")
Back to Journals » Drug Design, Development and Therapy » Volume 11
Paeoniflorin attenuates incipient diabetic nephropathy in streptozotocin-induced mice by the suppression of the Toll-like receptor-2 signaling pathway
Authors Shao YX , Xu XX, Wang K , Qi XM, Wu YG
Received 18 August 2017
Accepted for publication 7 October 2017
Published 9 November 2017 Volume 2017:11 Pages 3221—3233
DOI https://doi.org/10.2147/DDDT.S149504
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Qiongyu Guo
Yun-xia Shao,1,2 Xing-xin Xu,1 Kun Wang,1 Xiang-ming Qi,1 Yong-gui Wu1
1Department of Nephrology, The First Affiliated Hospital, Anhui Medical University, Hefei, 2Department of Nephrology, The Second People’s Hospital of Wuhu, Wuhu, Anhui, People’s Republic of China
Abstract: Toll-like receptors (TLRs) may be involved in diabetic nephropathy (DN). Paeoniflorin (PF) is an effective Chinese traditional medicine with anti-inflammatory and immunoregulatory effects that may inhibit the TLR2 signaling pathway. In this study, we investigated the effects of PF on the kidneys of mice with streptozotocin-induced type 1 diabetes mellitus using TLR2 knockout mice (TLR2-/-) and wild-type littermates (C57BL/6J-WT). After 12 weeks of intraperitoneal injection of PF at doses of 25, 50, and 100 mg/kg once a day, diabetic mice had significantly reduced albuminuria and attenuated renal histopathology. These changes were associated with substantially alleviated macrophage infiltration and decreased expression of TLR2 signaling pathway biomarkers. These data support a role of TLR2 in promoting inflammation and indicate that the effect of PF is associated with the inhibition of the TLR2 pathway in the kidneys of diabetic mice. PF thus shows therapeutic potential for the prevention and treatment of DN.
Keywords: paeoniflorin, Toll-like receptors 2, TLR2, diabetic nephropathy, DN, inflammation, macrophages
Introduction
Diabetic nephropathy (DN) continues to be the leading cause of chronic kidney disease and end-stage renal disease,1 accounting for nearly 50% of all cases requiring dialysis each year in many developed countries.2 However, with the increasing prevalence of DN, existing clinical interventions that strictly control hyperglycemia and hypertension and block the renin–angiotensin–aldosterone axis have been demonstrated only to delay DN progression, not to prevent or reverse the pathological state.3,4
Renal injury in diabetes is multifactorial, and numerous studies have suggested that macrophage infiltration and activation in diabetic kidneys during inflammation via the release of related factors, such as inflammatory cytokines, C-reactive protein, and nuclear factor-κB (NF-κB), subsequently lead to the development and progression of DN.5–8
Toll-like receptors (TLRs) are a family of germ line–encoded receptors responsible for innate immune responses that are expressed on antigen-presenting cells, such as monocytes, macrophages, and dendritic cells, as well as nonimmune cells, such as podocytes, endothelial cells, and renal tubular epithelial cells.9–14 The TLR ligands are categorized as pathogen-associated molecular patterns and endogenous danger signals (danger-associated molecular patterns). Upon binding with certain ligands, the signaling pathways of TLRs are activated through adaptor protein myeloid differentiation factor 88 (MyD88)-dependent or MyD88-independent downstream signaling pathways, resulting in the transcription of NF-κB, which contributes to the release of pro-inflammatory cytokines and chemokines.15 Recent studies have revealed a link between TLR signaling and the renal tissue pro-inflammatory state associated with diabetes.16–18 Devaraj et al showed in a type 1 diabetes model that TLR2 knockout significantly attenuated the pro-inflammatory state, altered incipient DN, and influenced albuminuria and podocyte loss.19 Studies on childhood-onset type 1 diabetic patients showed that increased expression of TLR2, pro-inflammatory cytokines in blood leukocytes, and TLR2 expression were associated with the occurrence of microalbuminuria.20 These data indicated that the activation of inflammation and the innate immune system via the TLR signaling pathway in the pathogenesis of diabetic mellitus and its complications were important. Thus, we hypothesized that the terminal renal injury known as DN was resulted from the TLR2-mediated inflammatory state triggered by certain diabetic microenvironments. Thus, the inhibition of the TLR2 signaling pathway could be a novel therapeutic approach for DN patients.
In recent years, DN patients have shown an increasing interest in the use of Chinese medicinal herbal therapies.21,22 Paeoniflorin (PF), a typical Chinese herbal medicine ingredient, is a major bioactive component of total glucosides of paeony (TGP) extracted from the dried peeled root of Paeonia lactiflora Pall. As a promising drug, PF has been shown to have the anti-inflammatory,23 antioxidative,24 and immunoregulatory effects.25 TGP has been assessed as a clinical treatment for rheumatoid arthritis,26 psoriasis,27 systemic lupus erythaematosus,28 and mesenteric hyperplastic nephritis,29,30 and its effects are related to PF-mediated improvement in the activity of immune regulatory cells and anti-inflammatory effects. Fu et al31 studied diabetic rat models and found that PF treatment resulted in decreased urinary albumin and blood glucose levels and attenuated glomerular hypertrophy, suppressing the expression of inflammatory factors and macrophage infiltration; these results indicated that PF might prevent the development of DN via the inactivation of inflammation. Our previous studies suggested that PF has strong anti-inflammatory effects on high-glucose-induced macrophage activation through the inhibition of the TLR2 signaling pathway.32 In this setting, we examined the therapeutic potential of PF on inflammation in the kidneys of mice with streptozotocin (STZ)-induced type 1 diabetes mellitus using TLR2 knockout mice (TLR2−/−) and wild-type littermates (C57BL/6J-WT) and investigated whether the effects are mediated through the TLR2 signaling pathway. The purpose of the present study was to investigate how TLR2 signaling is activated and triggers inflammation, resulting in DN, and the value of PF in modulating the development of DN.
