")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Pirfenidone-loaded liposomes for lung targeting: preparation and in vitro/in vivo evaluation
Received 5 March 2015
Accepted for publication 16 April 2015
Published 30 June 2015 Volume 2015:9 Pages 3369—3376
DOI https://doi.org/10.2147/DDDT.S84046
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Wei Duan
Hui Meng, Yong Xu
Department of Pharmaceuticals, 85th People’s Liberation Army Hospital, Shanghai, People’s Republic of China
Background: The purpose of this study was to develop novel pirfenidone (PFD)-loaded liposomes for targeting to the lung.
Methods: The liposomes were prepared by the film hydration method, and their in vitro/vivo characteristics were evaluated.
Results: The PFD liposomes appeared visually as green to yellowish suspensions and were spherical in shape. The particle size was 582.3±21.6 nm and the entrapment efficiency was relatively high (87.2%±5.7%). The liposomes showed typical sustained and prolonged drug-release behavior in vitro and fitted well with the Weibull distribution equation. The relatively slower time taken to reach a minimal plasma PFD concentration in vivo suggests that PFD liposomes have a sustained-release profile, which is consistent with the results of the in vitro release study. The PFD liposomes showed the largest area under the curve for the lung. The high distribution of PFD achieved in the lungs using this liposomal formulation may be explained by physical entrapment of the liposomes in the vascular network of the lung. Histopathological results indicated that liposomal PFD could alleviate pathological injury in lung tissue.
Conclusion: This liposomal formulation can enable sustained release of PFD and increase targeting to the lung.
Keywords: pirfenidone, liposomes, lung targeting, in vivo, histopathological
Introduction
Idiopathic pulmonary fibrosis is a specific fibrosing form of interstitial pneumonia that is chronic, progressive, and of unknown cause. It is limited to the lungs, occurs primarily in older adults, and is associated with the histological and/or radiological characteristics of usual interstitial pneumonia.1 Studies in Europe suggest an annual incidence of 0.22–7.4 cases per 100,000 and a prevalence of 1.25–23.4 cases per 100,000 in the population.2 The prognosis is poor, with a median survival time estimated at 2–5 years from the time of diagnosis.3 The 5-year survival rate is approximately 20%,4 and the mortality burden associated with idiopathic pulmonary fibrosis is higher than that of some cancers.5
Pirfenidone (Figure 1, 5-methyl-1-phenyl-2-[1H]-pyridone, PFD) is an orally bioavailable, synthetic pyridone compound indicated for the treatment of idiopathic pulmonary fibrosis in various countries worldwide,6 including Europe,7,8 the USA,9 and the People’s Republic of China. It is classified as an immunosuppressant.8 Although the mechanism of action is not fully understood, existing data from in vitro studies and animal models of pulmonary fibrosis suggest that PFD has antifibrotic, anti-inflammatory, and antioxidant properties.10–12 In experimental animal models, PFD has demonstrated an antifibrotic effect in several tissues, including the lung, liver, and kidney.13,14 The drug product is an immediate-release hard capsule containing 267 mg of PFD; the recommended daily dose is three 267 mg capsules three times a day with food, for a total of 2,403 mg/day. When taken as directed with a meal, PFD is absorbed slowly, and is predominantly (80%–85%) excreted via the urine, with a mean apparent terminal elimination half-life of 2.4 hours. Tissue distribution studies have provided little evidence to suggest accumulation of PFD and its metabolites in any tissues; adequate exposure has been shown in the presumptive target organ, the lung.
 | Figure 1 Structure of pirfenidone. |
Liposomes are being widely investigated as potential drug carriers due to their ability to protect and carry drug molecules.15 Application of liposomes could not only increase accumulation of a drug within the target organ, but also reduce the amount of drug that can penetrate into healthy tissues and thereby reduces systemic toxicity. Liposomes can carry hydrophilic and lipophilic as well as amphoteric drug molecules entrapped either in the core or in the liposome bilayer. It has been demonstrated that liposomes can increase drug solubility and stability, as well as providing good drug loading for both hydrophilic and lipophilic drugs.16 Thus, a number of studies have shown that the liposome carrier can improve the oral bioavailability of poorly bioavailable drugs and change the in vivo distribution of entrapped drugs.17,18
The objective of this study was to develop a novel PFD-loaded liposomal system using the thin film hydration technique. The PFD-loaded liposomes were characterized in terms of their morphology, size, zeta potential, encapsulation efficiency, and in vitro release profile. Pharmacokinetic and distribution studies were carried out in rats after intravenous administration of PFD-loaded liposomes, with PFD injection as the control preparation.
