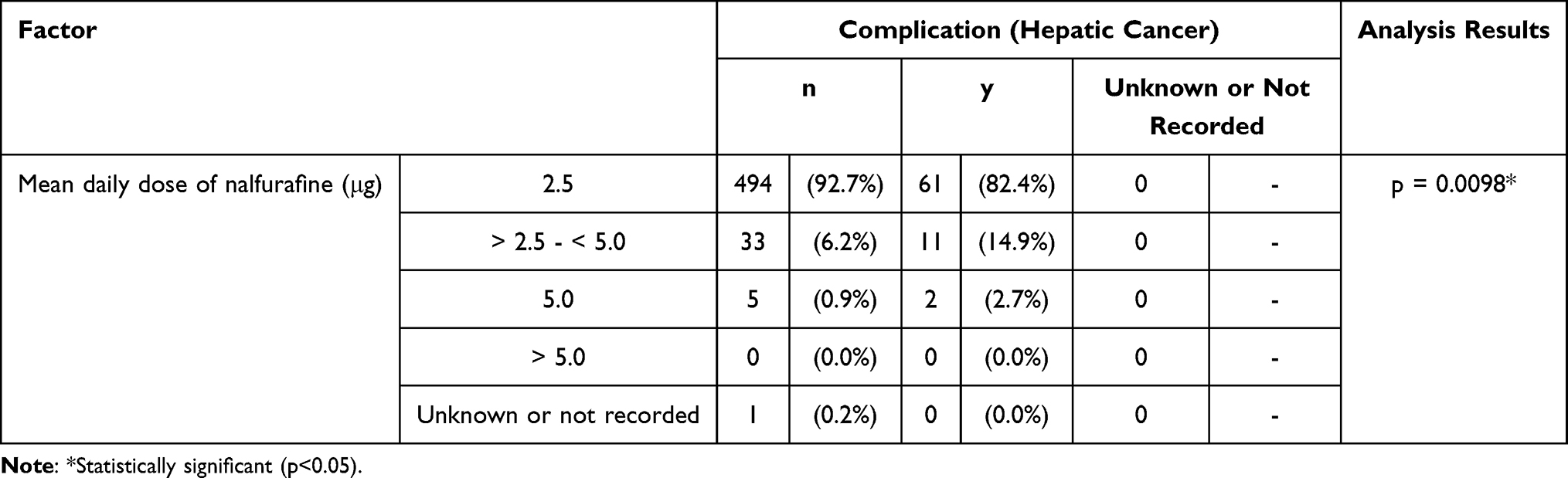 |
Table 7 Mean Daily Dose for Patients with or without Complication (Hepatic Cancer) |
Mean Daily Doses
According to the general assessment of itch improvement at 1 year, the response rate of 70.45% (31/44) in the population treated with the mean daily dose of > 2.5 µg to < 5.0 µg was low relative to other treatment groups (p = 0.0070). In fact, 43 patients, or all but a single exception in this treatment group, had received an elevated dose of 5.0 µg from the baseline 2.5 µg. Based on the results of a stratified analysis, separating the patients treated with and without dose elevation (Table 8), the statistically significant difference may be explained by the fact that the treatment group with mean daily dose of > 2.5 µg to < 5.0 µg comprises a good number of patients who received a higher dose specifically because they did not respond well at the baseline 2.5 µg.