Materials and methods
Drug and reagents
PF (C23H28O11; molecular weight =480.45; purity =98.78% [high-performance liquid chromatography]; lethal dose =9,530 mg/kg; Figure 1) was obtained from Nanjing Goren Bio-Technology Co., Ltd. (Nanjing, People’s Republic of China). STZ was obtained from Sigma-Aldrich Co. (St Louis, MO, USA). The microalbumin assay kit was acquired from Abcam Biotechnology (Abcam, Cambridge, UK). The immunohistochemistry kit (PV-9000) was purchased from Beijing Zhongshan Biotechnology, Inc. (Zhongshan, People’s Republic of China). The rabbit anti-TLR2, MyD88, Trif, and inducible nitric oxide synthase (iNOS) antibodies were bought from Abcam Biotechnology, and p-IRF3, NF-κB p65, and NF-κB p-p65 antibodies were purchased from Cell Signaling Technology (Beverly, MA, USA). The rabbit anti-p-IRAK1 and CD68 were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-β-actin antibodies, anti-mouse IgG, and anti-rabbit IgG horseradish-peroxidase were purchased from Wuhan Sanying Biotechnology, Inc. (Wuhan, People’s Republic of China). The mouse anti-MyD88 and Protein A/G PLUS agarose immunoprecipitation reagent were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The chemiluminescence kit was acquired from Amersham Life Science (Little Chalfont, UK). Bicinchoninic acid protein assay kit and crystal violet were obtained from Beyotime Institute of Biotechnology (Jiangsu, People’s Republic of China). TRIzol reagent and cDNA synthesis kit were from Invitrogen (Carlsbad, CA, USA) and Promega (Madison, WI, USA), respectively. SYBR Green PCR Master Mix Kit was purchased from Bio-Rad Laboratories (Hercules, CA, USA). GAPDH and tumor necrosis factor-alpha (TNF-α) primers were obtained from the Shanghai Sangon Company (Shanghai, People’s Republic of China). Primers for iNOS (MQP029793), IL-1β (MQP027422), and MCP-1 (MQP027672) were purchased commercially from GeneCopoeia, Inc. (Rockville, MD, USA).
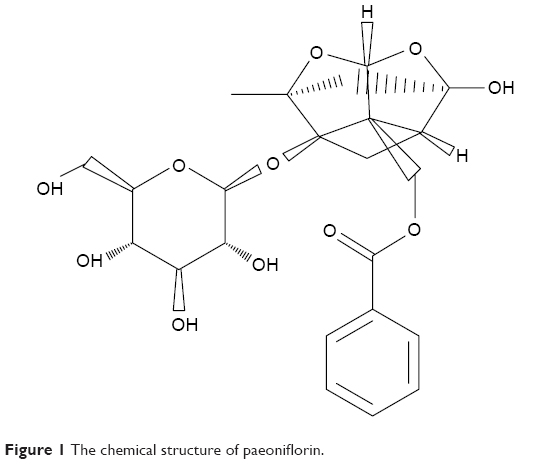 | Figure 1 The chemical structure of paeoniflorin. |
Animals and experimental design
The wild-type C57BL/6J littermates and TLR2−/− mice33 (male, 8–10 weeks of age) were purchased from the Model Animal Research Centre of Nanjing University and housed individually in cages under standard raising condition with a 12-hour light–dark cycle, free access to food and water, room temperature of 24°C, and humidity of 60%. After 7 days of acclimation, mice were administered STZ in citrate buffer (0.1 M; pH 4.5) daily at a dose of 50 mg/kg body weight for 5 days to establish the diabetic model. Then, the mice were randomly divided as follows: wild-type C57BL/6J group (WT, n=12); WT+STZ group (WT+STZ, n=12); PF treatment group (WT+STZ+PF, n=12, dose of 25, 50, or 100 mg/kg); TLR2−/− group (TLR2−/−, n=12); and TLR2−/−+STZ group (TLR2−/−+STZ, n=12). PF was administered by intraperitoneal injection once a day for 12 weeks to the WT+STZ+PF group at a dose of 25, 50, and 100 mg/kg. Other groups were administered equal amounts of normal saline. All animal experimental protocols were approved by the Ethical Committee of Animal Research of Anhui Medical University, and the mice were sacrificed according to the Guide for the Care and Use of Laboratory Animals recommendations.
Physical and biochemical analysis
Body weight, kidney weight, and blood glucose were measured. The 24-hour urine samples were collected from the mice at the end of 12 weeks in metabolic cages. Urinary albumin was assayed by using a mouse microalbumin ELISA kit (Abcam Biotechnology; Abcam, Cambridge, UK) according to the manufacturer’s instructions.
Histology analysis
For pathology and immunohistochemistry, 4% paraformaldehyde-fixed and paraffin-embedded fresh renal tissues were cut into 2 μm thickness. After deparaffinization, the slides were stained with periodic acid–Schiff (PAS) reagent to identify the kidney structure. The glomerular mesangial expansion index and the tubulointerstitial injury index were evaluated and graded in 10 visual fields randomly.14 The slides were treated with 3% hydrogen peroxide and then heated in a microwave to enclose the endogenous peroxidase and retrieve the antigen. The tissue slices were blocked with normal horse serum for 30 minutes at 37°C and then incubated with primary antibody (anti-CD68, 1:100; anti-TLR2, 1:50; anti-NF-κB p65, 1:100) overnight at 4°C followed by incubation with polyperoxidase-anti-mouse/rabbit IgG and 3,3-diaminobenzidine (Sigma-Aldrich, St. Louis, MO, USA) and hematoxylin staining. CD68-positive cells, representing the recruitment of macrophages in tissue sections, were counted in 10 random high-power (×400) fields, and the TLR2 and NF-κB p65 staining was detected by ImagePro Plus Systems (Media Cybernetics; Rockville, MD, USA).