Materials and methods
Materials
PFD (purity >99.6%, Figure 1) and carbamazepine (internal standard, purity >98.2%) were purchased from the National Institute for the Control of Pharmaceutical and Biological Products (Beijing, People’s Republic of China). PFD (purity >98%) was obtained from Kovii Chemical Co Ltd (Shanghai, People’s Republic of China). PFD injection was purchased from InterMune Co Ltd (Tokyo, Japan). Egg phosphatidyl choline and high-purity cholesterol were donated by Phospholipid Technology Co., Ltd. (Shanghai, People’s Republic of China). All other chemicals and reagents were of at least analytical grade. Alpha-tocopherol and stearamine were obtained from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, People’s Republic of China). Double-distilled water was produced by a Millipore water purification system (Millipore Corporation, Billerica, MA, USA). All other chemicals used in the study were of analytical grade and obtained commercially.
Animals
The experiments were performed on rats weighing 220±20 g and mice weighing 22±2 g. The animals were kept in cages in a room at a temperature of 25°C±2°C, with a 12:12-hour light-dark cycle. Food and water were freely available. All experiments were performed in strict accordance with the Guide for the Care and Use of Laboratory Animals as adopted by the Chinese National Institute of Health.
Preparation of PFD-loaded liposomes
PFD liposomes was prepared by the film hydration method reported by Ng et al19 with slight modifications. Briefly, egg phosphatidyl choline, cholesterol, and PFD (mass ratio 60:7.5:1.5) were dissolved in dichloromethane. A thin film of dry lipid was deposited on the inner wall of the flask by evaporating the solvent under vacuum at 40°C. The film was hydrated at 40°C with phosphate-buffered saline (pH 7.4) followed by sonication for six cycles at 1,200 bar with an ultrasound probe (EmulsiFlex-B15, Avestin, Ottawa, ON, Canada) to form the PFD liposomes. The final suspension volume was about 10 mL, which was then passed through a 1 μm mesh sieve to remove aggregates (recovery rate >85%). The liposomes were collected by filtration of 5 mm mixed cellulose eater membrane.
Physicochemical characterization
Electron microscopy
The morphology of the PFD liposomes was observed by transmission electron microscopy (JEM-1230, JEOL, Tokyo, Japan). A drop of the sample placed on a carbon-coated copper grid was negatively stained with 2% phosphotungstic acid, then viewed and photographed.
Particle size
The particle size and zeta potential of the liposomes were measured using a Zetasizer Nano ZS analyzer (Malvern Instruments, Malvern, UK). Next, 20 μL of the prepared liposomes were diluted in 1.5 mL of ultrapure water and shaken prior to measurement at 25°C. Dynamic light scattering data were collected using a helium laser as the light source and mean results were provided by photon correlation spectroscopy.
Entrapment efficiency
The entrapment efficiency was determined by equilibrium dialysis as described elsewhere20 using an equilibrium system with dialysis membranes (molecular weight 12,000, Shanghai Chemical Reagent Co Ltd., Shanghai, People’s Republic of China). The diluted liposome dispersion (100 μL to 1.5 mL) was dialyzed against phosphate-buffered saline (pH 7.4) for 4 hours. After precipitation by 10 mL ethanol, the concentration of PFD in the liposomes was determined by reverse phase HPLC. The encapsulation efficiency of the liposomes was calculated using the formula:
(M1 - M2)/M1 ×100% |
where M1 and M2 are the mass of the initially added PFD and non-encapsulated PFD, respectively.
In vitro release
In vitro release was investigated via a dialysis bag method. First, 1 mL each of PFD liposomes and PFD injection were placed in cellulose ester dialysis bags (molecular weight 12,000). These bags were then put in 26 mL of phosphate-buffered saline (pH 7.4) and heated at 37°C±0.5°C. At predetermined time points of 0.25, 0.5, 1, 2, 4, 6, 8, 10, 12, and 24 hours, 1 mL of dissolution medium was withdrawn and precipitated before analysis by HPLC. The supernatant (20 μL) was then directly injected into the HPLC system and analyzed for the PFD released. The release profiles were plotted and fitted using different in vitro release models.
