")
Back to Journals » Drug Design, Development and Therapy » Volume 12
Pharmacokinetic comparison of a fixed-dose combination versus concomitant administration of fimasartan, amlodipine, and rosuvastatin using partial replicated design in healthy adult subjects
Authors Oh M, Ghim JL, Park SE, Kim EY, Shin JG
Received 31 January 2018
Accepted for publication 27 March 2018
Published 8 May 2018 Volume 2018:12 Pages 1157—1164
DOI https://doi.org/10.2147/DDDT.S164215
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Qiongyu Guo
Minkyung Oh,1 Jong-Lyul Ghim,1,2 Sung-Eun Park,2 Eun-Young Kim,1,2 Jae-Gook Shin1,2
1Department of Pharmacology and PharmacoGenomics Research Center, Inje University College of Medicine, Busan, Republic of Korea; 2Department of Clinical Pharmacology, Inje University Busan Paik Hospital, Busan, Republic of Korea
Objective: The aim of this study was to compare the pharmacokinetics (PK) and safety profiles of a fixed-dose combination (FDC) formulation of fimasartan, amlodipine, and rosuvastatin with the co-administration of the two products by using a replicated crossover study design in healthy male subjects.
Materials and methods: This was an open-label, randomized, three-sequence, three-period replicated crossover study in healthy male subjects. The replicated crossover design was done because of high coefficient of variation of PK parameter for fimasartan, that is, >30%. With a 14 days washout period, an FDC tablet containing 60 mg fimasartan, 10 mg amlodipine, and 20 mg rosuvastatin was administered only once, and separate formulations of fimasartan/amlodipine 60 mg/10 mg FDC tablet and 20 mg rosuvastatin tablet administered twice. Blood samples were collected up to 72 hours following drug administration. The plasma concentrations of fimasartan, amlodipine, and rosuvastatin were measured by liquid chromatography tandem mass spectrometry. Safety was assessed by evaluating vital signs, clinical laboratory parameters, physical examinations, and medical interviews.
Results: The geometric mean ratios and 90% confidence intervals (CIs) for the maximum plasma concentration (Cmax) and area under the curve from time zero to the last measurable sampling time (AUCt) were 1.0776 (0.9201–1.2622) and 0.9978 (0.9538–1.0439) for fimasartan, 1.0038 (0.9782–1.0301) and 1.0055 (0.9828–1.0288) for amlodipine, and 1.0006 (0.9290–1.0776) and 0.9986 (0.9532–1.0461) for rosuvastatin, respectively. A total of 22 adverse events (AEs) were reported by 60 subjects; there were no significant differences in the incidence of AEs between the two groups.
Conclusion: The 90% CI of the Cmax of fimasartan was within the widened acceptance limit, ln(0.6984)–ln(1.4319). The 90% CIs of the other PK parameters for drugs were between ln(0.8) and ln(1.25). These results suggest that the FDC formulation is pharmacokinetically bioequivalent and has a similar safety profile, to the co-administration of its three constituent drugs.
Keywords: replicated crossover, bioequivalence, fixed-dose combination, pharmacokinetics, fimasartan
Introduction
Cardiovascular diseases, such as coronary heart disease and cerebrovascular disease, are one of the leading causes of death worldwide including in Korea. According to the statistics of deaths by the Korea National Statistical Office in 2013, the number of deaths attributed to the three major causes of death per 100,000 population was 149 for malignant neoplasm (cancer), 50.3 for cerebrovascular disease, and 50.2 for heart disease.1 Also, the Centers for Disease Control and Prevention reported that cardiovascular disease was the leading cause of death in 2011.2 Hypertension and dyslipidemia are major risk factors for cardiovascular diseases; indeed, the incidence of coronary artery disease is reduced by 36% when statin therapy is added, rather than simply controlling blood pressure in the presence of both the diseases.3,4
In patients with hypertension, the first-line treatment is one of the thiazide-type diuretics, angiotensin-converting enzyme inhibitors, angiotensin receptor blockers (ARB), or calcium channel blockers (CCB), and if blood pressure is not controlled, two or more drugs may be used concomitantly.5 Fimasartan is an ARB that blocks the angiotensin II AT1 receptor in the renin-angiotensin-aldosterone system, which maintains homeostasis of blood pressure and body fluids.6,7 It has a superior blood pressure lowering effect than valsartan and losartan and is not inferior to that of candesartan.8 The incidence rate of adverse events (AEs) related to the nervous system, digestive system, and infection at the recommended doses of fimasartan (60 and 120 mg) was 11%, 4.3%, and 5.5%, respectively. Moreover, the incidence of AEs was not significantly different from that associated with losartan (50 and 100 mg), with good tolerability and safety.8 Amlodipine, a dihydropyridine-based CCB with indications for hypertension and angina, relaxes the vascular smooth muscle and reduces peripheral resistance.9 Due to the excellent efficacy and safety of amlodipine, several derivative compounds have been developed. Rosuvastatin inhibits the synthesis of low-density lipoprotein cholesterol in the liver as an inhibitor of 3-hydroxy-3-methylglutaryl-coenyme A (HMG-CoA) reductase.10 Rosuvastatin can reduce the non-high-density lipoprotein cholesterol level by 50% and triglyceride by 30%, and its efficacy in this regard is superior to that of simvastatin and pravastatin.11,12
According to the guidelines from the European Medicines Agency (EMA)13 and the US Food and Drug Administration (FDA),14–16 a highly variable drug (HVD) is defined that the within-subject coefficient of variation (CV) is ≥30%, as determined using a replicate crossover study design with three or four periods. These guidelines state that if the within-subject CV of the maximum plasma concentration (Cmax) of a reference drug is ≥30%, the conventional bioequivalence (BE) limits, 0.80–1.25, can be widened. In previous drug–drug interaction studies, the intrasubject variability of the Cmax of fimasartan was 60.9% and 76.1%. Thus, fimasartan is an HVD, and hence, the widened BE limits can be used for comparison with the pharmacokinetic profiles. Thus, we designed a three-period replicate crossover study in which the reference drug was administered twice and the test drug only once.
