")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Pharmacoinformatics approach for investigation of alternative potential hepatitis C virus nonstructural protein 5B inhibitors
Authors Mirza MU , Ghori N, Ikram N, Adil AR, Manzoor S
Received 15 October 2014
Accepted for publication 24 November 2014
Published 27 March 2015 Volume 2015:9 Pages 1825—1841
DOI https://doi.org/10.2147/DDDT.S75886
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Shu-Feng Zhou
Muhammad Usman Mirza,1 Noor-Ul-Huda Ghori,2 Nazia Ikram,3 Abdur Rehman Adil,4 Sadia Manzoor3
1Centre for Research in Molecular Medicine (CRiMM), The University of Lahore, Lahore, 2Atta-ur-Rehman School of Applied Biosciences (ASAB), National University of Science and Technology, Islamabad, 3Institute of Molecular Biology and Biotechnology (IMBB), The University of Lahore, Lahore, Pakistan; 4Centre for Excellence in Molecular Biology (CEMB), The University of Punjab, Lahore, Pakistan
Abstract: Hepatitis C virus (HCV) is one of the major viruses affecting the world today. It is a highly variable virus, having a rapid reproduction and evolution rate. The variability of genomes is due to hasty replication catalyzed by nonstructural protein 5B (NS5B) which is also a potential target site for the development of anti-HCV agents. Recently, the US Food and Drug Administration approved sofosbuvir as a novel oral NS5B inhibitor for the treatment of HCV. Unfortunately, it is much highlighted for its pricing issues. Hence, there is an urgent need to scrutinize alternate therapies against HCV that are available at affordable price and do not have associated side effects. Such a need is crucial especially in underdeveloped countries. The search for various new bioactive compounds from plants is a key part of pharmaceutical research. In the current study, we applied a pharmacoinformatics-based approach for the identification of active plant-derived compounds against NS5B. The results were compared to docking results of sofosbuvir. The lead compounds with high-binding ligands were further analyzed for pharmacokinetic and pharmacodynamic parameters based on in silico absorption, distribution, metabolism, excretion, and toxicity (ADMET) profile. The results showed the potential alternative lead compounds that can be developed into commercial drugs having high binding energy and promising ADMET properties.
Keywords: hepatitis C, NS5B inhibitors, molecular docking, AutoDock Vina, ADMET, sofosbuvir, phytochemicals
Introduction
Hepatitis C virus (HCV) is a major cause of liver-related infectious diseases, including hepatitis C, liver cirrhosis, fibrosis, and hepatocellular carcinoma. Approximately 170 million people worldwide are affected by hepatitis C. About three to four million new infections are developing every year and turn into chronic infections.1 HCV is a single-stranded positive-sense RNA virus of family Flaviviridae. The HCV genome (~9.6 kb), with a single open-reading frame, encodes a single and long protein of about 3,010 amino acids which is further processed to make several active proteins by HCV serine proteases (NS3, NS4A). These active proteins include three structural (core, E1, and E2) and seven nonstructural proteins (p7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B). The HCV genome has conserved 5′ and 3′ untranslated regions (UTRs) where 5′ UTR possesses an internal ribosome entry site that recruits host ribosome to translate its genome and 3′ UTR is involved in viral replication and its packaging.2 Among all HCV proteins, NS3, NS5A, and NS5B are potential drug targets for the development of anti-HCV agents.3
Phylogenetically, HCV is classified into seven major groups and further subclassified into 67 subtypes.4 Among these variants, genotype 3 is rather dominant in Southeast Asia and less prevalent in certain European countries. In addition, fast fibrosis progression has been observed in genotype 3.5 Apart from genotype variation, HCV exists as a heterogeneous population of different viruses called quasispecies, within each infected individual.6,7 The variability of genomes is mainly due to rapid replication catalyzed by NS5B. NS5B is an error-prone RNA-dependent RNA polymerase (RdRp) with inefficient proofreading ability. NS5B belongs to a class of tail-anchored proteins with a confirmation resembling that of fingers, palm, and thumb of the right hand.8,9 The palm domain contains the active site of polymerase enzyme, and the finger and thumb modulate interactions with RNA template. The finger and thumb are considered to have multiple interactions with each other which encircle the active site and form a tunnel that guides RNA template directly to the active site. RNA synthesis is regulated by a flexible beta hairpin loop present in the palm domain.10,11 NS5B replicates RNA in a semiconservative manner and initiates de novo synthesis. Various NS5B inhibitors have been developed that mimic natural polymerase substrates and can inhibit their catalytic efficiency.12 These inhibitors can be classified as either nucleoside inhibitors or non-nucleoside inhibitors. Nucleoside inhibitors such as ribavirin cause premature termination of RNA synthesis and include purine, pyrimidine, or miscellaneous nucleoside inhibitor classes. The non-nucleoside inhibitors include allosteric, active site and miscellaneous non-nucleoside inhibitors.13 However, available drugs are associated with adverse side effects that pose a need for the development of an effective drug.
Recently, sofosbuvir has been approved by the US Food and Drug Administration as a novel oral antiviral drug to target NS5B for the treatment of HCV.14,15 Sofosbuvir, being a uridine nucleotide analog, targets the catalytic site of NS5B polymerase, thus serving as a non-obligate chain terminator.16 Common side effects associated with sofosbuvir treatment include fatigue, headache, nausea, rash, and irritability. Moreover, the drug is considered less effective in genotype 3, especially in the presence of cirrhosis.17 Despite its potential for HCV treatment, sofosbuvir is much highlighted for its pricing controversies. In fact, the drug is given in combination with other HCV drugs, and a 24-week-long treatment plan costs as much as $168,000 when used against genotype 3. This high cost makes it unaffordable in underdeveloped countries where genotype 3 is prevalent. Hence, there is a vital need to scrutinize alternate therapies against HCV with minimal side effects and low cost.
The search for various new bioactive compounds from plants is a key part of pharmaceutical research.18,19 Plants have been used to treat several human diseases for centuries. Not surprisingly, many modern drugs have also been developed from molecules that are isolated from natural sources.20 Plants are rich in phytochemicals such as phenolics, alkaloids, and flavonoids. These compounds, being natural, are associated with least side effects and cost. Hence, this mode of drug production may lead to the development of less expensive treatments. One approach of screening bioactive compounds is through molecular docking.21 Comparative studies of phytochemicals provides the best avenue for searching new economic plant for HCV,22,23 and some natural active compounds have been revealed to have antiviral activities against hepatitis B virus and HCV.24–26
In addition to high potency and good docking results, a drug should have an impressive absorption, distribution, metabolism, excretion, and toxicity (ADMET) profile. Thousands of potential compounds fail to reach drug status because of poor pharmacodynamic (PD) and pharmacokinetic (PK) properties each year.27 These characteristics depend upon the physiochemical properties of the compound that determine its ADMET. A drug’s absorption usually depends upon its lipophilicity and solubility.28 The more a drug is lipophilic, the greater is its rate of absorption in the bloodstream due to greater affinity of lipophilic compounds for metabolic enzymes. Once absorbed, the drug is distributed across the body. This is majorly determined by the affinity of the drug for blood and tissues. Lipophilicity also affects the binding of a drug with the protein.29 The more the drug is lipophilic, the greater is its distribution. A drug is metabolized and converted into a more water-soluble compound so that it can be easily eliminated from the body. The metabolism is generally carried out by cytochrome P450 (CYP450) enzymes.30 Interestingly, more than 60 isoforms of CYP450 are present in the body with hundreds of genetic variations possible.31 The variety accounts for their susceptibility toward various toxins. The kidney and liver are mainly involved in the excretion of drugs.32 The ADMET profiling is usually done in the end stages of drug development accounting for a high risk, greater expense, and time. Alternatively, in silico approach is applied to provide a more efficient route to drug development. The PK properties are now determined as a mathematical description of the rates of absorption, distribution, metabolism, and excretion (ADME) processes and of concentration–time relationships.32
Considering these critical requirements, the current in silico study was undertaken to find efficient NS5B polymerase inhibitors through comparative study of various phytochemicals found in the literature which possess anti-HCV properties. The potential compounds were also evaluated for their PK and PD properties. The efficacy of all compounds was compared with that of sofosbuvir with the sole purpose of providing a cheaper alternative HCV therapy.