Pharmacokinetics
The rats used in this study were randomly divided into two groups with ten animals each. Group 1 received the intravenous PFD liposomes at a PFD dose of 2.5 mg/kg. Group 2 received the same intravenous dose in the form of PFD injection as a control. For the pharmacokinetic studies, 0.5 mL samples of whole blood were collected from groups 1 and 2 at 0.25, 0.5, 1, 1.5, 2, 4, 6, 8, 12, and 24 hours following intravenous administration and immediately centrifuged at 4,000 rpm for 10 minutes to obtain plasma samples. The supernatant plasma (0.25 mL) was transferred to new tubes and stored at −80°C until analysis.
Biodistribution studies
One hundred mice were divided randomly into two groups, each containing 50 mice. The PFD liposomal formulation or PFD injection was injected into each mouse via the tail vein at a PFD dose of 2.5 mg/kg. At various time intervals after intravenous administration (0.6667, 0.25, 0.5, 1, 2, 4, 6, 8, 10, 12 hours), five mice in each group were selected randomly. Approximately 500 μL of blood were taken from the retro-orbital plexus of these mice and centrifuged (4,000 rpm, 4°C, 10 minutes) to obtain plasma. Each animal was euthanized, in accordance with the the ethics committee of 85 Hospital of People’s Liberation Army, then heart, liver, spleen, lung, and kidney were immediately removed, and approximately 100 mg of tissue slices were excised, weighed, and homogenized with a 10-fold volume of saline in a tissue homogenizer and centrifuged (4,000 rpm, 4°C, 10 minutes) immediately to collect the supernatant. The plasma and supernatant samples were stored at −80°C until analysis.
After the biodistribution studies, the mice from the last observation point (12 hours) were used to investigate the histopathological change of each tissue after intravenous administration of a single dose of the formulation. All the organs were pressed between filter pads, weighed, and then fixed in 10% neutral formalin using standard techniques and stained with hematoxylin and eosin for histopathological examination. All tissue samples were examined and graded under light microscopy with 500× magnification. PFD injection was investigated as a contrast and saline was the negative control.
Analytical conditions
Liquid chromatography tandem-mass spectrometry (LC-MS/MS) experiments were conducted with an API2000 tandem mass spectrometer product line of Thermo Fisher Scientific, Waltham, MA, USA equipped with a binary and a quaternary pump (series 1200, Agilent, Santa Barbara, CA, USA). Analyses were performed on a 5 μm ODS-3 column (100 mm ×4.6 mm) at a flow rate of 0.4 mL per minute. The mobile phase consisted of 0.1% formic acid and acetonitrile (45:55, v/v) and the column temperature was maintained at 30°C. LC-MS/MS was carried out using nitrogen to assist nebulization. The multiple reaction monitoring transitions are 186.2→92.1 for PFD and 237.1→194.2 for internal standard.
The mass spectrometer was run in positive ion mode and tuned for unit mass resolution in the mass range used in the experiments. The exact source conditions were: drying gas temperature 350°C, drying gas flow 10 L per minute, nebulizer pressure 30 psig, quadrupole temperature 100°C, and capillary voltage 4,000 eV. Nitrogen was used exclusively as the drying gas. After acquisition of the resultant selected ion monitoring, chromatograms were integrated using HP Chemstation software.
Next, 100 μL of the plasma sample were transferred to a 5 mL plastic test tube together with 10 μL of internal standard solution (0.5 mg/mL carbamazepine). After shaking for 30 seconds (5432 vortex mixer, Eppendorf, Hamburg, Germany), 300 μL of acetonitrile were added, and the mixture was vortexed for another 2 minutes. After centrifugation at 12,000 rpm for 10 minutes (Thermo Scientific IEC, Waltham, MA, USA), a 5 μL aliquot of the solution was injected into the LC-MS/MS system for analysis.
Tissue samples (200 mg) were homogenized in normal saline solution (0.5 mL). A 0.1 mL volume of the homogenized sample was transferred to a 5 mL plastic test tube. The rest of the preparation process was the same as for the plasma samples.
Statistical analysis
All data are presented as the mean ± the standard deviation. The statistical analysis was performed using the Student’s t-test. P-values <0.05 were considered to be statistically significant.
