")
Back to Journals » Open Access Rheumatology: Research and Reviews » Volume 15
Overview on the Link Between the Complement System and Auto-Immune Articular and Pulmonary Disease
Authors Triggianese P, Conigliaro P, De Martino E, Monosi B, Chimenti MS
Received 4 January 2023
Accepted for publication 5 May 2023
Published 15 May 2023 Volume 2023:15 Pages 65—79
DOI https://doi.org/10.2147/OARRR.S318826
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Chuan-Ju Liu
Paola Triggianese, Paola Conigliaro, Erica De Martino, Benedetta Monosi, Maria Sole Chimenti
Department of Systems Medicine, Rheumatology, Allergology and Clinical Immunology, University of Rome Tor Vergata, Rome, Italy
Correspondence: Maria Sole Chimenti, Rheumatology, Allergology and Immunology, University of Rome Tor Vergata, Via Montpellier 1, Rome, Italy, Tel +39 6 20900358, Email [email protected]
Abstract: Complement system (CS) dysregulation is a key factor in the pathogenesis of different autoimmune diseases playing a central role in many immune innate and adaptive processes. Rheumatoid arthritis (RA) is a chronic inflammatory disease characterized by ta breach of self-tolerance leading to a synovitis and extra-articular manifestations. The CS is activated in RA and seems not only to mediate direct tissue damage but also play a role in the initiation of RA pathogenetic mechanisms through interactions with citrullinated proteins. Interstitial lung disease (ILD) represents the most common extra-articular manifestation that can lead to progressive fibrosis. In this review, we focused on the evidence of CS dysregulation in RA and in ILD, and highlighted the role of the CS in both the innate and adaptive immune responses in the development of diseases, by using idiopathic pulmonary fibrosis as a model of lung disease. As a proof of concept, we dissected the evidence that several treatments used to treat RA and ILD such as glucocorticoids, pirfenidone, disease modifying antirheumatic drugs, targeted biologics such as tumor necrosis factor (TNF)-inhibitors, rituximab, tocilizumab, and nintedanib may act indirectly on the CS, suggesting that the CS might represent a potential therapeutic target in these complex diseases.
Keywords: complement system, rheumatoid arthritis, interstitial lung disease, target therapies
Introduction
The complement system (CS) is a component of the innate immune system and consists of several proteins that play a pivotal role in many protective immune processes.1 The primary aim of the CS is to protect the host against microbes, repair injuries, and contribute to the elimination of cellular debris or immune complexes (IC). Moreover, it has been described that CS components have immunoregulatory functions too through the modulation of both adaptive and innate immune responses.2 The detection of C3 receptors on lymphocytes provides the first evidence of CS participation in adaptive immunity.3 In the following years, it was documented that the CS is also crucial for the regulation of autoreactive B cells as well as for the development of T cell immunity and natural antibodies.4 Three different pathways which share a common terminal pathway activate the CS (Figure 1). C1q, C1r and C1s components are part of the classical pathway (CP) of the complement, while the lectin pathway (LP) includes complement components mannan-binding lectin (MBL), ficolins (FCNs) and collectins (CLs), along with three MBL-associated serum proteases (MASPs). The C3 convertase, made up of C2 and C4 components, is shared by both CP and LP. The alternative pathway (AP) includes C3, factor B (FB), factor D (FD) and Properdin (P). The C5 and the complex C5b-C9, also known as the membrane attack complex (MAC), represent the terminal pathway.5 Among CS components, C3a, C4a and C5a also known as anaphylatoxins have pro-inflammatory functions even at a very low concentration.1 They also drive other processes such as immune cell modulation and chemotaxis, cell survival, tissue repair and regeneration, vasodilatation, and smooth muscle contraction. The formation of IC represents the principal way of activating the CP of the CS.
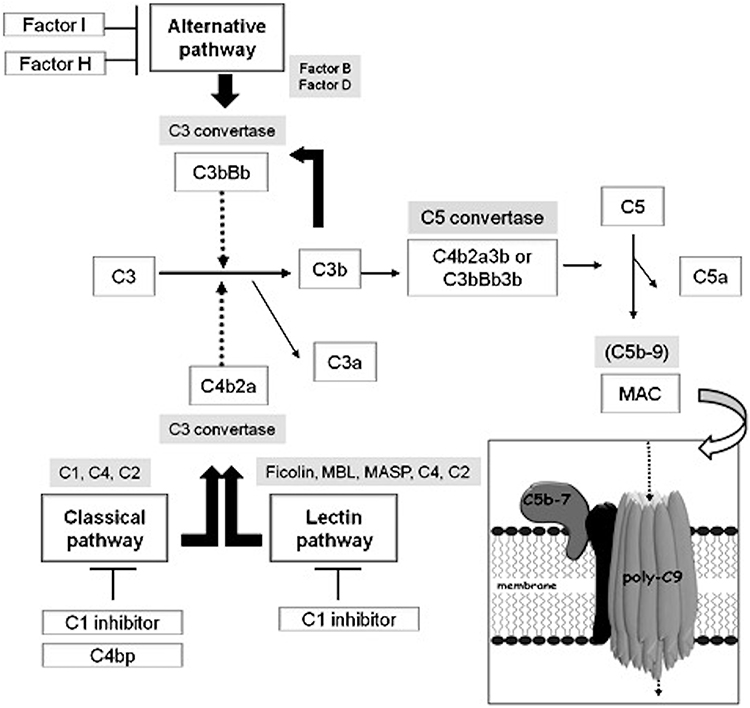 |
Figure 1 Activation of the complement system. Activation of the complement system (CS) occurs through three possible pathways: the classical, the alternative and the lectin pathway. All the pathways converge at the activation of C3 and C5 and lead to the formation of the membrane attack complex (MAC). Activation of the cascades is controlled by several regulatory proteins. C1 inhibitor, C4 binding protein, Factor I and Factor H represent the most important regulators and act at different points of the pathways. |
CS dysfunctions in terms of upregulation, downregulation, or dysregulation can create an imbalance of both host defence and inflammatory response leading to autoimmunity.6,7 As well described by evidence from the literature, the CS dramatically acts in the pathogenesis of several systemic autoimmune diseases including rheumatoid arthritis (RA).1 In this review, we aimed at focusing on the role of the CS in the pathogenesis of different autoimmune diseases with a particular focus on a specific extra-articular manifestation of RA, the RA-associated interstitial lung disease (ILD). With this purpose, we reviewed the main points of the CS-mediated mechanisms in the pathogenesis of autoimmune diseases such as Systemic Lupus Erythematosus (SLE), Anti-phospholipid Syndrome (APS), Systemic Sclerosis (SSc), Sjögren’s Syndrome (SjS), vasculitides, and Psoriatic Arthritis (PsA). We focused on the pathogenic role of CS dysregulation in parenchymal lung disorders of both known and unknown causes that can be classified into ILD and, thus, the interplay between CS, inflammation, and autoimmunity in ILD. Therefore, the intriguing challenge of modulating CS as a treatment strategy for ILD has been discussed.
What is so special about complement in RA-ILD? What about other innate pathways? Is there evidence of immune complex deposition in the lung to suggest what activates complement?
CS Dysregulation in Autoimmune Diseases and RA
As is well documented, the abnormal CS activity has a pathogenic role in several autoimmune diseases, mainly including RA and SLE, APS, SSc, SjS, vasculitides and PsA.4 The best “model” of the pathogenetic role of the CS in autoimmune diseases is in SLE where it seems to exert both a protective and a pathogenetic role. Indeed, the deficiency of early components C1q, C1r, C1s, C4, and C2 results in severe forms of SLE with an early onset.7 Moreover, anti-C1q autoantibodies can be found in 30–60% of SLE patients and their presence correlates with renal involvement and with the severity of lupus nephritis.8–10 In patients with APS, increasing data indicate that the CS is highly activated and acts as a cofactor in the pathogenesis of clinical manifestations promoting coagulation and mediating pregnancy morbidity. For instance, catastrophic APS seems to be associated with mutations in CS regulatory genes serving as a second hit in the pathogenesis.11,12 In SSc, the CS is locally activated in the skin of patients at immunohistochemistry level where complement C5b-9 and C5aR could be detected in involved skin samples from SSc patients, with a perivascular localization.13 Complement deposition has also been detected in renal biopsies from SSc patients with a peritubular capillary deposition of C4d and C1q and glomerular deposits of C3b.14–16 Another disease where hypocomplementemia has been observed is the SjS ranging between 2−25% for C3, 10–39.5% for C4 and 14–15% for CH50.17 Heterozygous C2 deficiency in combination with a low copy number of C4a substantially increases the risk of primary SjS and this genetic combination is associated with low age at diagnosis.18 The apparent interaction between C4a copy number and heterozygous C2 deficiency further strengthens the pivotal role of the classical complement pathway in the pathogenesis of SjS.18 In small-medium-vessel vasculitides, the role of the CS can be different in accordance with the specific diseases. In general, the pathogenesis of vasculitides is related to the presence of leukocytes in the vessels and the formation of IC with their local deposition; therefore, CS is activated and mediates directly the damage of vessel structures.2
Differently from autoimmune diseases, serum C3 and C4 levels have been reported to be higher in PsA patients compared with healthy controls.19 Likewise, complement activation has been detected at joint level where C3 levels in the synovial fluid from PsA patients were found to be higher compared with those from RA patients and osteoarthritis.20
The CS seems to play a role both in the initiation and evolution of RA, in particular in tissue damage in RA through interactions with citrullinated proteins from synovial neutrophils.1,21 Authors have shown that antibodies against citrullinated proteins (ACPA) activate the CS in vitro via both the classical (CP) and the alternative (AP) but not through the lectin pathway (LP): ACPA from all enrolled subjects activated the CS suggesting that complement activation could play a key role in ACPA-positive RA patients.22
Autoantibodies in RA patients target antigens in cartilage and the synovium, contributing to the formation of different types of circulating ICs (Circulating immune complexes (CICs), small, intermediate and large).23 Intermediate CICs typically cause the most damage as they get trapped in the tissues or in the joints.1 Crucially, these CICs can activate complement, which give rise to chronic destruction of the joint, via the initiation of innate as well as adaptive immune responses.23
The possible participation of C1s in cartilage remodelling is supported by a few studies, one of them demonstrating that C1s is intensely immunostained in the hypertrophic chondrocytes, but not in normal articular chondrocytes.24 The authors described the evidence for a negative C1 staining in normal articular cartilage biopsies together with its positivity in all degenerating cartilage biopsies from analyzed RA samples.25 Furthermore, IgM rheumatoid factor (RF) and IgA RF amplify the CS activation mediated by ACPA-containing IC.26 ACPA-IC, incorporating IgM or IgA RF may participate in the triggering of the inflammation promoting activation of complement cascades in RA joints. Both ACPA and RF have been shown to have a role in systemic bone loss in early RA patients due to the presence of citrullinated antigens on the surface of osteoclastic cells, making these cells the main targets of circulating ACPA leading to the induction of osteoclastogenesis.27,28