The combination therapy improves patient compliance, simplifies the regimen by reducing the number of drugs and the frequency of dosing, reduces costs, and may produce a superior blood pressure lowering effect in some patients.17,18 Therefore, the combination of fimasartan, amlodipine, and rosuvastatin is expected to be effective against both hypertension and dyslipidemia and to inhibit the development of cardiovascular disease. In this study, we aimed to compare the pharmacokinetics (PK) and safety profiles of fixed-dose combination (FDC) of fimasartan, amlodipine, and rosuvastatin with the co-administration of the three drugs in healthy male subjects.
Materials and methods
The study protocol was approved by the Institutional Review Board of Inje University Busan Paik Hospital and the Ministry of Food and Drug Safety (MFDS; ClinicalTrials.gov: NCT02995720). All procedures were conducted in compliance with the Good Clinical Practice guidelines and tenets of the Declaration of Helsinki.
Study population
All subjects submitted written informed consent prior to participation. Eligible subjects were healthy Korean male subjects aged 19–50 years and within 20% of their ideal body weight according to Broca’s formula. All subjects were confirmed to be healthy based on their medical history and the results of physical examination, electrocardiograph (ECG), vital sign measurements, and clinical laboratory tests. The following exclusion criteria were applied: a medical history that might affect drug absorption, distribution, metabolism, and/or excretion; a sitting diastolic blood pressure >90 or <65 mmHg, a systolic blood pressure >140 or <100 mmHg or a pulse rate of >100 beats/min; excessive consumption of caffeine (>400 mg/day), cigarettes (>10 cigarettes/day), or alcohol (>30 g/day) or drinking alcohol within 7 days before the first administration; taking any medication that induces or inhibits drug-metabolizing enzymes; a blood donation within 30 days of the first day of study drug administration or whole blood donation within 60 days; and participation in other clinical trials within 3 months prior to this study.
Study design
This study was conducted as a randomized, open-label, two-treatment, three-period, three-sequence, partial replicated crossover design with 14 days washout periods. Subjects were randomly assigned to one of three sequences (RTR, TRR, or RRT), in each of which the reference drug (R) was administered twice and the test drug (T) only once. The sample size required for each sequence was calculated to be 14 subjects to satisfy the BE criteria for HVDs with 80% statistical power at a 5% level of significance,13,19 assuming that the intrasubject variability of Cmax for fimasartan was 58.8% and the true geometric mean ratio (GMR) was 1.1 based on a previous unpublished study (Shin, unpublished data, 2015). After considering the dropouts and the failure rate, the total number of subjects was 60. This study was subdivided into two parts, A and B, 30 subjects per part, for the convenience of quality assurance and ease of performance because of the large number of subjects.
All subjects were administered an FDC tablet containing 60 mg fimasartan/10 mg amlodipine/20 mg rosuvastatin (Boryung Pharmaceutical Co., Ltd., Seoul, Republic of Korea) [test] or co-administered 60 mg fimasartan/10 mg amlodipine FDC tablet (Boryung Pharmaceutical Co., Ltd.) and 20 mg rosuvastatin (AstraZeneca, Seoul, Korea) [reference] with 240 mL of water after 10 PM overnight fasting.