Results and discussion
Physicochemical characterization
Among the various new drug delivery dosage formats and technologies, the liposomal formulation is considered to be a relatively nontoxic technology that can encapsulate both lipophilic and hydrophilic drugs.16,21,22 Liposomes are presently considered to be one of the most promising carriers for drug delivery.
In this study, PFD liposomes were prepared successfully by the film hydration method and visually appeared as a green to yellowish suspension, and morphologically were spherical in shape and multilamellar in nature (Figure 2). The particle size of the PFD liposomes was 582.3±21.6 nm. Relatively high entrapment efficiency (87.2%±5.7%) could be attributed to the lipophilic nature of the PFD in neutral conditions; it tends to interact with the hydrophobic domain of lipid membranes.23,24 The zeta potential of the PFD liposomes was −22.9±0.81 mV, indicating that there was a high number of negative charges on the surface of the liposomes. It was found from the previous studies that the higher the absolute value of the zeta potential (>15 mV), the stronger the electrostatic repulsion between particles, which makes it more difficult for these liposome particles to aggregate so they are more stable in the dispersal system.25
 | Figure 2 Transmission electron micrograph of the liposomal pirfenidone formulation. Magnification 1,000×. The scale bar represents 250 nm. |
In vitro release
The dialysis bag method was chosen for separation of free PFD from PFD liposomes in the in vitro release study, and the results are shown in Figure 3. The injectable formulation released PFD rapidly and almost completely within 6 hours. However, the PFD liposomes released 45.3%±6.7%, 62.6%±5.4%, and 74.7%±5.1% of PFD within 2, 8, and 24 hours of dialysis, respectively. The first 2 hours was an initial burst-release phase, which could be related to release of drug adsorbed on the surface of the liposomes. Thereafter, the release rate became slow, demonstrating typical sustained and prolonged drug-release behavior. From Table 1, the in vitro drug-release kinetic model for the PFD liposomes fitted well with the Weibull distribution equation, ie, lnln (1/(1–Q)) =−3.431lnt+2.226 (r=0.9963). Therefore, it was speculated that the sustained-release properties of the liposomes could enhance the absorption of PFD in the blood circulation.
 | Figure 3 Drug release profiles for PFD liposomes (Δ) and PFD injection (◊) in phosphate-buffered saline (pH 7.4) for 24 hours (n=6). |
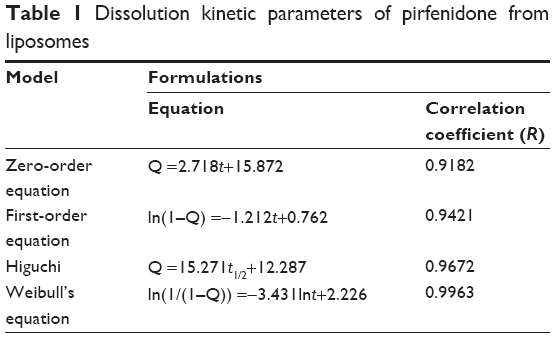 | Table 1 Dissolution kinetic parameters of pirfenidone from liposomes |
Validation of the analysis method
The linear range of the assay for animal plasma and tissues was 5–2,000 ng/mL and the standard curve for PFD was Y =2.651C – 1.712 (r=0.9993). The intraday and interday assay variability for all samples did not exceed 10% and 15%, respectively. The recoveries of the extract method were in the range of 90%–110%. The detection limits and quantitation limits of liquid chromatographic analysis were determined to be 5 ng/mL for the in vivo plasma/tissue samples. Further, there were no interfering peaks observed in any of the chromatograms. It should be noted that the LC-MS/MS technique, although simple, is an effective method for analyzing PFD in a liposomal system.
Pharmacokinetic studies
The mean plasma concentration-time profiles after oral administration of free PFD and liposomal PFD are presented in Figure 4. The pharmacokinetic parameters are shown in Table 2. Significant differences in the pharmacokinetic parameters of PLD were observed between the PFD injection and the PFD-loaded liposomal formulation. Liposomes provided a higher mean residence time (2.93-fold), clearance half-life (t1/2α; 2.13-fold), and distribution half-life (t1/2b; 3.42-fold) when compared with the PFD injection. The peak plasma concentration reached was 453.3±65.7 ng/mL for free PFD compared with 324.7±36.9 ng/mL for the liposomal PFD formulation. The area under the curve to the last measurable point (AUC0–t) for PFD in the liposomal formulation was significantly increased, being 2.06-fold that of the PFD injection. The increased peak plasma concentration and AUC0–t indicate increased absorption. The absolute bioavailability of the encapsulated PFD was 206%, ie, significantly (P<0.05) enhanced when compared with the free drug. However, compared with the PFD injection, the clearance of PFD was reduced in the case of the liposomal PFD formulation, which might possibly be due to the fact that the injection had a larger volume of distribution than the liposomal formulation. The relatively slower time taken to reach a minimal plasma concentration of PFD suggests that the liposomes have a sustained-release profile in vivo, which is consistent with the results of the in vitro release study.