Although increased CS activation is potentially related to the occurrence and/or exacerbation of inflammation in RA, a status of complement deficiency may predispose to RA. The association between CS defects and RA has been reported for deficiencies of several complement components, including C1r and C1s, C1q, C4, C7, C9 and factor I.29–31 Association between CS activation and disease activity in RA has been documented in the literature. According to Wouters et al, a correlation between plasma levels of C1q-C4 complexes and disease activity score was shown in RA patients.32 Makinde et al, have shown that the ratios C3d/C3 and C4d/C4 may provide a sensitive assessment of disease activity in RA.33 Doherty et al reported raised synovial C3d levels in active compared with inactive RA.34 Moreover, Nguyen et al demonstrated that antirheumatic therapy is associated with reduced complement activation in RA.35
However, other potential triggers for CS activation in RA should be considered such as C-reactive protein (CRP).36–38 In 2001, Moleenar et al demonstrated that plasma levels of activated CS components and CRP–complement complexes are increased in most patients with RA and correlated with disease activity, pointing to the role of CRP-mediated complement activation in RA pathogenesis.39
Lung Involvement in RA
Pulmonary nodules, pleural effusion, bronchiectasis, and ILD are the most common extra-articular manifestations of respiratory disorders that are RA-correlated.40,41 ILD is the most common among these, representing the second reason of mortality in RA patients due to progressive fibrosis of the lung parenchyma.42 Moreover, RA patients can develop lung complications due to immunosuppressive therapy, such as drug toxicity and/or infections.43,44
The epidemiology of lung involvement in RA has been difficult to define over time in terms of morbidity and symptoms: according to some authors, radiological evidence of the disease has been observed from 19–67% of RA patients, clinical prevalence is estimated to be 3–5% while some studies report an elevated post mortem incidence of RA-ILD.45,46
Recent evidence defines RA-ILD as the second leading cause of death in RA patients, overcoming the risk of death from malignancy.47 Though RA is itself a known risk factor for ILD, only a subset of patients develops the lung disease.42,48 Sex and/or age, environmental agents, serological variables, clinical features, genetic background, and drug-related pathways have been reported as potential risk factors by several authors49–51 but an international consensus has not yet been reached.52 According to radiological data in conventional chest radiography studies, the prevalence of ILD in RA patients (RA-ILD) varies from 1–6%, while from 5–67.3% in high-resolution computed tomography (HRCT) studies.53,54
HRCT has emerged as an important tool for the detection of ILD in RA patients55,56: the most common HRCT pattern in RA-ILD is the Usual Interstitial Pneumonia (UIP) while Nonspecific Interstitial Pneumonia (NSIP) is described less frequently.57,58 RA-associated UIP (RA-UIP) is difficult to differ from the idiopathic one (e.g. idiopathic pulmonary fibrosis [IPF]) on HRCT scans. The crucial features include the peripheral- and basal-predominant reticulation and the honeycombing with or without bronchiectasis. In a recent study, investigators found that HRCT-UIP significantly correlates with histological-UIP in patients with RA-ILD, potentially obviating the need for a lung biopsy in individuals showing specific HRCT finding.59 Limited data suggest the prognostic utility of the HRCT in RA-ILD: the HRCT-UIP appears to predict a worse survival compared with the other ILD patterns including NSIP in RA patients.60,61 In addition, the occurrence of traction bronchiectasis and honeycombing, documented by HRCT, seems to confer additional survival information in RA-patients.57 In accordance with the most recent American Thoracic Society (ATS)/European Respiratory Society (ERS)/Japanese Respiratory Society (JRS)/Latin American Thoracic Society (ALAT) guidelines, three defined HRCT patterns of fibrosing lung disease can be described in the setting of an IPF: definite UIP, possible UIP, and inconsistent with UIP. Definite UIP and possible UIP differ for the presence of honeycombing.62 However, studies from selected IPF patients have documented no significant difference in survival according to the type of HRCT findings.63–65 Broader studies on idiopathic interstitial pneumonias (IIPs) reported that a definite UIP on HRCT scans is able to predict a worse survival with the respect to an indeterminate UIP.66 Nevertheless, the prognostic value of HRCT-UIP classifications has not been definitely analysed in RA-ILD patients. As described in the literature, the impairment in respiratory functions and/or the worsening in general respiratory functions over time have been proposed as poor prognostic factors in connective tissue disease (CTD)-ILD. In addition, patients showing an UIP pattern on HRCT were also suggested to have a worse prognosis than those with a different HRCT pattern regardless of the specific disease diagnosis.60,67,68
CS Dysregulation in ILD
As described, ILD comprises several types of parenchymal lung disorders that can be categorized into four subtypes: ILDs with a known association (e.g., treatments, CTD), granulomatous (e.g., sarcoidosis), idiopathic interstitial pneumonias (IIPs) and rare ILDs.69 Despite the different aetiologies, some ILDs share common pathophysiological aspects: the proliferation of fibroblasts and myofibroblasts, the accumulation in the extracellular matrix, and the consequent interstitial fibrosis.70 The CS may act with a functional duality at the lung level, in host defence and lung injury.71,72 As known, the CS plays a key role in the innate immune response to pathogens at the lung level.73 Pathogens can escape the complement-mediated eradication by inhibiting CP or AP genes.74 Interestingly, recent evidence documents a localized production of complement components in peripheral tissues and mucosal sides including the lung.75–77 Recently, authors documented that complement-related genes might contribute to the pro-fibrotic inflammatory lung responses in the setting of autoimmune arthritis and could be both targeted and predictive for disease burden (Figure 2).78 Data described C3 in mesothelial cells and fibroblasts in both murine and human lungs as well as in goblet, mucous and alveolar epithelial type (AT)2 cells in humans; the component C5 was mainly documented in AT2 cells.71 In addition, human Properdin (CFP) expression has been reported in epithelial cells and alveolar macrophages.73,79 The constitutive expression of C5 has been described to affect the functions of immunoregulation and repair of AT2 cells during infection and lung injury.80
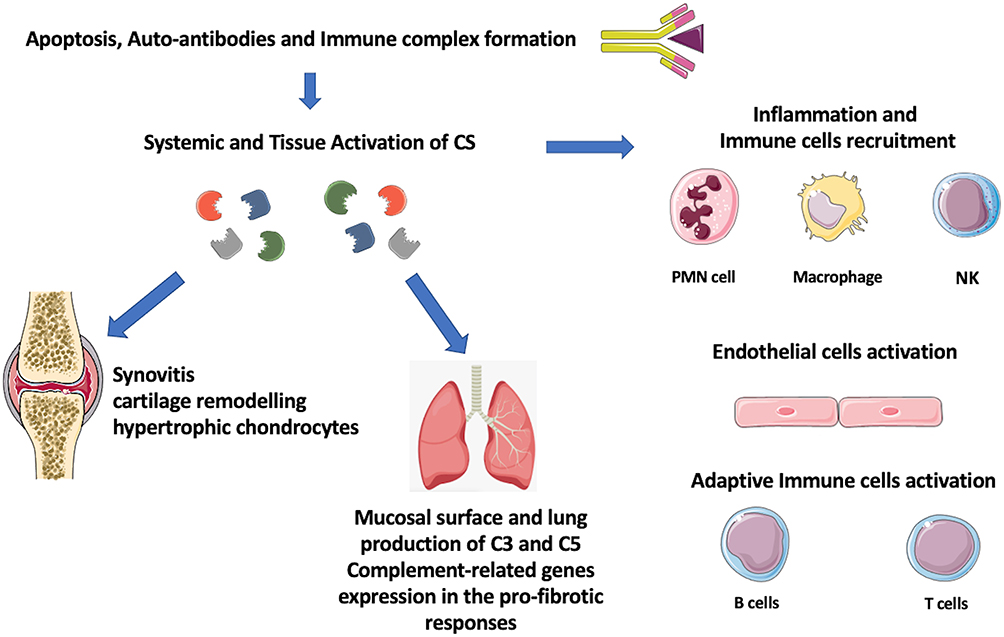 |
Figure 2 Link between autoimmunity, inflammation and tissue injury and complement system (CS) activation in rheumatoid arthritis and interstitial lung disease. |
Gu et al have shown that patients with IPF showed greater levels of local and systemic C3a and C5a.81 C3aR is expressed ubiquitously, including in the lung.82 Interestingly, mechanisms of cellular damage mediated by C3a and C5a have been observed in acute lung injury and the highly fatal acute respiratory distress syndrome.72,83–85 Among the CS components, C1q can interact with fibronectin and lead the fibroblasts’ adhesion to IC with resulting collagen synthesis.86 A potential role of the CS in lung fibrosis was previously suggested by the bleomycin-induced fibrosis in mice without the specific identification of individual proteins involved in the establishment of fibrosis.87 Authors indicated a crucial role for C5 which was related with the expression of TGF-β1 and matrix metalloproteinase-3, key profibrotic mediators in murine models.88 C5-deficient mice showed elevated inflammatory markers compared with C5-sufficient mice during acute bleomycin-induced lung injury suggesting the anti-inflammatory role for C5; in contrast, during chronic stages of bleomycin-induced injury, C5 had a potential profibrotic role.88 Studies described in both the sera and the bronchoalveolar fluid from IPF patients increased levels of IC as well as CS fragments supporting a relevant activation of CS pathways and its potential association with pulmonary fibrosis in IPF.81,89–91 An impaired clearance of apoptotic debris might promote an inflammatory status: the epithelial injuries are able to trigger and enhance the mesenchymal activation and, thus, might affect the alveolar epithelial mechanisms of repair.92 In the past years, authors reported that IC could activate the CS in IPF.89 However, more recent evidence described the presence of CS components in patients with IPF and documented that products by the AP were clinically significant as indicators of disease severity. An increased risk of lung diseases has been associated with low serum levels of mannose- binding lectin (MBL).93 MBL deficiency is also associated with CTDs (SLE, cystic fibrosis, and primary immunodeficiencies).94 In addition, a case control study described the occurrence of MBL deficiency in early onset IPF and familial cases.95 The possible involvement of the LP in the pathogenesis of ILD has been suggested by evidence reporting the significantly higher level of CS products in the bronchoalveolar lavage fluid (BALF) in a cohort of patients with ILD compared with controls.96 Authors hypothesized that, in SSc-ILD, the recurrent injuries, which recognized a lectin-mediated ischemic-reperfusion mechanism, might cause an endothelial dysfunction potentially leading to endothelial and epithelial apoptosis and the fibrotic response in the lung.97 In a recent study from Pellicano et al the total complement activity was associated with ILD, assessed by both diffusing capacity for carbon monoxide (DLco) and HRCT, suggesting its pathogenetic role in ILD.98 The potential link between innate immune response, including CS, and ILD has been thus proposed and documented. In this context, the CS appears to accelerate the pathogenesis of IPF.99,100
May the CS be a Treatment Target in RA?
