")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Phenolic indeno[1,2-b]indoles as ABCG2-selective potent and non-toxic inhibitors stimulating basal ATPase activity
Authors Gozzi GJ, Bouaziz Z, Winter E, Daflon-Yunes N, Honorat M, Guragossian N, Marminon C, Valdameri G, Bollacke A, Guillon J, Pinaud N, Marchivie M, Cadena SM, Jose J, Le Borgne M, Di Pietro A
Received 19 March 2015
Accepted for publication 2 May 2015
Published 3 July 2015 Volume 2015:9 Pages 3481—3495
DOI https://doi.org/10.2147/DDDT.S84982
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Shu-Feng Zhou
Gustavo Jabor Gozzi,1,2 Zouhair Bouaziz,3 Evelyn Winter,1,4 Nathalia Daflon-Yunes,1 Mylène Honorat,1 Nathalie Guragossian,3 Christelle Marminon,3 Glaucio Valdameri,1,2 Andre Bollacke,5 Jean Guillon,6 Noël Pinaud,7 Mathieu Marchivie,8 Silvia M Cadena,2 Joachim Jose,5 Marc Le Borgne,3 Attilio Di Pietro1
1Equipe Labellisée Ligue 2014, BMSSI UMR5086 CNRS/Lyon I University, IBCP, Lyon, France; 2Department of Biochemistry and Molecular Biology, Federal University of Paraná, Curitiba, Paraná, Brazil; 3Faculty of Pharmacy – ISPB, EA 4446 Biomolecules, Cancer and Chemoresistance, Health SFR of East Lyon CNRS UMS3453 - INSERM US7, University of Lyon, Lyon I University, Lyon Cedex 8, France; 4Department of Pharmaceutical Sciences, PGFAR, Federal University of Santa Catarina, Florianopolis, Santa Catarina, Brazil; 5Institute of Pharmaceutical and Medicinal Chemistry, University of Münster, Münster, Germany; 6ARNA Laboratory, Pharmaceutical Sciences UFR, INSERM U869, University of Bordeaux, Bordeaux Cedex, France; 7ISM – CNRS UMR 5255, University of Bordeaux Cedex, France; 8ICMCB CNRS-UPR 9048, University of Bordeaux, Pessac Cedex, France
Abstract: Ketonic indeno[1,2-b]indole-9,10-dione derivatives, initially designed as human casein kinase II (CK2) inhibitors, were recently shown to be converted into efficient inhibitors of drug efflux by the breast cancer resistance protein ABCG2 upon suited substitutions including a N5-phenethyl on C-ring and hydrophobic groups on D-ring. A series of ten phenolic and seven p-quinonic derivatives were synthesized and screened for inhibition of both CK2 and ABCG2 activities. The best phenolic inhibitors were about threefold more potent against ABCG2 than the corresponding ketonic derivatives, and showed low cytotoxicity. They were selective for ABCG2 over both P-glycoprotein and MRP1 (multidrug resistance protein 1), whereas the ketonic derivatives also interacted with MRP1, and they additionally displayed a lower interaction with CK2. Quite interestingly, they strongly stimulated ABCG2 ATPase activity, in contrast to ketonic derivatives, suggesting distinct binding sites. In contrast, the p-quinonic indenoindoles were cytotoxic and poor ABCG2 inhibitors, whereas a partial inhibition recovery could be reached upon hydrophobic substitutions on D-ring, similarly to the ketonic derivatives.
Keywords: multidrug resistance, cancer cells, ABCG2/BCRP, indenoindole inhibitors, structure–activity relationships, ATPase activity
Introduction
A major obstacle of tumor treatment by chemotherapy is cancer cell multidrug resistance, which may be caused by several factors including overexpression of multidrug ATP-binding cassette (ABC) transporters. These transmembrane proteins work as efflux pumps reducing the intracellular concentration of drugs.1 Among the 48 human genes encoding ABC transporters, only three are recognized to be associated with low prognostic in cancer patients: ABCB1/Pgp (P-glycoprotein), ABCC1/MRP1 (multidrug resistance protein 1) and ABCG2/BCRP (breast cancer resistance protein).2 Pgp was the first multidrug ABC transporter to be discovered, and has been extensively studied,3 while MRP1 was later associated with multidrug resistance,4 and ABCG2 was more recently identified.5–7
One of the strategies aimed at eliminating resistant tumors is to use inhibitors of the multidrug ABC transporters. Combination of inhibitors with anticancer drugs should increase the intracellular accumulation of drugs and their availability to cellular targets. A number of Pgp inhibitors have been optimized in vitro, up to third-/fourth-generation compounds, but their intrinsic toxicity and low in vivo activity prevented the achievement of clinical trials.8,9
ABCG2 is a “half-transporter” of 655 amino acids, containing one cytosolic nucleotide-binding domain and one transmembrane domain with six α-helical spans, which needs to at least dimerize to be functional (Figure 1). This transporter is present in various membrane barriers protecting sensitive organs, as well as in many types of cancer cells.10,11 Selective inhibitors are interesting for two main reasons: 1) in vitro to study the specific role of the transporter and 2) in vivo, hopefully up to clinical trials, to use very low concentrations to inhibit each targeted transporter because most potent Pgp inhibitors, such as elacridar or tariquidar, also inhibited ABCG2 but at much higher concentrations expected to induce cytotoxicity and related side effects. The first specific ABCG2 inhibitor, of natural origin, was fumitremorgin C that unfortunately displayed serious neurotoxicity.12 Synthetic derivatives were developed, resulting in the highly potent Ko143 inhibitor, which however still retained significant residual toxicity.13 Screening of different classes of flavonoids identified interesting inhibitors such as hydrophobic flavones, acridones, chromones, asymmetric chalcones, and symmetric bis-chalcones,14 some of them being active in vivo in mouse models.15,16
 | Figure 1 Structural arrangement of ABCG2. |
A different type of ABCG2-selective inhibitors was recently developed as a series of ketonic indenoindoles, upon appropriate substitutions of potent inhibitors of casein kinase II (CK2), such as the replacement of isopropyl by phenethyl at N5 position of C-ring, and the addition of hydrophobic substituents on D-ring.17 The present work was aimed at further modifying the D-ring, by replacing the ketone by either a phenol or a p-quinone, and reinvestigating the effects of substituents (Figure 2). The results showed that phenolic indenoindoles, independently of hydrophobic substituents, were better inhibitors of ABCG2-mediated drug efflux than the previously described ketonic derivatives, through higher potency and selectivity, and the ability to strongly stimulate ATPase activity. In contrast, the p-quinonic derivatives displayed reduced inhibition capacity and significant cytotoxicity.
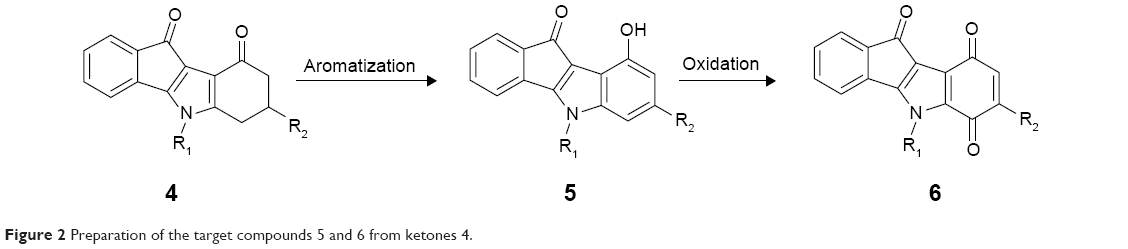 | Figure 2 Preparation of the target compounds 5 and 6 from ketones 4. |
Materials and methods
Chemistry
Melting points were determined on an Electrothermal 9200 capillary apparatus. The infrared (IR) spectra were recorded on a Perkin Elmer Spectrum Two IR Spectrometer. The 1H and 13C nuclear magnetic resonance (NMR) spectra were recorded at 400 MHz on a Bruker DRX 400 spectrometer. Chemical shifts are expressed in ppm (δ) downfield from internal tetramethylsilane and coupling constants J are reported in hertz (Hz). The following abbreviations are used: s: singlet; bs: broad singlet; d: doublet; t: triplet; dd: doubled doublet; dt: doubled triplet; q: quartet; m: multiplet; Cquat: quaternary carbons. The mass spectra were performed by direct ionization (electron ionization or chemical ionization) on a ThermoFinnigan MAT 95 XL apparatus. Chromatographic separations were performed on silica gel columns by column chromatography (Kieselgel 300–400 mesh). All reactions were monitored by thin-layer chromatography on GF254 plates that were visualized under a UV lamp (254 nm). Evaporation of solvent was performed in vacuum with rotating evaporator. The purity of the final compounds (>95%) was determined by ultra high performance liquid chromatography/mass spectrometry on an Agilent 1290 system using a Agilent 1290 Infinity ZORBAX Eclipse Plus C18 column (2.1×50 mm, 1.8 μm particle size) with a gradient mobile phase of H2O/CH3CN (90:10, v/v) with 0.1% of formic acid to H2O/CH3CN (10:90, v/v) with 0.1% of formic acid at a flow rate of 0.5 mL/min, with UV monitoring at the wavelength of 254 nm with a runtime of 10 minutes.
General procedure for the synthesis of compounds 5
To a solution of compound 4 (2.4 mmol) in Ph2O (15 mL) was added 0.48 g of 10% Pd-C. Then, the mixture was heated to reflux for 6 hours. After cooling, 25 mL of MeOH was added and the solution filtered on celite. Evaporation of the solvent left a residue which was purified by silica gel column chromatography using ethyl acetate (EtOAc)/cyclohexane (1:2, v/v) as the eluent.