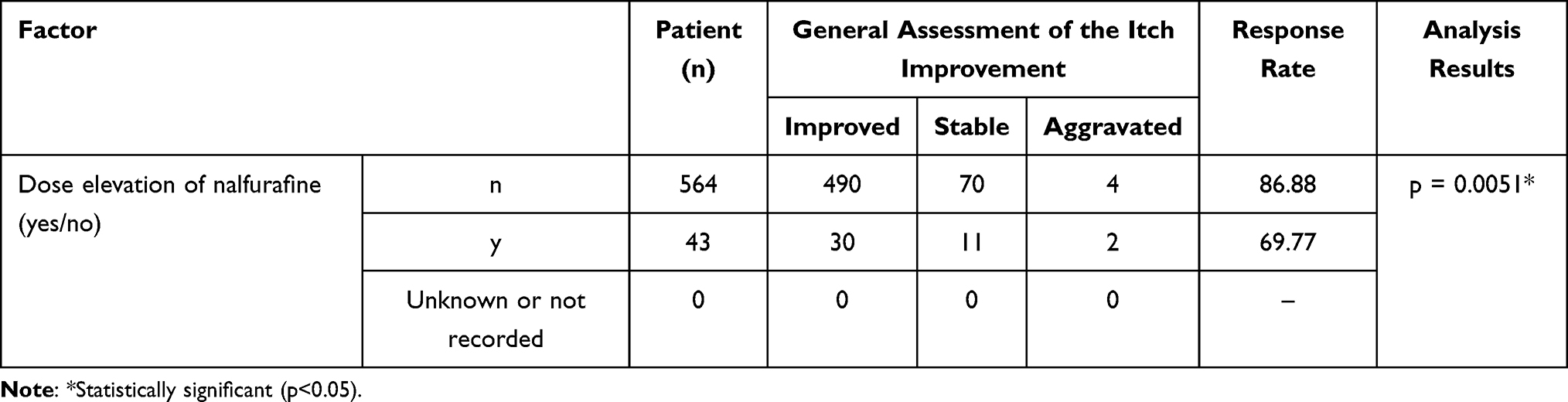 |
Table 8 Response Rate of Patients Treated with or without Dose Elevation of Nalfurafine (General Assessment of the Itch Improvement at 1 Year) |
Total Dose and Treatment Duration (Number of Days Nalfurafine Was Given)
In both general assessments of itch improvement at 12 weeks and 1 year, the population that received a larger dose and that was treated for a long term (more actual days administered) showed a higher response rate (p < 0.0001, p = 0.0001, and p < 0.0001, p < 0.0001, respectively). The results imply that those patients who benefited from the higher efficacy of nalfurafine might have continued treatment.
Concomitant Treatment and Symptoms Other Than Pruritus
The general assessments of itch improvement at 1 year showed a low response rate of 84.62% (462/546) in the population with concomitant treatment for symptoms other than pruritus compared to 95.08% (58/61) in the population without treatment (p = 0.0318). The reason for the difference in the response rate is still unknown, but the response rate was not remarkably low in the population that received concomitant treatment against symptoms other than pruritus. Hence, the results do not appear to suggest a lack of efficacy.
Concomitant Treatment (with or without)
The general assessment of itch improvement at 12 weeks showed a lower response rate of 72.37% (749/1035) in the population that received concomitant treatments, in contrast with the 83.54% (66/79) response rate in the rest of the population that did not take any other drugs concomitantly (p = 0.0344). Although the reason for this difference is unknown, the response rate in the population that received concomitant treatment is not remarkably low, and it is no sign of a lack of efficacy.
Specific Patient Subgroups
Regarding specific patient subgroups, the proportion of patients in the efficacy analysis set (overall assessment at 12 weeks) was 0% for children (< 15 years old, data not collected), 70.20% (782/1114 patients) for the elderly (≥ 65 years), 0% for pregnant or postpartum women (data not collected), 9.25% (103/1114) for patients with renal impairment, and 27.92% (311/1114) for patients with long-term treatment (≥ 365 days). The proportion of patients in the efficacy analysis in specific subgroups (overall assessment at 1 year) was 0% for children (< 15 years old, data not collected), 69.36% (421/607) for the elderly (≥ 65 years), 0% for pregnant or postpartum women (data not collected), 8.57% (52/607) for patients with renal impairment, and 50.74% (308/607) for long-term treatment. According to the assessment at 1 year, the response rate was 84.68% (470/555) in the population without renal impairment, which was relatively lower than the rate of 96.15% (50/52) in those with renal impairment (p = 0.0215). Although the reason for the significant difference remains unknown, the response rate of the population without renal impairment is not extremely low, which therefore does not suggest a lack of efficacy. The response rate to nalfurafine analyzed by the population for long-term treatment was higher in both general assessments of itch improvement at 12 weeks and 1 year. Cases of demonstrated efficacy were thought to be the result of continued administration of nalfurafine.
VAS
The change in the mean VAS value was tested at 12 weeks and 1 year after the first dose with nalfurafine. The test results provided in Tables 9 and 10 show the post-treatment values at 12 weeks and 1 year after the first dose were significantly lower than the baseline (p < 0.0001 and p < 0.0001, respectively).
 |
Table 9 VAS Assessments (12 Weeks After First Dose) |
 |
Table 10 VAS Assessments (1 Year After First Dose) |
Kawashima’s Severity Score
The change in the mean score was tested at 12 weeks and 1 year after the first dose with nalfurafine in comparison to baseline. The test results provided in Tables 11 and 12 show that the mean scores decreased significantly both at 12 weeks and 1 year after the first dose (p < 0.0001 and p < 0.0001, respectively).