Genome-wide studies in RA patients have shown the occurrence of specific CS polymorphisms, including C5 and C3 gene polymorphisms that resulted in an efficient cleavage into the proinflammatory anaphylatoxins.101,102 Recently, a positive correlation between disease activity [by disease activity score (DAS)-28] and specific complement mRNA expression levels was demonstrated in the synovium from patients with early RA.103 Elevated levels of CS activation fragments have been detected in blood, synovial fluid, and tissue from RA patients supporting the possible inadequate control of CS activation in disease pathogenesis.1,104–107 Murine models of arthritis such as collagen-induced arthritis demonstrated that a compromised CS may be associated with a less severe disease.108,109 Likewise, in K/BxN mouse model of arthritis, mediated by T-cell dependent autoantibody response to glucose-6-phosphate isomerase (GPI), GPI deposits localized in the joints with IgG and C3 complement.110,111 Circulating C3 is necessary and sufficient for the induction of arthritis in this model.111 A trigger of complement activation in humans could be autoantibodies such as RF and ACPA that interact locally in the joint with antigens, resulting in IC that triggers classical local CS activation.107 ACPA were demonstrated to activate complement in a dose-dependent manner via the CP and also the AP.112 Moreover, RA patients have been reported to have higher concentrations of terminal complement complex (TCC) in the blood than negative controls.5,113 Indeed, synovial tissue seems a complement factor rich environment, since different cells such as synovial fibroblasts, macrophages, and endothelial cells produce complement factors locally in the joints.104,114,115 Recent findings highlight that, in inflammatory tissues, synovial fibroblasts express high amounts of C3 and C3a receptor which drive metabolic reprogramming of fibroblasts, and shift to a pro-inflammatory state.116 In arthritis, tissue priming may provide an explanation for relapses and for the evolution from self-limited arthritis to chronic inflammation. Furthermore, complement C3 and C5 and its receptors are produced intracellularly in T cells and in turn activate the cells in an autocrine manner.117–120
Upregulation of complement synovial tissue and tendons has also been previously associated with exacerbation of arthritis caused by mechanical stress.121,122 The increased observed plasma levels of C3 and C4 in active RA patients may reflect increased hepatic synthesis induced by interleukin (IL)-6, as for other acute phase reactants.32 Indeed, local complement activation within affected joints could be triggered by extracellular DNA, dead cells and CRP.112 Several conventional synthetic disease-modifying antirheumatic drugs (csDMARDs) and biological (b)DMARDs that are used in RA treatment have been demonstrated to affect CS. Few studies suggested direct effects of methotrexate (MTX) on CS.123 The MTX, via its metabolite adenosine, exerts an inhibitory effect on production of Tumor necrosis factor (TNF) and promotes the IL-6 production, thus potentially attenuating the complement cascade.124 MTX exposure has been associated with an elevated expression of complement genes, and increased C3 and C5 in liver of patients with inflammatory arthritis.125 The effects of MTX and TNF inhibitors (TNFi), such as adalimumab, etanercept or infliximab, on CS activation have been assessed recently using soluble TCC levels in RA.35 Patients with active RA had elevated baseline TCC levels, indicating an increased complement activation and TCC decreased quickly with MTX, while a sustained reduction was observed with combination treatment of adalimumab, etanercept or infliximab plus MTX during a 6-month follow-up period. Other studies previously showed an effect of adalimumab and etanercept on CS with a significant reduction of C3 and C4 levels during treatment while complement activity correlated with RF in a cohort of RA patients.126–128 Higher levels of complement C3 were associated with a worse EULAR response and normalization of C3 complement levels was associated with improvement of disease activity in RA patients treated with adalimumab or etanercept.126 Similar results were obtained in PsA patients treated with TNFi agents suggesting that elevated C3 and C4 levels could be considered a negative predictive factor of response influencing the outcome of TNFi therapy in treated patients.19,129 Elevated complement C3 levels may reflect the presence of the inflammatory process contributing to the acute phase response. High plasma amounts of component C3 could be due to spill over from the joints or to an abnormal production. The mechanisms underlining complement activation in response to TNFi may have several explanations: complement components are produced by hepatocytes, and hepatic synthesis may be upregulated in response to TNF.130,131 TNFi might revert the increased soluble complement products. Second, antirheumatic therapy can reduce CRP levels which is able to interact with CS with subsequent activation of the classical complement cascade.39,132 Third, most antirheumatic therapies exert their effect partly by inhibition of IL-6, which in turn causes a downregulation of CRP synthesis by hepatocytes and subsequently reduced activation of the CP.133 Of note, also tocilizumab (TCZ), an anti-IL6 receptor monoclonal antibody, has been described to be able to reduce levels of CRP, C3 and C4 as early as 4 weeks after the first treatment in a cohort of RA patients.134 This effect has been demonstrated with other biological disease-modifying anti-rheumatic drug (bDMARD) such as rituximab (RTX), an anti-CD20 monoclonal antibody used to treat RA: RTX was found to affect complement C3 levels in RA patients reinforcing the concept to represent a potential useful serological marker of disease activity and response to treatment.135 However, evidence on potential effects of both TCZ and RTX via CS describe mainly indirect mechanisms.134,135
A direct proof of efficacy of specific complement-targeted therapy in human arthritis is still lacking.1,112 In animal models, the inhibition of C5, administered systemically or at intra-articular level, was associated with good results.112,136 The C5a-C5aR axis may be a relevant target for treatment of arthritis.137 However, both the inhibitory anti-C5 antibody eculizumab and the oral C5aR inhibitor PMX-53 resulted unsuccessful in clinical trials for RA treatment.138,139 Eculizumab, the first approved CS antagonist, is an IgG-kappa humanized monoclonal antibody, that binds C5 protein inhibiting its cleavage, thus preventing the generation of active C5a and C5b, and the C5b-C9. It has been developed to treat RA and SLE and obtained approval for the treatment of paroxysmal nocturnal hemoglobinuria (PNH).140 Of note, both the eculizumab and the oral C5aR inhibitor act downstream of C3 cleavage rather than targeting intracellular autocrine C3 function. Treatment with PMX-53 did not result in a reduction of synovial inflammation nor in a clinical improvement in RA patients.138 Therefore, an intriguing strategy might be to target C3- or C3a preventing flares due to its effect on tissue priming rather than to decrease inflammatory response interfering with cytokines and anaphylatoxins.
May the CS be a Treatment Target in ILD?