 | Figure 4 Mean plasma concentration-time profiles for pirfenidone after intravenous administration of a single 2.5 mg/kg dose of the PFD injection and the liposomal PFD formulation to rats. |
 | Table 2 Pharmacokinetic parameters of the two formulations |
Biodistribution studies
A lung targeting study was done to compare the PFD injection and the liposomal PFD formulation and determine the magnitude of change in the redistribution profile of the drug when administered in liposomal form. The total amount of drug accumulated in each tissue or plasma (AUC0–t) was calculated, and the results are shown in Table 3. The ratio between liposomes and injection of AUC value of plasma was consistent with the results of the pharmacokinetic study. The largest AUC for the liposomal PFD formulation was found in the lung. The high drug distribution achieved in the lungs using the liposomal formulation may be the result of physical trapping of the liposomes in the vascular network of the lung. The AUC of PFD in reticuloendothelial system-rich organs (liver and spleen) also increased after administration of the liposomal formulation. However, this increase was smaller in the liver and spleen than in the lung, indicating that the liposomal PFD used in this study preferentially targets the lung.
 | Table 3 Area under the curve from 0 to 12 hours for PFD in the heart, liver, spleen, lung, kidney, and plasma after intravenous administration of the injection and liposomal formulations to rats (n=5) |
Histopathological examination of the heart, liver, spleen, lung, and kidney was carried out to check for any tissue damage. After hematoxylin and eosin staining, sections of tissue were observed under a light microscope (Figure 5), and myocardial toxicity was identified for the PFD injection. However, myocardial cells in the liposome group showed no obvious damage. The toxicity of heart reduced because the liposome of PFD could change the biodistribution of PFD in vivo. Slices from other organs indicated no obvious pathological features between the treatment group and the saline group. In the normal control group, the structure of the alveoli and epithelium of the bronchi were intact and the alveolar walls were normal without any neutrophilic infiltration. In the PFD injection group, both interstitial edema and neutrophilic infiltration could be found. However, the liposomal PFD formulation caused much less damage, with no distinct interstitial edema but a small amount of neutrophilic infiltration. These results indicate that a PFD liposomal formulation could alleviate pathological injury in lung tissue.
 | Figure 5 Hematoxylin-eosin stained tissue sections from different tissue of mice after intravenous injection of the 2.5 mg/kg PFD formulations. |
Conclusion
PFD liposomes were prepared successfully by the film hydration method. The particle size was 582.3±21.6 nm and the entrapment efficiency was relatively high (87.2%±5.7%). These liposomes showed typical sustained and prolonged drug-release behavior in vitro. The relatively longer time taken for PFD to reach minimal plasma concentration suggests a sustained-release profile for the liposomes in vivo. The largest AUC value for the liposomal formulation was in the lung, which may be the result of physical trapping of the liposomes in the vascular network of the lung. Histopathology results showed that liposomal PFD could alleviate pathological injury in the lung.
Disclosure
The authors report no conflicts of interest in this work.