Materials and methods
Proteins and ligands
Several crystal structures of viral protein NS5B have been solved with their potential inhibitors and are present in structural databases. The crystallographic structures of NS5B in complex with its inhibitors were taken from Brookhaven Protein Data Bank (PDB). The crystal structures of NS5B are present in the Protein Data Bank with 2WCX,33 2BRL, 2BRK,34 2XWY,35 and 2DXS36 codes. The residues making binding pockets for the inhibitor were investigated. The binding site of NS5B was further validated by using COACH,37 Castp,38 and 3DLigandSite servers.39 An extensive literature review was done to know about the ligands that act as inhibitors. Compounds found in various plants, reported in the literature to possess activity against HCV, were selected. These compounds belong to different classes of phytochemicals such as flavonoids, lignans, alkaloids, and coumarins (Table S1). A total of 84 ligands were short listed, and their structures were downloaded in SDF format from different small-compound databases including PubChem,40 Chemspider,41 Zinc,42 and DrugBank.43 The SDF format was converted to 3D Mol format using ChemOffice 2012 (Cambridge Soft Corp., Cambridge, MA, USA, http://www.cambridgesoft.com).
In silico analysis of drug likeness and ADMET properties
Selected compounds were subjected to further selection on the basis of Lipinski’s rule of five (Ro5),44 and compounds without any Ro5 violations were eliminated. This was simply done by using Mcule,45 Cresset, and Molinspiration server (http://www.molinspiration.com/cgi-bin/properties) for calculation of their physicochemical properties. The drug likeness of screened compounds was further confirmed by using Marvinsketch (http://www.chemaxon.com/marvin/sketch/index.jsp). The ADMET properties of the filtered compounds were found to know about the rate and extent of ADMET threats of compounds through blood, body fluids, tissues, and excretory material of the body. A wide range of tools was used for ADMET assessment. These include OSIRIS property explorer (http://www.organic-chemistry.org/prog/peo/), Discovery Studio 4.0 (Accelrys Software Inc.), Cresset, admetSAR (http://1mmd.ecust.edu.cn:8000/),27 ADMET prediction suite by ACD/Labs, and Molsoft (http://molsoft.com/mprop/). Ligands were selected by calculating ligands drug likeness based upon Lipinski’s Ro5. Selected ligands were docked in to the binding pocket of NS5B protein.
Pre-docking procedure
In order to prepare the selected compounds for docking, hydrogens and Gasteiger charges were added. Charges were merged, and nonpolar hydrogens, lone pairs, water molecules, and nonstandard residues were removed. Energy minimization and geometry optimization of all structures, ligands, and proteins were performed using general purpose semiempirical molecular orbital package (MOPAC) version 2012 with improved accuracy of semiempirical method PM7. A grid with a dimension of 30 Å ×30 Å ×30 Å was centered covering the binding pocket of NS5B protein. AutoDockVina (Scripps Research Institute, La Jolla, CA, USA) was employed in order to perform molecular docking.46 Standard autodock protocol was used with ligands. AutoDockVina was set up on a DELL Inspiron Core i7 workstation under operating system as Ubuntu 12. Protein complexes of all filtered ligands were further analyzed using UCSF Chimera 1.9 (Resource for Biocomputing, Visualization and Informatics, University of California, San Francisco, CA, USA),47 Discovery Studio 4.0 (Accelrys Software Inc.),48 and Pymol (The PyMOL Molecular Graphics System, Version 1.5.0.4, Schrödinger, LLC).49
Results and discussion
The aim of the current study was to elucidate alternative inhibitory compounds against NS5B protein and to find the binding pocket against viral RdRp. A total number of 84 plant-derived compounds were chosen from the literature. In silico virtual screening of selected compounds was performed on the basis of Lipinski’s Ro5 and physicochemical properties, to assess the drug likeness (Table 1). The compounds showing strong binding affinity for NS5B viral protein were selected for further investigations. Binding affinity calculations for all 30 filtered compounds were carried out using AutoDockVina. Out of 30 filtered compounds, ten showed strong binding affinity for the target. These results were compared to the docking result of the recently approved drug against NS5B, namely sofosbuvir. Further analysis includes PK and PD properties of screened compounds as early drug discovery process. The ADMET profile was analyzed to infer the suitable lead compound. The criteria for selection of best compounds were high binding affinity compared to sofosbuvir and good pharmacological properties. In the present study, only the result of these ten compounds will be discussed. Further, we reported inhibitory effect of compounds that will be the potential lead compounds for alternative drug therapy of HCV patients providing cheap and effective therapy in comparison to expensive sofosbuvir treatment.
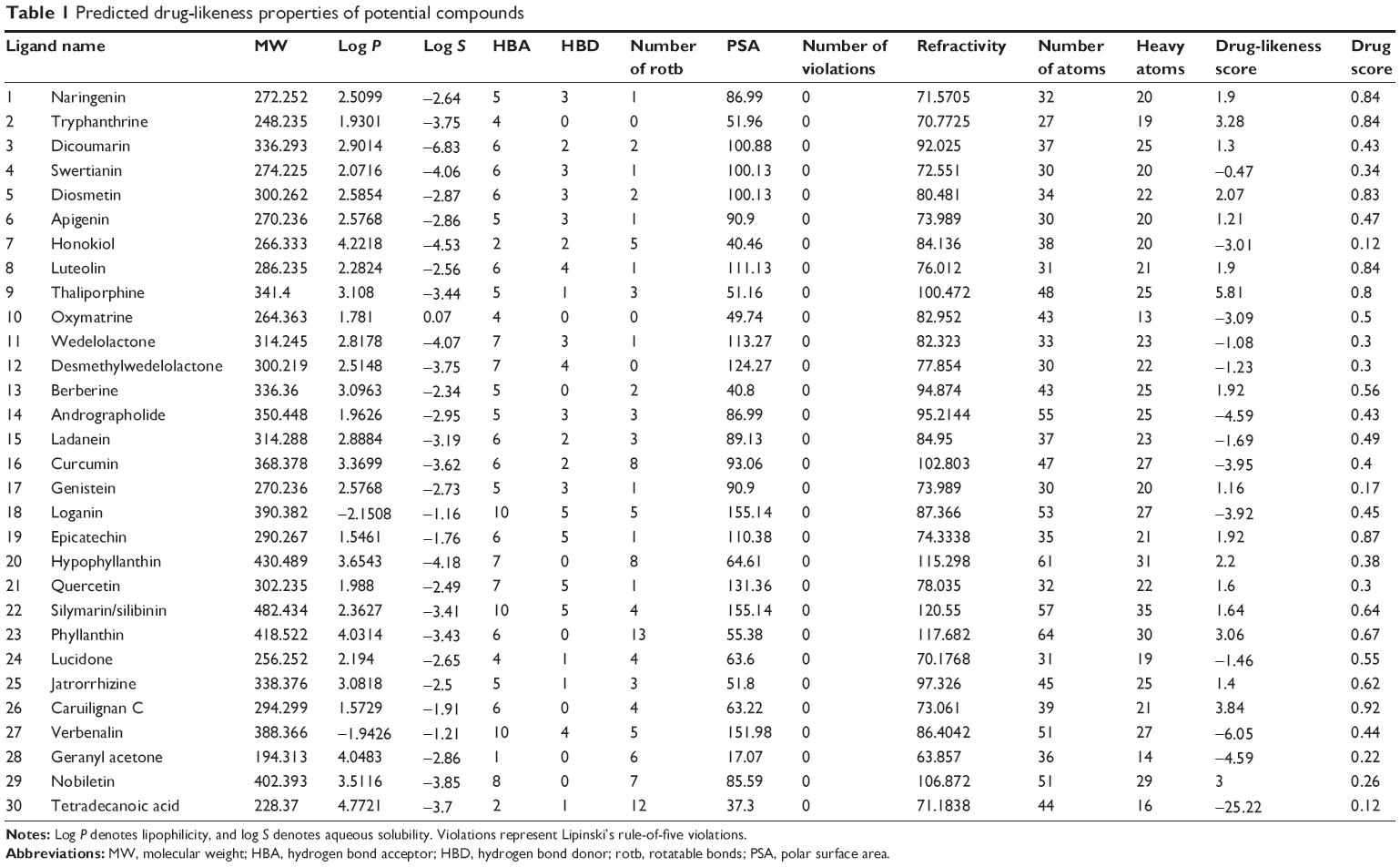 | Table 1 Predicted drug-likeness properties of potential compounds |
Binding site analysis
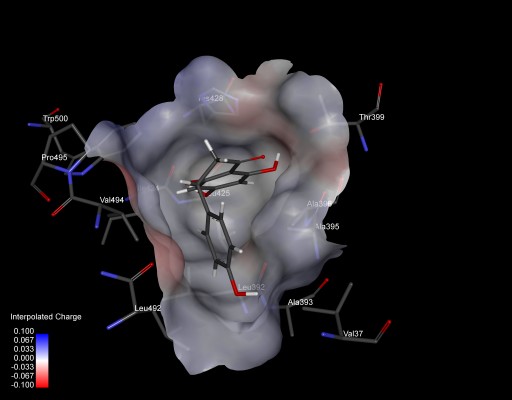
The protein-binding site prediction servers validated the binding site residues of target protein. A superimposition of all five complexes further confirmed the most conserved interacting amino acids of NS5B (Figure 1). It was found that nine amino acids were actually involved in interactions with HCV-NS5B inhibitors. These interacting residues include Val37, Leu492, His428, Ala395, Leu392, Val494, Ala396, Arg503, Ile424, and Pro495, constituting the active site for HCV-NS5B. Furthermore, a deep groove, encompassing the binding pocket, was observed, thus providing space for inhibitors to strongly bind with the active site of NS5B (Figure 2). It was assumed that binding of drug in this deep groove will inhibit virus from replication, and it seems to be a promising mode of action to be chosen for designing drug candidates against HCV.