The current knowledge on the role of CS dysregulation in the pathogenesis of ILD remains not completely defined. Furthermore, direct evidence of its role in the pathogenesis of RA-ILD is missing. However, different treatments for patients with ILD, including RA-ILD, might indirectly act on CS-mediated inflammation (Table 1).112
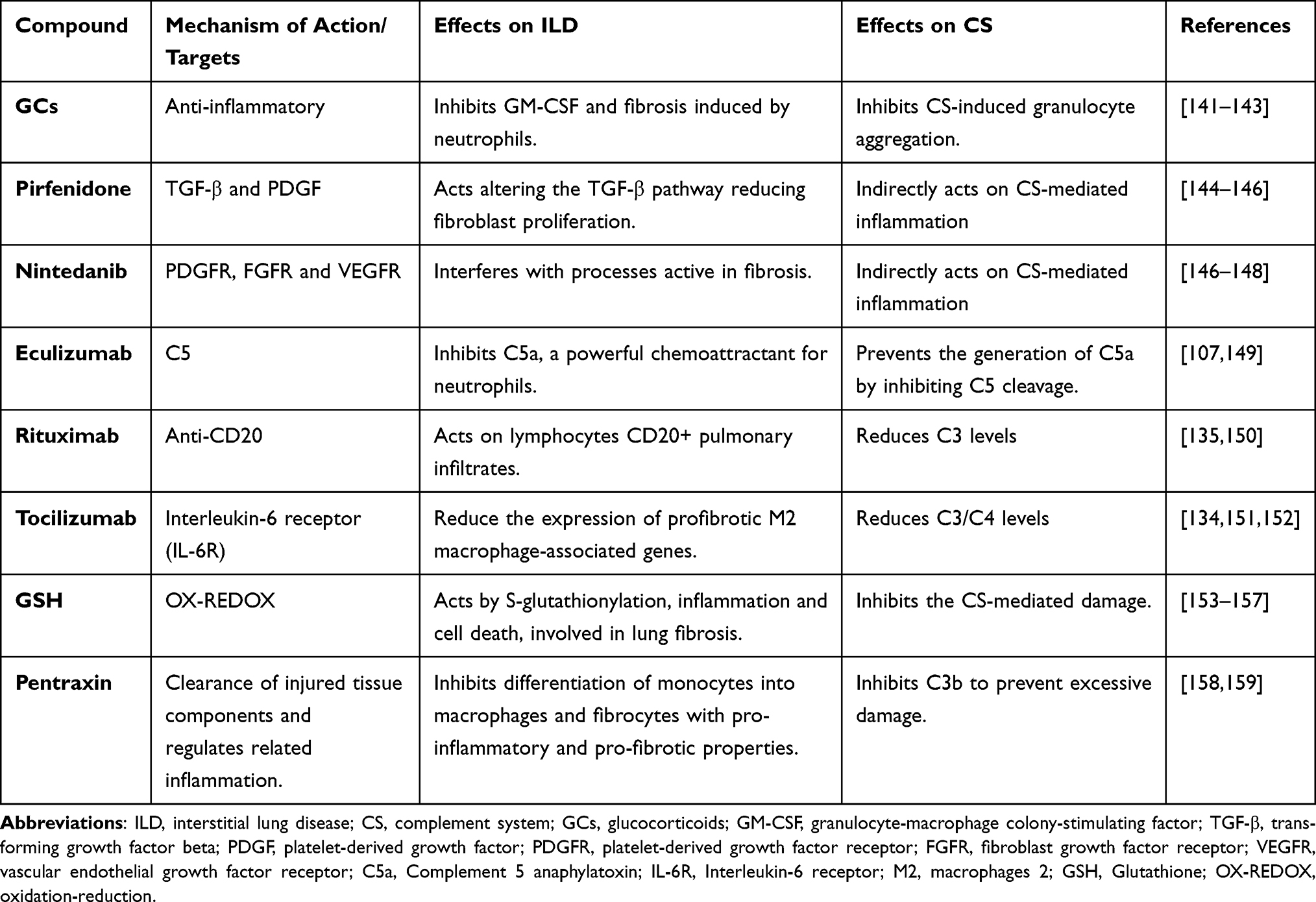 |
Table 1 Effect of Therapeutic Agents in Rheumatoid Arthritis and Interstitial Lung Disease |
Glucocorticoids (GCs) are anti-inflammatory agents used to treat ILD. The mechanism of their action is complex and includes the regulation of gene expression, from signal transduction to post-translational variations.160,161 GCs induce genes of anti-inflammatory factors thus raising innate immunity actions.162 GCs are powerful inhibitors of key pro-inflammatory genes including ILs and chemokines as well as granulocyte/macrophage colony stimulating factor (GM-CSF) and TNF-α.141 As documented, neutrophils have a crucial role in the acute phase of inflammation: however, aggregated neutrophils could promote fibrosis in lung damage.142 GCs inhibit complement-induced granulocyte aggregation.143 However, the potential prognostic value of the neutrophilic inflammation in IPF lungs remains unclear. C5a is a chemoattractant for neutrophils, and the link C5a-C5aR/CD88 activates neutrophils;149 in addition, the feedback mechanism between activation of neutrophils and the CS participates in the progress of tissue damage in autoimmune and IC diseases characterized “as having” pulmonary involvement.163
In 2014, pirfenidone (PFD) and nintedanib were approved by the US Food and Drug Administration and recommended for the treatment of IPF, for their effectiveness in reducing disease progression.164 PFD is a non-peptide synthetic chemical that inhibits the production of cytokines including TGF-β1, TNF-α, and platelet-derived growth factor (PDGF).144,145 It thus can slow or inhibit the progressive fibrosis.165 Nintedanib inhibits in vitro a distinctive spectrum of kinases at pharmacologically significant concentrations that comprises the vascular endothelial growth factor receptor (VEGFR) subtypes, the fibroblast growth factor receptor (FGFR) types 1, 2 and 3, and PDGF receptors (R)-α and -β with a key antifibrotic effect.147,148 Evidence of a direct CS regulation by PFD is not yet described as well as for nintedanib. However, as documented, PDGFs activate CS through both the CP and AP and, through CS, increase the levels of several mediators, trigger polarization of macrophages, thereby promoting mechanisms of pathological neovascularization and their functions of tissue-regeneration together with anti-/pro-fibrotic effects.146 PDGFs are, thus, involved in the advancement of inflammation and increase the migration of mononuclear cells in tissue damage.166 It can be hypothesized that by modulating PDGFs, both PFD and nintedanib can be able to indirectly act on CS-mediated inflammatory pathways. In addition, nintedanib inhibits profibrotic mechanisms also acting on VEGFR subtypes.148,167 The interaction between the CS and VEGF is suggested by its role in neovascular age-related macular degeneration (nAMD) mainly relying on the evidence of both the uncontrolled activation of CS and the upregulation of VEGF in nAMD.168 However, recent findings suggest an association between polymorphisms in genes related to the CS with the severity of macular lesions as well as with the response to antiangiogenic therapy in patients with nAMD.169 In IPF, pulmonary endothelial cells might be the source of pulmonary fibroblasts through endothelial mesenchymal transition (EndoMT), which participates in pulmonary fibrosis.170 Interestingly, both in vivo and in vitro studies documented that nintedanib inhibits EndoMT, probably by modulating the VEGF signaling pathway.170
In autoimmune associated-ILD, as reported by data from the literature, the lung progression during treatment with Conventional synthetic (cs)DMARDs represents the most frequent rationale for starting targeted therapies: RTX seems to be the most available option together with nintedanib and TCZ, and the combination therapy represents the most frequent therapeutic scheme for nintedanib and RTX.171 In addition, RTX therapy appears to be effective in stabilising lung function deterioration and ILD involvement in RA patients with UIP.172 In patients with CTD-NSIP, the reasons for RTX use rely on the evidence of immunoglobulins, complement depots, and lymphocytes CD20+ infiltrates in the pulmonary capillaries in patients with NSIP.150 The antitumor molecular mechanisms of anti-CD20 antibodies include a potent complement activation: nevertheless, complement depletion significantly reduced the antitumor activity of RTX.173 However, RTX activates the CS in vitro, and there is an ongoing debate on the exact role of this mechanism of action in vivo.174 Results of randomized controlled and clinical studies also support the use of TCZ for SSc-ILD.171 Specifically, TCZ is able to reduce C3 and C4 serum levels during treatment by inhibiting IL-6-mediated signal transduction.134 As reported, IL-6 dysregulated expression is implicated in the pathogenesis of interstitial pneumonias and IPF.175,176 In addition, decreased complement levels have been associated with the TCZ treatment longevity without specific adverse outcomes.151
Khanna et al in a phase 3 trial, have shown that TCZ is able to preserve lung function through the reduction in expression of genes associated to profibrotic M2 macrophages, thus supporting its antifibrotic effects.152 Furthermore, ongoing studies are designed to compare efficacy and safety of TCZ versus regular treatments, in patients with severe rapidly progressive-ILD secondary to systemic diseases.177 As described, IL-6 activates the JAK/STAT signalling pathway, initiating the cellular changes observed in ILDs.178 The JAK/STAT axis is also activated by growth factors such as TGF-β1, FGF, PDGF, and VEGF: in this context, TCZ by acting on IL-6/JAK/STAT signalling could also represent an indirect JAK/STAT inhibitor.179
To date, several trials are ongoing on different treatment strategies in IPF. Abnormalities in redox homeostasis have been described in both patients with IPF and animal models of fibrosis.180 A complex network includes redox-dependent processes including S-glutathionylation, inflammation, and cell death, all mechanisms involved in lung fibrosis.154 Among novel strategies under investigation in IPF, antioxidant agents have been proposed: they may act on the CS because of the known effects of CS on intracellular glutathione (GSH) and its disulphide forms leading to oxidative damage.164 Authors documented that GSH can inhibit the CS and suggested the possibility for designing additional therapeutic interventions modulating GSH metabolism in order to inhibit CS-mediated damage in autoimmune diseases.155,156
Pentraxins are soluble receptors of innate immunity and include sensing danger molecules, protect against infections, regulate inflammation and the clearance of injured tissue components.158,181 They have a dual link with the CS. Initially, the pentraxins activate CS by binding C1q. Therefore, pentraxins inhibit the CS at the C3b stage to avoid excessive damage. However, the emerging inflammation needs to be limited to the target area.158,159 A completed phase 2 trial has recently shown a significant reduction in modifications of respiratory volumes and 6-min walk distance from baseline in patients with ILD.182
Animal models described potential beneficial effects of CS inhibition in bleomycin-induced lung fibrosis: the blockade of C3aR and C5aR resulted in the reduction of the progression of fibrosis by alleviating the local CS activation and suggested an encouraging treatment strategy for IPF.100 However, recent evidence did not confirm the role of eculizumab in lung fibrosis induced by bleomycin.183 Case reports describe efficacy of eculizumab in rapidly inducing remission in aggressive patients with antineutrophil cytoplasmic antibody (ANCA)–associated vasculitis (AAV) with respiratory syndromes.184
More recently, authors interestingly provided evidence supporting a therapeutic strategy by targeting the CS in lung damage in an acute setting.
Conclusion
Overall, the activation of the CS is critical in different autoimmune-mediated diseases such as RA where it can amplify tissue injury directly or indirectly. Its dysregulation can create an imbalance of both host defence and inflammatory response inducing the development of autoimmunity. Autoantibodies such as RF and ACPA activate the CS in vitro via both the CP and the AP, contributing to the formation of IC. Autoantibodies may have a detrimental effect not only at joint levels but also in systemic manifestations, such as lung involvement. RA-ILD is typically associated with the presence of ACPA and the CS and may act at this level with a role in favouring lung injury. Local and systemic activation of CS has been demonstrated in IPF with increased levels of IC in BALF and sera of patients. Murine models of lung fibrosis explored the role of CS and C5 has been associated with profibrotic mediators. Current therapeutic strategies in RA and ILD take advantage of the molecular mechanisms implicated in the inflammatory process as highlighted by the advent of biological treatments that can slow down the progression of the diseases. To date, there is no direct evidence strongly supporting that any of the current treatments for RA-ILD act via the CS pathway but indirect mechanisms link CS and several therapeutic options of both conditions. Nevertheless, therapeutic agents for ILD, such as nintedanib and pirfenidone, mainly show direct mechanisms of action that are independent of their effect on the CS but they both might indirectly act on CS-mediated inflammation: the idea of potential interplay between anti-fibrotic pathways and CS certainly needs further investigations.
RA is a complex disease with different tissues being the target of inflammation and damage. Key molecules such as those in the CS bridging innate and adaptive immune responses might be future therapeutic targets in those conditions where local activation at tissue site, IC deposition, and anaphylatoxins exert pro-inflammatory activities.