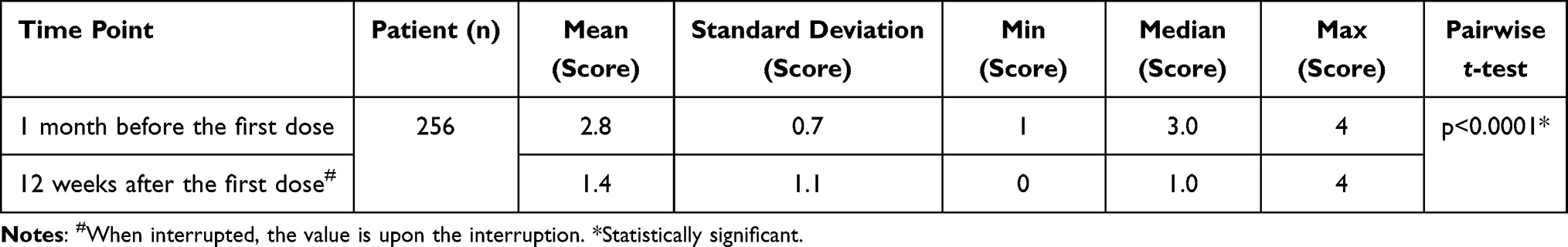 |
Table 11 Kawashima’s Severity Score Results (12 Weeks After First Dose) |
 |
Table 12 Kawashima’s Severity Score Results (1 Year After First Dose) |
Discussion
This surveillance aimed to review the safety and efficacy of treatment with nalfurafine for the improvement of pruritus in patients with chronic liver disease (for use only when existing treatments bring insufficient efficacy). By the end of the re-examination period, 1195 cases were registered at 210 institutions, and 1190 case report forms were collected from 206 institutions.
The frequency of ADRs was 9.4% (112/1186), which is lower than the 60.0% in the 2.5 µg group and 54.1% in the 5.0 µg group reported in the pre-approval surveillance, a placebo-controlled double-blind phase III study of patients with chronic liver disease-associated refractory pruritus that had not been well controlled by antihistamines and anti-allergic therapy.23 In terms of serious ADRs, 0.8% (10/1186) in this study was similar to 1.0% in the 2.5 µg group and 1.8% in the 5.0 µg group reported in the pre-approval surveillance study.
There was no relatively common ADR observed in the present surveillance. The most common serious ADR reported was hepatic encephalopathy, which may have resulted from a chronic liver disease, complications or concomitant treatment, and thus no clear association with nalfurafine was suspected. No ADRs were observed that are inconsistent with the warnings and precautions stated in the current package insert.
The factorial analysis of 22 baseline characteristics of patients showed significant difference in 7 factors, but further investigation suggested that none of them influenced the frequency of ADRs. The reviewed safety specifications were insomnia, somnolence, dizziness, aggravated hepatic function, increased blood prolactin and other endocrine dysfunction, concurrent use of sleep drugs, antianxiety drugs, antidepressants, antipsychotics, or antiepileptics, and use in patients with moderate to severe (Child-Pugh grades B and C) liver disease. No substantive issues were identified in the surveillance. Importantly, no cases of concern were found following screening for suspected cases of dependence.
The general assessment of the itch improvement was 73.16% (815/1114) of 1114 cases in the efficacy (general assessment of the itch improvement at Week 12) analysis set, and 85.67% (520/607) of 607 cases in the efficacy (general assessment of the itch improvement at 1 year) analysis set. As the general assessment of the itch improvement rate was not a measure of efficacy in the pre-approval surveillance, no comparison was made between the two surveillances. Based on the factorial analysis of 22 items in the baseline patient characteristics, 4 factors in the efficacy (general assessment of itch improvement at Week 12) analysis set and 8 factors (general assessment of itch improvement at 1 year) showed significant difference.
The response rate was lower among the patients classified Child-Pugh grade C before the first dose, but not to such a degree to be evaluated as ineffective. Some patients did not respond to a mean daily dose of >2.5 µg to <5 µg. This may have contributed to the statistically significant difference in the mean daily dose, as well as to the significant difference between the populations with and without a complication (hepatic cancer). Larger doses and longer treatment duration (number of days nalfurafine was given) appeared to result in better response rate, which may simply reflect those responders continued the treatment with nalfurafine. Despite significant differences in the response rate between with and without concomitant treatment, none of the differences suggested a lack of efficacy. Likewise, although the reason remains unknown for the difference in response rate between with and without renal impairment, the response rate was as high as 84.68% in patients without renal impairment, which similarly did not suggest a lack of efficacy. In summary, none of the factors examined affected the general assessment of itch improvement.