A 14 days washout period was sufficient to eliminate the effects of the drugs, as this was more than fivefold the half-life of the plasma concentration of all the three drugs (the half-life of fimasartan is 5.8 hours;20 amlodipine 37.08–48.04 hours;9,21,22 rosuvastatin 17.94–24.4 hours10,23,24). Blood samples for the PK analysis of fimasartan were collected at 0, 0.25, 0.5, 0.75, 1, 1.5, 2, 3, 4, 5, 6, 8, 12, 24, and 48 hours after drug administration. Blood samples for the PK analysis of amlodipine and rosuvastatin were taken at 0, 1, 2, 3, 4, 5, 6, 8, 12, 24, 48, and 72 hours after drug administration. The blood samples (5–10 mL) were collected in a heparinized vacutainer tube for each blood sampling point, centrifuged at 4°C and 200 g for 10 minutes and stored at −70°C until analysis.
Safety assessment
The vital signs, physical examinations, and clinical laboratory test of all subjects who took the study drug at least once were evaluated. AEs were monitored through spontaneous reporting and by asking subjects general health-related questions, and their severity, frequency, and relationship with the treatment were also recorded.
Bioanalysis
The plasma concentrations of fimasartan, amlodipine, and rosuvastatin were measured by using liquid chromatography-tandem mass spectrometry (LC-MS/MS) method by Kyunghee Drug Analysis Center of Kyung Hee University College of Pharmacy (Seoul, Republic of Korea).
In the case of fimasartan, sample preparation was performed by liquid–liquid extraction methods. Briefly, plasma samples (50 μL) and internal standard (20 μL of 500 ng/mL BR-A-563) were added to a tube containing 50 μL of 1% formic acid and 1 mL of n-hexane:ethylacetate (2:8, volume/volume [v/v]). After vortex mixing for 10 minutes and centrifuging at 4°C and 3,500 rpm for 10 minutes, the organic layer was separated and evaporated to dryness under a stream of nitrogen. The residue was reconstituted in 1 mL of 0.05% formic acid in water and 0.05% formic acid in acetonitrile (9:1, v/v), and 3 μL of the sample was injected into the LC-MS/MS system. LC-MS/MS was performed by using an API 4000 triple-quadrupole mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA) coupled to an Agilent 1200 series HPLC system (Agilent Technologies, Santa Clara, CA, USA). The separation was performed with a Luna C18 column (50×2.0 mm, 3 μm; Phenomenex, Torrance, CA, USA) by using a mobile phase of 0.05% formic acid and 0.05% formic acid in acetonitrile (20:80, v/v) at a flow rate of 0.25 mL/min. Electrospray ionization (ESI) was performed in the positive ion mode. The multiple-reaction monitoring mode (MRM) using specific precursor/product ion transition was employed for quantification. Detection of the ions was performed by monitoring the m/z transitions of 502.415→207.100 for fimasartan and 526.478→207.200 for BR-A-563.
For amlodipine, sample preparation was performed by liquid–liquid extraction methods. Briefly, plasma samples (100 μL) and internal standard (20 μL of 20 ng/mL amlodipine-d4) were added to a tube containing 5 mL of methyl tertiary butyl ether:methyl chloride (8:2, v/v). After vortex mixing for 15 minutes and centrifuging at 4°C and 3,500 rpm for 15 minutes, the organic layer was separated and evaporated to dryness under a stream of nitrogen. The residue was reconstituted in 800 μL of 50% methanol, and 10 μL of the sample was injected into the LC-MS/MS system. LC-MS/MS was performed by using an API 4000 triple-quadrupole mass spectrometer (Thermo Fisher Scientific) coupled to an Agilent 1200 series HPLC system (Agilent Technologies, Santa Clara, CA, USA). The separation was performed with a Luna C18 column (50×2.0 mm, 3 μm; Phenomenex) by using a mobile phase of 0.1% formic acid and 100% methanol (40:60, v/v) at a flow rate of 0.15 mL/min. ESI was performed in the positive ion mode. The MRM using specific precursor/product ion transition was employed for quantification. Detection of the ions was performed by monitoring the m/z transitions of 409.155→238.000 for amlodipine and 413.041→238.000 for amlodipine-d4.
For rosuvastatin, sample preparation was performed by liquid–liquid extraction methods. Briefly, plasma samples (100 μL) and internal standard (20 μL of 70 ng/mL rosuvastatin-d6) were added to a tube containing 10 μL of 1% HCl and 1.5 mL of methyl tertiary butyl ether. After vortex mixing for 10 minutes and centrifuging at 4°C and 14,000 rpm for 10 minutes, the organic layer was separated and evaporated to dryness under a stream of nitrogen. The residue was reconstituted in 300 μL of 50% methanol, and 7 μL of the sample was injected into the LC-MS/MS system. LC-MS/MS was performed by using an API 4000 triple-quadrupole mass spectrometer (Thermo Fisher Scientific) coupled to a Shimadzu Nexera X2 HPLC system (Shimadzu Corporation, Tokyo, Japan). The separation was performed with a Halo C18 column (100×2.1 mm, 2.7 μm; Advanced Materials Technology, Wilmington, DE, USA) by using a mobile phase of 0.1% formic acid and 100% methanol (20:80, v/v) at a flow rate of 0.2 mL/min. ESI was performed in the positive ion mode. The MRM using specific precursor/product ion transition was employed for quantification. Detection of the ions was performed by monitoring the m/z transitions of 482.142→257.900 for rosuvastatin and 488.165→264.200 for rosuvastatin-d6.