RNA extraction and real-time PCR
The RNA extracted from the fresh renal tissue via TRIzol reagent was used for reverse transcription of cDNA with a reverse transcription kit for RT-PCR using Power SYBR Green PCR Master Mix and the GAPDH primers (GAPDH primers: forward primer 5′-ACCCCAGCAAGGACACTGAGCAAG-3′; reverse primer 5′-GGCCCCTCCTGTTATTATGGGGGT-3′). The forward and reverse primers for the detected RNA sequence were as follows: TNF-α: 5′- CCCTCCTGGCCAACGGCATG-3′ and 5′-TCGGGGCAGCCTTGTCCCTT-3′; iNOS (MQP029793), IL-1β (MQP027422), and MCP-1 (MQP027672) primers were purchased from GeneCopoeia, Inc. Finally, the relative expression levels of genes were analyzed by using the 2−ΔΔCt method with values normalized to the reference gene GAPDH.
Western blot and coimmunoprecipitation (co-IP)
Proteins from the homogeneous renal samples were lysed and combined with a phosphatase inhibitor cocktail in radioimmunoprecipitation assay buffer. The protein concentration was detected with a Bio-Rad protein assay kit. After boiling, the protein was separated by 8%–12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE). Proteins were electroblotted onto a nitrocellulose membrane and incubated with primary antibody overnight at 4°C after blocking with 5% defatted milk for 2 hours. The horseradish peroxidase-labeled secondary antibody was added once the membrane was washed. Finally, the bound secondary antibody was detected by enhanced chemiluminescence, and the protein content was quantified and analyzed using the Leica Q500IW image analysis system (Leica Ltd., Cambridge, UK). Proteins from homogeneous renal samples were removed by centrifugation at 1,000× g for 25 minutes at 4°C, and the supernatants were precleared with 30 μL Protein A/G-PLUS agarose beads for 1 hour at 4°C. Immunoprecipitation with 3 μg/mL anti-MyD88 was performed overnight at 4°C, and then, 50 μL of Protein A/G-PLUS agarose beads were added and incubated for 2 hours at 4°C. Equivalent amounts of protein were analyzed for each condition. The immune complexes were boiled in SDS sample buffer and loaded on SDS-PAGE gels for immunoblot analysis.
Statistical analysis
Data were analyzed using SPSS Version 16.0 (SPSS Inc., Chicago, IL, USA). Normally distributed data are expressed as the mean ± SD, and all the data were assessed by using one-way analysis of variance. The difference between groups was tested by least significant difference and the Levene method was used for homogeneity test of variance, in which a p-value <0.05 was declared significant.
Results
Effects of PF on physical and biochemical markers and pathology in the different groups