Funding
There is no funding to report.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Holers VM, Banda NK. Complement in the initiation and evolution of rheumatoid arthritis. Front Immunol. 2018;9:1057. doi:10.3389/fimmu.2018.01057
2. Chimenti MS, Ballanti E, Triggianese P, Perricone R. Vasculitides and the complement system: a comprehensive review. Clin Rev Allergy Immunol. 2015;49:333–346. doi:10.1007/s12016-014-8453-8
3. Lay WH, Nussenzweig V. Receptors for complement of leukocytes. J Exp Med. 1968;128(5):991–1009. doi:10.1084/jem.128.5.991
4. Ballanti E, Perricone C, Greco E, et al Complement and autoimmunity. Immunol Res. 2013;56(2–3):477–491. doi:10.1007/s12026-013-8422-y
5. Bemis EA, Norris JM, Seifert J, et al Complement and its environmental determinants in the progression of human rheumatoid arthritis. Mol Immunol. 2019;112:256–265. doi:10.1016/j.molimm.2019.05.012
6. Walport MJ. Complement. Second of two parts. N Engl J Med. 2001;344(15):1140–1144. doi:10.1056/NEJM200104123441506
7. Conigliaro P, Triggianese P, Ballanti E, Perricone C, Perricone R, Chimenti MS. Complement, infection, and autoimmunity. Curr Opin Rheumatol. 2019;31(5):532–541. doi:10.1097/BOR.0000000000000633
8. Jia C, Tan Y, Zhao M. The complement system and autoimmune diseases. Chronic Dis Transl Med. 2022;8(3):184–190. doi:10.1002/cdt3.24
9. Yuan ZC, Xu WD, Lan YY, et al Association of MBL2 gene polymorphisms and systemic lupus erythematosus susceptibility: a meta-analysis. Int J Rheum Dis. 2021;24(2):147–158. doi:10.1111/1756-185X.14017
10. Mahto H, Pati A, Sahu SK, Sharma HP, Padhi A, Panda AK. Association of MBL-2 gene polymorphisms with systemic lupus erythematosus: an updated meta-analysis and trial sequential analysis. Lupus. 2020;29(10):1227–1237. doi:10.1177/0961203320939156
11. Gropp K, Weber N, Reuter M, et al β2-glycoprotein I, the major target in antiphospholipid syndrome, is a special human complement regulator. Blood. 2011;118(10):2774–2783. doi:10.1182/blood-2011-02-339564
12. Chaturvedi S, Braunstein EM, Yuan X, et al Complement activity and complement regulatory gene mutations are associated with thrombosis in APS and CAPS. Blood. 2020;135:
13. Sprott H, Müller-Ladner U, Distler O, et al Detection of activated complement complex C5b-9 and complement receptor C5a in skin biopsies of patients with systemic sclerosis (scleroderma). J Rheumatol. 2000;27(2):402–404.
14. Devresse A, Aydin S, Le Quintrec M, et al Complement activation and effect of eculizumab in scleroderma renal crisis. Medicine. 2016;95(30):e4459. doi:10.1097/MD.0000000000004459
15. Batal I, Domsic RT, Shafer A, et al Renal biopsy findings predicting outcome in scleroderma renal crisis. Hum Pathol. 2009;40(3):332–340. doi:10.1016/j.humpath.2008.08.001
16. Okrój M, Johansson M, Saxne T, Blom AM, Hesselstrand R. Analysis of complement biomarkers in systemic sclerosis indicates a distinct pattern in scleroderma renal crisis. Arthritis Res Ther. 2016;18(1):267. doi:10.1186/s13075-016-1168-x
17. Brito-Zerón P, Retamozo S, Ramos-Casals M. Phenotyping Sjögren’s syndrome: towards a personalised management of the disease. Clin Exp Rheumatol. 2018;36(3):198–209.
18. Lundtoft C, Sjöwall C, Rantapää-Dahlqvist S, et al Strong association of combined genetic deficiencies in the classical complement pathway with risk of systemic lupus erythematosus and primary Sjögren’s syndrome. Arthritis Rheumatol. 2022;74(11):1842–1850. doi:10.1002/art.42270
19. Chimenti MS, Perricone C, Graceffa D, et al Complement system in psoriatic arthritis: a useful marker in response prediction and monitoring of anti-TNF treatment. Clin Exp Rheumatol. 2012;30(1):23–30.
20. Partsch G, Bauer K, Bröll H, Petera P, Dunky A, Merétey K. Complement C3 cleavage product in synovial fluids detected by immunofixation. Z Rheumatol. 1991;50(2):82–85.
21. Bugatti S, Bogliolo L, Vitolo B, Manzo A, Montecucco C, Caporali R. Anti-citrullinated protein antibodies and high levels of rheumatoid factor are associated with systemic bone loss in patients with early untreated rheumatoid arthritis. Arthritis Res Ther. 2016;18:226. doi:10.1186/s13075-016-1116-9
22. Trouw LA, Haisma EM, Levarht EW, et al Anti-cyclic citrullinated peptide antibodies from rheumatoid arthritis patients activate complement via both the classical and alternative pathways. Arthritis Rheum. 2009;60(7):1923–1931. doi:10.1002/art.24622
23. Dijkstra DJ, Joeloemsingh JV, Bajema IM, Trouw LA. Complement activation and regulation in rheumatic disease. Semin Immunol. 2019;45:101339. doi:10.1016/j.smim.2019.101339
24. Sakiyama H, Inaba N, Toyoguchi T, et al Immunolocalization of complement C1s and matrix metalloproteinase 9 (92kDa gelatinase/type IV collagenase) in the primary ossification center of the human femur. Cell Tissue Res. 1994;277(2):239–245.
25. Nakagawa K, Sakiyama H, Tsuchida T, et al Complement C1s activation in degenerating articular cartilage of rheumatoid arthritis patients: immunohistochemical studies with an active form specific antibody. Ann Rheum Dis. 1999;58(3):175–181. doi:10.1136/ard.58.3.175
26. Anquetil F, Clavel C, Offer G, Serre G, Sebbag M. IgM and IgA rheumatoid factors purified from rheumatoid arthritis sera boost the Fc receptor- and complement-dependent effector functions of the disease-specific anti-citrullinated protein autoantibodies. J Immunol. 2015;194(8):3664–3674. doi:10.4049/jimmunol.1402334
27. Harre U, Georgess D, Bang H, et al Induction of osteoclastogenesis and bone loss by human autoantibodies against citrullinated vimentin. J Clin Invest. 2012;122(5):1791–1802. doi:10.1172/JCI60975
28. Malmström V, Catrina AI, Klareskog L. The immunopathogenesis of seropositive rheumatoid arthritis: from triggering to targeting. Nat Rev Immunol. 2017;17(1):60–75. doi:10.1038/nri.2016.124
29. Mizuno M. A review of current knowledge of the complement system and the therapeutic opportunities in inflammatory arthritis. Curr Med Chem. 2006;13(14):1707–1717. doi:10.2174/092986706777441959
30. D’Cruz D, Taylor J, Ahmed T, Asherson R, Khamashta M, Hughes GR. Complement factor 2 deficiency: a clinical and serological family study. Ann Rheum Dis. 1992;51(11):1254–1256. doi:10.1136/ard.51.11.1254
31. Trouw LA, Daha N, Kurreeman FA, et al Genetic variants in the region of the C1q genes are associated with rheumatoid arthritis. Clin Exp Immunol. 2013;173(1):76–83. doi:10.1111/cei.12097
32. Wouters D, Voskuyl AE, Molenaar ET, Dijkmans BA, Hack CE. Evaluation of classical complement pathway activation in rheumatoid arthritis: measurement of C1q-C4 complexes as novel activation products. Arthritis Rheum. 2006;54(4):1143–1150. doi:10.1002/art.21729
33. Makinde VA, Senaldi G, Jawad AS, Berry H, Vergani D. Reflection of disease activity in rheumatoid arthritis by indices of activation of the classical complement pathway. Ann Rheum Dis. 1989;48(4):302–306. doi:10.1136/ard.48.4.302
34. Doherty M, Richards N, Hornby J, Powell R. Relation between synovial fluid C3 degradation products and local joint inflammation in rheumatoid arthritis, osteoarthritis, and crystal associated arthropathy. Ann Rheum Dis. 1988;47(3):190–197. doi:10.1136/ard.47.3.190
35. Nguyen THP, Hokstad I, Fagerland MW, et al Antirheumatic therapy is associated with reduced complement activation in rheumatoid arthritis. PLoS One. 2022;17(2):e0264628. doi:10.1371/journal.pone.0264628
36. Volanakis JE. Complement activation by C-reactive protein complexes. Ann N Y Acad Sci. 1982;389:235–250. doi:10.1111/j.1749-6632.1982.tb22140.x
37. Wolbink GJ, Brouwer MC, Buysmann S, ten Berge IJ, Hack CE. CRP-mediated activation of complement in vivo: assessment by measuring circulating complement-C-reactive protein complexes. J Immunol. 1996;157(1):473–479. doi:10.4049/jimmunol.157.1.473
38. Lagrand WK, Niessen HW, Wolbink GJ, et al C-reactive protein colocalizes with complement in human hearts during acute myocardial infarction. Circulation. 1997;95(1):97–103. doi:10.1161/01.CIR.95.1.97
39. Molenaar ET, Voskuyl AE, Familian A, van Mierlo GJ, Dijkmans BA, Hack CE. Complement activation in patients with rheumatoid arthritis mediated in part by C-reactive protein. Arthritis Rheum. 2001;44(5):997–1002. doi:10.1002/1529-0131(200105)44:5<997::AID-ANR178>3.0.CO;2-C
40. Conigliaro P, D’Antonio A, Pinto S, et al Autoimmune thyroid disorders and rheumatoid arthritis: a bidirectional interplay. Autoimmun Rev. 2020;19(6):102529. doi:10.1016/j.autrev.2020.102529
41. Turesson C, O’Fallon WM, Crowson CS, Gabriel SE, Matteson EL. Extra-articular disease manifestations in rheumatoid arthritis: incidence trends and risk factors over 46 years. Ann Rheum Dis. 2003;62(8):722–727. doi:10.1136/ard.62.8.722
42. Fazeli MS, Khaychuk V, Wittstock K, et al Rheumatoid arthritis-associated interstitial lung disease: epidemiology, risk/prognostic factors, and treatment landscape. Clin Exp Rheumatol. 2021;39(5):1108–1118. doi:10.55563/clinexprheumatol/h9tc57