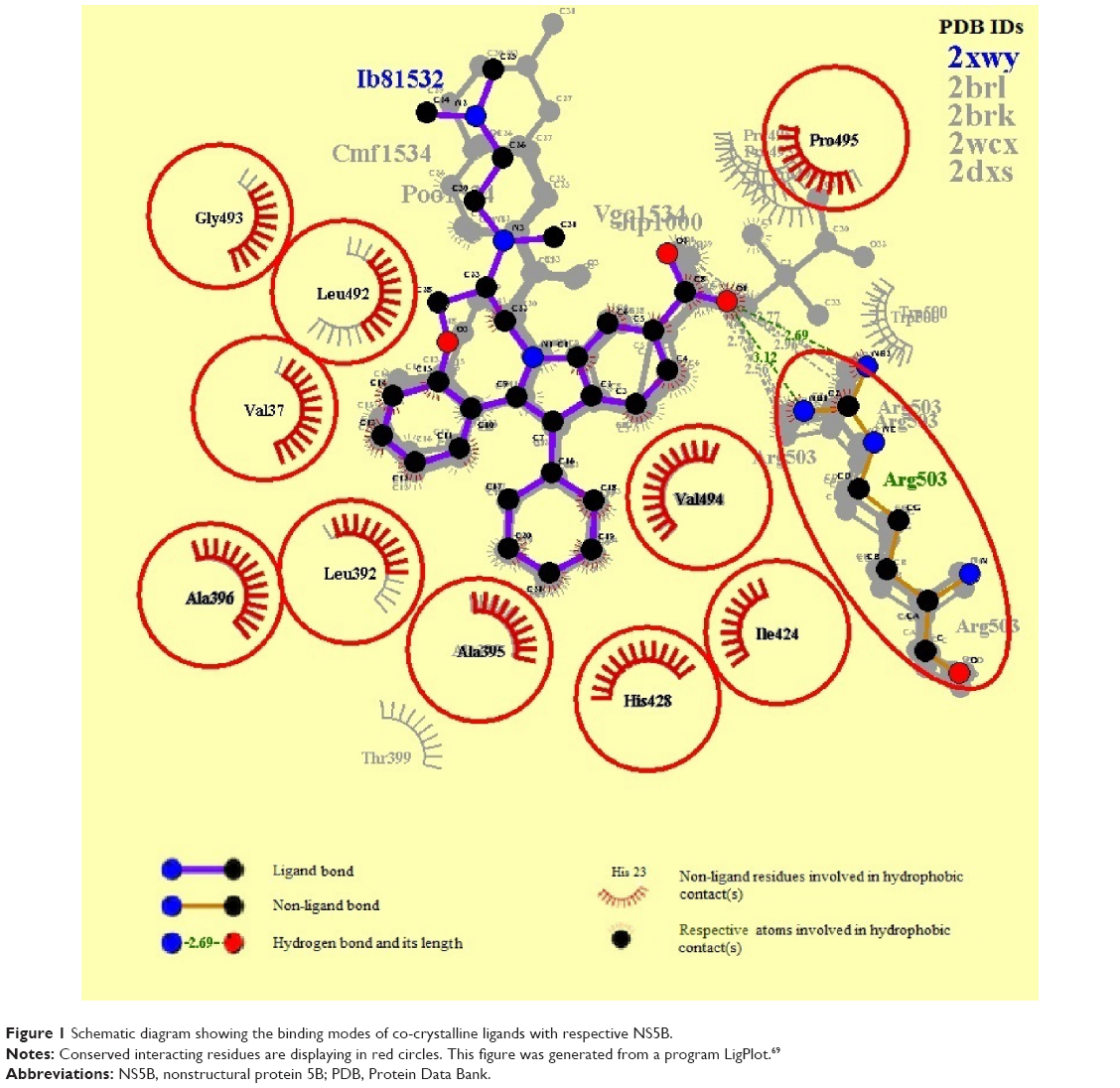 | Figure 1 Schematic diagram showing the binding modes of co-crystalline ligands with respective NS5B. |
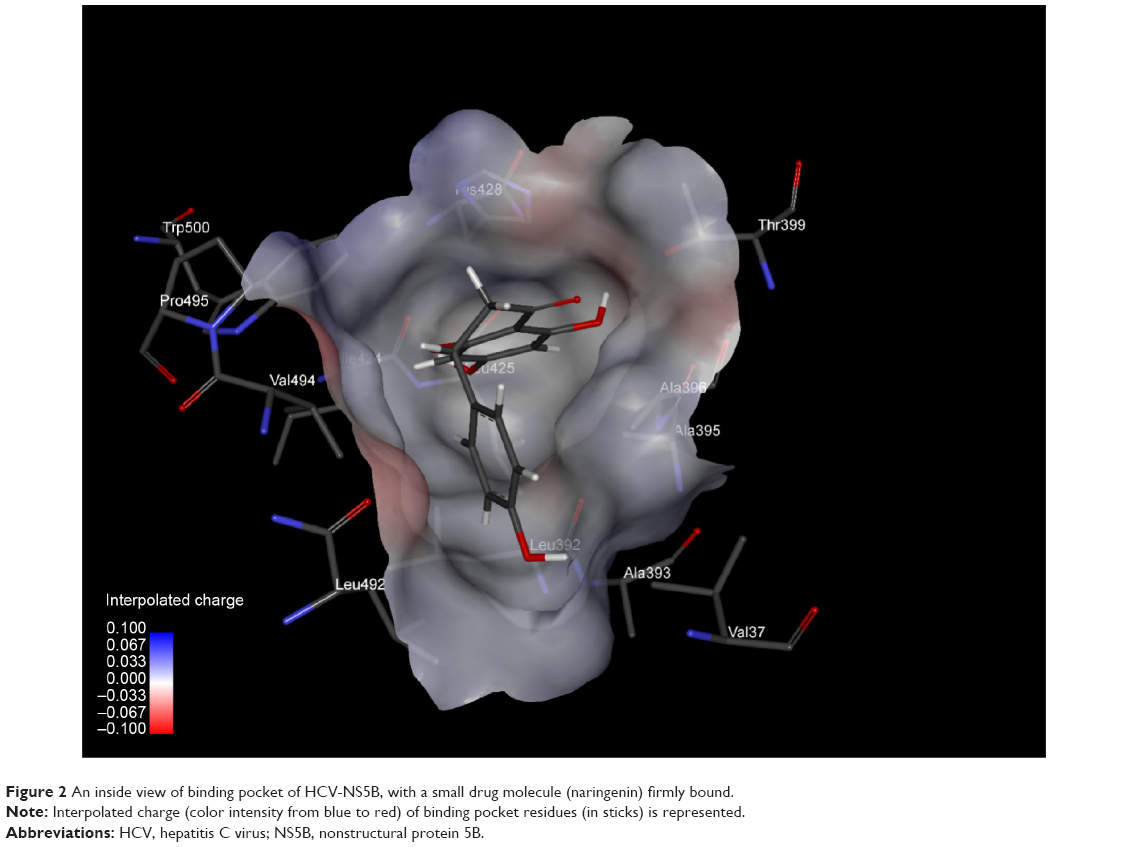 | Figure 2 An inside view of binding pocket of HCV-NS5B, with a small drug molecule (naringenin) firmly bound. |
Molecular docking study
Molecular docking of two molecules, the ligand and target, predicts the best ways of their interactions.50 In the current study, NS5B was docked with various plant-derived compounds to find the best candidate that inhibits viral replication. A total of 84 phytochemicals having inhibitory effects against NS5B were screened for their maximum probable activity.