To test the effect of PF on DN progression, we measured physical and biochemical markers and pathology of mice in different groups. As shown in Figure 2A, STZ injection substantially increased blood glucose levels compared with those of the WT and TLR2−/− groups, but PF treatment did not significantly reduce blood glucose levels, which differed from the results of Fu et al31 in diabetic rats. Compared with nondiabetic WT and TLR2−/− mice, mice injected with STZ showed increased urine albumin excretion and kidney/body weight (Figure 2B and C). PF treatment reduced urine albumin excretion levels and the kidney/body weight ratio of STZ-induced diabetic mice (WT-STZ-PF treatment groups), although their levels were still higher than those of the WT and TLR2−/− mouse groups. Figure 2D shows the histological analysis of kidney sections stained with PAS. In the model group, the mouse kidney volume was increased; PAS staining of the glomerular mesangium, inner walls of the renal tubules, and basement membrane were dark red; and the glomerular mesangial expansion index and the tubulointerstitial damage index were significantly increased, which is consistent with the early manifestation of DN. In contrast, PAS analysis of the PF intervention and TLR2 gene knockout groups showed that the glomerular mesangial expansion index and the tubulointerstitial damage index were significantly decreased. Previous studies have clearly demonstrated that the mesangial dilatation index and tubulointerstitial damage index are closely related to renal function.34 These data indicated that PF could significantly prevent the progression of DN.
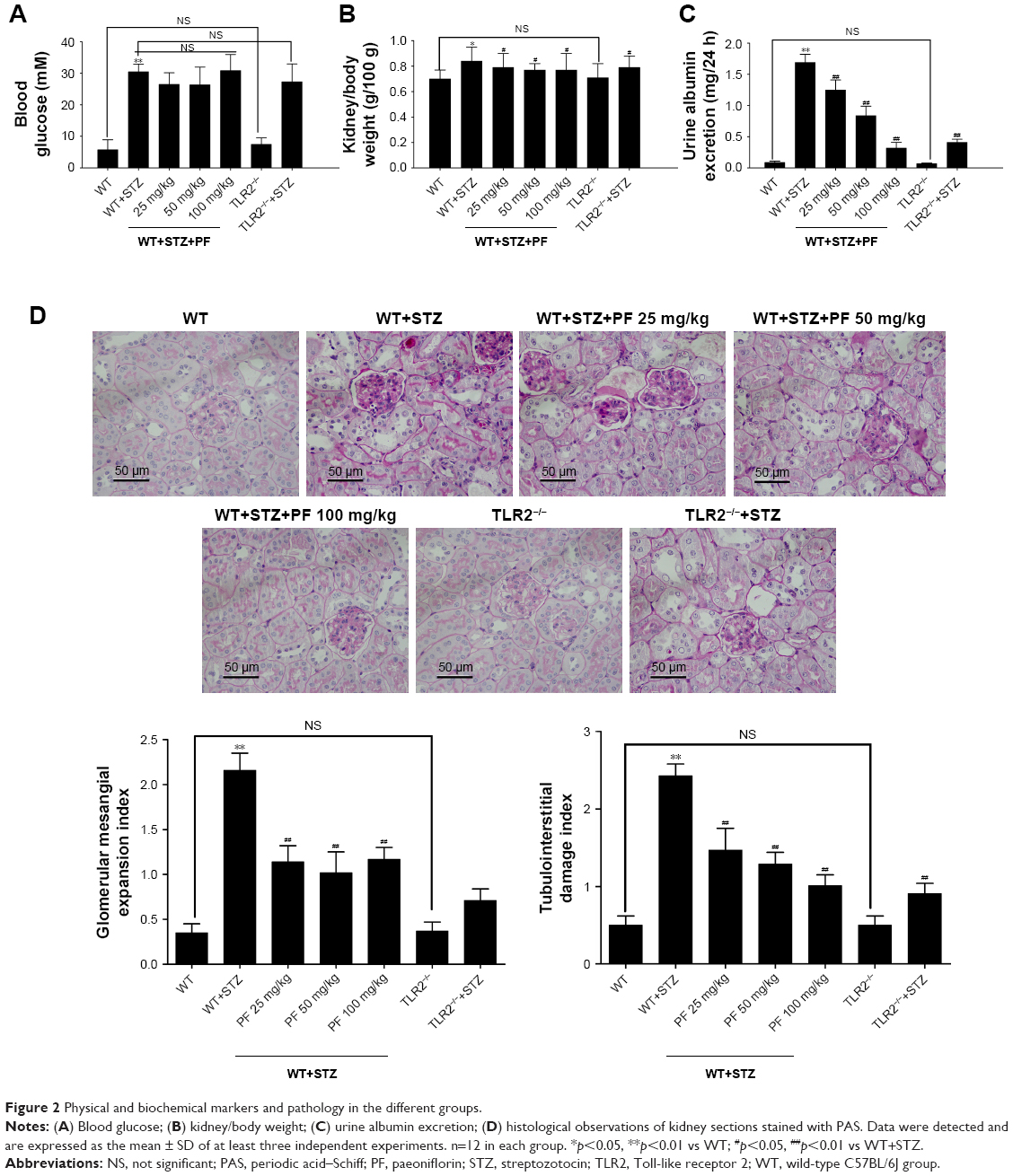 | Figure 2 Physical and biochemical markers and pathology in the different groups. |
Immunohistochemical analysis for the distribution of TLR2, NF-κB p65, and CD-68 in the different groups
Considering the relationship between TLR2 and the renal tissue pro-inflammatory state associated with diabetes, we examined the expression profile of TLR2 in mouse kidney tissues. As shown in Figure 3A, TLR2 was predominantly expressed in the tubulointerstitium. As expected, TLR2 immunohistochemical staining was barely observed in the kidneys of TLR2−/− and TLR2−/−+STZ mice. However, compared with WT mice, the WT+STZ mice showed increased expression of TLR2 (Figure 3A; WT group vs WT+STZ group). After PF treatment, the intensity of TLR2 immunostaining was strikingly decreased in a PF dose-dependent manner (Figure 3A; WT+STZ+PF treatment groups), demonstrating that this treatment can decrease TLR2 expression in diabetic models.
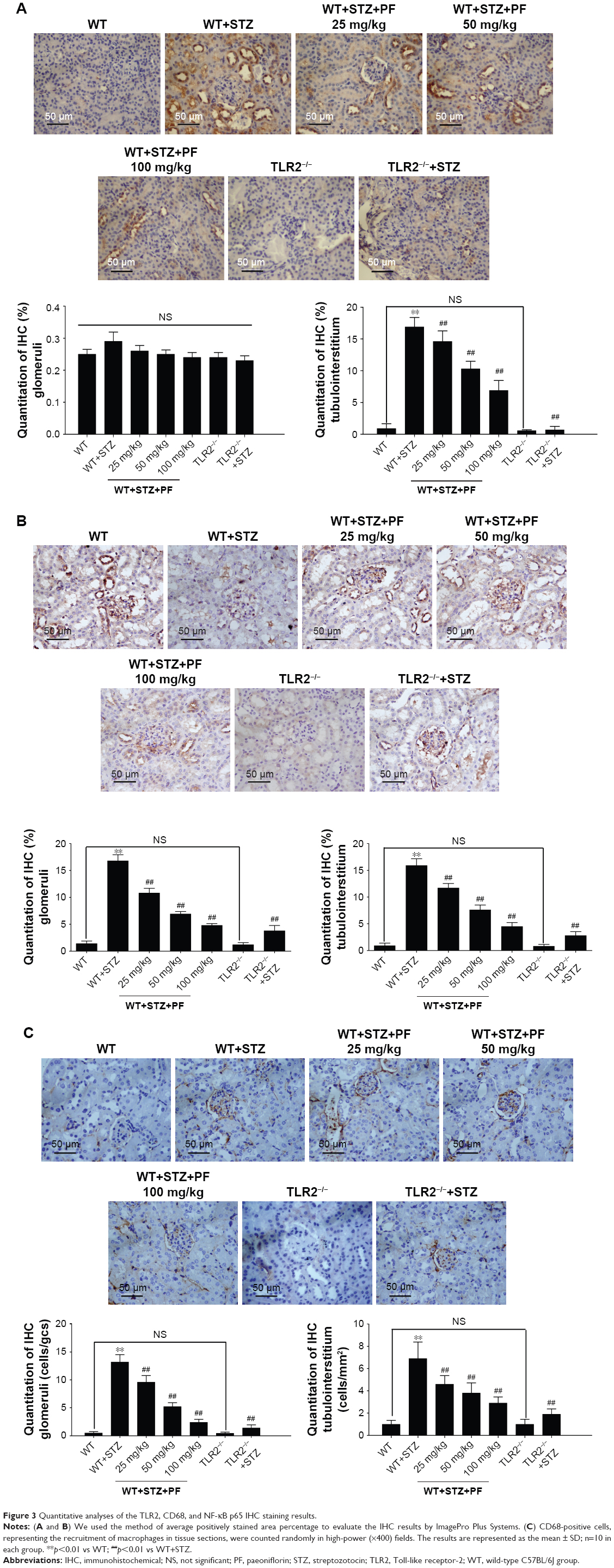 | Figure 3 Quantitative analyses of the TLR2, CD68, and NF-κB p65 IHC staining results. |
Next, we investigated the expression pattern of NF-κB p65. As shown in Figure 2B, NF-κB p65 was significantly overexpressed in the nucleus and cytoplasm of glomerular cells and renal tubular cells from WT+STZ mice compared with WT mice. However, NF-κB p65 expression was reduced following PF treatment or knockout of TLR2 (Figure 3B).