43. Fragoulis GE, Nikiphorou E, Larsen J, Korsten P, Conway R. Methotrexate-associated pneumonitis and rheumatoid arthritis-interstitial lung disease: current concepts for the diagnosis and treatment. Front Med. 2019;6:238. doi:10.3389/fmed.2019.00238
44. Brown KK. Rheumatoid lung disease. Proc Am Thorac Soc. 2007;4(5):443–448. doi:10.1513/pats.200703-045MS
45. Sihvonen S, Korpela M, Laippala P, Mustonen J, Pasternack A. Death rates and causes of death in patients with rheumatoid arthritis: a population-based study. Scand J Rheumatol. 2004;33(4):221–227. doi:10.1080/03009740410005845
46. Bongartz T, Nannini C, Medina-Velasquez YF, et al Incidence and mortality of interstitial lung disease in rheumatoid arthritis: a population-based study. Arthritis Rheum. 2010;62(6):1583–1591. doi:10.1002/art.27405
47. Kim D, Cho SK, Choi CB, et al Impact of interstitial lung disease on mortality of patients with rheumatoid arthritis. Rheumatol Int. 2017;37(10):1735–1745. doi:10.1007/s00296-017-3781-7
48. Iqbal K, Kelly C. Treatment of rheumatoid arthritis-associated interstitial lung disease: a perspective review. Ther Adv Musculoskelet Dis. 2015;7(6):247–267. doi:10.1177/1759720X15612250
49. Weyand CM, Schmidt D, Wagner U, Goronzy JJ. The influence of sex on the phenotype of rheumatoid arthritis. Arthritis Rheum. 1998;41(5):817–822. doi:10.1002/1529-0131(199805)41:5<817::AID-ART7>3.0.CO;2-S
50. Kelly CA, Saravanan V, Nisar M, et al; British Rheumatoid Interstitial Lung (BRILL) Network. Rheumatoid arthritis-related interstitial lung disease: associations, prognostic factors and physiological and radiological characteristics--a large multicentre UK study. Rheumatology. 2014;53(9):1676–1682. doi:10.1093/rheumatology/keu165
51. Gochuico BR, Avila NA, Chow CK, et al Progressive preclinical interstitial lung disease in rheumatoid arthritis. Arch Intern Med. 2008;168(2):159–166. doi:10.1001/archinternmed.2007.59
52. Salaffi F, Carotti M, Di Carlo M, Tardella M, Giovagnoni A. High-resolution computed tomography of the lung in patients with rheumatoid arthritis: prevalence of interstitial lung disease involvement and determinants of abnormalities. Medicine. 2019;98(38):e17088. doi:10.1097/MD.0000000000017088
53. Norton S, Koduri G, Nikiphorou E, Dixey J, Williams P, Young A. A study of baseline prevalence and cumulative incidence of comorbidity and extra-articular manifestations in RA and their impact on outcome. Rheumatology. 2013;52(1):99–110. doi:10.1093/rheumatology/kes262
54. Zou YQ, Li YS, Ding XN, Ying ZH. The clinical significance of HRCT in evaluation of patients with rheumatoid arthritis-associated interstitial lung disease: a report from China. Rheumatol Int. 2012;32(3):669–673. doi:10.1007/s00296-010-1665-1
55. Yunt ZX, Chung JH, Hobbs S, et al High resolution computed tomography pattern of usual interstitial pneumonia in rheumatoid arthritis-associated interstitial lung disease: relationship to survival. Respir Med. 2017;126:100–104. doi:10.1016/j.rmed.2017.03.027
56. Caples SM, Utz JP, Allen MS, Ryu JH. Thoracic surgical procedures in patients with rheumatoid arthritis. J Rheumatol. 2004;31(11):2136–2141.
57. Kim EJ, Elicker BM, Maldonado F, et al Usual interstitial pneumonia in rheumatoid arthritis-associated interstitial lung disease. Eur Respir J. 2010;35(6):1322–1328. doi:10.1183/09031936.00092309
58. Tanaka N, Kim JS, Newell JD, et al Rheumatoid arthritis-related lung diseases: CT findings. Radiology. 2004;232(1):81–89. doi:10.1148/radiol.2321030174
59. Assayag D, Elicker BM, Urbania TH, et al Rheumatoid arthritis-associated interstitial lung disease: radiologic identification of usual interstitial pneumonia pattern. Radiology. 2014;270(2):583–588. doi:10.1148/radiol.13130187
60. Solomon JJ, Ryu JH, Tazelaar HD, et al Fibrosing interstitial pneumonia predicts survival in patients with rheumatoid arthritis-associated interstitial lung disease (RA-ILD). Respir Med. 2013;107(8):1247–1252. doi:10.1016/j.rmed.2013.05.002
61. Tsuchiya Y, Takayanagi N, Sugiura H, et al Lung diseases directly associated with rheumatoid arthritis and their relationship to outcome. Eur Respir J. 2011;37(6):1411–1417. doi:10.1183/09031936.00019210
62. Raghu G, Collard HR, Egan JJ, et al An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183(6):788–824. doi:10.1164/rccm.2009-040GL
63. Lynch DA, Godwin JD, Safrin S, et al; Idiopathic Pulmonary Fibrosis Study Group. High-resolution computed tomography in idiopathic pulmonary fibrosis: diagnosis and prognosis. Am J Respir Crit Care Med. 2005;172(4):488–493. doi:10.1164/rccm.200412-1756OC
64. Sumikawa H, Johkoh T, Colby TV, et al Computed tomography findings in pathological usual interstitial pneumonia: relationship to survival. Am J Respir Crit Care Med. 2008;177(4):433–439. doi:10.1164/rccm.200611-1696OC
65. Yagihashi K, Huckleberry J, Colby TV, et al; Idiopathic Pulmonary Fibrosis Clinical Research Network (IPFnet). Radiologic-pathologic discordance in biopsy-proven usual interstitial pneumonia. Eur Respir J. 2016;47(4):1189–1197. doi:10.1183/13993003.01680-2015
66. Flaherty KR, Thwaite EL, Kazerooni EA, et al Radiological versus histological diagnosis in UIP and NSIP: survival implications. Thorax. 2003;58(2):143–148. doi:10.1136/thorax.58.2.143
67. Yıldırım F, Türk M, Bitik B, et al Comparison of clinical courses and mortality of connective tissue disease-associated interstitial pneumonias and chronic fibrosing idiopathic interstitial pneumonias. Kaohsiung J Med Sci. 2019;35(6):365–372. doi:10.1002/kjm2.12066
68. Nurmi HM, Purokivi MK, Kärkkäinen MS, Kettunen HP, Selander TA, Kaarteenaho RL. Variable course of disease of rheumatoid arthritis-associated usual interstitial pneumonia compared to other subtypes. BMC Pulm Med. 2016;16(1):107. doi:10.1186/s12890-016-0269-2
69. Shigemitsu H, Azuma A. Sarcoidosis and interstitial pulmonary fibrosis; two distinct disorders or two ends of the same spectrum. Curr Opin Pulm Med. 2011;17(5):303–307. doi:10.1097/MCP.0b013e3283486d52
70. Bagnato G, Harari S. Cellular interactions in the pathogenesis of interstitial lung diseases. Eur Respir Rev. 2015;24(135):102–114. doi:10.1183/09059180.00003214
71. Chaudhary N, Jayaraman A, Reinhardt C, Campbell JD, Bosmann M. A single-cell lung atlas of complement genes identifies the mesothelium and epithelium as prominent sources of extrahepatic complement proteins. Mucosal Immunol. 2022;15(5):927–939. doi:10.1038/s41385-022-00534-7
72. Pandya PH, Wilkes DS. Complement system in lung disease. Am J Respir Cell Mol Biol. 2014;51(4):467–473. doi:10.1165/rcmb.2013-0485TR
73. Kulkarni HS, Liszewski MK, Brody SL, Atkinson JP. The complement system in the airway epithelium: an overlooked host defense mechanism and therapeutic target? J Allergy Clin Immunol. 2018;141(5):1582–1586.e1. doi:10.1016/j.jaci.2017.11.046
74. Andre GO, Converso TR, Politano WR, et al Role of Streptococcus pneumoniae proteins in evasion of complement-mediated immunity. Front Microbiol. 2017;8:224. doi:10.3389/fmicb.2017.00224
75. Ackerman SK, Friend PS, Hoidal JR, Douglas SD. Production of C2 by human alveolar macrophages. Immunology. 1978;35(2):369–372.
76. Angelidis I, Simon LM, Fernandez IE, et al An atlas of the aging lung mapped by single cell transcriptomics and deep tissue proteomics. Nat Commun. 2019;10(1):963. doi:10.1038/s41467-019-08831-9
77. Travaglini KJ, Nabhan AN, Penland L, et al A molecular cell atlas of the human lung from single-cell RNA sequencing. Nature. 2020;587(7835):619–625. doi:10.1038/s41586-020-2922-4
78. Gaurav R, Mikuls TR, Thiele GM, et al High-throughput analysis of lung immune cells in a combined murine model of agriculture dust-triggered airway inflammation with rheumatoid arthritis. PLoS One. 2021;16(2):e0240707. doi:10.1371/journal.pone.0240707
79. Mangogna A, Varghese PM, Agostinis C, et al Prognostic value of complement properdin in cancer. Front Immunol. 2021;11:614980. doi:10.3389/fimmu.2020.614980
80. Luo L, Li X, Hu X, et al Anaphylatoxins enhance recruitment of nonclassical monocytes via chemokines produced by pleural mesothelial cells in tuberculous pleural effusion. Am J Respir Cell Mol Biol. 2019;60(4):454–464. doi:10.1165/rcmb.2018-0075OC
81. Gu H, Mickler EA, Cummings OW, et al Crosstalk between TGF-β1 and complement activation augments epithelial injury in pulmonary fibrosis. FASEB J. 2014;28(10):4223–4234. doi:10.1096/fj.13-247650
82. Drouin SM, Kildsgaard J, Haviland J, et al Expression of the complement anaphylatoxin C3a and C5a receptors on bronchial epithelial and smooth muscle cells in models of sepsis and asthma. J Immunol. 2001;166(3):2025–2032. doi:10.4049/jimmunol.166.3.2025
83. Bosmann M, Ward PA. Role of C3, C5 and anaphylatoxin receptors in acute lung injury and in sepsis. Adv Exp Med Biol. 2012;946:147–159.
84. Bosmann M. Complement control for COVID-19. Sci Immunol. 2021;6(59):eabj1014. doi:10.1126/sciimmunol.abj1014
85. Conigliaro P, Triggianese P, Perricone C, Chimenti MS, Perricone R. COVID-19: disCOVering the role of complement system. Clin Exp Rheumatol. 2020;38(4):587–591.