Compound 5c: 9-Hydroxy-5-(2-phenylethyl)-5H-indeno[1,2-b]indole-10-one
Red solid. Yield 53%. mp 144°C. IR (ν cm−1): 3,232, 1,660, 1,602, 1,581. 1H NMR (CDCl3, 400 MHz) δ: 7.31 (m, 1H, Harom), 7.25–7.12 (m, 3H, Harom), 7.10–7.03 (m, 5H, Harom), 6.75–6.72 (m, 2H, Harom), 6.67 (dd, 1H, J1=0.4 Hz, J2=7.8 Hz, Harom), 6.45 (bs, 1H, OH), 4.36 (t, 2H, J=7.1 Hz, CH2N), 3.14 (t, 2H, J=7.1 Hz, CH2Ph). 13C NMR + DEPT (CDCl3, 100 MHz) δ: 186.20 (Cquat), 157.04 (Cquat), 150.29 (Cquat), 143.65 (Cquat), 140.55 (Cquat), 137.40 (Cquat), 135.91 (Cquat), 132.41 (CH), 129.62 (CH), 129.19 (2CH), 129.10 (2CH), 127.55 (CH), 125.72 (CH), 123.56 (CH), 118.72 (CH), 115.14 (Cquat), 113.48 (Cquat), 108.33 (CH), 103.41 (CH), 47.92 (CH2), 36.71 (CH2). HRMS calcd for C23H17NNaO2 [M + Na]+ 362.1151; found, 362.1140.
Compound 5d: 9-Hydroxy-5-(3-phenylpropyl)-5H-indeno[1,2-b]indole-10-one
Red solid. Yield 64%. mp 146°C. IR (ν cm−1): 3,441, 1,665, 1,603. 1H NMR (DMSO-d6, 400 MHz) δ: 9.53 (bs, 1H, OH), 7.34–7.23 (m, 8H, Harom), 7.10–6.96 (m, 3H, Harom), 6.62 (m, 1H, Harom), 4.39 (t, 2H, J=7.3 Hz, NCH2CH2CH2Ph), 2.74 (t, 2H, J=7.5 Hz, NCH2CH2CH2Ph), 2.12 (m, 2H, NCH2CH2CH2Ph). 13C NMR + DEPT (DMSO-d6, 100 MHz) δ: 183.62 (Cquat), 157.76 (Cquat), 152.13 (Cquat), 144.96 (Cquat), 141.82 (Cquat), 141.17 (Cquat), 134.98 (Cquat), 133.25 (CH), 130.36 (CH), 129.36 (2CH), 129.31 (2CH), 126.98 (CH), 125.34 (CH), 123.31 (CH), 119.76 (CH), 114.35 (Cquat), 113.59 (Cquat), 109.01 (CH), 103.67 (CH), 45.28 (CH2), 32.98 (CH2), 32.61 (CH2). HRMS calcd for C24H19NNaO2 [M + Na]+ 376.1308; found, 376.1299.
Compound 5f: 9-Hydroxy-7-methyl-5-(2-phenylethyl)-5H-indeno[1,2-b]indole-10-one
Dark red solid. Yield 44%. mp 154°C. IR (ν cm−1): 3,433, 1,667, 1,646, 1,602. 1H NMR (DMSO-d6, 400 MHz) δ: 9.41 (s, 1H, OH), 7.27–7.12 (m, 9H, Harom), 6.82 (s, 1H, H-6 or H-8), 6.45 (s, 1H, H-6 or H-8), 4.57 (t, 2H, J=7.0 Hz, CH2N), 3.11 (t, 2H, J=7.0 Hz, CH2Ph), 2.54 (s, 3H, CH3-7). 13C NMR + DEPT (DMSO-d6, 100 MHz) δ: 183.70 (Cquat), 157.84 (Cquat), 151.60 (Cquat), 144.88 (Cquat), 140.92 (Cquat), 138.58 (Cquat), 135.11 (Cquat), 134.98 (Cquat), 132.98 (CH), 129.97 (2CH), 129.90 (CH), 129.24 (2CH), 127.52 (CH), 122.91 (CH), 119.45 (CH), 114.17 (Cquat), 111.51 (Cquat), 110.56 (CH), 103.97 (CH), 47.30 (CH2), 36.45 (CH2), 22.48 (CH3). HRMS calcd for C24H19NNaO2 [M + Na]+ 376.1308; found, 376.1295.
Compound 5g: 9-Hydroxy-7-phenyl-5-(2-phenylethyl)-5H-indeno[1,2-b]indole-10-one
Dark red solid. Yield 55%. mp 168°C. IR (ν cm−1): 3,428, 1,665, 1,633, 1,600. 1H NMR (CDCl3, 400 MHz) δ: 7.53 (d, 2H, J=7.3 Hz, Harom), 7.44 (t, 2H, J=7.3 Hz, Harom), 7.38–7.03 (m, 9H, Harom), 6.91 (s, 1H, H-6 or H-8), 6.82 (s, 1H, H-6 or H-8), 6.72 (d, 1H, J=6.5 Hz, Harom), 6.42 (bs, 1H, OH), 4.36 (t, 2H, J=7.1 Hz, CH2N), 3.14 (t, 2H, J=7.1 Hz, CH2Ph). 13C NMR + DEPT (CDCl3, 100 MHz) δ: 186.11 (Cquat), 157.42 (Cquat), 150.08 (Cquat), 144.05 (Cquat), 141.75 (Cquat), 140.47 (Cquat), 139.64 (Cquat), 137.41 (Cquat), 135.82 (Cquat), 132.44 (CH), 129.59 (CH), 129.17 (2CH), 129.12 (2CH), 129.06 (2CH), 127.55 (4CH), 123.50 (CH), 118.73 (CH), 114.91 (Cquat), 112.58 (Cquat), 108.09 (CH), 102.29 (CH), 47.81 (CH2), 36.74 (CH2). HRMS calcd for C29H22NO2 [M + H]+ 416.1645; found, 416.1635.
Compound 5h: 9-Hydroxy-5-[2-(2-methoxyphenyl)ethyl]-5H-indeno[1,2-b]indole-10-one
Red solid. Yield 51%. mp 166°C. IR (ν cm−1): 3,408, 1,666, 1,642, 1,603. 1H NMR (CDCl3, 400 MHz) δ: 7.34 (dd, 1H, J1=1.0 Hz, J2=7.0 Hz, Harom), 7.21–6.99 (m, 6H, Harom), 6.85–6.80 (m, 3H, Harom), 6.67 (d, 1H, J=7.8 Hz, Harom), 6.45 (bs, 1H, OH), 4.37 (t, 2H, J=7.6 Hz, CH2N), 3.86 (s, 3H, OCH3), 3.16 (t, 2H, J=7.6 Hz, CH2Ph). 13C NMR + DEPT (CDCl3, 100 MHz) δ: 186.19 (Cquat), 157.83 (Cquat), 157.16 (Cquat), 150.24 (Cquat), 143.95 (Cquat), 140.82 (Cquat), 136.09 (Cquat), 132.39 (CH), 131.04 (CH), 129.65 (CH), 129.03 (CH), 125.60 (CH), 125.53 (Cquat), 123.55 (CH), 121.20 (CH), 118.96 (CH), 115.01 (Cquat), 113.49 (Cquat), 110.67 (CH), 108.22 (CH), 103.51 (CH), 55.57 (CH3), 46.12 (CH2), 32.12 (CH2). HRMS calcd for C24H19NNaO3 [M + Na]+ 392.1257; found, 392.1248.
Compound 5i: 9-Hydroxy-5-[2-(3-methoxyphenyl)ethyl]-5H-indeno[1,2-b]indole-10-one
Red solid. Yield 50%. mp 138°C. IR (ν cm−1): 3,461, 1,671, 1,604, 1,582. 1H NMR (CDCl3, 400 MHz) δ: 7.32 (d, 1H, J=6.2 Hz, Harom), 7.16–7.04 (m, 4H, Harom), 6.75–6.65 (m, 5H, Harom), 6.55 (bs, 1H, OH), 6.45 (s, 1H, Harom), 4.36 (t, 2H, J=7.1 Hz, CH2N), 3.67 (s, 3H, OCH3), 3.10 (t, 2H, J=7.1 Hz, CH2Ph). 13C NMR + DEPT (CDCl3, 100 MHz) δ: 186.18 (Cquat), 160.26 (Cquat), 157.10 (Cquat), 150.32 (Cquat), 143.66 (Cquat), 140.52 (Cquat), 138.93 (Cquat), 135.93 (Cquat), 132.40 (CH), 130.27 (CH), 129.61 (CH), 125.74 (CH), 123.55 (CH), 121.37 (CH), 118.75 (CH), 115.12 (Cquat), 115.06 (CH), 113.48 (Cquat), 112.77 (CH), 108.36 (CH), 103.43 (CH), 55.52 (CH3), 47.84 (CH2), 36.67 (CH2). HRMS calcd for C24H19NNaO3 [M + Na]+ 392.1257; found, 392.1258.