CD68, which indicates renal macrophage accumulation,35 was occasionally observed in WT and TLR2−/− mouse kidneys, whereas CD68-positive macrophage infiltration was substantially increased in WT+STZ mice. Immunostaining indicated a significant reduction in CD68 expression in kidneys from PF-treated and TLR2−/−+STZ mice compared with WT+STZ mice, which verifies the effects of PF on macrophage accumulation and infiltration and the influence of TLR2 knockout on the same (Figure 3C).
mRNA expression of iNOS, TNF-α, IL-1β, and MCP-1 in different groups
Increasing evidence has demonstrated that the phenotype of the infiltrated macrophages is the major characteristic that ultimately determines the sequelae of DN.36 To determine whether macrophage infiltration and inflammation are induced in the kidneys of diabetic mice, we measured the expression of iNOS and inflammatory genes in the kidneys of different groups. M1 macrophages promote inflammatory responses and tissue injury, while M2 macrophages induce anti-inflammatory and tissue-protective effects.37 As shown in Figure 3, low levels of iNOS, TNF-α, IL-1β, and MCP-1 were observed, consistent with the transcriptional regulation of the mRNA levels in WT mice, and were further decreased in TLR2−/− mice. In contrast, the mRNA levels of iNOS, TNF-α, IL-1β, and MCP-1 were significantly elevated in WT+STZ mice and decreased strikingly following PF treatment and TLR2 knockout (Figure 4).
 | Figure 4 Quantitative real-time PCR analysis of PF on macrophage activation and inflammatory cytokine expression. |
Effects of PF on TLR2 and downstream signaling pathway expression in the different groups
TLR2, together with the expression of downstream signaling proteins in different groups, was further detected by using Western blot analysis, which showed a significant upregulation of TLR2, MyD88, p-IRAK1, Trif, p-IRF3, NF-κB p-p65, NF-κB p65, and iNOS in the WT+STZ group compared with the WT or TLR2−/− mice. By comparison, the expression of the above proteins significantly decreased following PF treatment in a dose-dependent manner (Figure 5A and B). A co-IP assay also indicated that PF could weaken the interaction between TLR2 and MyD88 (Figure 5C). However, the TLR2−/−+STZ group showed notable attenuation of MyD88, p-IRAK1, NF-κB p-p65, NF-κB p65, and iNOS, but there was no significant suppression of the MyD88-independent downstream signaling pathway, including Trif and p-IRF3, compared with the diabetic model mice, which indicated that TLR2 knockout had no effect on the protein expression of Trif and p-IRF3 (Figure 5A and B).
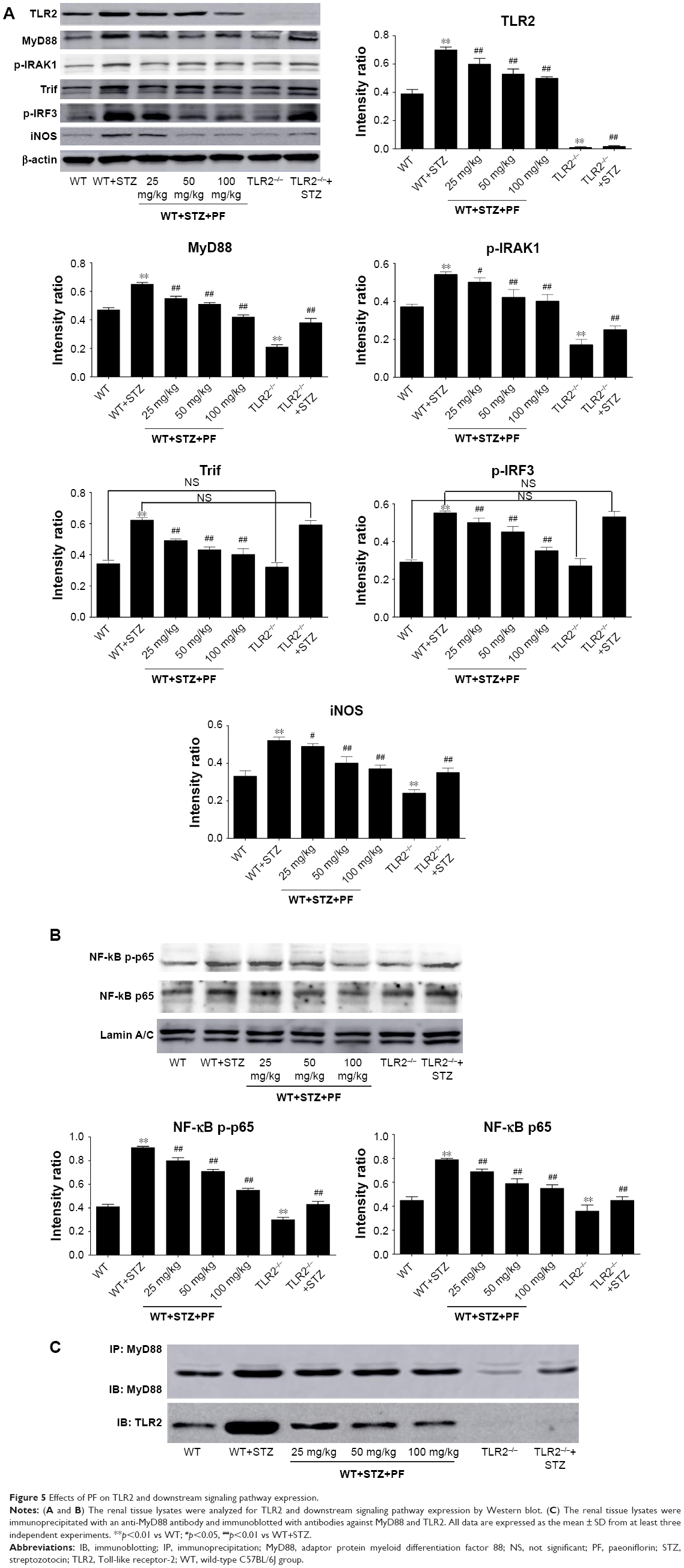 | Figure 5 Effects of PF on TLR2 and downstream signaling pathway expression. |
Discussion
DN is a long-term complication, and the prevention of its progression remains a challenge. Accumulating evidence has indicated that albuminuria is a leading risk factor for the progression of renal disease.38,39 One of the major manifestations of renal involvement of incipient DN is renal hypertrophy, defined as an increased kidney/body weight ratio.38,40 In the present study, although we demonstrated that PF intervention and TLR2 knockout had no effect on blood glucose, they attenuated urine albumin and ameliorated the kidney/body weight ratio, which prevented DN progression. Glycemic control is critical for DN treatment. However, our results demonstrated that PF had no effect on blood glucose, which differed from the findings of Fu et al in a study using 5–20 mg/kg in diabetic rats.31 Another report demonstrated that PF can strongly reduce plasma glucose levels at 1 mg/kg, but the effect was less pronounced when the doses were increased.41 Therefore, the remission of the experimental DN mice due to a hypoglycemic effect was controversial. Next, we focused on the mechanisms of PF intervention and TLR2 knockout in delaying the development of DN.