Data were acquired by Analyst 1.6.2 software (Thermo Fisher Scientific) for fimasartan, amlodipine, and rosuvastatin. The calibration curves were established in the concentration range of 1–1,000 ng/mL for fimasartan, 0.2–20 ng/mL for amlodipine, and 0.2–200 ng/mL for rosuvastatin; these had coefficients of determination (R2) greater than 0.9996, 0.9977 and 0.9996, respectively. The CVs for assay precision were less than 8.6%, 9.5%, and 8.5%, and the accuracy values were greater than 93.3%, 88.3%, and 95.9%, respectively. There was no relevant cross-talk or matrix effect.
PK analysis
The PK parameters of fimasartan, amlodipine, and rosuvastatin were assessed by non-compartmental analysis with WinNonlin software (ver. 6.1; Pharsight Corp., Mountain View, CA, USA). The Cmax and the time to reach Cmax (Tmax) were obtained from time–concentration curves. The area under the plasma concentration time curve from zero to the last sampling time (AUCt) was estimated by using the linear trapezoidal rule. The area under the plasma concentration time curve from infinity (AUCinf) was calculated by the formula: AUCinf = AUCt + Ct/k, where Ct is the last measured plasma concentration and k is the terminal elimination rate constant. The elimination rate constant (k) is obtained by the least squares method of regression analysis using the log concentration of the terminal data from a concentration–time plot and the terminal elimination half-life (t1/2β) was calculated by the equation t1/2β = ln(2)/k.
Statistical analysis
Statistical analysis was performed by using SAS 9.4 (SAS Institute Inc., Cary, NC, USA). Continuous variables are expressed as mean ± SD, and categorical variables are presented as counts and percentages.
To compare the PK profiles, the analysis of variance (ANOVA) with treatment, period, sequence, and part as fixed effects and subjects nested within the part–sequence interaction as the random effect using the PROC MIXED procedure was conducted by using the log-transformed Cmax and AUCt. The GMR and 90% confidence intervals (CIs) of GMR for Cmax and AUCt of fimasartan, amlodipine, and rosuvastatin were estimated from the results of the ANOVA.
The BE criteria for the Cmax of fimasartan used the widened acceptance interval in the BE guidelines for HVDs of the MFDS,19 which are identical to reference-scaled average BE of the EMA.13 For other PK parameter of fimasartan, amlodipine, and rosuvastatin, the standard BE criteria, 0.80–1.25, was used.
Results
Subject characteristics
Sixty healthy male subjects were enrolled in this study (mean age, 25.7±3.67 years; mean weight, 70.6±8.04 kg; mean height, 174.5±6.02 cm). Seven subjects did not complete the study: three withdrew their consent, three dropped out due to abnormal clinical laboratory results, and one dropped out due to investigational judgment. Safety profiles were assessed in the 60 subjects who had administered study drug at least once. PK analyses were evaluated in 53 subjects who completed the scheduled study.
PK
The mean plasma concentration–time profiles of fimasartan, amlodipine, and rosuvastatin are shown in Figures 1–3, respectively. The results of the ANOVA indicated that there was no statistically significant difference in part, treatment, period, or sequence. The statistics of PK parameters for test and reference drug are summarized in Table 1. The PK profiles of fimasartan, amlodipine, and rosuvastatin for test were similar to those of the reference drugs.