Compound 5j: 9-Hydroxy-5-[2-(4-methoxyphenyl)ethyl]-5H-indeno[1,2-b]indole-10-one
Red solid. Yield 47%. mp 150°C. IR (ν cm−1): 3,422, 1,667, 1,601, 1,512. 1H NMR (CDCl3, 400 MHz) δ: 7.31 (m, 1H, Harom), 7.10–7.03 (m, 3H, Harom), 6.96–6.93 (m, 2H, Harom), 6.76–6.66 (m, 5H, Harom), 6.45 (bs, 1H, OH), 4.31 (t, 2H, J=7.0 Hz, CH2N), 3.69 (s, 3H, OCH3), 3.07 (t, 2H, J=7.0 Hz, CH2Ph). 13C NMR + DEPT (CDCl3, 100 MHz) δ: 186.20 (Cquat), 159.20 (Cquat), 157.13 (Cquat), 150.28 (Cquat), 143.65 (Cquat), 140.56 (Cquat), 135.92 (Cquat), 132.35 (CH), 130.13 (2CH), 129.56 (CH), 129.44 (Cquat), 125.69 (CH), 123.50 (CH), 118.77 (CH), 115.06 (Cquat), 114.61 (2CH), 113.50 (Cquat), 108.32 (CH), 103.46 (CH), 55.62 (CH3), 48.17 (CH2), 35.81 (CH2). HRMS calcd for C24H19NNaO3 [M + Na]+ 392.1257; found, 392.1252.
General procedure for the synthesis of compound 6
A solution containing 0.63 mmol of 5 and 0.013 g of salcomine (Co-Salen) in 13 mL of dimethylformamide was stirred under oxygen atmosphere at room temperature for 24 hours. The solution was then poured into 100 mL of ice and water and stirred for 1 hour. The resulting precipitate was filtered and washed with water and dried to give a first quantity of 6. The filtrate was extracted with CH2Cl2. The organic phase was dried over sodium sulfate and evaporated in vacuum to give a second quantity of 6, which was purified by silica gel column chromatography with EtOAc/cyclohexane (1:2, v/v) as the eluent.
Compound 6c: 5-(2-Phenylethyl)-5H-indeno[1,2-b]indole-6,9,10-trione
Dark red solid. Yield 70%. mp 263°C. IR (ν cm−1): 1,709, 1,663, 1,645, 1,594, 1,526. 1H NMR (CDCl3, 400 MHz) δ: 7.58 (dd, 1H, J1=1.0 Hz, J2=7.2 Hz, Harom), 7.34–7.16 (m, 7H, Harom), 7.04 (dd, 1H, J1=1.0 Hz, J2=6.9 Hz, Harom), 6.63 (AB, 2H, H-7 and H-8), 4.77 (t, 2H, J=7.2 Hz, CH2N), 3.18 (t, 2H, J=7.2 Hz, CH2Ph). 13C NMR + DEPT (CDCl3, 100 MHz) δ: 186.40 (Cquat), 183.79 (Cquat), 181.95 (Cquat), 178.45 (Cquat), 155.80 (Cquat), 140.04 (Cquat), 137.47 (CH), 136.81 (Cquat), 136.70 (CH), 133.74 (Cquat), 133.58 (CH), 130.47 (CH), 129.26 (2CH), 129.23 (2CH), 127.68 (CH), 124.96 (CH), 122.96 (Cquat), 120.79 (Cquat), 119.44 (CH), 49.76 (CH2), 37.28 (CH2). HRMS calcd for C23H15NNaO3 [M + Na]+ 376.0944; found, 376.0936.
Compound 6d: 5-(3-Phenylpropyl)-5H-indeno[1,2-b]indole-6,9,10-trione
Red solid. Yield 30%. mp 205°C. IR (ν cm−1): 1,709, 1,662, 1,647, 1,589, 1,526. 1H NMR (CDCl3, 400 MHz) δ: 7.55 (dd, 1H, J1=0.9 Hz, J2=7.2 Hz, Harom), 7.36–7.18 (m, 7H, Harom), 6.61 (AB, 2H, H-7 and H-8), 6.52 (d, 1H, J=7.0 Hz, Harom), 4.52 (t, 2H, J=7.9 Hz, CH2N), 2.82 (t, 2H, J=7.1 Hz, CH2Ph), 2.24 (m, 2H, CH2CH2CH2). 13C NMR + DEPT (CDCl3, 100 MHz) δ: 183.69 (Cquat), 181.84 (Cquat), 178.50 (Cquat), 155.46 (Cquat), 140.47 (Cquat), 140.11 (Cquat), 137.44 (CH), 136.65 (CH), 133.87 (Cquat), 133.74 (CH), 136.61 (Cquat), 130.43 (CH), 129.07 (2CH), 128.97 (2CH), 126.88 (CH), 124.94 (CH), 122.80 (Cquat), 120.82 (Cquat), 119.71 (CH), 47.41 (CH2), 33.04 (CH2), 32.21 (CH2). HRMS calcd for C24H17NNaO3 [M + Na]+ 390.1101; found, 390.1094.
Compound 6e: 5-Benzyl-7-phenyl-5H-indeno[1,2-b]indole-6,9,10-trione
Red solid. Yield 40%. mp 255°C. IR (ν cm−1): 1,715, 1,646, 1,585. 1H NMR (CDCl3, 400 MHz) δ: 7.61 (d, 1H, J=6.8 Hz, Harom), 7.43–7.27 (m, 12H, Harom), 7.16 (d, 1H, J=7.1 Hz, Harom), 6.73 (s, 1H, H-8), 5.88 (s, 2H, NCH2Ph). 13C NMR + DEPT (CDCl3, 100 MHz) δ: 183.78 (Cquat), 181.71 (Cquat), 177.86 (Cquat), 156.49 (Cquat), 146.86 (Cquat), 140.20 (Cquat), 135.14 (Cquat), 134.29 (Cquat), 133.86 (Cquat), 133.85 (CH), 133.71 (CH), 133.34 (Cquat), 130.62 (CH), 130.10 (CH), 129.81 (2CH), 129.49 (2CH), 128.65 (2CH), 128.63 (CH), 126.85 (2CH), 125.02 (CH), 123.26 (Cquat), 120.66 (Cquat), 119.96 (CH), 51.21 (CH2). HRMS calcd for C28H17NNaO3 [M + Na]+ 438.1101; found, 438.1086.
Compound 6f: 7-Methyl-5-(2-phenylethyl)-5H-indeno[1,2-b]indole-6,9,10-trione
Red solid. Yield 80%. mp 228°C. IR (ν cm−1): 1,717, 1,645, 1,609, 1,532. 1H NMR (CDCl3, 400 MHz) δ: 7.53 (dd, 1H, J1=0.7 Hz, J2=7.1 Hz, Harom), 7.32–7.15 (m, 7H, Harom), 7.01 (d, 1H, J=7.3 Hz, Harom), 6.48 (q, 1H, J=1.6 Hz, H-8), 4.74 (t, 2H, J=7.3 Hz, CH2N), 3.17 (t, 2H, J=7.3 Hz, CH2Ph), 2.07 (d, 3H, J=1.6 Hz, Me-7). 13C NMR + DEPT (CDCl3, 100 MHz) δ: 183.81 (Cquat), 181.95 (Cquat), 178.89 (Cquat), 155.72 (Cquat), 146.31 (Cquat), 140.04 (Cquat), 136.91 (Cquat), 133.99 (Cquat), 133.85 (Cquat), 133.51 (CH), 133.45 (CH), 130.28 (CH), 129.27 (2CH), 129.17 (2CH), 127.58 (CH), 124.79 (CH), 123.30 (Cquat), 120.39 (Cquat), 119.35 (CH), 49.78 (CH2), 37.25 (CH2), 16.19 (CH3). HRMS calcd for C24H17NNaO3 [M + Na]+ 390.1102; found, 390.1097.
Compound 6g: 7-Phenyl-5-(2-phenylethyl)-5H-indeno[1,2-b]indole-6,9,10-trione
Red solid. Yield 74%. mp 190°C. IR (ν cm−1): 1,713, 1,655, 1,583. 1H NMR (CDCl3, 400 MHz) δ: 7.59 (d, 1H, J=7.3 Hz, Harom), 7.52–7.21 (m, 12H, Harom), 7.10 (d, 1H, J=7.1 Hz, Harom), 6.76 (s, 1H, H-8), 4.85 (t, 2H, J=7.3 Hz, CH2N), 3.25 (t, 2H, J=7.3 Hz, CH2Ph). 13C NMR + DEPT (CDCl3, 100 MHz) δ: 183.87 (Cquat), 181.83 (Cquat), 177.91 (Cquat), 156.17 (Cquat), 146.93 (Cquat), 140.11 (Cquat), 137.01 (Cquat), 134.13 (Cquat), 133.86 (Cquat), 133.75 (CH), 133.67 (CH), 133.53 (Cquat), 130.48 (CH), 130.20 (CH), 129.91 (2CH), 129.40 (2CH), 129.25 (2CH), 128.79 (2CH), 127.69 (CH), 124.91 (CH), 123.19 (Cquat), 120.34 (Cquat), 119.59 (CH), 49.94 (CH2), 37.29 (CH2). HRMS calcd for C29H20NO3 [M + H]+ 430.1438; found, 430.1426.
X-ray data
The structure of compound 5c has been established by X-ray crystallography at 170 K. Red single crystal (0.25×0.25×0.03 mm3) of 5c was obtained after 20 hours at 17°C from a CS2/chloroform (70/30) solution: monoclinic, space group P1 21/c 1, a=10.4901(3) Å, b=10.1187(3) Å, c=15.8195(4) Å, α=90°, β=91.6571(11)°, γ=90°, V=1,678.48(8) Å3, Z=4, δ(calcd)=1.343 Mg·m−3, formula weight=339.38 for C23H17NO2, F(000)=712. Crystallographic data were acquired at ICMCB (UPR 9048) on a Bruker K-CCD APEX 2. Full crystallographic results have been deposited at the Cambridge Crystallographic Data Centre (CCDC-991361), UK, as supplementary material.18 The data were corrected for Lorentz and polarization effects and for empirical absorption correction.19 The structure was solved by direct methods Shelx 2013 and refined using Shelx 2013 suite of programs20 found in the integrated OLEX2 package.21
Biology and biochemistry
Compounds
Mitoxantrone was purchased from Sigma-Aldrich (Saint-Quentin Fallavier, France). All commercial reagents were of the highest available purity grade. The compounds were dissolved in DMSO, and then diluted in Dulbecco’s Modified Eagle’s Medium (DMEM) (high-glucose DMEM). The stock solutions were stored at -20°C, and warmed to 25°C just before use.