The binding pocket was determined by various crystalline structures and binding site prediction servers. A total of 30 ligands with high binding affinities for NS5B were obtained. The docking scores were represented along with hydrogen bonds, direct contacts based on van der Waals (vdW) radii, and interacting residues profiled in Table 2. Binding energies were the representative of how precisely the drug (ligand) binds to the target molecule (protein), and thus were taken as baseline comparison for selection of lead compounds in drug designing. Ninety-three percent of the ligands showed a binding score stronger than 8 kcal/mol on docking with NS5B. None of the ligands showed binding score weaker than −7.4 kcal/mol. Ligands with high affinity scores were naringenin, tryphanthrine, dicoumarin, swertianin, diosmetin, apigenin, honokiol, luteolin, thaliporphine, and oxymatrine. Binding energies of these compounds ranged from −9.7 kcal/mol to −9 kcal/mol, which were stronger as compared to sofosbuvir (−6.2 kcal/mol). These ligands were found to interact mostly with NS5B via Leu392, Ala395, Ala396, His428, and Leu492 residues forming hydrogen and VdW interactions. It is inferred that these interactions stabilize the protein–ligand complex and lead to inhibitory activity on NS5B active site. Among molecular interactions, compounds, namely, naringenin, tryphanthrine, swertianin, diosmetin, luteolin, and thaliporphine were observed to form three hydrogen bonds, each mainly with Ala396 and Arg503. They were also seen to form VdW interactions mainly with His428, Val494, Leu492, Leu392, Pro495, and Ala395 (Table 2). Interestingly, all filtered compounds bind within a narrow groove line with their nonpolar and positively charged residues, and these ligands commonly interact with Val37, Leu392, Ala395, Ala396, His428, Leu492, and Val494 located in this groove (Figure 3). Comprehensively, it can be deduced that Leu492, Leu392, Val494, and Pro495 residues are involved in VdW interactions with NS5B, while Cys146, Ala395, Ala396, His428, and Arg503 are largely involved in forming hydrogen bonds.
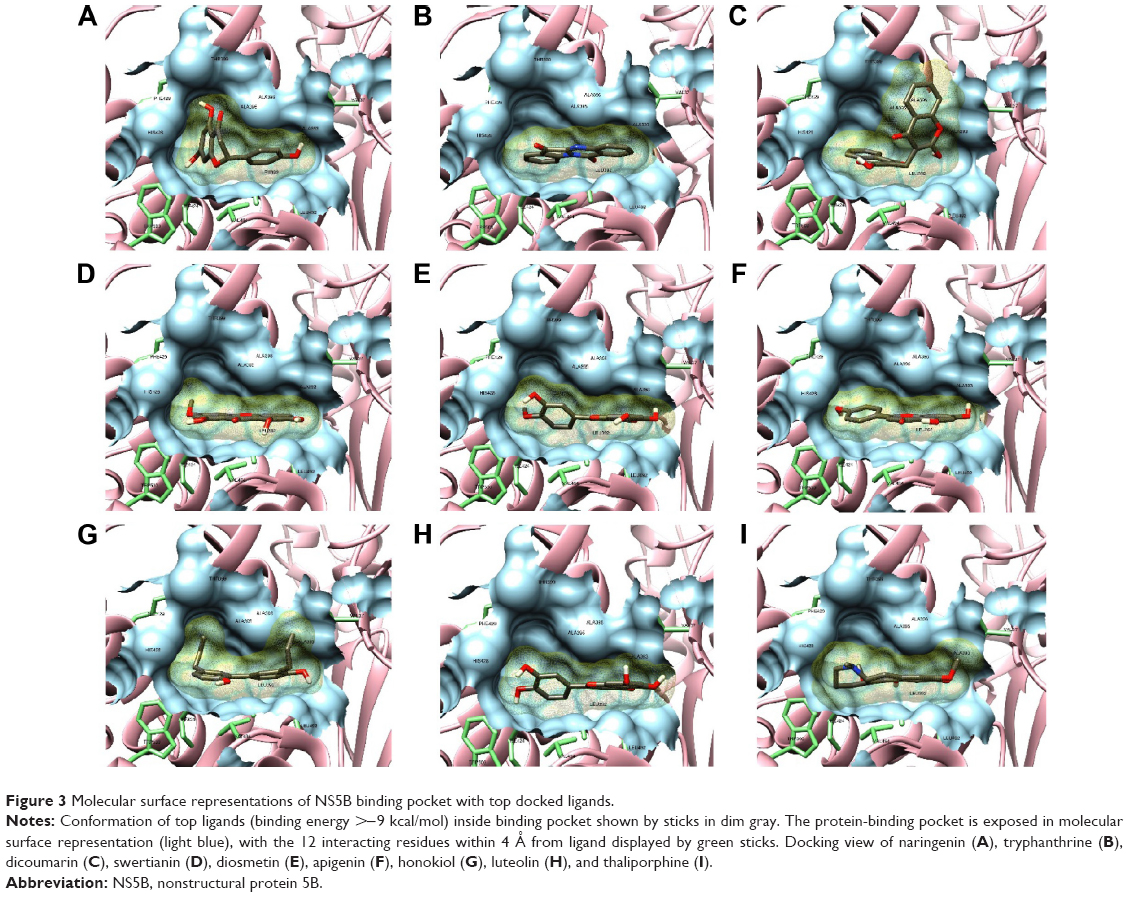 | Figure 3 Molecular surface representations of NS5B binding pocket with top docked ligands. |
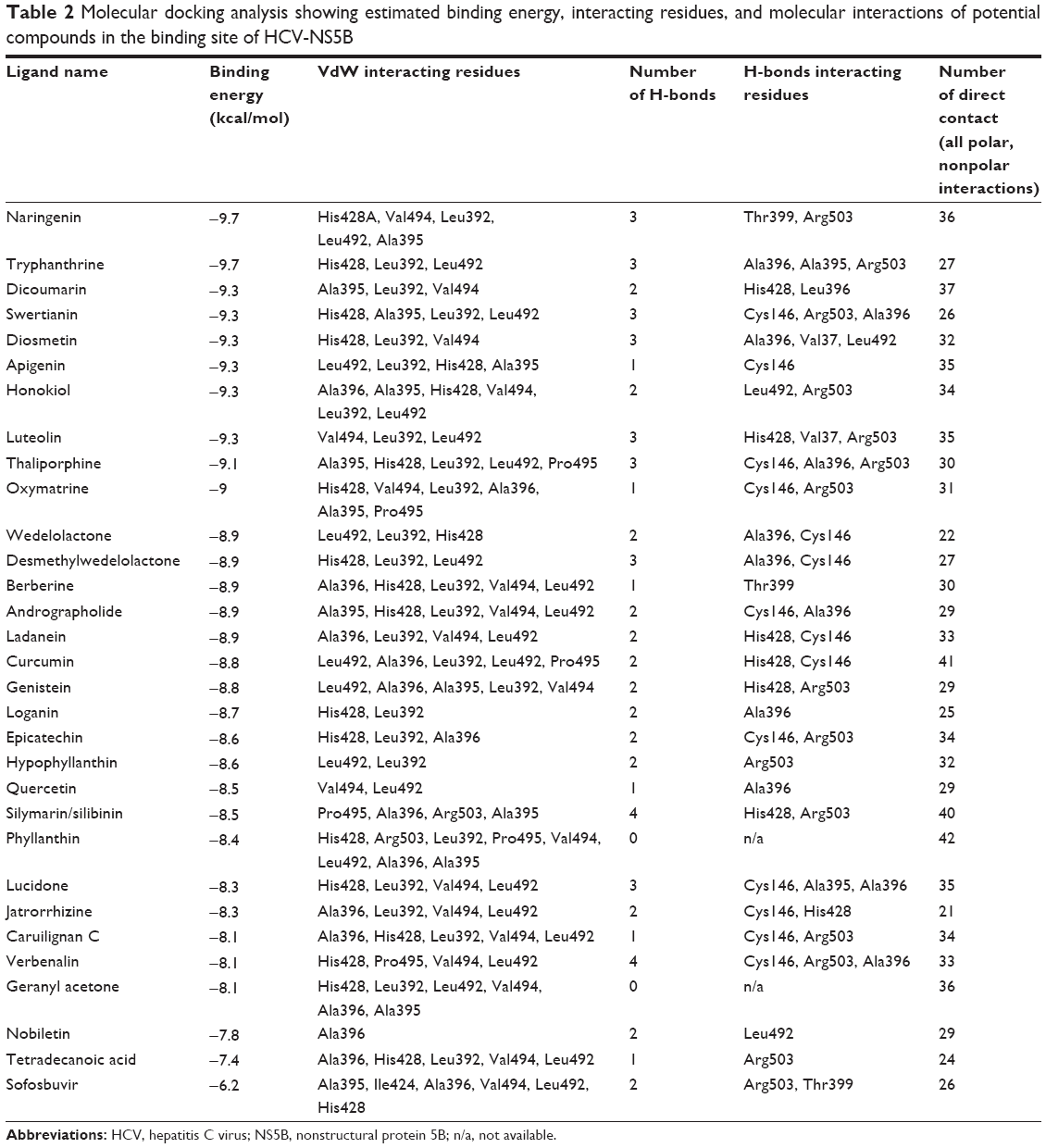 | Table 2 Molecular docking analysis showing estimated binding energy, interacting residues, and molecular interactions of potential compounds in the binding site of HCV-NS5B |
From the tabulated results (Table 2), it is clearly evident that all the 30 filtered compounds were successfully docked with the viral NS5B protein, with a binding affinity ranging between −7.4 kcal/mol and −9.7 kcal/mol as compared to sofosbuvir.