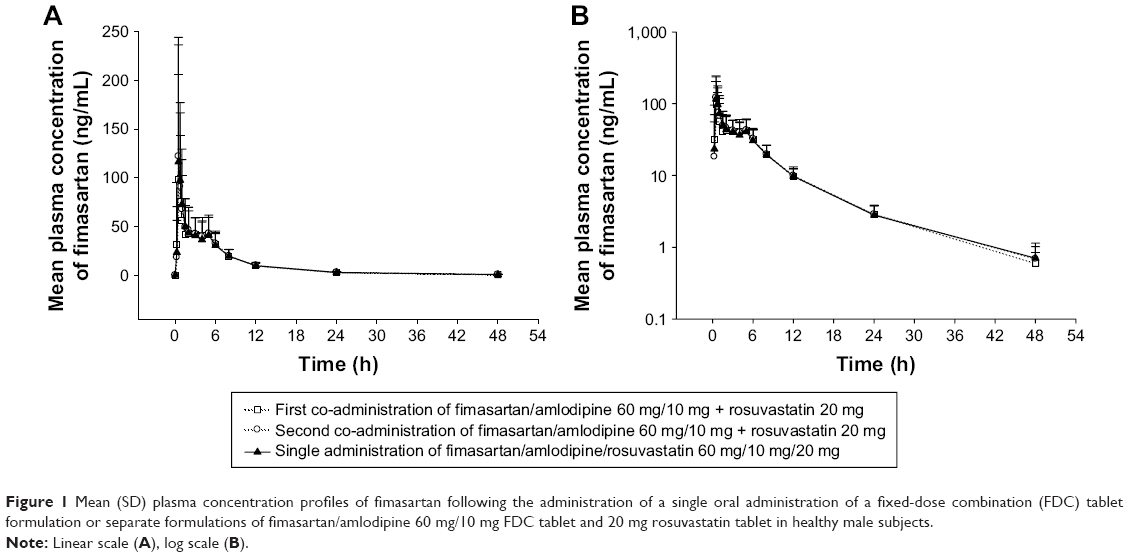 | Figure 1 Mean (SD) plasma concentration profiles of fimasartan following the administration of a single oral administration of a fixed-dose combination (FDC) tablet formulation or separate formulations of fimasartan/amlodipine 60 mg/10 mg FDC tablet and 20 mg rosuvastatin tablet in healthy male subjects. |
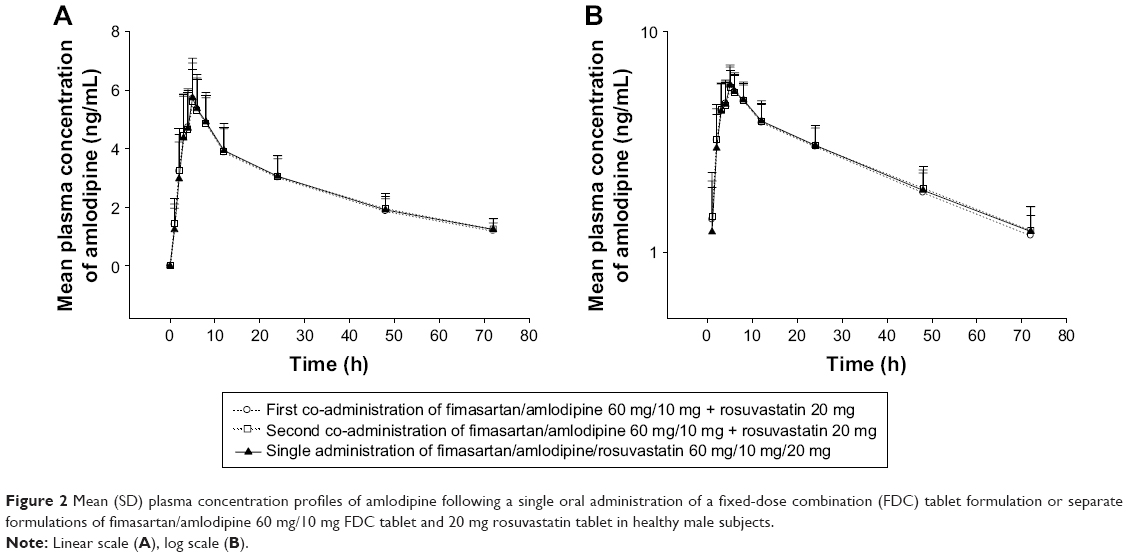 | Figure 2 Mean (SD) plasma concentration profiles of amlodipine following a single oral administration of a fixed-dose combination (FDC) tablet formulation or separate formulations of fimasartan/amlodipine 60 mg/10 mg FDC tablet and 20 mg rosuvastatin tablet in healthy male subjects. |
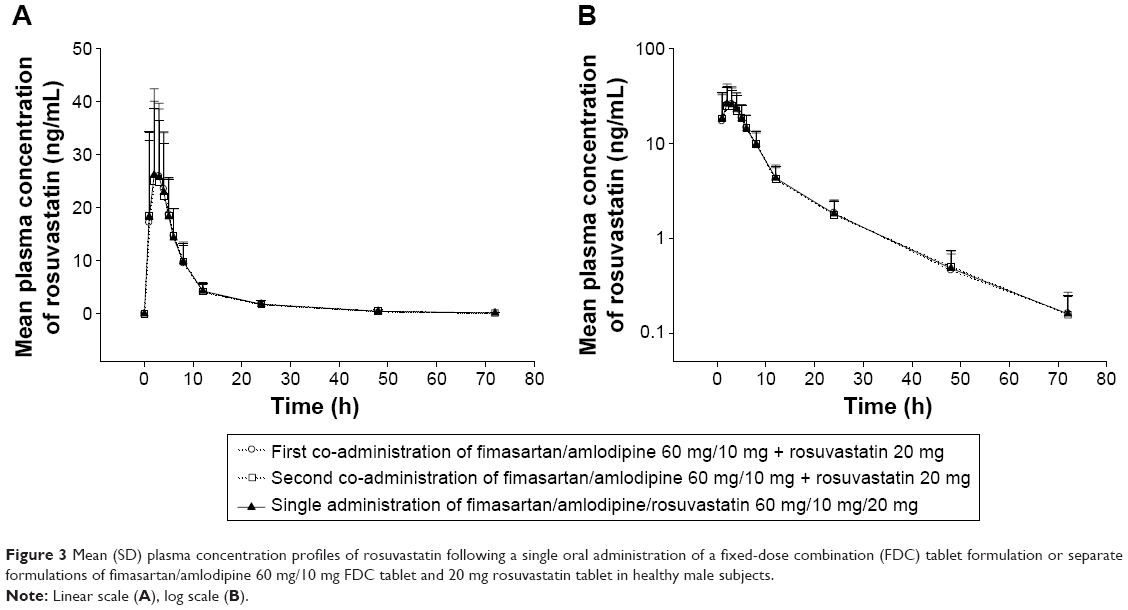 | Figure 3 Mean (SD) plasma concentration profiles of rosuvastatin following a single oral administration of a fixed-dose combination (FDC) tablet formulation or separate formulations of fimasartan/amlodipine 60 mg/10 mg FDC tablet and 20 mg rosuvastatin tablet in healthy male subjects. |
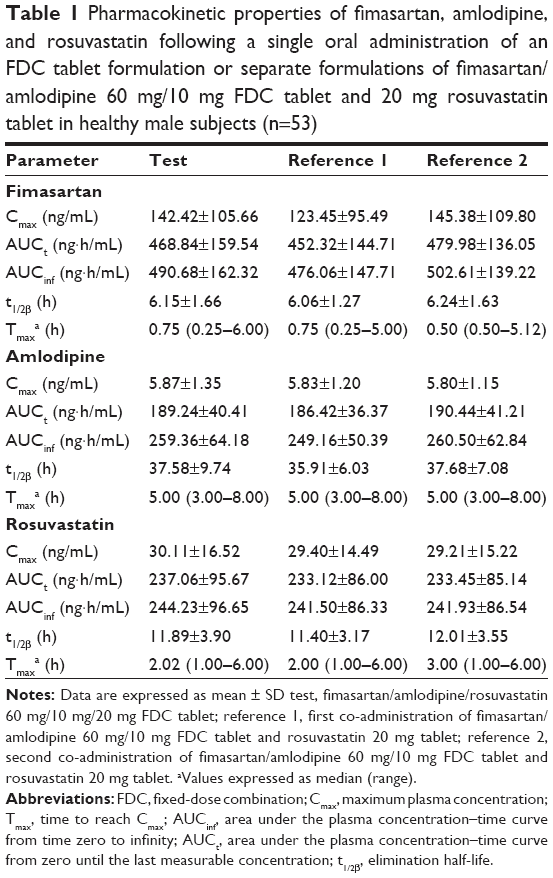 | Table 1 Pharmacokinetic properties of fimasartan, amlodipine, and rosuvastatin following a single oral administration of an FDC tablet formulation or separate formulations of fimasartan/amlodipine 60 mg/10 mg FDC tablet and 20 mg rosuvastatin tablet in healthy male subjects (n=53) |