Cell cultures
The human fibroblast human embryonic kidney 293 (HEK293) cell line was transfected with ABCG2 (HEK293-ABCG2) or its empty vector (HEK293-pcDNA3),22 as well as MRP1 (HEK293-ABCC1) and its empty vector (HEK293-pcDNA5). The cells were maintained in high-glucose DMEM supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin at 37°C, 5% CO2 under controlled humidity. The mouse embryonic fibroblast wild-type (NIH3T3) and Pgp-overexpressing (NIH3T3-ABCB1) were maintained under the same conditions. The cell culture medium was supplemented with 0.75 mg/mL G418 (HEK293ABCG2), 200 μg/mL hygromycin B (HEK293pcDNA5 and HEK293ABCC1), or 60 ng/mL colchicin (NIH3T3).
ABCG2-mediated mitoxantrone efflux and inhibition
As previously described,22 cells were seeded at a density of 1.0×105 cells/well into 24-well culture plates. After 72 hours incubation, the cells were exposed to 5 μM mitoxantrone for 30 minutes at 37°C, in the presence or absence of each compound, and then washed with phosphate buffer saline (PBS and trypsinized. The intracellular fluorescence was monitored with a FACS Calibur cytometer (Becton Dickinson, Franklin Lakes, NJ, USA) equipped with a 635-nm red laser, using the FL4 channel and at least 10,000 events were collected. The percentage of inhibition was calculated by using the following equation: % inhibition = (C-M)/(Cev-M) ×100, where C is the intracellular fluorescence of resistant cells (HEK293-ABCG2) in the presence of compounds and mitoxantrone, M is the intracellular fluorescence of resistant cells with only mitoxantrone, and Cev the intracellular fluorescence of control cells (the same HEK293-ABCG2 cells 100% inhibited with 1 μM Ko143).
Intrinsic cytotoxicity of the inhibitory compounds
Cell viability was evaluated through the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) colorimetric assay.23 Wild-type HEK293 cells were seeded at a density of 1×104 cells/well, into 96-well culture plates. After overnight incubation, the cells were treated with the compounds (0–250 μM) for 72 hours. To assess the viability, the cells were exposed to 0.5 mg/mL of MTT and incubated for 4 hours at 37°C. The culture medium was discarded, and 100 μL of a DMSO/ethanol (1:1) solution was added into each well and mixed by gently shaking for 10 minutes. Absorbance was measured at 570 nm using a microplate reader at 570 nm, and the value measured at 690 nm was subtracted. Data are the mean ± SD of at least three independent experiments.
Inhibition of Pgp- and MRP1-mediated drug efflux
NIH3T3-ABCB1 were seeded at a density of 6×104 cells/well into 24-well culture plates and incubated for 48 hours at 37°C, whereas HEK293 cells transfected with ABCC1 were seeded at 2.5×105 cells/well for 72 hours. The cells were respectively exposed to rhodamine 123 (0.5 μM) or calcein-AM (0.2 μM) for 30 minutes at 37°C, in the presence or absence of each compound, then washed with PBS and trypsinized. The intracellular fluorescence was monitored with a FACS Calibur cytometer (Becton Dickinson) using the FL1 channel and at least 10,000 events were collected. The percentage of inhibition was calculated relatively to 5 μM GF120918 or 35 μM verapamil, respectively, using similar equation as demonstrated to ABCG2 inhibition.
Effects on ABCG2 ATPase activity
Vanadate-sensitive ATPase activity was measured colorimetrically by determining the liberation of inorganic phosphate from ATP, the Sf9 membranes were prepared as previously and loaded with cholesterol.24 The incubation was performed in 96-well plates. Sf9 insect cell membranes (1 mg/mL) were incubated in a 50 mM Tris/HCl, 50 mM NaCl buffer (pH 8.0) containing sodium azide (3.3 mM) in the absence (with or without sodium orthovanadate at 0.33 mM) or in the presence of tested compounds (2 μM). The reaction was started by the addition of ATP-Mg (3.9 mM) and the plates were incubated for 30 minutes at 37°C. The reaction was stopped with sodium dodecylsulfate (10%) and revealed with a mixture of ammonium molybdate reagent and 10% ascorbic acid (1:4). The absorbance was measured after 30-minute incubation at 880 nm using a reader plate.
Preparation of recombinant human CK2 holoenzyme and assay of inhibitors activity
The human protein kinase CK2 holoenzyme was prepared as previously described.25,26 In brief, human CK2α (CSNK2A1) and CK2β subunits (CSNK2B) were expressed separately in Escherichia coli BL21(DE3) cells using the pT7-7 vector. Freshly-transformed cultures were grown overnight at 37°C in lysogeny broth (LB) medium until the stationary phase was reached. LB medium was inoculated with the starter cultures (1:100), and protein expression was induced by isopropyl β-D-1-thiogalactopyranoside addition (1 mM final concentration) when an OD500 of 0.6 was reached. The cultures were further incubated at 30°C during 5–6 hours for the CK2α subunit, or 3 hours for the CK2β subunit. After harvesting the bacterial cells by centrifugation (6,000× g for 10 minutes at 4°C) and disruption by sonication (3×30 seconds on ice), cell debris was removed by another centrifugation at 15,000× g (10 minutes, 4°C). Both extracts were combined and CK2 holoenzyme was purified by a three-column procedure. Fractions were analyzed by sodium dodecyl sulphate-polyacrylamide gel electrophoresis and western blot. Those containing CK2 and showing CK2 activity were pooled and stored at -80°C as aliquots, which obtained CK2 holoenzyme with a purity higher than 99%.27
For testing the compounds on CK2 inhibition, a capillary electrophoresis-based CK2 activity assay28 was used. Briefly, 2 μL of the inhibitor solutions in DMSO was mixed with 78 μL of kinase buffer (50 mM Tris/HCl (pH 7.5), 100 mM NaCl, 10 mM MgCl2 and 1 mM DTT) containing 1 μg CK2, and preincubated at 37°C for 10 minutes. The CK2 reaction was initiated by the addition of preincubated assay buffer (25 mM Tris/HCl (pH 8.5), 150 mM NaCl, 5 mM MgCl2, 1 mM DTT, 100 μM ATP and 190 μM of the CK2 substrate peptide RRRDDDSDDD), carried on for 15 minutes at 37°C and was stopped by addition of 4 μL of 0.5 M ethylenediaminetetraacetic acid. Subsequently, the reaction samples were fed to a PA800 plus capillary electrophoresis system (Beckman Coulter, Krefeld, Germany) using acetic acid (2 M, adjusted to pH 2.0 with concentrated HCl) as the background electrolyte. Detection of the separated CK2 reaction substrate and product peptide at 214 nm was accomplished by a DAD detector. As controls for 0% and 100% inhibition, samples containing pure DMSO instead of inhibitor and samples additionally lacking the CK2 holoenzyme respectively, were treated under identical conditions. Compounds that showed more than 50% inhibition in the initial test at a final inhibitor concentration of 10 μM were subjected to half-maximal inhibitory concentration (IC50) determination. For this purpose, nine final inhibitor concentrations in suitable intervals ranging from 0.001 μM to 100 μM were tested. IC50 values were calculated from the resulting dose–response curves using GraphPadPrism 5.02 (GraphPad Software, Inc., La Jolla, CA, USA).
Results
Chemistry
The access to 5,6,7,8-tetrahydroindeno[1,2-b]indole-9,10-diones 4 was previously detailed.17 Their syntheses started by the reaction of a cyclohexane-1,3-dione with the appropriate primary amine to form the enaminone derivatives, which were then condensed with ninhydrin. The resulting vic-dihydroxyindeno[1,2-b]indole-9,10-diones were deoxygenated with tetraethylthionylamide to afford compound 4.
9-Hydroxy-5H-indeno[1,2-b]indole-10-ones 5 were prepared by dehydrogenation of 4 with 10% Pd-C in refluxing diphenyl ether.29 Subsequent oxidation with molecular dioxygen in the presence of salcomine30 (Co-Salen), at room temperature, gave 5H-indeno[1,2-b]indole-6,9,10-triones 6 (Figure 3).
 | Figure 3 Reagents and conditions. |
It is known that oxidation of phenols with Fremy’s salt represents an excellent synthetic method for the preparation of p-quinones under mild conditions and usually in good yield.31 For this reason, we applied these conditions to oxidize compounds 5c and 5g (Figure 4), but very low yields of the desired products were obtained. This was presumably due to steric effects of R1 and R2, or/and electronic destabilization of the radical intermediates. Accordingly, we preferred the method of oxidation with O2/salcomine that we applied for the synthesis of all p-quinones mentioned in this work.
 | Figure 4 Structures of the investigated indenoindole series 5 and 6. |
In total, ten phenols (series 5) and seven quinones (series 6) have been synthesized, and their structures are shown in Figure 4. The 3D spatial determinations of 5c were established by X-ray crystallography, and confirmed the structure in the solid state as anticipated on the basis of IR and 1H NMR data (Figure 5). The key bond lengths and angles of this indeno[1,2-b]indolone are very similar to those given in the literature for other substituted indenoindole derivatives.32,33 The indeno[1,2-b]indolone system of 5c is nearly planar with a mean out-of-plane deviation of 0.027 Å and the largest deviation of 0.062(2) Å for atom C7. The C4=O1 double bond was noticed at 1.231(2) Å.