Li et al42 reported that TLR2 was prominently expressed in both the glomeruli and tubulointerstitium in human renal biopsies of DN. In addition, macrophages accumulated in renal biopsy samples of DN patients and were shown to play a pathogenic role in DN progression.43 In addition, in a previous report, we showed that TLR2 gene deficiency alleviated inflammatory cytokine production in high-glucose-induced bone marrow–derived macrophages (BMDMs).32 However, until now, limited evidence has been reported on the functional role of TLR2 in macrophages during DN progression. Based on the above experiments, we examined inflammation, the TLR2 signaling pathway, and critical macrophage biomarkers in our animal experiments using diabetic mice. Our experiment showed that in the kidneys of WT+STZ mice, TLR2 and NF-κB p65 signaling was activated, along with a large number of infiltrating macrophages. Meanwhile, the mRNA expression of iNOS (as a macrophage activation marker) was significantly overexpressed, and macrophage activation was predominant in the M1 phenotype; furthermore, high expression of pro-inflammatory mediators (TNF-α, IL-1β, and MCP-1) was observed. M1 phenotype macrophages promoted the production of inflammatory mediators and tissue damage, while TLR2 gene knockout significantly attenuated the above changes. In addition, the analysis of the TLR2 signaling pathway showed that the expression levels of TLR2, MyD88, p-IRAK1, Trif, p-IRF3, NF-κB p-p65, and NF-κB p65 were substantially increased. TLR2 resulted in a significant reduction in the expression of MyD88, p-IRAK1, NF-κB p-p65, and NF-κB p65, while there was no inhibition of non-MyD88-dependent signaling proteins, such as Trif and p-IRF3. Although we did not detect the expression of other TLRs in this experiment and we did not define whether other TLRs were involved in this process, we hypothesized that TLR2 regulates macrophage aggregation and activation by a MyD88-dependent pathway. A similar conclusion was reported in our study of high-glucose-induced BMDMs.32 Thus, TLR2 gene deficiency attenuated the inflammatory state in the kidneys of DN mice, reduced proteinuria, and inhibited renal hypertrophy. Our data are consistent with the published research of Devaraj et al.19
While some reports support a positive role for PF in preventing DN, the underlying mechanisms of its effect are still unclear despite intense investigation. However, compelling evidence has shown that targeted inhibition of abnormal TLR2 overexpression is valuable for alleviating DN progression.32 Therefore, the purpose of this study was to explore whether the beneficial effect of PF was mediated, at least in part, through changes in the expression of the TLR2 signaling pathway. In the present study, PF intervention showed the same therapeutic effect as TLR2 knockout in our experiments. We observed that PF intervention decreased albuminuria and alleviated renal hypertrophy in WT+STZ mice. We also blocked NF-κB p65 activation and macrophage recruitment, together with the suppression of inflammatory cytokines and chemokines (TNF-α, IL-1β, and MCP-1), in the PF group, and the results are highly consistent with those reported by Fu et al.31 Meanwhile, we showed that the increased TLR2 levels in DN were suppressed by PF. PF could also weaken the interaction between TLR2 and MyD88 in co-IP experiments, as it may inhibit the binding of TLR2 to MyD88 in the renal tissue of DM mice. Notably, PF treatment reduced the expression of MyD88 and p-IRAK1 and inhibited Trif and p-IRF3. Thus, the expression of non-MyD88-dependent signaling proteins was also inhibited, which was different from that of TLR2−/− mice, indicating that the inhibitory effect of PF in diabetic mice may be partly correlated with other TLR signaling pathways, which should to be further elucidate.
TLRs are activated through a MyD88-dependent or a MyD88-independent downstream signaling pathway.15 TLR2 signaling is primarily mediated through the MyD88-dependent pathway to activate inflammation. Here, we provide novel evidence using STZ-induced diabetic mice that TLR2 deficiency substantially decreased inflammation, MyD88, and p-IRAK1, but MyD88-independent signaling proteins (Trif and p-IRF3) were unaltered. Similar results were found when TLR2 was knocked out in BMDMs under high-glucose conditions.32 PF intervention was not consistent with the results of TLR2 knockout, which decreased the expression of the MyD88-independent signaling pathway. Therefore, we hypothesized that the therapeutic effect of PF in mediating inflammation in DN might be mediated through other TLR signaling pathways. In support of our data, our previous animal studies suggested that the protective effect of TGP on DN was associated with the blockade of TLR2 and TLR4 activation in tubular and glomerulus cells and macrophages.44,45 In addition, Zhang et al recently showed that PF abrogates DSS-induced colitis by decreasing the expression of TLR4 signaling pathways.46 Based on these observations, we concluded that PF can improve the inflammatory status of DN by inhibiting the TLR2-MyD88-NFκB pathway, and its anti-inflammatory mechanism is also associated with other signaling pathways.