The GMRs (90% CIs) of Cmax and AUCt were 1.0776 (0.9201–1.2622) and 0.9978 (0.9538–1.0439) for fimasartan, 1.0038 (0.9782–1.0301) and 1.0055 (0.9828–1.0288) for amlodipine, and 1.0006 (0.9290–1.0776) and 0.9986 (0.9532–1.0461) for rosuvastatin, respectively (Table 2). The within-subject CV of the Cmax for fimasartan was 67.95% and the expanded BE limit was 0.6894–1.4319. The 90% CIs of the Cmax and AUCt for fimasartan, amlodipine, and rosuvastatin fell within the predefined BE limit.
 | Table 2 GMRs (90% CI) of the PK properties of fimasartan, amlodipine, and rosuvastatin following a single oral administration of an FDC tablet formulation or separate formulations of fimasartan/amlodipine 60 mg/10 mg FDC tablet and 20 mg rosuvastatin tablet in healthy male subjects (n=53) |
Safety
During the study, a total of 22 AEs occurred in 19 subjects who were administered study drugs at least once. The number of AEs was 10 in nine subjects who received the test drug, 6 in six subjects who received reference 1, and 6 in five subjects who received reference 2. There was no statistically significant difference among the treatment groups in the rate of AEs.
The most frequently reported AE was headache (seven cases recorded in seven subjects) followed by dizziness (four cases in three subjects); blood creatine phosphokinase elevation (four cases in four subjects); nausea (two cases in two subjects); and oropharyngeal pain, diarrhea, epigastric discomfort, white blood cells in urine, and hypotension (each in one subject).
The severity of all AEs was mild and resolved spontaneously; there was no serious AE. Of the AEs, headache, dizziness, nausea, epigastric discomfort, and hypotension were considered to possibly be related to the study drugs.