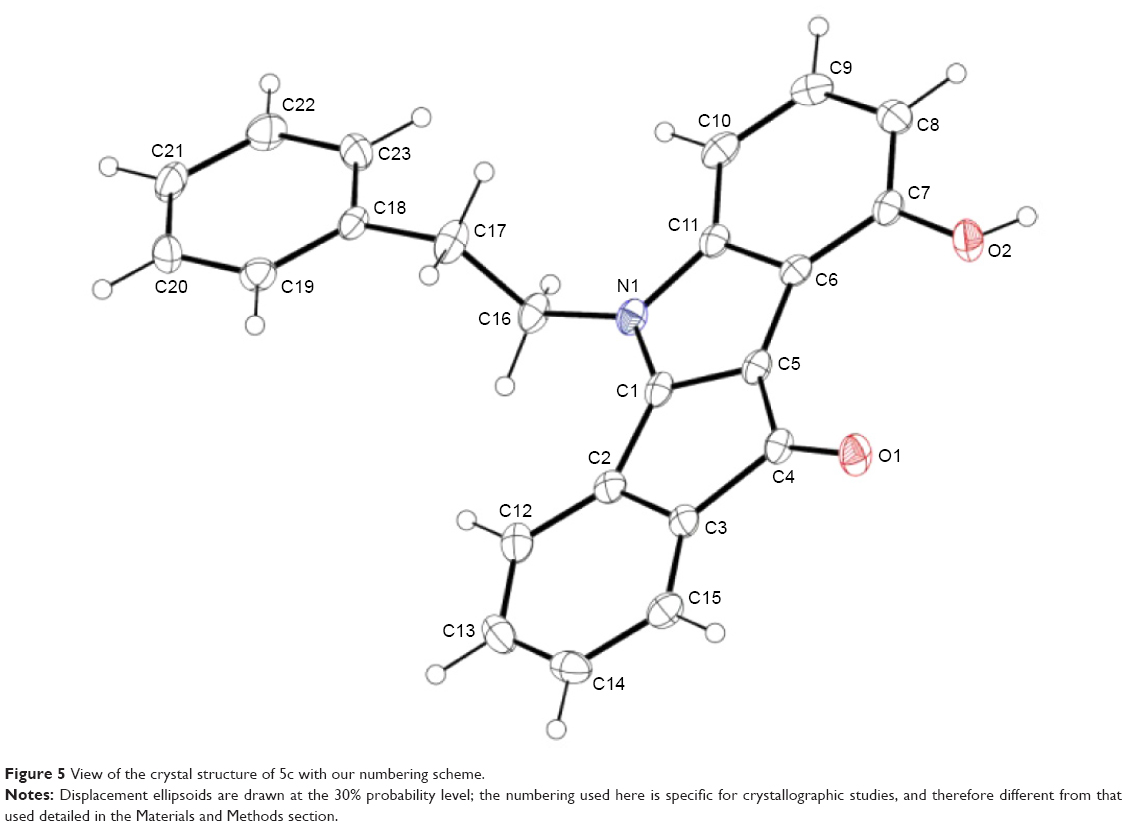 | Figure 5 View of the crystal structure of 5c with our numbering scheme. |
Biological evaluation and structure–activity relationships
The newly synthesized phenolic and p-quinonic indenoindoles were assayed for their capacity to modulate ABCG2 and CK2 activities, and for their cytotoxicity.
Inhibition of ABCG2-mediated mitoxantrone efflux, and cytotoxicity
The new series of compounds 5 and 6 were first screened for their ABCG2-inhibitory activities using mitoxantrone as a substrate, and the inhibitory potency was compared with the recently-characterized ketonic derivative 4c17 (Figure 6). Other reference inhibitors of mitoxantrone efflux, used here as positive controls, were chromone 6g and Ko143 with IC50 values of 0.11–0.13 μM and 0.09 μM, respectively.22,34
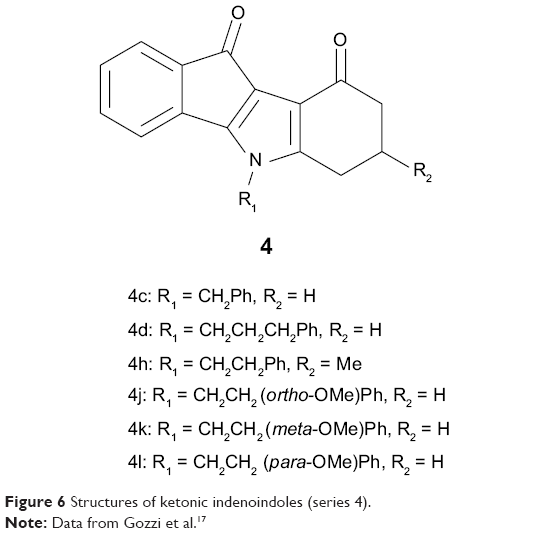 | Figure 6 Structures of ketonic indenoindoles (series 4). |
Table 1 shows that the corresponding phenolic derivative 5c, without any substituent on both D-ring and N5-phenethyl, was about threefold more potent (IC50=0.16 μM versus 0.43 μM). In contrast to ketonic indenoindoles,17 the addition of hydrophobic methyl and phenyl substituents on D-ring did not further increase inhibition, producing either no effect or a negative contribution, as in 5f (0.16 μM) and 5g (0.51 μM) versus 5c. Replacement of N5-phenethyl by a shorter linker drastically decreased the inhibitory potency, as evidenced from 5g (full inhibition, with IC50=0.51 μM) to 5e (only 56% inhibition at 10 μM, with IC50 around 9 μM), as well as from 5c (0.16 μM) to 5b (68% inhibition). However, changing the N5-phenethyl substituent by a longer linker only slightly reduced the inhibitory potency, in 5d (0.64 μM) versus 5c (0.16 μM). Methoxy substitution of the N5-phenethyl phenolic-indenoindole core structure (5c), at position ortho in 5h (0.15 μM), para in 5j (0.20 μM), or meta in 5i (0.37 μM) did not show substantial improvement on activity. Phenyl substitution of D-ring was not able to recover any activity for N5-benzyl derivatives (5e versus 5b). In sharp contrast, p-quinonic derivatives were very poor inhibitors when comparing 6c (18% inhibition) with 5c (and 4c), 6d (33% inhibition) with 5d, and 6b (10% inhibition) with 5b. However, hydrophobic substitutions of D-ring allowed a partial recovery of inhibition potency, as illustrated for methyl and phenyl additions in 6f (0.84 μM) and 6g (0.43 μM) versus 6c, and in 6e (41% inhibition) versus 6b.
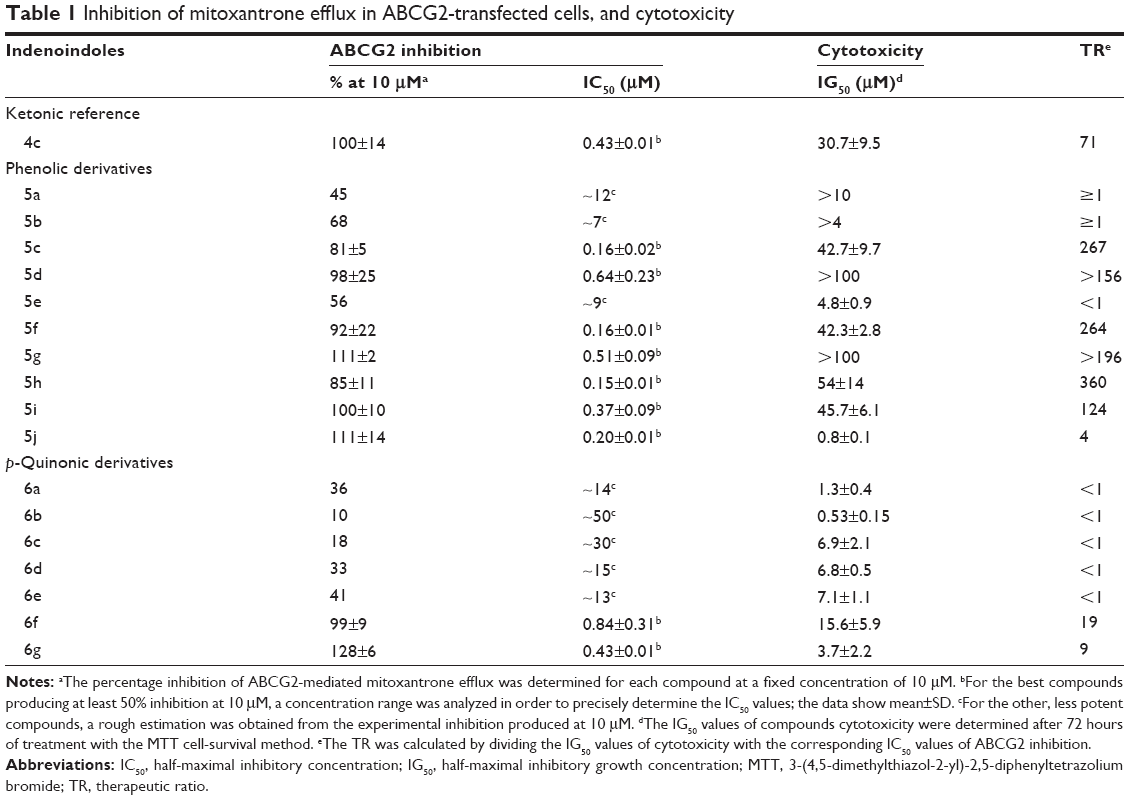 | Table 1 Inhibition of mitoxantrone efflux in ABCG2-transfected cells, and cytotoxicity |
Since the root cause of in vivo and preclinical trial failures of potent ABCG2 inhibitors are their intrinsic cytotoxic effects, one valuable complementary approach is to evaluate the in vitro cytotoxicity of new inhibitors. The ratio between cytotoxicity and inhibitory potency of the inhibitor, defined here as therapeutic ratio (TR), gives a valuable information to guide future in vivo trials. The phenolic leads, 5c, 5f, and 5h (IC50=0.15–0.16 μM), as well as 5d, 5g, and 5i (IC50=0.37–0.64 μM), displayed a low cytotoxicity, with half-maximal inhibitory growth concentrations (IG50) values >42 μM, and even >100 μM in two cases. Such IG50 values were similar to those previously observed with ketonic derivatives.17 This gave high TR values, up to 360, assumed to be quite promising for further investigations. The only exception was 5j, which displayed an unexpectedly high cytotoxicity, possibly due to strong interaction with unknown critical cellular target(s). Similarly, shortening the phenethyl substituent into either benzyl (in 5b and 5e) or isopropyl (in 5a) drastically increased cytotoxicity, leading to very low TR values (around 1). All p-quinonic derivatives were much more cytotoxic than phenolic compounds, except for 6e where the already high toxicity of 5e was not further increased, with IG50 values in the 0.5–15 μM range, and gave very low TR values (<20, and often <1). No cross-resistance was ever observed in ABCG2-transfected cells versus the control cell line (data not shown), indicating that both phenolic and p-quinonic indenoindoles were apparently not transported by ABCG2.