References
Raghu G, Collard HR, Egan JJ, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183(6):788–824. | ||
Nalysnyk L, Cid-Ruzafa J, Rotella P, Esser D. Incidence and prevalence of idiopathic pulmonary fibrosis: review of the literature. Eur Respir Rev. 2012;21(126):355–361. | ||
Meltzer EB, Noble PW. Idiopathic pulmonary fibrosis. Orphanet J Rare Dis. 2008;3:8. | ||
European Respiratory Society. Interstitial lung diseases. In: Gibson GJ, Loddenkemper R, Sibille Y, Lundback B, editors. European Lung White Book: Respiratory Health and Disease in Europe. Sheffield, UK: European Respiratory Society; 2013. | ||
Vancheri C, Failla M, Crimi N, Raghu G. Idiopathic pulmonary fibrosis: a disease with similarities and links to cancer biology. Eur Respir J. 2010;35(3):496–504. | ||
Cottin V, Wijsenbeek M, Bonella F, et al. Slowing progression of idiopathic pulmonary fibrosis with pirfenidone: from clinical trials to real-life experience. Clin Investig. 2014;4:313–326. | ||
Carter NJ. Pirfenidone in idiopathic pulmonary fibrosis. Drugs. 2011; 71(13):1721–1732. | ||
European Medicines Agency. Esbriet (pirfenidone) hard capsules: EU summary of product characteristics. 2014. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002154/WC500103049.pdf. Accessed January 5, 2015. | ||
US Food and Drug Administration. Esbriet (pirfenidone) capsules: US prescribing information. 2014. Available from: http://www.accessdata.fda.gov. Accessed January 5, 2015. | ||
Oku H, Shimizu T, Kawabata T, et al. Antifibrotic action of pirfenidone and prednisolone: different effects on pulmonary cytokines and growth factors in bleomycin-induced murine pulmonary fibrosis. Eur J Pharmacol. 2008;590(1–3):400–408. | ||
Cottin V. The role of pirfenidone in the treatment of idiopathic pulmonary fibrosis. Respir Res. 2013;14:S5. | ||
Schaefer CJ, Ruhrmund DW, Pan L, Seiwert SD, Kossen K. Antifibrotic activities of pirfenidone in animal models. Eur Respir Rev. 2011;20(120):85–97. | ||
Zhao XY, Zeng X, Li XM, Wang TL, Wang BE. Pirfenidone inhibits carbon tetrachloride and albumin complex-induced liver fibrosis in rodents by preventing activation of hepatic stellate cells. Clin Exp Pharmacol Physiol. 2009;36(10):963–968. | ||
Miric G, Dallemagne C, Endre Z, Margolin S, Taylor SM, Brown L. Reversal of cardiac and renal fibrosis by pirfenidone and spironolactone in streptozotocin-diabetic rats. Br J Pharmacol. 2001;133(5):687–694. | ||
Allen TM, Moase EH. Therapeutic opportunities for targeted liposomal drug delivery. Adv Drug Deliv Rev. 1996;21:117–133. | ||
Ramos-Cabrer P, Campos F. Liposomes and nanotechnology in drug development: focus on neurological targets. Int J Nanomedicine. 2013;8:951–960. | ||
Gradauer K, Barthelmes J, Vonach C, et al. Liposomes coated with thiolated chitosan enhance oral peptide delivery to rats. J Control Release. 2013;172(3):872–878. | ||
Jain S, Patil SR, Swarnakar NK, Agrawal AK. Oral delivery of doxorubicin using novel polyelectrolyte-stabilized liposomes (layersomes). Mol Pharm. 2012;9(9):2626–2635. | ||
Ng HL, Lu A, Lin G, Qin L, Yang Z. The potential of liposomes with carbonic anhydrase IX to deliver anticancer ingredients to cancer cells in vivo. Int J Mol Sci. 2014;16(1):230–255. | ||
Schneider T, Sachse G, Röβling G, Brandl M. Generation of contrast-carrying liposomes of defined size with a new continuous high pressure extrusion method. Int J Pharm. 1995;117:1–12. | ||
Wei Y, Zhao L. Passive lung-targeted drug delivery systems via intravenous administration. Pharm Dev Technol. 2014;19(2): 129–136. | ||
Zhao L, Wei Y, Li W, et al. Solid dispersion and effervescent techniques used to prepare docetaxel liposomes for lung-targeted delivery system: in vitro and in vivo evaluation. J Drug Target. 2011;19(3):171–178. | ||
Betageri GV, Dipali SR. Partitioning and thermodynamics of dipyridamole in the n-octanol/buffer and liposome systems. J Pharm Pharmacol. 1993;45(10):931–933. | ||
Patricia MN, Luis EA, Marcel T. Binding of dipyridamole to phospholipid vesicles: a fluorescence study. Biochim Biophys Acta. 1997;1328:140–150. | ||
Dong Y, Feng SS. Methoxy poly(ethylene glycol)-poly(lactide) (MPEG-PLA) nanoparticles for controlled delivery of anticancer drugs. Biomaterials. 2004;25(14):2843–2849. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.