Conclusion
In summary, our findings suggest that TLR2 activation initiates macrophage infiltration and M1 polarization, resulting in the release of inflammatory cytokines and chemokines, which in turn exacerbate inflammation and ultimately aggravate DN. The analyses of PF treatment in vivo for the first time demonstrate that PF prevents macrophage activation via the inhibition of TLR2 expression in diabetic mice and provide support for PF therapeutic strategies in DN patients.
Acknowledgment
This project was financially supported by the Natural Science Foundation of China (numbers 81374034 and 81470965).
Disclosure
The authors report no conflicts of interest in this work.
References
Gilbertson DT, Liu J, Xue JL, et al. J. Projecting the number of patients with end-stage renal disease in the United States to the year 2015. J Am Soc Nephrol. 2005;16(12):3736–3741. | ||
Atkins RC, Zimmet P. Diabetic kidney disease: act now or pay later. Nephrol Dial Transplant. 2010;25(2):331–333. | ||
Leoncini G, Viazzi F, Pontremoli R. RAAS inhibition and renal protection. Curr Pharm Des. 2012;18(7):971–980. | ||
Taler SJ, Agarwal R, Bakris GL, et al. KDOQI US commentary on the 2012 KDIGO clinical practice guideline for management of blood pressure in CKD. Am J Kidney Dis. 2013;62(2):201–213. | ||
Devaraj S, Glaser N, Griffen S, Wang-Polagruto J, Miguelino E, Jialal I. Increased monocytic activity and biomarkers of inflammation in patients with type 1 diabetes. Diabetes. 2006;55(3):774–779. | ||
Devaraj S, Dasu MR, Jialal I. Diabetes is a proinflammatory state: a translational perspective. Expert Rev Endocrinol Metab. 2010;5(1):19–28. | ||
Libby P, Nathan DM, Abraham K, et al. Report of the National Heart, Lung, and Blood Institute-National Institute of Diabetes and Digestive and Kidney Diseases Working Group on cardiovascular complications of type 1 diabetes mellitus. Circulation. 2005;111(25):3489–3493. | ||
Devaraj S, Cheung AT, Jialal I, et al. Evidence of increased inflammation and microcirculatory abnormalities in patients with type 1 diabetes and their role in microvascular complications. Diabetes. 2007;56(11):2790–2796. | ||
Brown HJ, Lock HR, Sacks SH, Robson MG. TLR2 stimulation of intrinsic renal cells in the induction of immune-mediated glomerulonephritis. J Immunol. 2006;177(3):1925–1931. | ||
Banas MC, Banas B, Hudkins KL, et al. TLR4 links podocytes with the innate immune system to mediate glomerular injury. J Am Soc Nephrol. 2008;19(4):704–713. | ||
Brown HJ, Lock HR, Wolfs TG, Buurman WA, Sacks SH, Robson MG. Toll-like receptor 4 ligation on intrinsic renal cells contributes to the induction of antibody-mediated glomerulonephritis via CXCL1 and CXCL2. J Am Soc Nephrol. 2007;18(6):1732–1739. | ||
Kokkinopoulos I, Jordan WJ, Ritter MA. Toll-like receptor mRNA expression patterns in human dendritic cells and monocytes. Mol Immunol. 2005;42(8):957–968. | ||
Shigeoka AA, Holscher TD, King AJ, et al. TLR2 is constitutively expressed within the kidney and participates in ischemic renal injury through both MyD88-dependent and -independent pathways. J Immunol. 2007;178(10):6252–6258. | ||
Wolfs TG, Buurman WA, van Schadewijk A, et al. In vivo expression of Toll-like receptor 2 and 4 by renal epithelial cells: IFN-gamma and TNF-alpha mediated up-regulation during inflammation. J Immunol. 2002;168(3):1286–1293. | ||
Miggin SM, O’Neill LA. New insights into the regulation of TLR signaling. J Leukoc Biol. 2006;80(2):220–226. | ||
Mudaliar H, Pollock C, Panchapakesan U. Role of Toll-like receptors in diabetic nephropathy. Clin Sci (Lond). 2014;126(10):685–694. | ||
Navarro-Gonzalez JF, Mora-Fernandez C, Muros de Fuentes M, Garcia-Perez J. Inflammatory molecules and pathways in the pathogenesis of diabetic nephropathy. Nat Rev Nephrol. 2011;7(6):327–340. | ||
Shirali AC, Goldstein DR. Tracking the toll of kidney disease. J Am Soc Nephrol. 2008;19(8):1444–1450. | ||
Devaraj S, Tobias P, Kasinath BS, Ramsamooj R, Afify A, Jialal I. Knockout of toll-like receptor-2 attenuates both the proinflammatory state of diabetes and incipient diabetic nephropathy. Arterioscler Thromb Vasc Biol. 2011;31(8):1796–1804. | ||
Ururahy MA, Loureiro MB, Freire-Neto FP, et al. Increased TLR2 expression in patients with type 1 diabetes: evidenced risk of microalbuminuria. Pediatr Diabetes. 2012;13(2):147–154. | ||
Li X, Yang JL, Ma DF, Lin HQ, Xu XD, Jiang Yue H. [Effects of different compatibilities of Ramulus Cinnamomi and Peony in Guizhi Decoction on diabetic cardiac autonomic neuropathy]. Zhongguo Zhong Xi Yi Jie He Za Zhi. 2015;35(6):741–745. Chinese. | ||