Selectivity toward ABCG2 inhibition
The most potent indenoindole inhibitors of ABCG2 were tested against the two other major multidrug ABC transporters of cancer cells, namely Pgp/ABCB1 and MRP1/ABCC1. Figure 7 shows that none of the selected compounds, including phenolic, p-quinonic, and ketonic derivatives were able to induce any inhibition of Pgp-mediated rhodamine 123 efflux, as compared with the reference inhibitor GF120918/elacridar. A differential pattern was obtained with MRP1-mediated calcein efflux: while the phenolic derivatives (5c, 5f, and 5h) and p-quinonic derivatives (6f and 6g) produced a very limited, if any, inhibition, in contrast to the ketonic derivatives (4h, 4j, and 4k)17 (Figure 6) significantly inhibited at 2 μM and, in two cases, as efficiently as verapamil at 10 μM. Therefore, phenolic and p-quinonic, but not ketonic, inhibitors could be considered as ABCG2 selective over MRP1.
 | Figure 7 Ability of leads of the different series of indenoindoles to inhibit drug efflux by ABCB1 or ABCC1. |
A higher selectivity of phenolic indenoindoles, relatively to the ketonic derivatives, was also observed toward the CK2 protein kinase (Table 2). Indeed, the phenolic derivatives inhibited CK2 activity with a limited potency, never reaching a complete inhibition at 10 μM. The high values of the ABCG2/CK2 ratio, in the range of 70–75 for 5f and 5j, were 7- to 12-fold higher than those previously obtained (in the range of 6–11) for the corresponding ketonic derivatives (4h and 4l)17 (Figure 6). A significant effect was also observed with 5d and 5i, giving a ratio value around 31, which was two- to fourfold higher than for the respective ketonic indenoindoles 4d and 4k (Figure 6). In the case of p-quinonic derivatives, the potency to inhibit CK2 was also lower, by about twofold, as compared with the ketonic ones, such as for 6c (IC50 ~13 μM) versus 4c (7.0 μM), and 6f (4.1 μM) versus 4h (2.5 μM),17 but their very low capacity to inhibit ABCG2 gave extremely low ABCG2/CK2 ratio values (<1), except for the two more active compounds, 6f and 6g, containing hydrophobic substituents on D-ring (5–27).
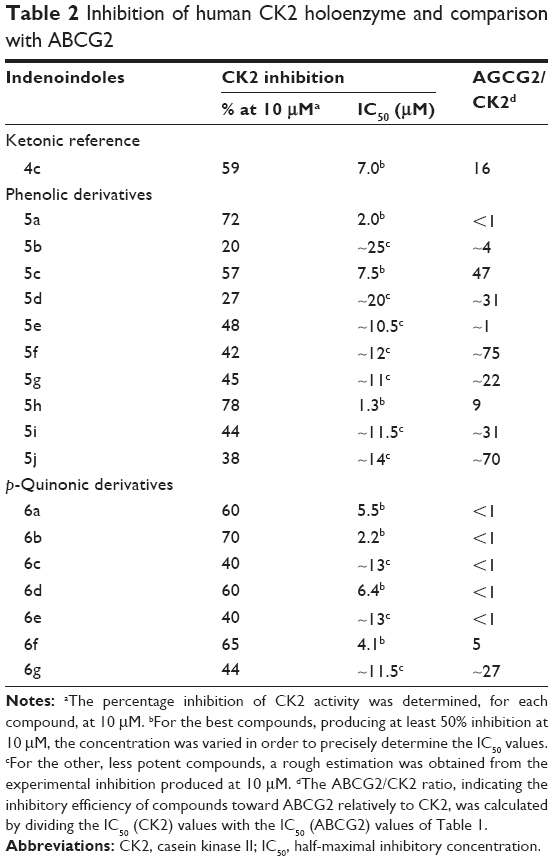 | Table 2 Inhibition of human CK2 holoenzyme and comparison with ABCG2 |
The interest of phenolic indenoindoles as ABCG2 inhibitors, versus ketonic and p-quinonic derivatives, is illustrated in Figure 8, where five phenolic leads, namely 5c, 5d, 5f, 5g, and 5i, display both a good selectivity, with a lower interaction toward CK2 than ketonic derivatives such as 4c (Figure 8A), and a low cytotoxicity by difference with all p-benzoquinonic derivatives (Figure 8B).
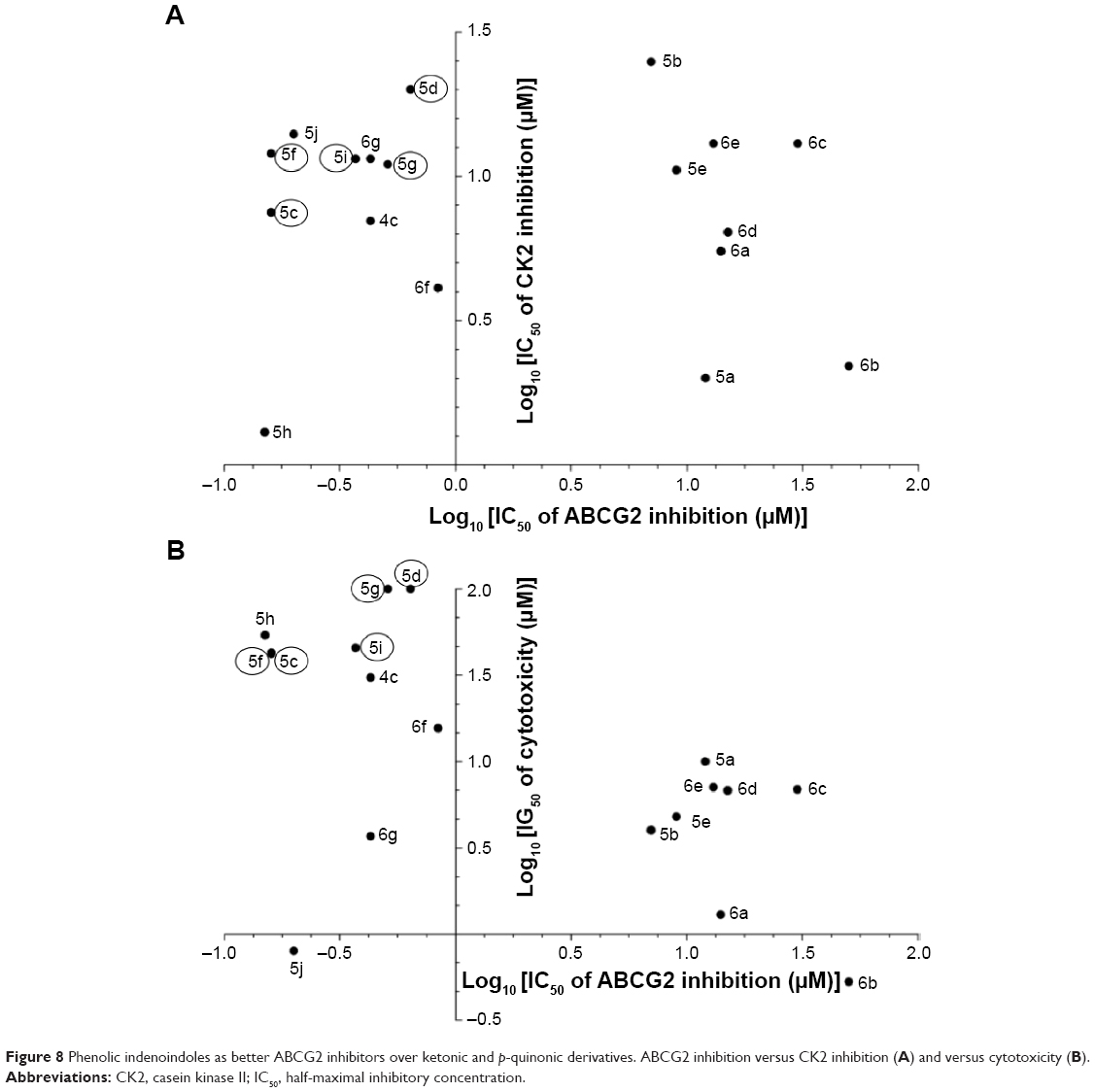 | Figure 8 Phenolic indenoindoles as better ABCG2 inhibitors over ketonic and p-quinonic derivatives. ABCG2 inhibition versus CK2 inhibition (A) and versus cytotoxicity (B). |
Effects on ABCG2 basal ATPase activity
The phenolic indenoindoles could also be distinguished from both ketonic and p-quinonic derivatives on the basis of their differential modulation of ABCG2 ATPase activity, which provided indirect information about the binding sites of the different compounds. Figure 9 shows that the vanadate-sensitive ATPase activity of insect-cell plasma membranes overexpressing human ABCG2 was strongly stimulated by the most potent phenolic inhibitors of mitoxantrone efflux: a 2- to 4.5-fold stimulation was observed with 5c, 5d, 5f, 5g, 5h, 5i, and 5j, in contrast to the very low stimulation observed with much less active 5e (Table 1). Addition of hydrophobic methoxy substitution on N5-phenethyl at position ortho did not further stimulate the ATPase activity, as in 5c versus 5h. However, shifting the methoxy substituent to position meta significantly increased the ATPase activity (5i), a maximal stimulation being observed for compound 5j, with a methoxy substituent at position para. A similar lack of positive effects was observed by lengthening the linker at N5-phenethyl when comparing 5c with 5d. However, changing the N5-phenethyl substituent into a shorter linker, from 5g to 5e, was quite detrimental to stimulation. Methyl substitution of D-ring was not able to increase the ATPase stimulation activity (5c versus 5f); however, changing the methyl into phenyl (5f versus 5g) induced a twofold increased stimulation.