86. Rennard SI, Chen YF, Robbins RA, Gadek JE, Crystal RG. Fibronectin mediates cell attachment to C1q: a mechanism for the localization of fibrosis in inflammatory disease. Clin Exp Immunol. 1983;54(1):239–247.
87. Phan SH, Thrall RS. Inhibition of bleomycin-induced pulmonary fibrosis by cobra venom factor. Am J Pathol. 1982;107(1):25–28.
88. Addis-Lieser E, Köhl J, Chiaramonte MG. Opposing regulatory roles of complement factor 5 in the development of bleomycin-induced pulmonary fibrosis. J Immunol. 2005;175(3):1894–1902. doi:10.4049/jimmunol.175.3.1894
89. Dreisin RB, Schwarz MI, Theofilopoulos AN, Stanford RE. Circulating immune complexes in the idiopathic interstitial pneumonias. N Engl J Med. 1978;298(7):353–357. doi:10.1056/NEJM197802162980701
90. Haslam PL, Allan F, Watling AF, Barrett C, Morris L, Turner-Warwick M. Impaired antibody-dependent cell-mediated cytotoxicity in cryptogenic fibrosing alveolitis (synonym: idiopathic pulmonary fibrosis). Clin Exp Immunol. 1982;49(1):59–66.
91. Jansen HM, Schutte AJ, Elema JD, et al Local immune complexes and inflammatory response in patients with chronic interstitial pulmonary disorders associated with collagen vascular diseases. Clin Exp Immunol. 1984;56(2):311–320.
92. Morimoto K, Janssen WJ, Terada M. Defective efferocytosis by alveolar macrophages in IPF patients. Respir Med. 2012;106(12):1800–1803. doi:10.1016/j.rmed.2012.08.020
93. Eisen DP. Mannose-binding lectin deficiency and respiratory tract infection. J Innate Immun. 2010;2(2):114–122. doi:10.1159/000228159
94. Kilpatrick DC. Mannan-binding lectin: clinical significance and applications. Biochim Biophys Acta. 2002;1572(2–3):401–413. doi:10.1016/S0304-4165(02)00321-5
95. Arney V. The deficiency of serum mannose binding lectin in early onset idiopathic pulmonary fibrosis and familial cases. J Clin Cell Immunol. 2013;3:127.
96. Vogt S, Trendelenburg M, Tamm M, Stolz D, Hostettler KE, Osthoff M. Local and systemic concentrations of pattern recognition receptors of the lectin pathway of complement in a cohort of patients with interstitial lung diseases. Front Immunol. 2020;11:562564. doi:10.3389/fimmu.2020.562564
97. Osthoff M, Ngian GS, Dean MM, et al Potential role of the lectin pathway of complement in the pathogenesis and disease manifestations of systemic sclerosis: a case-control and cohort study. Arthritis Res Ther. 2014;16(6):480. doi:10.1186/s13075-014-0480-6
98. Pellicano C, Miglionico M, Romaggioli L, et al Increased complement activation in systemic sclerosis patients with skin and lung fibrosis. J Pers Med. 2022;12(2):284. doi:10.3390/jpm12020284
99. Okamoto T, Mathai SK, Hennessy CE, et al The relationship between complement C3 expression and the MUC5B genotype in pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2018;315(1):L1–L10. doi:10.1152/ajplung.00395.2017
100. Gu H, Fisher AJ, Mickler EA, et al Contribution of the anaphylatoxin receptors, C3aR and C5aR, to the pathogenesis of pulmonary fibrosis. FASEB J. 2016;30(6):2336–2350. doi:10.1096/fj.201500044
101. Korczowska I. Rheumatoid arthritis susceptibility genes: an overview. World J Orthop. 2014;5(4):544–549. doi:10.5312/wjo.v5.i4.544
102. Sena L, Oliveira-Toré CF, Skare T, de Messias-Reason IJ, Andrade FA. C3 gene functional polymorphisms and C3 serum levels in patients with rheumatoid arthritis. Immunol Invest. 2021;50(8):1027–1041. doi:10.1080/08820139.2020.1800726
103. Banda NK, Deane KD, Bemis EA, et al Analysis of complement gene expression, clinical associations, and biodistribution of complement proteins in the synovium of early rheumatoid arthritis patients reveals unique pathophysiologic features. J Immunol. 2022;208(11):2482–2496. doi:10.4049/jimmunol.2101170
104. Neumann E, Barnum SR, Tarner IH, et al Local production of complement proteins in rheumatoid arthritis synovium. Arthritis Rheum. 2002;46(4):934–945. doi:10.1002/art.10183
105. Brodeur JP, Ruddy S, Schwartz LB, Moxley G. Synovial fluid levels of complement SC5b-9 and fragment Bb are elevated in patients with rheumatoid arthritis. Arthritis Rheum. 1991;34(12):1531–1537. doi:10.1002/art.1780341209
106. Konttinen YT, Ceponis A, Meri S, et al Complement in acute and chronic arthritides: assessment of C3c, C9, and protectin (CD59) in synovial membrane. Ann Rheum Dis. 1996;55(12):888–894. doi:10.1136/ard.55.12.888
107. Galindo-Izquierdo M, Pablos Alvarez JL. Complement as a therapeutic target in systemic autoimmune diseases. Cells. 2021;10(1):148. doi:10.3390/cells10010148
108. Banda NK, Kraus D, Vondracek A, et al Mechanisms of effects of complement inhibition in murine collagen-induced arthritis. Arthritis Rheum. 2002;46(11):3065–3075. doi:10.1002/art.10591
109. Hietala MA, Jonsson IM, Tarkowski A, Kleinau S, Pekna M. Complement deficiency ameliorates collagen-induced arthritis in mice. J Immunol. 2002;169(1):454–459. doi:10.4049/jimmunol.169.1.454
110. Matsumoto I, Maccioni M, Lee DM, et al How antibodies to a ubiquitous cytoplasmic enzyme may provoke joint-specific autoimmune disease. Nat Immunol. 2002;3(4):360–365. doi:10.1038/ni772
111. Monach PA, Verschoor A, Jacobs JP, et al Circulating C3 is necessary and sufficient for induction of autoantibody-mediated arthritis in a mouse model. Arthritis Rheum. 2007;56(9):2968–2974. doi:10.1002/art.22859
112. Trouw LA, Pickering MC, Blom AM. The complement system as a potential therapeutic target in rheumatic disease. Nat Rev Rheumatol. 2017;13(9):538–547. doi:10.1038/nrrheum.2017.125
113. Mollnes TE, Lea T, Mellbye OJ, Pahle J, Grand O, Harboe M. Complement activation in rheumatoid arthritis evaluated by C3dg and the terminal complement complex. Arthritis Rheum. 1986;29(6):715–721. doi:10.1002/art.1780290603
114. Moffat GJ, Lappin D, Birnie GD, Whaley K. Complement biosynthesis in human synovial tissue. Clin Exp Immunol. 1989;78(1):54–60.
115. Linton SM, Morgan BP. Complement activation and inhibition in experimental models of arthritis. Mol Immunol. 1999;36(13–14):905–914. doi:10.1016/S0161-5890(99)00113-3
116. Friščić J, Böttcher M, Reinwald C, et al The complement system drives local inflammatory tissue priming by metabolic reprogramming of synovial fibroblasts. Immunity. 2021;54(5):1002–1021.e10. doi:10.1016/j.immuni.2021.03.003
117. Kolev M, Dimeloe S, Le Friec G, et al Complement regulates nutrient influx and metabolic reprogramming during Th1 cell responses. Immunity. 2015;42(6):1033–1047. doi:10.1016/j.immuni.2015.05.024
118. Laumonnier Y, Karsten CM, Köhl J. Novel insights into the expression pattern of anaphylatoxin receptors in mice and men. Mol Immunol. 2017;89:44–58. doi:10.1016/j.molimm.2017.05.019
119. Liszewski MK, Kolev M, Le Friec G, et al Intracellular complement activation sustains T cell homeostasis and mediates effector differentiation. Immunity. 2013;39(6):1143–1157. doi:10.1016/j.immuni.2013.10.018
120. Arbore G, West EE, Spolski R, et al T helper 1 immunity requires complement-driven NLRP3 inflammasome activity in CD4+ T cells. Science. 2016;352(6292):aad1210. doi:10.1126/science.aad1210
121. Cambré I, Gaublomme D, Burssens A, et al Mechanical strain determines the site-specific localization of inflammation and tissue damage in arthritis. Nat Commun. 2018;9(1):4613. doi:10.1038/s41467-018-06933-4
122. Cambré I, Gaublomme D, Schryvers N, et al Running promotes chronicity of arthritis by local modulation of complement activators and impairing T regulatory feedback loops. Ann Rheum Dis. 2019;78(6):787–795. doi:10.1136/annrheumdis-2018-214627
123. Marchi LF, Paoliello-Paschoalato AB, Oliveira RDR, et al Activation status of peripheral blood neutrophils and the complement system in adult rheumatoid arthritis patients undergoing combined therapy with infliximab and methotrexate. Rheumatol Int. 2018;38(6):1043–1052. doi:10.1007/s00296-018-3997-1
124. Cutolo M, Seriolo B, Pizzorni C, Craviotto C, Sulli A. Methotrexate in psoriatic arthritis. Clin Exp Rheumatol. 2002;20(6 Suppl 28):S76–80.