Prediction of ADME profile
Earlier, scanning of ADME properties was carried out at the end stage of drug discovery or during preclinical trials. As a result, more and more drugs failed to reach final stage because of their poor PK and PD profiles. In silico, a rather new approach to analyze ADMET properties is time- and cost-effective. It also provides a less risky route for bringing new therapies to patients.51 Nowadays, ADME analysis is performed at the early stages of drug discovery and is a part of high-throughput drug screening.52 ADMET predictions are based on molecular descriptor values according to Lipinski’s Ro5. Ligands having suitable pharmacological and drug-like properties are considered vital for drug discovery process.44 They include properties such as blood–brain barrier (BBB) penetration,53 human intestinal absorption (HIA),54 aqueous solubility,55 and CYP450 inhibition.56 Analysis of PK and PD properties is a crucial step for rational drug designing.32 In the current study, in silico method was employed using 30 filtered ligands that were selected upon drug-likeness properties assessed using Lipinski’s Ro5. The rule states that most orally administered drugs have a molecular weight of less than 500 Da, and their log P-value is less than five. Also, they possess five or less hydrogen bond donor sites and ten or less hydrogen bond acceptor sites, hence predicting their oral bioavailability.57 The in silico models of ADMET properties use the quantitative structure–activity relationship, in which physicochemical properties to describe molecules are used to search for a relationship with an ADMET property graphically represented in Figure 4. The analysis was performed by submitting the molecular structures of ligands on the admetSAR server and OSIRIS property explorer, and by using ADME descriptors of Discovery Studio 4.0 for predicting various ADME properties.
 | Figure 4 The journey of an oral drug candidate depicting the quantitative structure–activity relationship of physicochemical properties with absorption, distribution, metabolism, excretion, and toxicity. |
The ADME profile of high-scoring ligands (>–9 kcal/mol) in docking is precisely showing in Table 3. It is seen that diosmetin and luteolin have side effects on absorption through BBB and only oxymatrine exhibits poor absorption through human intestine. The tight junctions present between endothelial cells of brain capillaries are highly resistant and prevent uptake and delivery of polar drugs by the brain.58 Penetration of BBB is required to minimize central nervous system-related consequences. It depends on factors such as lipophilicity, molecular size, desolvation potential, pKa-to-charge ratio, and hydrogen bond.59,60 Similar results were obtained for HIA and Caco-2 permeability where most of the high-scoring ligands showed positive result, with the only exception of oxymatrine and luteolin showing negative results. Thaliporphine and oxymatrine were observed to show inhibitory side effect for renal organic cation transporter. For P-gp substrate, only half of the ligands showed positive result, whereas all ten ligands were non-inhibitors for P-gp inhibitor.
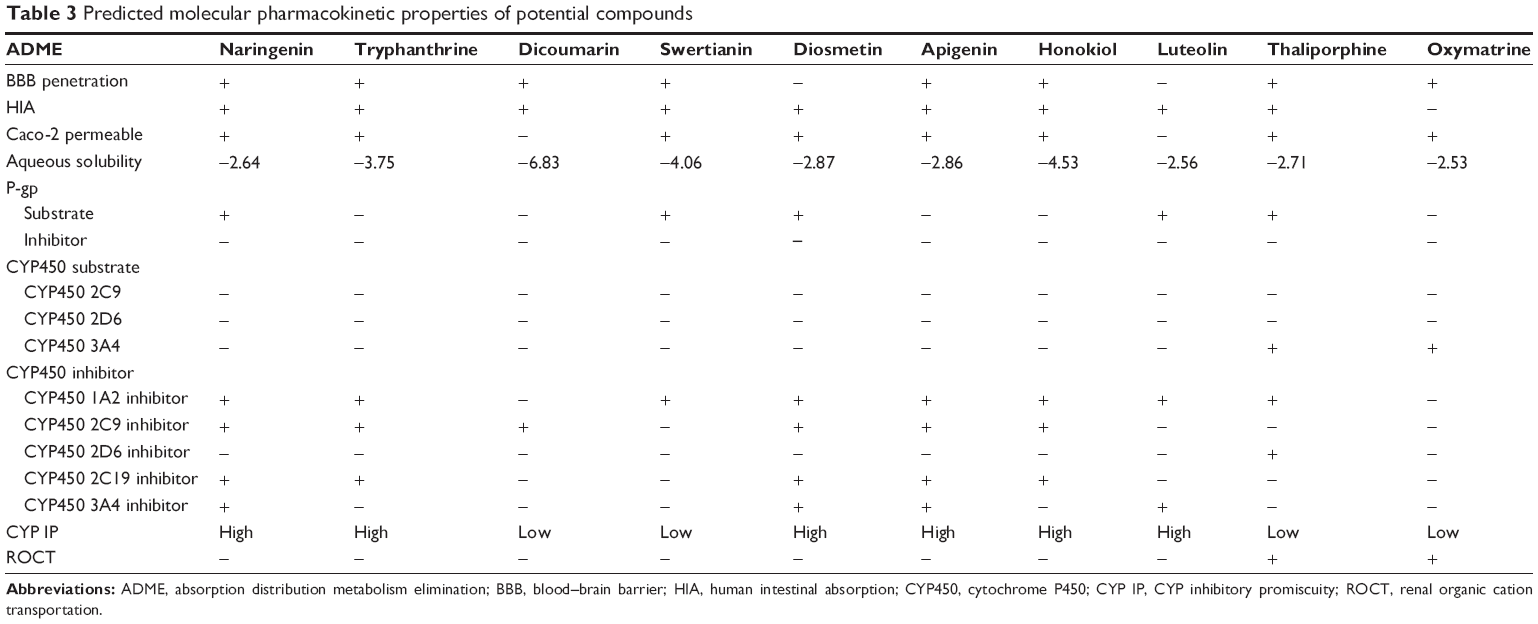 | Table 3 Predicted molecular pharmacokinetic properties of potential compounds |
CYP450 belongs to superfamily of hemoproteins and is a group of isozymes that are involved in the metabolism of carcinogens, fatty acids, bile acids, steroids, and importantly of drugs and other xenobiotics.61 Thus, CYP enzymes play a major role in drug metabolism. Therefore, various CYP450 substrate (2C9, 2D6, and 3A4) and CYP450 inhibitor (1A2, 2C9, 2C19, 2D6, and 3A4) models are calculated during ADME profiling. All ten ligands were non-substrate for all CYP450 substrates (2C9, 2D6, 3A4), whereas oxymatrine and thaliporphine were found to be substrate for CYP450 3A4 enzyme. The analysis showed that naringenin, tryphanthrine, diosmentin, apigenin, honokiol, and luteolin revealed high CYP inhibitory promiscuity, as these ligands inhibited most of the CYP450 inhibitors (1A2, 2C9, 2C19, 2D6, and 3A4).30 The inhibition of CYP450 isoforms affects the drug metabolism and causes the accumulation of toxic levels.62 It can be summarized from the results that dicoumarin and swertianin have the best ADME profile with good BBB, HIA, and non-inhibitors of CYP450 isoforms, which help in drug metabolism and its clearance from the body.30 The predicted ADME profile of all 30 filtered compounds is tabulated in Table S2.