There was no clinically significant finding or clinically relevant change in vital signs, clinical laboratory test results, physical examinations, or ECG profiles.
Discussion
This study showed the PKs and safety profiles of an FDC tablet formulation containing 60 mg fimasartan/10 mg amlodipine/20 mg rosuvastatin and separate formulations of fimasartan/amlodipine 60 mg/10 mg FDC tablet and 20 mg rosuvastatin tablet. And the 90% CI for the GMRs of Cmax and AUCt of an FDC formulation and co-administration of fimasartan, amlodipine, and rosuvastatin were within the accepted limit of BE based on partial replicated crossover study design. No serious or unexpected AEs occurred, and there was no clinically significant difference among the treatment groups.
About 228 subjects were required to detect a 20% difference between the test and reference drugs with 80% statistical power at a 5% level of significance based on the intrasubject variability of the Cmax for fimasartan, 58.8%, in the conventional 2×2 crossover study design. However, we performed a replicated crossover study design according to the guidelines for HVDs of the FDA and EMA. The study involved 60 subjects after considering a 30% dropout rate or compliance to the treatment groups. The largest intraindividual CV of the Cmax for fimasartan was 67.95%.
Two-by-two crossover studies of HVDs typically require a large number of healthy subjects in order to satisfy the statistical power. However, this required many ethical and financial considerations. And high within-formulation variability may lead to problem than high within-subject variability. Therefore, HVDs may consider the extended BE criteria and, FDA and EMA suggested the guidelines for the method of adjusting the acceptance range of equivalence in HVDs followed by the intraindividual variability in reference drug.13,14 Also, MFDS, South Korea, adopted these guidelines and added new rule in the regulation documents. This is similar to the EMA’s way of expanding the range of BE criteria according to the variation of intraindividual and not expanding beyond 50%.9,19–25
According to the ESH/ESC 2013 guidelines, monotherapy has a blood pressure lowering effect in limited patients, and most patients require two or more concomitant therapies. In patients with stage 1 hypertension, if the patient is at risk of cardiovascular disease, or if there is a risk of cardiovascular disease, or if the patient maintains the same blood pressure after a certain period of lifetime, the patient is advised to start treatment with antihypertensive agents. If the target blood pressure is not reached, a combination therapy is recommended.26,27
We assumed that the half-life of fimasartan was 5.8 hours and the washout period was 14 days. As a result, fimasartan is absorbed rapidly and its maximum concentration occurred within 1 hour (0.75 hours, 0.25–5.00), and it had a relatively short terminal elimination half-life (6.06±1.27 hours). When the variability of drug is high and the elimination half-life of it is relatively short, replicated crossover study design with three or four periods would be appropriate to assess the BE.
Therefore, we can expect that it will be possible to reduce the failure burden of the clinical trial and to conduct clinical trials designed with three or four periods with appropriate number of subjects if a study is intended to demonstrate BE in the case of a HVD with a short half-life.
Conclusion
This study showed that the PK profile of an FDC formulation and co-administration of fimasartan, amlodipine, and rosuvastatin in partial replicated study design were within the BE acceptance range by using reference-scaled average BE method. There was no significant difference for safety assessment between the FDC formulation and co-administration of the two products.
Acknowledgments
This study was funded by Boryung Pharmaceutical Co. Ltd., Seoul, Republic of Korea, and in part from the Global Center of Excellence, a grant from the Korean Health Technology R&D Project, Ministry of Health & Welfare, Republic of Korea (grant number: HI14C1063).
Disclosure
All authors report no conflicts of interest in this work.