Efficacy was measured by the VAS assessment and Kawashima’s severity criteria. Compared to the baseline before the first dose of nalfurafine, mean VAS values, as well as the average severity scores, were significantly lower both at 12 weeks and 1 year after the first dose of nalfurafine. In the pre-approval surveillance, the primary endpoint for the evaluation of efficacy was the change in the VAS value before and after the start of the drug treatment. After 12 weeks, which is the longest observation period in pre-approval surveillance, the change in the placebo group was 32.03 mm (n = 96; 95% CI, 26.58–37.47), the 2.5 µg group was 41.62 mm (n = 98; 95% CI, 36.23–47.01), and the 5.0 µg group was 39.30 mm (n = 98; 95% CI, 33.91–44.69). The results of this study showed a variation of 42.5 mm (from 60.0 mm to 17.5 mm: median) after 12 weeks of initiation, which we believe is equivalent in efficacy to the results of the clinical trials before approval.
Thus, we reviewed the safety and efficacy of specific subgroups of patients, including the elderly (≥ 65 years), patients with renal impairment, and those with long-term treatment (≥ 365 days). No noteworthy issues were identified for each of these subgroups or for the remainder of patients surveyed. Neither pregnant nor postpartum women nor children (< 15 years) were registered in the surveillance.
Limitations
This surveillance is a prospective observation based on predetermined survey items. It lacks a control arm. As such, the interpretation of the survey results has certain limitations inherent in this standard approach. The limitation of this surveillance is that the number of the patients between safety analysis and efficacy analysis was very different.
Conclusion
In conclusion, no additional safety concerns or lack of efficacy were identified by the Specific Drug Use Survey of nalfurafine for the treatment of pruritus in patients with chronic hepatitis.
Ethics Approval and Consent to Participate
In accordance with the Japanese Ministry of Health, Labor and Welfare, this surveillance was conducted in compliance with the Good Post-marketing Study Practice (GPSP) and the Standard for Conducting Post-marketing Surveillance and Trials of Drugs, which is an ordinance enacted under Article 14, Section 4, Clause 4 and Article 14, Section 6, Clause 4 of the Japanese Law for Ensuring the Quality, Efficacy, and Safety of Drugs and Medical Devices. At the time approval was obtained for the present surveillance, separate ethics approval was not required under Japanese law. It should also be noted that all original data have been completely anonymized such that there are no risks for deteriorating the privacy of patients or facilities involved.
Acknowledgments
This surveillance was sponsored by Toray Industries, Inc. in association with Sumitomo Dainippon Pharma Co., Ltd. The statistical analyses were jointly planned by Toray Industries, Inc. and EPS Corporation, and performed by the EPS Corporation. Medcore Associates, Inc., Fumiko Matsumura and Dr Martin Guppy provided editorial support. Toray Industries, Inc. covered all financial costs for the above. We would like to thank the physicians, nurses and staff of the participating institutions for their cooperation and assistance in this post-marketing surveillance.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work. All coauthors read and approved the final version of the manuscript.
Funding
This post-marketing surveillance was funded by Toray Industries, Inc.
Disclosure
Hiroshi Yoshitani, Junko Ito and Hideki Kozono, are employees of Toray Industries, Inc. The authors report no other conflicts of interest in this work.
References
1. Ministry of Health Labor and Welfare patient surveillance; 2017. Available from: https://www.mhlw.go.jp/toukei/saikin/hw/kanja/10syoubyo/dl/h29syobyo.pdf.
2. Tanaka J, Koyama T, Mizui M, et al. Total numbers of undiagnosed carriers of hepatitis C and B viruses in Japan estimated by age- and area-specific prevalence on the national scale. Intervirology. 2011;54(4):185–195. doi:10.1159/000324525