Prediction of toxicity profile
Along with PK, a lead compound must also not show a threat to toxicity risks upon consumption; that is, it must have good PD properties.63 In the present study, ten qualitative classification models were used, which included ligand’s biodegradability, developmental toxicity potentials, AMES toxicity, mutagenicity, carcinogenicity, irritancy, tumorigenicity, fish toxicity, honey bee toxicity, Tetrahymena pyriformis toxicity, and reproductive effectiveness (Table 4). Toxicity profile revealed that most of the compounds were not mutagenic, carcinogenic, and tumorigenic, were negative for AMES toxicity, and had no significant toxicity properties that can produce harmful effects in humans. However, there were a few exceptions. Also, only a few ligands showed toxicity effect among all 30 filtered compounds (Table S3).
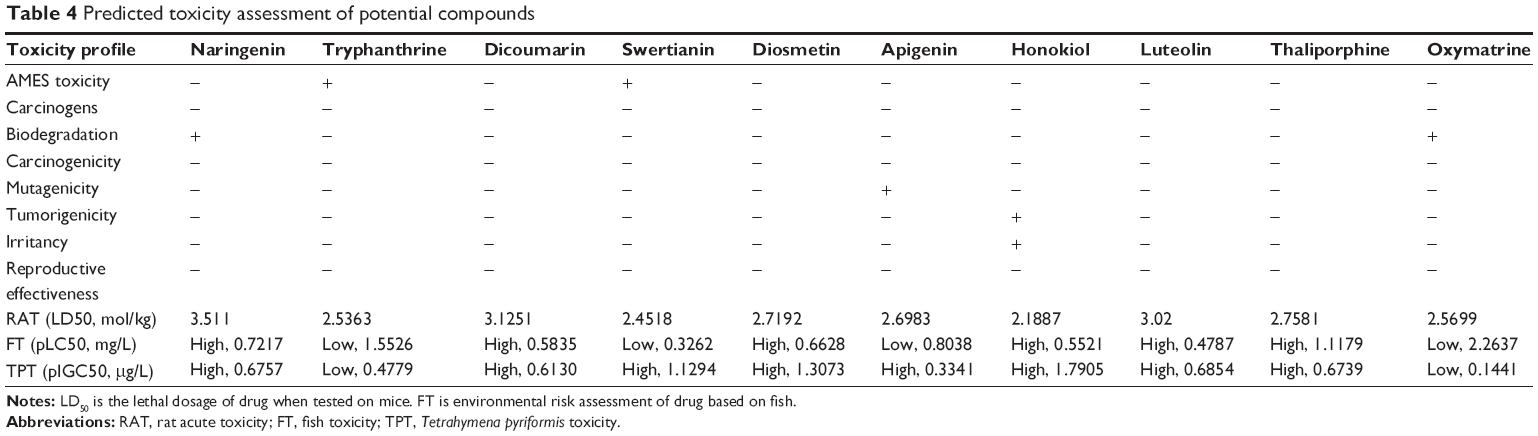 | Table 4 Predicted toxicity assessment of potential compounds |
Conclusion
In this past decade there has been a great interest in exploitation of phytochemicals for pharmaceutical use.64 It is not surprising that such studies has been carried out for hepatitis C also. HCV is a highly variable virus, having a rapid reproduction rate.65 Inhibition of NS5B, an RdRp, is therefore the principal option for the treatment of the disease caused by this virus. Treatments providing inhibitory activity include nucleotide analog inhibitors such as sofosbuvir. It should, however, be noted that sofosbuvir is used in combination with ribavirin and is considered as an add-on therapy. Such multidrug regimen is not only high in cost but also increases the side effects.66 Ribavirin and interferon therapies already have well-known consequences, including fatigue, influenza-like symptoms, hematologic abnormalities, and neuropsychiatric symptoms.67 Therefore, there is a need to find cheaper and efficient drugs for HCV treatment.
The present in silico studies provide insight into the inhibition of NS5B viral protein by plant-derived compounds which are more or equally potent to sofosbuvir. Many of the compounds also fulfill the ADMET criteria and thus can be formulated into a commercial drug. In the current investigation, a total of 84 compounds were screened and analyzed for their inhibitory action against NS5B protein. Molecular docking studies suggested ten highly active compounds, namely, naringenin, tryphanthrine, dicoumarin, swertianin, diosmetin, apigenin, honokiol, luteolin, thaliporphine, and oxymatrine as HCV inhibitory drugs. These compounds showed strong binding affinity (greater than −9 kcal/mol) when docked against NS5B active site. The binding energies are much higher than the binding energy of the FDA-approved drug, sofosbuvir (−6.2 kcal/mol). These inhibitors also presented good molecular interactions (hydrogen and VdW) with the active site of NS5B. After ADMET analysis, it has been observed that among high-scoring ligands, dicoumarin and swertianin are fit for human consumption. They have a favorable druggability and good ADMET properties. Nowadays, in silico, in vitro, and in vivo ADMET tools constitute an integrated part of modern medicinal chemistry and drug discovery in terms of the early assessment of drug-exposure profile and safety performance. The full awareness of the benefits and limitations of each tool assures the right questions to be answered using right tools at the right time.68 The active compounds identified in this study can be used as an alternative treatment for HCV, especially in countries with poor socioeconomic conditions.
Disclosure
The authors have no conflicts of interest to disclose.