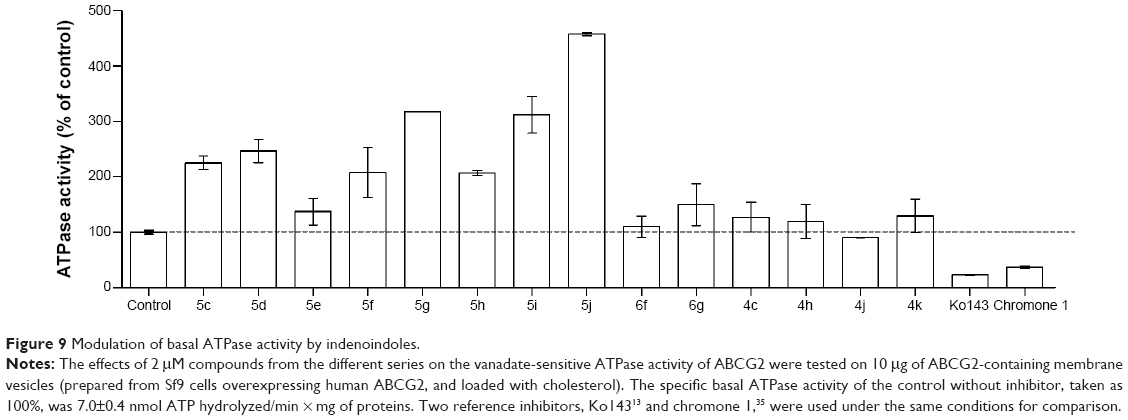 | Figure 9 Modulation of basal ATPase activity by indenoindoles. |
In addition, the p-quinonic derivative 6f, as well as 6c and 6e (not shown here), did not produce any effect, while a very limited stimulation was observed for compound 6g (Figure 9). This lack of effect on ATPase activity was also observed with the four ketonic derivative leads, 4c, 4h, 4j, and 4k, which did not induce any significant stimulation, suggesting either distinct binding sites or different induced conformational changes associated with inhibition of ABCG2-mediated mitoxantrone efflux. All indenoindole derivatives behaved differently from the reference inhibitors, Ko143 and chromone 1, which strongly inhibited ATPase activity.
Discussion
This paper shows that phenolic indenoindoles constitute quite interesting ABCG2 modulators as potent and selective inhibitors, with low cytotoxicity. They strongly stimulate ATPase activity, by difference with ketonic derivatives and reference inhibitors.
Phenolic indenoindoles as better ABCG2 inhibitors than ketonic derivatives
The advantages of the phenolic, over the ketonic, derivatives are related to three main aspects. Firstly, a higher potency (about threefold) in inhibition of drug-efflux activity: such an increase might be due to a higher reactivity of the phenol versus the ketone group rather than to the planarity of the indenoindole core, since the inhibition was markedly altered in planar p-quinonic derivatives. Secondly, a higher selectivity for ABCG2 among MDR transporters and CK2: this was monitored through the absence of interaction with MRP1, and a threefold lower interaction with CK2 giving a 3–7 higher ABCG2/CK2 ratio; this further confirms the possibility to convert CK2 inhibitors into ABCG2 inhibitors upon appropriate substitutions, as previously observed with ketonic derivatives,17 and extends the differences to high ABCG2/CK2 ratio values, of 70–75, for 5f and 5j. Thirdly, a strong modulation, up to 4.5-fold, of the basal ATPase activity: this allows considering the possibility of depleting intracellular ATP from ABCG2-overexpressing cells, with the aim of promoting selective cell death, as proposed for the Pgp-dependent collateral sensitivity observed in the presence of verapamil.35–37 Such a stimulation of ATPase activity contrasts with the lack of effect observed with both ketonic and p-quinonic derivatives, and the inhibition produced by the reference inhibitors chromone 1 and Ko143,22 as well as fumitremorgin C.38
Among the six best phenolic derivatives, toward both potency of ABCG2 inhibition (IC50 values in the range of 0.16–0.64 μM) and selectivity (ABCG2/CK2 ratio values in the range of 22–75), one compound, namely 5j, appeared to be cytotoxic. The five remaining leads, namely 5c, 5d, 5f, 5g, and 5i, which displayed high TR values, in the range of 124–267, constitute good candidates for additional experiments to check their suitability in future assays in animal models.
Molecular mechanism and polyspecificity
The different modulation of ABCG2 basal ATPase activity by phenolic indenoindoles versus non-stimulatory ketonic derivatives and inhibitory reference inhibitors suggests distinct binding sites. A similar situation was previously observed with methoxy trans-stilbenes which, in combination with GF120918, prazosin, or nilotinib, produced additive inhibitory effects on drug efflux.39 Although the inhibitor binding sites are probably distant from the cytosolic nucleotide-binding domain, they are indeed able to modulate the ATPase activity; this suggests the existence of allosteric interactions, between transmembrane and cytosolic domains of the transporter, controlling the strict coupling between ATP binding/hydrolysis and drug transport.
The common N5-phenethyl substituent found in all active indenoindole derivatives, as illustrated in Figure 10, should however induce partial overlapping of the binding sites. The hydrophobic substituents shown to increase the binding affinity of ketonic indenoindoles, produced similar effects in p-quinonic derivatives, where they partially compensated the strongly-negative contribution of the second ketone group, at para position versus the first one, within the quinone moiety. In contrast, the phenol group appeared to interact with a distinct, although likely overlapping, subsite associated with ATPase-stimulation effects. A similar overlapping of binding sites was previously observed for ABCG2-selective acridones in comparison with GF120918/elacridar, a dual inhibitor strongly interacting with Pgp.40
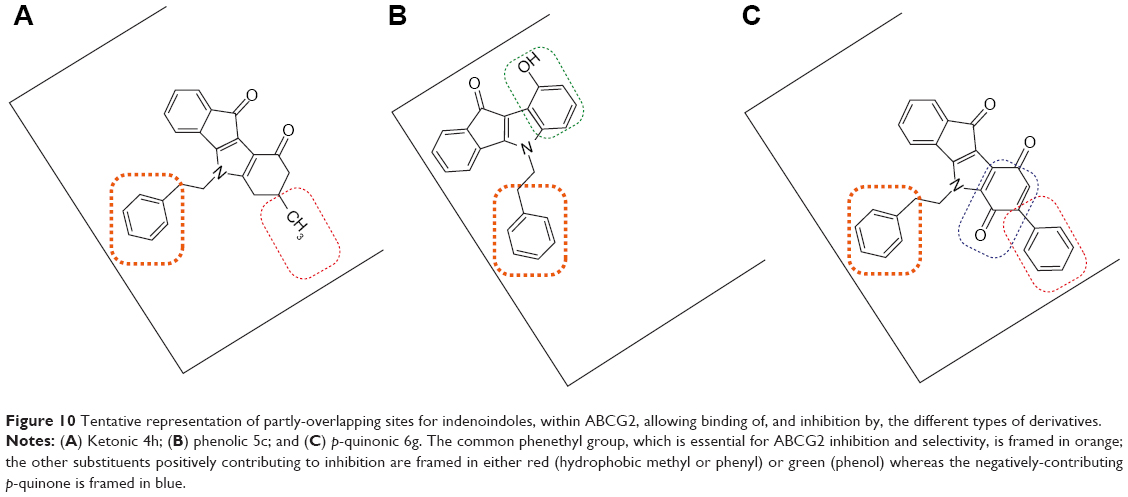 | Figure 10 Tentative representation of partly-overlapping sites for indenoindoles, within ABCG2, allowing binding of, and inhibition by, the different types of derivatives. |
Finally, these diverse binding sites, located close each other and partly overlapping, constitute a new demonstration of ABCG2 polyspecificity toward inhibitors, similarly to that better known for substrates.