3. Tohda C. Itching in hepatic disorder and opioid. J Clin Experiment Med. 2001;197:616–617.
4. Jones EA, Bergasa NV. The pruritus of cholestasis. Hepatology. 1999;29:1003–1006. doi:10.1002/hep.510290450
5. Bergasa NV. Pruritus and fatigue in primary biliary cirrhosis. Clin Liver Dis. 2003;7:879–900. doi:10.1016/S1089-3261(03)00105-3
6. Rishe E, Azarm A, Bergasa NV. Itch in primary biliary cirrhosis: a patients’ perspective. Acta Derm Venereol. 2008;88(1):34–37. doi:10.2340/00015555-0350
7. Koulentaki M, Ioannidou D, Stefanidou M, et al. Dermatological manifestations in primary biliary cirrhosis patients: a case control study. Am J Gastroenterol. 2006;101(3):541–546. doi:10.1111/j.1572-0241.2006.00423.x
8. Bonacini M. Pruritus in patients with chronic human immunodeficiency virus, hepatitis B and C virus infections. Dig Liver Dis. 2000;32(7):621–625. doi:10.1016/S1590-8658(00)80847-6
9. Dega H, Francès C, Dupin N, et al. Pruritus and the hepatitis C virus. The MULTIVIRC Unit. Ann Dermatol Venereol. 1998;125(1):9–12.
10. Lebovics E, Seif F, Kim D, et al. Pruritus in chronic hepatitis C: association with high serum bile acids, advanced pathology, and bile duct abnormalities. Dig Dis and Sci. 1997;42(5):1094–1099. doi:10.1023/A:1018865809556
11. Cacoub P, Poynard T, Ghillani P, et al. Extrahepatic manifestations of chronic hepatitis C. MULTIVIRC Group. Multidepartment Virus C. Arthritis Rheum. 1999;42(10):2204–2212. doi:10.1002/1529-0131(199910)42:10<2204::AID-ANR24>3.0.CO;2-D
12. Cribier B, Samain F, Vetter D, et al. Systematic cutaneous examination in hepatitis C virus infected patients. Acta Derm Venereol. 1998;78(5):355–357. doi:10.1080/000155598443051
13. Gillespie DA, Vickers CR. Pruritus and cholestasis: therapeutic options. J Gastroenterol Hepatol. 1993;8:168–173. doi:10.1111/j.1440-1746.1993.tb01510.x
14. Umeuchi H, Togashi Y, Honda T, et al. Involvement of central μ-opioid system in the scratching behavior in mice, and the suppression of it by the activation of κ-opioid system. Eur J Pharmacol. 2003;477:29–35. doi:10.1016/j.ejphar.2003.08.007
15. Jones EA, Bergasa NV. The pruritus of cholestasis: from bile acids to opiate agonists. Hepatology. 1990;11:884–887. doi:10.1002/hep.1840110526
16. Nakao K, Ikeda K, Kurokawa T, et al. Effect of TRK-820, a selective kappa-opioid receptor agonist, on scratching behavior in animal model of atopic dermatitis. Jpn J Neuropsychopharmacol. 2008;28:75–83.
17. Togashi Y, Umeuch H, Okano K, et al. Antipruritic activity of the kappa-opioid receptor agonist, TRK-820. Eur J Pharmacol. 2002;435(2–3):259–264. doi:10.1016/S0014-2999(01)01588-6
18. Nakao K, Hirakata M, Miyamoto Y, et al. Nalfurafine hydrochloride, a selective κ opioid receptor agonist, has no reinforcing effect on intravenous self-administration in rhesus monkeys. J Pharmacol Sci. 2016;130(1):8–14. doi:10.1016/j.jphs.2015.11.008
19. Kawai K, Hayakawa J, Miyamoto T, et al. Design, synthesis, and structure-activity relationship of novel opioid kappa-agonists. Bioorg Med Chem. 2008;16:9188–9201. doi:10.1016/j.bmc.2008.09.011
20. Nagase H, Hayakawa J, Kawamura K, et al. Discovery of a structurally novel opioid kappa-agonist derived from 4,5-epoxymorphinan. Chem Pharm Bull. 1998;46(2):366–369. doi:10.1248/cpb.46.366
21. Tsuji M, Takeda H, Matsumiya T, et al. The novel kappa-opioid receptor agonist TRK-820 suppresses the rewarding and locomotor-enhancing effects of morphine in mice. Life Sci. 2001;68(15):1717–1725. doi:10.1016/S0024-3205(01)00957-2
22. Nagase H, Fujii H. Essential structure of the κ opioid receptor agonist nalfurafine for binding to the κ receptor. Curr Pharm Des. 2013;19(42):7400–7414. doi:10.2174/138161281942140105165011
23. Kumada H, Miyakawa H, Muramatsu T, et al. Efficacy of nalfurafine hydrochloride in patients with chronic liver disease with refractory pruritus: a randomized, double-blind trial. Hepatol Res. 2017;47:972–982. doi:10.1111/hepr.12830
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.