Liu IM, Tzeng TF, Liou SS, Chang CJ. Beneficial effect of traditional Chinese medicinal formula danggui-shaoyao-san on advanced glycation end-product-mediated renal injury in streptozotocin-diabetic rats. Evid Based Complement Alternat Med. 2012;2012:140103. | ||
Jiang D, Chen Y, Hou X, Xu J, Mu X, Chen W. Influence of Paeonia lactiflora roots extract on cAMP-phosphodiesterase activity and related anti-inflammatory action. J Ethnopharmacol. 2011;137(1):914–920. | ||
Su J, Zhang P, Zhang JJ, Qi XM, Wu YG, Shen JJ. Effects of total glucosides of paeony on oxidative stress in the kidney from diabetic rats. Phytomedicine. 2010;17(3–4):254–260. | ||
Zhou Z, Lin J, Huo R, et al. Total glucosides of paeony attenuated functional maturation of dendritic cells via blocking TLR4/5 signaling in vivo. Int Immunopharmacol. 2012;14(3):275–282. | ||
Li PP, Liu DD, Liu YJ, et al. BAFF/BAFF-R involved in antibodies production of rats with collagen-induced arthritis via PI3K-Akt-mTOR signaling and the regulation of paeoniflorin. J Ethnopharmacol. 2012;141(1):290–300. | ||
Wang YN, Zhang Y, Wang Y, et al. The beneficial effect of total glucosides of paeony on psoriatic arthritis links to circulating Tregs and Th1 cell function. Phytother Res. 2014;28(3):372–381. | ||
Zhao M, Liang GP, Tang MN, et al. Total glucosides of paeony induces regulatory CD4(+)CD25(+) T cells by increasing Foxp3 demethylation in lupus CD4(+) T cells. Clin Immunol. 2012;143(2):180–187. | ||
Ding ZX, Yang SF, Wu QF, et al. [Therapeutic effect of total glucosides of paeony on lupus nephritis in MRL/lpr mice]. Nan Fang Yi Ke Da Xue Xue Bao. 2011;31(4):656–660. Chinese. | ||
Yun BR, Weon JB, Lee J, Eom MR, Ma CJ. Simultaneous determination of 11 bioactive compounds in Jaeumganghwa-tang by high performance liquid chromatography-diode array detection. Pharmacogn Mag. 2014;10 (Suppl 2):S256–S263. | ||
Fu J, Li Y, Wang L, Gao B, Zhang N, Ji Q. Paeoniflorin prevents diabetic nephropathy in rats. Comp Med. 2009;59(6):557–566. | ||
Shao YX, Xu XX, Li YY, et al. Paeoniflorin inhibits high glucose-induced macrophage activation through TLR2-dependent signal pathways. J Ethnopharmacol. 2016;193:377–386. | ||
Werts C, Tapping RI, Mathison JC, et al. Leptospiral lipopolysaccharide activates cells through a TLR2-dependent mechanism. Nat Immunol. 2001;2(4):346–352. | ||
Moriya T, Tanaka K, Hosaka T, Hirasawa Y, Fujita Y. Renal structure as an indicator for development of albuminuria in normo- and microalbuminuric type 2 diabetic patients. Diabetes Res Clin Pract. 2008;82(3):298–304. | ||
Nishikawa K, Iwaya K, Kinoshita M, et al. Resveratrol increases CD68(+) Kupffer cells colocalized with adipose differentiation-related protein and ameliorates high-fat-diet-induced fatty liver in mice. Mol Nutr Food Res. 2015;59(6):1155–1170. | ||
Zhang XL, Guo YF, Song ZX, Zhou M. Vitamin D prevents podocyte injury via regulation of macrophage M1/M2 phenotype in diabetic nephropathy rats. Endocrinology. 2014;155(12):4939–4950. | ||
Ricardo SD, van Goor H, Eddy AA. Macrophage diversity in renal injury and repair. J Clin Invest. 2008;118(11):3522–3530. | ||
Bakris GL. Slowing nephropathy progression: focus on proteinuria reduction. Clin J Am Soc Nephrol. 2008;3 (Suppl 1):S3–S10. | ||
Amor A, Jimenez A, Moize V, et al. Weight loss independently predicts urinary albumin excretion normalization in morbidly obese type 2 diabetic patients undergoing bariatric surgery. Surg Endosc. 2013;27(6):2046–2051. | ||
White KE, Bilous RW; Diabiopsies Study Group. Structural alterations to the podocyte are related to proteinuria in type 2 diabetic patients. Nephrol Dial Transplant. 2004;19(6):1437–1440. | ||
Hsu FL, Lai CW, Cheng JT. Antihyperglycemic effects of paeoniflorin and 8-debenzoylpaeoniflorin, glucosides from the root of Paeonia lactiflora. Planta Med. 1997;63(4):323–325. | ||
Li F, Yang N, Zhang L, et al. Increased expression of toll-like receptor 2 in rat diabetic nephropathy. Am J Nephrol. 2010;32(2):179–186. | ||
Nguyen D, Ping F, Mu W, Hill P, Atkins RC, Chadban SJ. Macrophage accumulation in human progressive diabetic nephropathy. Nephrology. 2006;11(3):226–231. | ||
Xu XX, Qi XM, Zhang W, et al. Effects of total glucosides of paeony on immune regulatory toll-like receptors TLR2 and 4 in the kidney from diabetic rats. Phytomedicine. 2014;21(6):815–823. | ||
Zhang W, Zhao L, Su SQ, Xu XX, Wu YG. Total glucosides of paeony attenuate renal tubulointerstitial injury in STZ-induced diabetic rats: role of Toll-like receptor 2. J Pharmacol Sci. 2014;125(1):59–67. | ||
Zhang J, Dou W, Zhang E, et al. Paeoniflorin abrogates DSS-induced colitis via a TLR4-dependent pathway. Am J Physiol Gastrointest Liver Physiol. 2014;306(1):G27–G36. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.