Acknowledgments
EW and GV were recipients of mobility doctoral fellowships from the Brazilian CNPq-CAPES (Science without Borders Program 245762/2012-4) and CAPES (Process numbers 8792127 and 2303/10-8), respectively. NDY was recipient of a postdoctoral fellowship for the Control of Cancer-CNPq (Science without Borders Program). Financial support was provided by the CNRS and Université Lyon 1 (UMR 5086), the Ligue Nationale Contre le Cancer (Equipe labellisée Ligue 2014), an international grant from French ANR and Hungarian NIH (2010-INT-1101-01). The Aquitaine Region is thanked for supporting equipment set up in CESAMO and at ICMCB. C Ozvegy-Laczka and B Sarkadi are acknowledged for providing the insect cell membranes overexpressing human ABCG2, L Luku Tchamabe for technical assistance, and Dr A Coleman for improving the English.
Author contributions
Gustavo Jabor Gozzi and Zouhair Bouaziz contributed equally to the experiments. Senior investigators, Marc Le Borgne and Attilio Di Pietro contributed equally to the work supervision. All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Gottesman MM. Mechanisms of cancer drug resistance. Annu Rev Med. 2002;53:615–627. | ||
Szakács G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Targeting multidrug resistance in cancer. Nat Rev Drug Discov. 2006;5(3):219–234. | ||
Juliano RL, Ling V. A surface glycoprotein modulating drug in chinese hamster ovary cell mutants. Biochim Biophys Acta. 1976;455(1):152–162. | ||
Cole SP, Bhardwaj G, Gerlach JH, et al. Overexpression of a transporter gene in a multidrug-resistant human lung cancer cell line. Science. 1992;258(5088):1650–1654. | ||
Allikmets R, Schriml LM, Hutchinson A, Romano-Spica V, Dean M. A human placenta-specific ATP-binding cassette gene (ABCP) on chromosome 4q22 that is involved in multidrug resistance. Cancer Res. 1998;58(23):5337–5339. | ||
Doyle LA, Yang W, Abruzzo LV, et al. A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc Natl Acad Sci U S A. 1998;95(26):15665–15670. | ||
Miyake K, Mickley L, Litman T, et al. Molecular cloning of cDNAs which are highly overexpressed in mitoxantrone-resistant cells: demonstration of homology to ABC transport genes. Cancer Res. 1999;59(1):8–13. | ||
Cripe LD, Uno H, Paietta EM, et al. Zosuquidar, a novel modulator of P-glycoprotein does not improve the outcome of older patients with newly diagnosed acute myeloid leukemia: a randomized, placebo-controlled trial of the Eastern Cooperative Oncology Group 3999. Blood. 2010;116(20):4077–4085. | ||
Darby RA, Callaghan R, Mc Mahon RM. P-glycoprotein inhibition: the past, the present and the future. Curr Drug Metab. 2011;12(8):722–731. | ||
Vlaming ML, Lagas JS, Schinkel AH. Physiological and pharmacological roles of ABCG2 (BCRP): recent findings in Abcg2 knockout mice. Adv Drug Deliv Rev. 2009;61(1):14–25. | ||
Natarajan K, Xie Y, Baer MR, Ross DD. Role of breast cancer resistance protein (BCRP/ABCG2) in cancer drug resistance. Biochem Pharmacol. 2012;83(8):1084–1103. | ||
Rabindran SK, He H, Singh M, et al. Reversal of a novel multidrug resistance mechanism in human colon carcinoma cells by fumitremorgin C. Cancer Res. 1998;58(24):5850–5858. | ||
Allen JD, van Loevezijn A, Lakhai JM, et al. Potent and specific inhibition of the breast cancer resistance protein multidrug transporter in vitro and in mouse intestine by a novel analogue of fumitremorgin C. Mol Cancer Ther. 2002;1(6):417–425. | ||
Winter E, Devantier Neuenfeldt P, Chiaradia-Delatorre LD, et al. Symmetric bis-chalcones as a new type of breast cancer resistance protein inhibitors with a different mechanism than chromones. J Med Chem. 2014;57(7):2930–2941. | ||
Arnaud O, Boumendjel A, Geze A, et al. The acridone derivative MBLI-87 reverses breast cancer resistance protein-mediated resistance to irinotecan in xenografts. Eur J Cancer. 2011;47(4):640–648. | ||
Honorat M, Guitton J, Gauthier C, et al. MBL-II-141, a chromone derivative, enhances irinotecan anticancer efficiency in ABCG2-positive xenografts. Oncotarget. 2014;5(23):11957–11970. | ||
Gozzi GJ, Bouaziz Z, Winter E, et al. Converting potent indeno[1,2-b]indole inhibitors of protein kinase CK2 into selective inhibitors of the breast cancer resistance protein ABCG2. J Med Chem. 2015;58(1):265–277. | ||
Supplementary X-ray crystallographic data: Cambridge Crystallographic Data Centre, University Chemical Lab, Lensfield Road, Cambridge CB2 1EW, UK; Email: [email protected] | ||
Sheldrick GM. SADABS. Göttingen: University of Göttingen; 1996. | ||
Sheldrick GM. A short history of SHELX. Acta Crystallogr A. 2008;64(Pt 1):112–122. | ||
Dolomanov OV, Bourhis LJ, Gildea RJ, Howard JAK, Puschmann H. OLEX2: a complete structure solution, refinement and analysis program. J Appl Cryst. 2009;42:339–341. | ||
Winter E, Lecerf-Schmidt F, Gozzi GJ, et al. Structure-activity relationships of chromone derivatives toward mechanism of interaction with, and inhibition of, breast cancer resistance protein ABCG2. J Med Chem. 2013;56(24):9849–9860. | ||
Mosmann T. Rapid colorimetric assay for cellular growth and survival. Application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65(1–2):55–63. | ||
Telbisz A, Muller M, Ozvegy-Laczka C, et al. Membrane cholesterol selectively modulates the activity of the human ABCG2 multidrug transporter. Biochim Biophys Acta. 2007;1768(11):2698–2713. | ||
Olgen S, Götz C, Jose J. Synthesis and biological evaluation of 3-(substituted-benzylidene)-1,3-dihydroindolin derivatives as human protein kinase CK2 and p60(c-Src) tyrosine kinase inhibitors. Biol Pharm Bull. 2007;30(4):715–718. | ||
Grankowski N, Boldyreff B, Issinger OG. Isolation and characterization of recombinant human casein kinase II subunits α and β from bacteria. Eur J Biochem. 1991;198(1):25–30. | ||
Guillon J, Le Borgne M, Rimbault C, et al. Synthesis and biological evaluation of novel substituted pyrrolo[1,2-a]quinoxaline derivatives as inhibitors of the human protein kinase CK2. Eur J Med Chem. 2013;65:205–222. | ||
Gratz A, Götz C, Jose J. A CE-based assay for human protein kinase CK2 activity measurement and inhibitor screening. Electrophoresis. 2010;31(4):634–640. | ||
Poumaroux A, Bouaziz Z, Domard M, Fillion H. Regiospecific hetero Diels-Alder synthesis of pyrido[2,3-b]- and [3,2-b]carbazole-5,11-diones. Heterocycles. 1997;45:585–596. | ||
Hundsdörfer C, Hemmerling HJ, Hamberger J, et al. Novel indeno[1,2-b]indoloquinones as inhibitors of the human protein kinase CK2 with antiproliferative activity towards a broad panel of cancer cell lines. Biochem Biophys Res Commun. 2012;424(1):71–75. | ||
Zimmer H, Lankin DC, Horgan SW. Oxidations with potassium nitrosodisulfonate (Fremy’s radical). Teuber reaction. Chem Rev. 1971;71(2):229–246. | ||
Janreddy D, Kavala V, Bosco JWJ, Kuo C-W, Yao C-F. An easy access to carbazolones and 2,3-disubstituted indoles. Eur J Org Chem. 2011;12:2360–2365. | ||
Bill K, Black GG, Falshaw CP, Sainsbury M. The coupling reactions of 3-acylindoles and proof of structure of the palladium (II) acetate mediated cyclisation reaction product of 3-benzoyl-1-methylindole. Heterocycles. 1983;20:2433–2436. | ||
Valdameri G, Genoux-Bastide E, Peres B, et al. Substituted chromones as highly potent nontoxic inhibitors, specific for the breast cancer resistance protein. J Med Chem. 2012;55(2):966–970. | ||
Karwatsky J, Lincoln MC, Georges E. A mechanism for P-glycoprotein-mediated apoptosis as revealed by verapamil hypersensitivity. Biochemistry. 2003;42(42):12163–12173. | ||
Hall MD, Handley MD, Gottesman MM. Is resistance useless? Multidrug resistance and collateral sensitivity. Trends Pharmacol Sci. 2009;30(10):546–556. | ||
Szakacs G, Hall M, Gottesman MM, et al. Targeting the Achilles heel of multidrug-resistant cancer by exploiting the fitness cost of resistance. Chem Rev. 2014;114(11):5753–5774. | ||
Ozvegy C, Litman T, Szakacs G, et al. Functional characterization of the human multidrug transporter, ABCG2, expressed in insect cells. Biochem Biophys Res Commun. 2001;285(1):111–117. | ||
Valdameri G, Pereira Rangel L, Spatafora C, et al. Methoxy stilbenes as potent, specific, untransported, and nontoxic inhibitors of breast cancer resistance protein. ACS Chem Biol. 2012;7(2):322–330. | ||
Boumendjel A, Macalou S, Valdameri G, et al. Targeting the multidrug ABCG2 transporter with flavonoidic inhibitors: in vitro optimization and in vivo validation. Curr Med Chem. 2011;18(22):3387–3401. | ||
Hundsdörfer C, Hemmerling HJ, Götz C, et al. Indeno[1,2-b]indole derivatives as novel class of potent human protein kinase CK2 inhibitors. Bioorg Med Chem. 2012;20(7):2282–2289. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.