References
Ashfaq UA, Idress S. Medicinal plants against hepatitis C virus. World J Gastroenterol. 2014;20(11):2941–2947. | ||
Moradpour D, Penin F, Rice CM. Replication of hepatitis C virus. Nat Rev Microbiol. 2007;5(6):453–463. | ||
Khan M, Qasim M, Ashfaq UA, Idrees S, Shah M. Computer aided screening of Accacia nilotica phyto chemicals against HCV NS3/4a. Bioinformation. 2013;9(14):710–714. | ||
Smith DB, Bukh J, Kuiken C, et al. Expanded classification of hepatitis C virus into 7 genotypes and 67 subtypes: updated criteria and genotype assignment web resource. Hepatology. 2014;59:318–327. | ||
Ampuero J, Gomez MR, Reddy KR. Review article: HCV genotype 3 – the new treatment challenge. Aliment Pharmacol Ther. 2014;39:686–698. | ||
Murakawa K, Esumi M, Kato T, Kambara H, Shikata T. Heterogeneity within the nonstructural protein 5-encoding region of hepatitis C viruses from a single patient. Gene. 1992;117:229–232. | ||
Okamoto H, Kojima M, Okada S, et al. Genetic drift of hepatitis C virus during an 8.2-year infection in a chimpanzee: variability and stability. Virology. 1992;190:894–899. | ||
Bressanelli S, Tomei L, Roussel A, et al. Crystal structure of the RNA dependent RNA polymerase of hepatitis C virus. Proc Natl Acad Sci U S A. 1999;96:13034–13039. | ||
Lesburg CA, Cable MB, Ferrari E, Hong Z, Mannarino AF, Weber PC. Crystal structure of the RNA-dependent RNA polymerase from hepatitis C virus reveals a fully encircled active site. Nat Struct Biol. 1999;6:937–943. | ||
Appel N, Schaller T, Penin F, Bartenschlager R. From structure to function: new insights into hepatitis C virus RNA replication. J Biol Chem. 2006;281:9833–9836. | ||
Dubuisson J. Hepatitis C virus proteins. World J Gastroenterol. 2007;13(17):2406–2415. | ||
Srinivasan P, Sudha A, Hameed AS, Kumar SP, Karthikeyan M. Screening of medicinal plant compounds against NS5B polymerase of hepatitis C virus (HCV) using molecular docking studies. J Pharm Res. 2010;4(1):136–140. | ||
Deore RR, Chern JW. NS5B RNA dependent RNA polymerase inhibitors: the promising approach to treat hepatitis C virus infections. Curr Med Chem. 2010;17:3806–3826. | ||
Lowery M. Sovaldi approved to treat chronic hepatitis C. 2013. | ||
Keating GM, Vaidya A. Sofosbuvir: first global approval. Drugs. 2014;74(2):273–282. | ||
Gentile I, Borgia F, Buonomo R, Castaldo G, Borgia G. A novel promising therapeutic option against hepatitis C virus: an oral nucleotide NS5B polymerase inhibitor sofosbuvir. Curr Med Chem. 2013;20:3733–3742. | ||
Koff RS. Review article: the efficacy and safety of sofosbuvir, a novel, oral nucleotide NS5B polymerase inhibitor, in the treatment of chronic hepatitis C virus infection. Aliment Pharmacol Ther. 2014;39:478–487. | ||
Clark AM. Natural products as a resource for new drugs. Pharm Res. 1996;13(8):1133–1141. | ||
Harvey AL. Natural products in drug discovery. Drug Discov Today. 2008;13(19):894–901. | ||
Balunas MJ, Kinghorn AD. Drug discovery from medicinal plants. Life Sci. 2005;78(5):431–441. | ||
Rollinger JM, Stuppner H, Langer T. Virtual screening for the discovery of bioactive natural products. In: Petersen F, Amstutz R, editors. Natural Compounds as Drugs Volume I. Basel: Birkhäuser; 2008:211–249. | ||
Manvar D, Mishra M, Kumar S, Pandey VN. Identification and evaluation of antihepatitis C virus phytochemicals from Eclipta alba. J Ethnopharmacol. 2012;144(3):545–554. | ||
Calland N, Dubuisson J, Rouillé Y, Séron K. Hepatitis C virus and natural compounds: a new antiviral approach? Viruses. 2012;4(10):2197–2217. | ||
Hudson JB. Plant photosensitizers with antiviral properties. Antiviral Res. 1989;12(2):55–74. | ||
Kitazato K, Wang Y, Kobayashi N. Viral infectious disease and natural products with antiviral activity. Drug Discov Ther. 2007;1(1):14–22. | ||
Kohler JJ, Nettles JH, Amblard F, et al. Approaches to hepatitis C treatment and cure using NS5A inhibitors. Infect Drug Resist. 2014;7:41. | ||
Cheng F, Li W, Zhou Y, et al. admetSAR: a comprehensive source and free tool for assessment of chemical ADMET properties. J Chem Inf Model. 2012;52(11):3099–3105. | ||
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2012;64:4–17. | ||
Olson RE, Christ DD. Plasma protein binding of drugs. Annu Rep Med Chem. 1996;31:327–336. | ||
Vasanthanathan P, Taboureau O, Oostenbrink C, Vermeulen NP, Olsen L, Jørgensen FS. Classification of cytochrome P450 1A2 inhibitors and noninhibitors by machine learning techniques. Drug Metab Dispos. 2009;37:658–664. | ||
Cupp MJ, Tracy TS. Cytochrome P450: new nomenclature and clinical implications. Am Fam Physician. 2009;57(1):107–116. | ||
Alavijeh MS, Chishty M, Qaiser MZ, Palmer AM. Drug metabolism and pharmacokinetics, the blood–brain barrier, and central nervous system drug discovery. Neuro Rx. 2005;2(4):554–571. | ||
Martin Hernando JI, Ontoria JM, Malancona S, et al. Optimization of thienopyrrole-based finger-loop inhibitors of the hepatitis C virus NS5B polymerase. Chem Med Chem. 2009;4(10):1695–1713. | ||
Di Marco S, Volpari C, Tomei L, et al. Interdomain communication in hepatitis C virus polymerase abolished by small molecule inhibitors bound to a novel allosteric site. J Biol Chem. 2005;280(33):29765–29770. | ||