References
Yeon OK, Yun JHS. The Statistics of deaths in 2013. The Korean National Statistical Office (KNSO); 2013. | ||
Centers for Disease Control and Prevention. National Vital Statistics Reports - Deaths: Final Data for 2015; 2018. Available from: https://www.cdc.gov/nchs/data/nvsr/nvsr66/nvsr66_06_tables.pdf. Accessed April 23, 2018. | ||
Kostis JB. The importance of managing hypertension and dyslipidemia to decrease cardiovascular disease. Cardiovasc Drugs Ther. 2007;21(4):297–309. | ||
Sever PS, Dahlöf B, Poulter NR, et al. Prevention of coronary and stroke events with atorvastatin in hypertensive patients who have average or lower-than-average cholesterol concentrations, in the Anglo-Scandinavian Cardiac Outcomes Trial – Lipid Lowering Arm (ASCOT-LLA): a multicentre randomised controlled trial. Lancet. 2003;361(9364):1149–1158. | ||
James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA. 2014;311(5):507–520. | ||
Lee SE, Kim Y-J, Lee H-Y, et al. Efficacy and tolerability of fimasartan, a new angiotensin receptor blocker, compared with losartan (50/100 mg): a 12-week, phase III, multicenter, prospective, randomized, double-blind, parallel-group, dose escalation clinical trial with an optional 12-week extension phase in adult Korean patients with mild-to-moderate hypertension. Clin Ther. 2012;34(3):552–568, 568.e559. | ||
Atlas SA. The renin-angiotensin aldosterone system: pathophysiological role and pharmacologic inhibition. J Manag Care Pharm. 2007;13(8 Suppl B):9–20. | ||
BORYUNG. The Investigator’s Brochure for Kanarb(R). BORYUNG; 2014. | ||
Pfizer. Prescribing Information for Norvasc. 2014. Available from: http://www.pfizer.ca/sites/g/files/g10017036/f/201505/Norvasc_PM_E_177714_30_Jan_2015.pdf. | ||
Trabelsi F, Bartunek A, Vlavonou R, et al. Single-dose, 2-way crossover, bioequivalence study of two rosuvastatin formulations in normal healthy subjects under fasting conditions. Int J Clin Pharmacol Ther. 2012;50(10):741–750. | ||
Jones PH, Davidson MH, Stein EA, et al. Comparison of the efficacy and safety of rosuvastatin versus atorvastatin, simvastatin, and pravastatin across doses (STELLAR Trial). Am J Cardiol. 2003;92(2):152–160. | ||
Jones PH, Hunninghake DB, Ferdinand KC, et al. Effects of rosuvastatin versus atorvastatin, simvastatin, and pravastatin on non-high-density lipoprotein cholesterol, apolipoproteins, and lipid ratios in patients with hypercholesterolemia: additional results from the STELLAR trial. Clin Ther. 2004;26(9):1388–1399. | ||
EMA. Guideline on the Investigation of Bioequivalence. London: European Medicines Agency (EMA); 2010. | ||
FDA. Office of Generic Drugs, Draft Guidance for Industry on Bioequivalence Recommendations for Progesterone Capsules; 2011. Available from: https://www.federalregister.gov/documents/2015/11/02/2015-27816/bioequivalence-recommendations-for-progesterone-draft-guidance-for-industry-availability. Accessed April 23, 2018. | ||
Haidar SH, Davit B, Chen ML, et al. Bioequivalence approaches for highly variable drugs and drug products. Pharm Res. 2008;25(1):237–241. | ||
Haidar SH, Makhlouf F, Schuirmann DJ, et al. Evaluation of a scaling approach for the bioequivalence of highly variable drugs. AAPS J. 2008;10(3):450–454. | ||
Sakima A, Ohshiro K, Nakada S, et al. Switching therapy from variable-dose multiple pill to fixed-dose single-pill combinations of angiotensin II receptor blockers and thiazides for hypertension. Clin Exp Hypertens. 2011;33(5):309–315. | ||
Sherrill B, Halpern M, Khan S, Zhang J, Panjabi S. Single-pill vs free-equivalent combination therapies for hypertension: a meta-analysis of health care costs and adherence. J Clin Hypertens (Greenwich). 2011;13(12):898–909. | ||
MFDS. Guideline for Bioequivalence Studies for Drugs. MFDS (Ministry of Food and Drug Safety); 2017. | ||
Lee H-Y, Oh B-H. Fimasartan: a new angiotensin receptor blocker. Drugs. 2016;76(10):1015–1022. | ||
Setiawati E, Sukmayadi, Yunaidi DA, et al. Comparative bioavailability cf two amlodipine formulation in healthy volunteers. Arzneimittelforschung. 2007;57(7):467–471. | ||
Shentu J, Fu L, Zhou H, et al. Determination of amlodipine in human plasma using automated online solid-phase extraction HPLC–tandem mass spectrometry: Application to a bioequivalence study of Chinese volunteers. J Pharm Biomed Anal. 2012;70:614–618. | ||
Martin PD, Mitchell PD, Schneck DW. Pharmacodynamic effects and pharmacokinetics of a new HMG-CoA reductase inhibitor, rosuvastatin, after morning or evening administration in healthy volunteers. Br J Clin Pharmacol. 2002;54(5):472–477. | ||
AstraZeneca. Prescribing Information for Rosuvastatin; 2014. Available from: https://www.azpicentral.com/crestor/crestor.pdf. Accessed April 23, 2018. | ||
Karalis V, Symillides M, Macheras P. On the leveling-off properties of the new bioequivalence limits for highly variable drugs of the EMA guideline. Eur J Pharm Sci. 2011;44(4):497–505. | ||
Mancia G, De Backer G, Dominiczak A, et al. 2007 ESH-ESC Practice Guidelines for the Management of Arterial Hypertension: ESH-ESC Task Force on the Management of Arterial Hypertension. J Hypertens. 2007;25(9):1751. | ||
Mancia G, Fagard R, Narkiewicz K, et al. Task Force Members. 2013 ESH/ESC Guidelines for the management of arterial hypertension: the task force for the management of arterial hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). J Hypertens. 2013;31(7):1281–1357. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.