Narjes F, Crescenzi B, Ferrara M, Habermann J, Colarusso S, et al. Discovery of (7 R)-14-cyclohexyl-7-{[2-(dimethylamino) ethyl](methyl) amino}-7, 8-dihydro-6 H-indolo [1, 2-e][1, 5] benzoxazocine-11-carboxylic acid (MK-3281), a potent and orally bioavailable finger-loop inhibitor of the hepatitis C virus NS5B polymerase. J Med Chem. 2010;54(1):289–301. | ||
Ikegashira K, Oka T, Hirashima S, et al. Discovery of conformationally constrained tetracyclic compounds as potent hepatitis C virus NS5B RNA polymerase inhibitors. J Med Chem. 2006;49(24):6950–6953. | ||
Yang J, Roy A, Zhang Y. Protein–ligand binding site recognition using complementary binding-specific substructure comparison and sequence profile alignment. Bioinformatics. 2013;29(20):2588–2595. | ||
Dundas J, Ouyang Z, Tseng J, Binkowski A, Turpaz Y, Liang J. CASTp: computed atla1s of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res. 2006;34:W116–W118. | ||
Wass MN, Kelley LA, Sternberg MJ. 3DLigandSite: predicting ligand-binding sites using similar structures. Nucleic Acids Res. 2010;38:W469–W473. | ||
Wang Y, Xiao J, Suzek TO, Zhang J, Wang J, Bryant SH. PubChem: a public information system for analyzing bioactivities of small molecules. Nucleic Acids Res. 2009;37:W623–W633. | ||
Pence HE, Williams A. ChemSpider: an online chemical information resource. J Chem Educ. 2010;87(11):1123–1124. | ||
Irwin JJ, Shoichet BK. ZINC-a free database of commercially available compounds for virtual screening. J Chem Inf Model. 2005;45(1):177–182. | ||
Wishart DS, Knox C, Guo AC, et al. DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006;34:D668–D672. | ||
Lipinski CA. Lead-and drug-like compounds: the rule-of-five revolution. Drug Discov Today Technol. 2004;1(4):337–341. | ||
Kiss R, Sandor M, Szalai FA. http://Mcule.com: a public web service for drug. Drug Discov. 2012;4(1):17. | ||
Trott O, Olson AJ. AutoDockVina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455–461. | ||
Pettersen EF, Goddard TD, Huang CC, et al. UCSF chimera – a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–1612. | ||
Accelrys Software Inc. Discovery Studio Modeling Environment, Release 4.0. San Diego: Accelrys Software Inc; 2013. | ||
DeLano WL. The PyMOL Molecular Graphics System. 2002. | ||
Kitchen DB, Decornez H, Furr JR, Bajorath J. Docking and scoring in virtual screening for drug discovery: methods and applications. Nat Rev Drug Discov. 2004;3(11):935–949. | ||
Rosen DM, Grimes K. Discovery and preclinical work. A Practical Guide to Drug Development in Academia. Springer International Publishing; 2014:31–77. | ||
Kassel DB. Applications of high-throughput ADME in drug discovery. Curr Opin Chem Biol. 2004;8(3):339–345. | ||
Oldendorf WH. Lipid solubility and drug penetration of the blood–brain barrier. Exp Biol Med. 1974;147(3):813–816. | ||
Egan WJ, Merz KM, Baldwin JJ. Prediction of drug absorption using multivariate statistics. J Med Chem. 2000;43(21):3867–3877. | ||
Cheng A, Merz KM. Prediction of aqueous solubility of a diverse set of compounds using quantitative structure-property relationships. J Med Chem. 2003;46(17):3572–3580. | ||
Susnow RG, Dixon SL. Use of robust classification techniques for the prediction of human cytochrome P450 2D6 inhibition. J Chem Inf Comput Sci. 2003;43(4):1308–1315. | ||
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 1997;23:3–25. | ||
Stamatovic SM, et al. Curr Neuropharmacol. 2008;6:179. | ||
Atkinson F, et al. Curr Med Chem CNS Agents. 2002;2:229. | ||
Abraham MH. The factors that influence permeation across the blood–brain barrier. Eur J Med Chem. 2004;39:235–240. | ||
Anzenbacher P, Anzenbacherova E. Cytochromes P450 and metabolism of xenobiotics. Cell Mol Life Sci. 2001;58(5–6):737–747. | ||
Lynch T, Price A. The effect of cytochrome P450 metabolism on drug response, interactions, and adverse effects. Am Fam Physician. 2007;76:391–396. | ||
Tuntland T, Ethell B, Zang R, et al. Implementation of pharmacokinetic and pharmacodynamic strategies in early research phases of drug discovery and development at Novartis Institute of Biomedical Research. Front Pharmacol. 2014;5:174. | ||
Mirza MU, Mirza AH, Ghori NUH, Ferdous S. Glycyrrhetinic acid and E. resveratroloside act as potential plant derived compounds against dopamine receptor D3 for Parkinson’s disease: a pharmacoinformatics study. Drug design, development and therapy. 2015;9:187. | ||
Varshney J, Sharma PK, Sharma A. A review on an update of NS5B polymerase hepatitis C virus inhibitors. Eur Rev Med Pharmacol Sci. 2012;16:667–671. | ||
Lin MV, Chung R. Recent FDA approval of sofosbuvir and simeprevir. Implications for current HCV treatment. Clin Liver Dis. 2014;3(3):65–68. | ||
Fried MW. Side effects of therapy of hepatitis C and their management. Hepatology. 2002;36:237–244. | ||
Grime KH, Barton P, McGinnity DF. Application of in silico, in vitro and preclinical pharmacokinetic data for the effective and efficient prediction of human pharmacokinetics. Mol Pharm. 2013;10(4):1191–1206. | ||
Wallace AC, Laskowski RA, Thornton JM. LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 1995;8(2):127–134. |
Supplementary materials
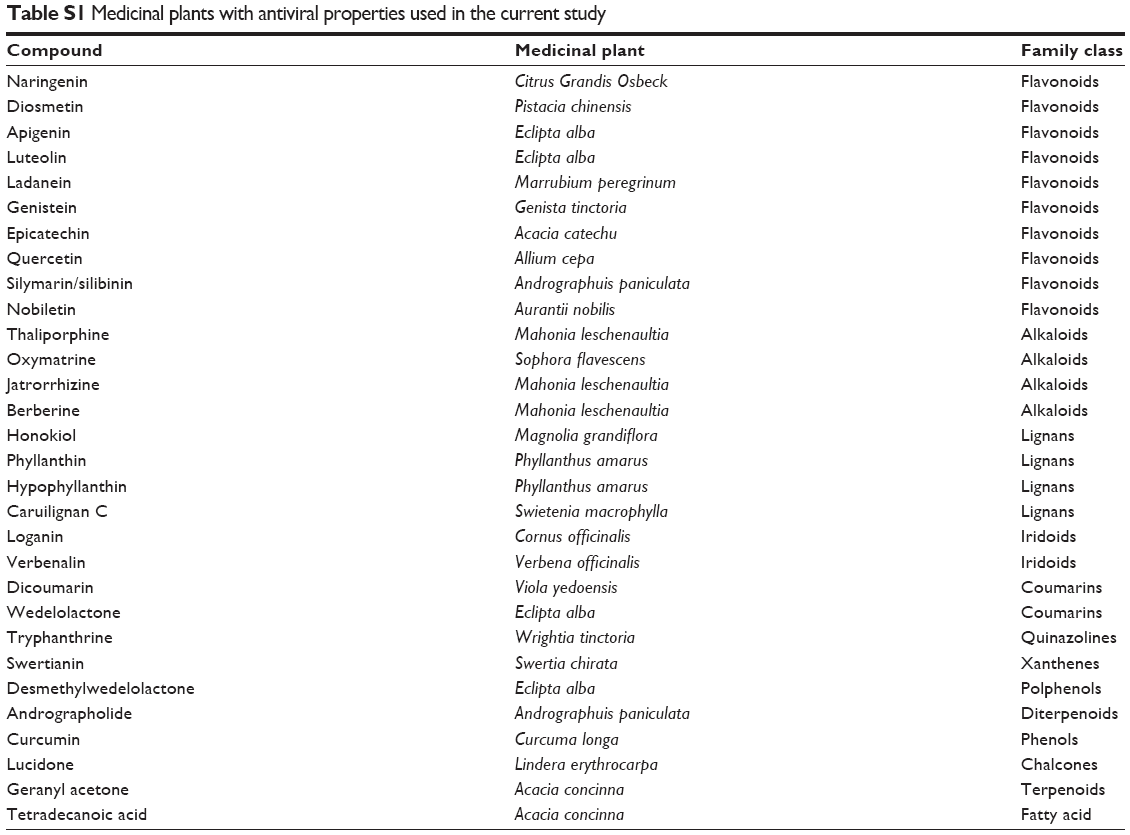 | Table S1 Medicinal plants with antiviral properties used in the current study |
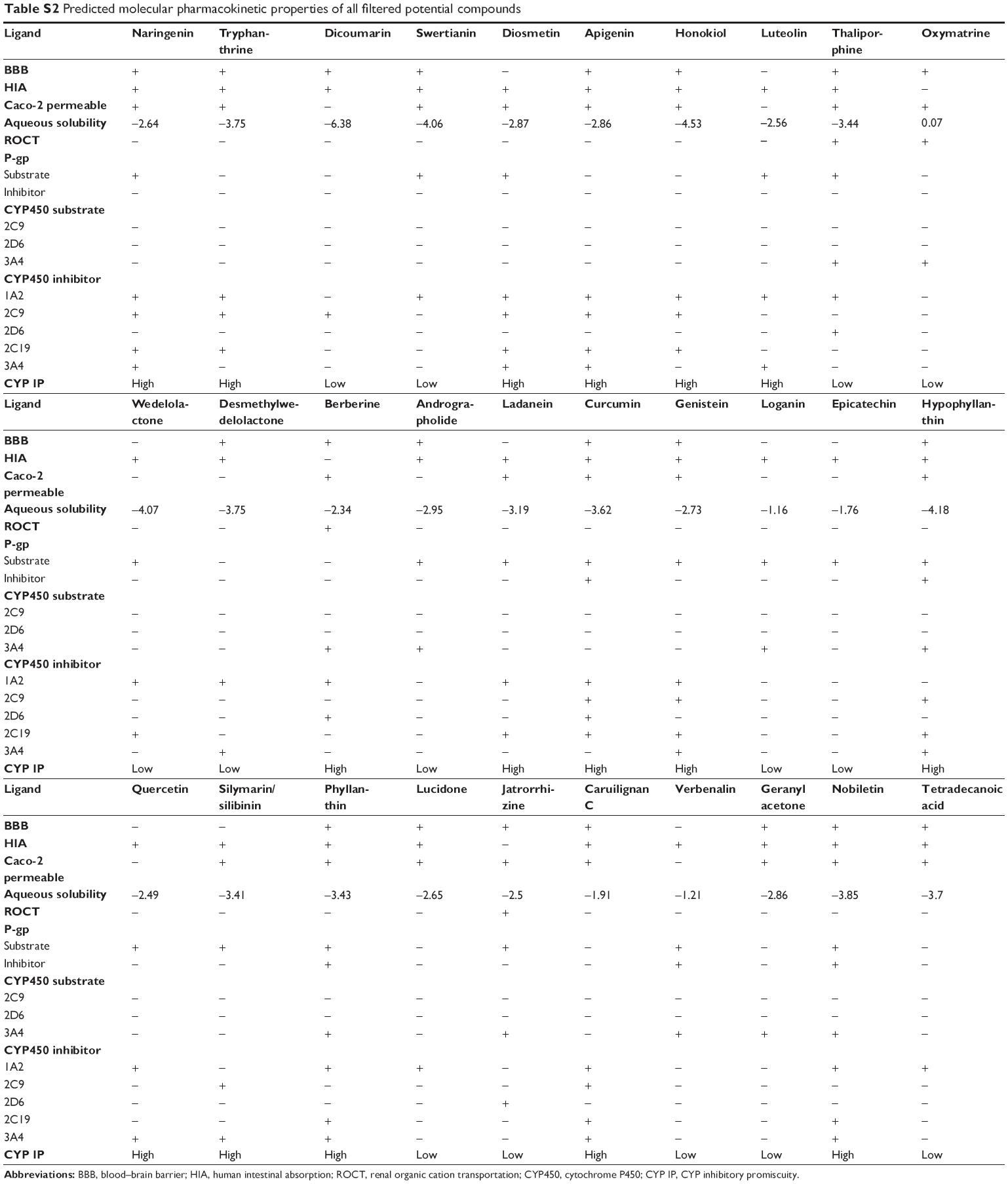 | Table S2 Predicted molecular pharmacokinetic properties of all filtered potential compounds |
 | Table S3 Predicted toxicity assessment of all filtered potential compounds |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.