125. Belinsky GS, Parke AL, Huang Q, et al The contribution of methotrexate exposure and host factors on transcriptional variance in human liver. Toxicol Sci. 2007;97(2):582–594. doi:10.1093/toxsci/kfm067
126. Di Muzio G, Perricone C, Ballanti E, et al Complement system and rheumatoid arthritis: relationships with autoantibodies, serological, clinical features, and anti-TNF treatment. Int J Immunopathol Pharmacol. 2011;24(2):357–366. doi:10.1177/039463201102400209
127. Ballanti E, Perricone C, Di Muzio G, et al Role of the complement system in rheumatoid arthritis and psoriatic arthritis: relationship with anti-TNF inhibitors. Autoimmun Rev. 2011;10(10):617–623. doi:10.1016/j.autrev.2011.04.012
128. Familian A, Voskuyl AE, van Mierlo GJ, et al Infliximab treatment reduces complement activation in patients with rheumatoid arthritis. Ann Rheum Dis. 2005;64(7):1003–1008. doi:10.1136/ard.2004.029124
129. Hokstad I, Deyab G, Wang Fagerland M, et al Tumor necrosis factor inhibitors are associated with reduced complement activation in spondylarthropathies: an observational study. PLoS One. 2019;14(7):e0220079. doi:10.1371/journal.pone.0220079
130. Qin X, Gao B. The complement system in liver diseases. Cell Mol Immunol. 2006;3(5):333–340.
131. Perissutti S, Tedesco F. Effect of cytokines on the secretion of the fifth and eighth complement components by HepG2 cells. Int J Clin Lab Res. 1994;24(1):45–48. doi:10.1007/BF02592409
132. Mold C, Gewurz H, Du Clos TW. Regulation of complement activation by C-reactive protein. Immunopharmacology. 1999;42(1–3):23–30. doi:10.1016/S0162-3109(99)00007-7
133. Eng GP, Bouchelouche P, Bartels EM, Bliddal H, Bendtzen K, Stoltenberg M. Anti-drug antibodies, drug levels, interleukin-6 and soluble TNF receptors in rheumatoid arthritis patients during the first 6 months of treatment with adalimumab or infliximab: a descriptive cohort study. PLoS One. 2016;11(9):e0162316. doi:10.1371/journal.pone.0162316
134. Romano C, Del Mastro A, Sellitto A, Solaro E, Esposito S, Cuomo G. Tocilizumab reduces complement C3 and C4 serum levels in rheumatoid arthritis patients. Clin Rheumatol. 2018;37(6):1695–1700. doi:10.1007/s10067-018-3992-7
135. Conigliaro P, Triggianese P, Chimenti MS, Lucchetti R, Kroegler B, Perricone R. Serological markers associated with disease activity in patients with rheumatoid arthritis treated with rituximab. J Int Med Res. 2016;44(1 suppl):53–57. doi:10.1177/0300060515593240
136. Blom AM, Nandakumar KS, Holmdahl R. C4b-binding protein (C4BP) inhibits development of experimental arthritis in mice. Ann Rheum Dis. 2009;68(1):136–142. doi:10.1136/ard.2007.085753
137. Hornum L, Hansen AJ, Tornehave D, et al C5a and C5aR are elevated in joints of rheumatoid and psoriatic arthritis patients, and C5aR blockade attenuates leukocyte migration to synovial fluid. PLoS One. 2017;12(12):e0189017. doi:10.1371/journal.pone.0189017
138. Vergunst CE, Gerlag DM, Dinant H, et al Blocking the receptor for C5a in patients with rheumatoid arthritis does not reduce synovial inflammation. Rheumatology. 2007;46(12):1773–1778. doi:10.1093/rheumatology/kem222
139. Barilla-Labarca ML, Toder K, Furie R. Targeting the complement system in systemic lupus erythematosus and other diseases. Clin Immunol. 2013;148(3):313–321. doi:10.1016/j.clim.2013.02.014
140. Hillmen P, Hall C, Marsh JC, et al Effect of eculizumab on hemolysis and transfusion requirements in patients with paroxysmal nocturnal hemoglobinuria. N Engl J Med. 2004;350(6):552–559. doi:10.1056/NEJMoa031688
141. Necela BM, Cidlowski JA. Mechanisms of glucocorticoid receptor action in noninflammatory and inflammatory cells. Proc Am Thorac Soc. 2004;1:239–246. doi:10.1513/pats.200402-005MS
142. Gregory AD, Kliment CR, Metz HE, et al Neutrophil elastase promotes myofibroblast differentiation in lung fibrosis. J Leukoc Biol. 2015;98(2):143–152. doi:10.1189/jlb.3HI1014-493R
143. Hammerschmidt DE, White JG, Craddock PR, Jacob HS. Corticosteroids inhibit complement-induced granulocyte aggregation. A possible mechanism for their efficacy in shock states. J Clin Invest. 1979;63(4):798–803. doi:10.1172/JCI109365
144. Conte E, Gili E, Fagone E, Fruciano M, Iemmolo M, Vancheri C. Effect of pirfenidone on proliferation, TGF-β-induced myofibroblast differentiation and fibrogenic activity of primary human lung fibroblasts. Eur J Pharm Sci. 2014;58:13–19. doi:10.1016/j.ejps.2014.02.014
145. Lopez-de la Mora DA, Sanchez-Roque C, Montoya-Buelna M, et al Role and new insights of pirfenidone in fibrotic diseases. Int J Med Sci. 2015;12(11):840–847. doi:10.7150/ijms.11579
146. Langer HF, Chung KJ, Orlova VV, et al Complement-mediated inhibition of neovascularization reveals a point of convergence between innate immunity and angiogenesis. Blood. 2010;116:4395–4403. doi:10.1182/blood-2010-01-261503
147. Roth GJ, Binder R, Colbatzky F, et al Nintedanib: from discovery to the clinic. J Med Chem. 2015;58(3):1053–1063. doi:10.1021/jm501562a
148. Wollin L, Wex E, Pautsch A, et al Mode of action of nintedanib in the treatment of idiopathic pulmonary fibrosis. Eur Respir J. 2015;45(5):1434–1445. doi:10.1183/09031936.00174914
149. Guo RF, Ward PA. Role of C5a in inflammatory responses. Annu Rev Immunol. 2005;23:821–852. doi:10.1146/annurev.immunol.23.021704.115835
150. Bejan-Angoulvant T, Naccache JM, Caille A, et al Evaluation of efficacy and safety of rituximab in combination with mycophenolate mofetil in patients with nonspecific interstitial pneumonia non-responding to a first-line immunosuppressive treatment (EVER-ILD): a double-blind placebo-controlled randomized trial. Respir Med Res. 2020;78:100770. doi:10.1016/j.resmer.2020.100770
151. Bieber A, Markovits D, Toledano K, et al Hypocomplementemia during tocilizumab treatment: long-term follow-up results. Medicine. 2022;101(24):e29528. doi:10.1097/MD.0000000000029528
152. Khanna D, Lin CJF, Furst DE, et al Tocilizumab in systemic sclerosis: a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Respir Med. 2020;8(10):963–974. doi:10.1016/S2213-2600(20)30318-0
153. Teramoto S, Fukuchi Y, Uejima Y, Shu CY, Orimo H. Superoxide anion formation and glutathione metabolism of blood in patients with idiopathic pulmonary fibrosis. Biochem Mol Med. 1995;55(1):66–70. doi:10.1006/bmme.1995.1033
154. Perricone C, De Carolis C, Giacomelli R, et al Inhibition of the complement system by glutathione: molecular mechanisms and potential therapeutic implications. Int J Immunopathol Pharmacol. 2011;24(1):63–68. doi:10.1177/039463201102400108
155. Bianchi L, Chimenti MS, Giunta A. Treatment of Hailey-Hailey disease with topical calcitriol. J Am Acad Dermatol. 2004;51(3):475–476. doi:10.1016/j.jaad.2003.10.668
156. Haapasalo K, Meri S. Regulation of the complement system by pentraxins. Front Immunol. 2019;10:1750. doi:10.3389/fimmu.2019.01750
157. Yang Z, Nicholson SE, Cancio TS, Cancio LC, Li Y. Complement as a vital nexus of the pathobiological connectome for acute respiratory distress syndrome: an emerging therapeutic target. Front Immunol. 2023;14:1100461. doi:10.3389/fimmu.2023.1100461
158. Du Clos TW. Pentraxins: structure, function, and role in inflammation. ISRN Inflamm. 2013;2013:379040. doi:10.1155/2013/379040
159. Raghu G, van den Blink B, Hamblin MJ, et al Effect of recombinant human pentraxin 2 vs placebo on change in forced vital capacity in patients with idiopathic pulmonary fibrosis: a randomized clinical trial. JAMA. 2018;319(22):2299–2307. doi:10.1001/jama.2018.6129
160. Hench PS, Kendall EC, Slocumb CH. The effect of a hormone of the adrenal cortex (17-hydroxy-11-dehydrocorticosterone; compound E) and of pituitary adrenocorticotropic hormone on rheumatoid arthritis. Mayo Clin Proc. 1949;24:181–197.
161. Ishmael FT, Fang X, Galdiero MR, et al Role of the RNA-binding protein tristetraprolin in glucocorticoid-mediated gene regulation. J Immunol. 2008;180(12):8342–8353. doi:10.4049/jimmunol.180.12.8342
162. Schleimer RP. Glucocorticoids suppress inflammation but spare innate immune responses in airway epithelium. Proc Am Thorac Soc. 2004;1:222–230. doi:10.1513/pats.200402-018MS
163. Mohammad AJ, Mortensen KH, Babar J, et al Pulmonary involvement in antineutrophil cytoplasmic antibodies (ANCA)-associated vasculitis: the influence of ANCA subtype. J Rheumatol. 2017;44(10):1458–1467. doi:10.3899/jrheum.161224
164. Heukels P, Moor CC, von der Thüsen JH, Wijsenbeek MS, Kool M. Inflammation and immunity in IPF pathogenesis and treatment. Respir Med. 2019;147:79–91. doi:10.1016/j.rmed.2018.12.015
165. Iyer SN, Margolin SB, Hyde DM, Giri SN. Lung fibrosis is ameliorated by pirfenidone fed in diet after the second dose in a three-dose bleomycin-hamster model. Exp Lung Res. 1998;24(1):119–132. doi:10.3109/01902149809046058
166. Xiong Z, Wang Q, Li W, et al Platelet-derived growth factor-D activates complement system to propagate macrophage polarization and neovascularization. Front Cell Dev Biol. 2021;9:686886. doi:10.3389/fcell.2021.686886
167. Fukihara J, Kondoh Y. Nintedanib (OFEV) in the treatment of idiopathic pulmonary fibrosis. Expert Rev Respir Med. 2016;10(12):1247–1254. doi:10.1080/17476348.2016.1249854
168. Omori T, Oguchi Y, Machida T, et al Evidence for activation of lectin and classical pathway complement components in aqueous humor of neovascular age-related macular degeneration. Ophthalmic Res. 2019;63:252–258. doi:10.1159/000503258
169. Kozhevnikova OS, Fursova AZ, Derbeneva AS, et al Association between polymorphisms in CFH, ARMS2, CFI, and C3 genes and response to anti-VEGF treatment in neovascular age-related macular degeneration. Biomedicines. 2022;10(7):1658. doi:10.3390/biomedicines10071658
170. Yu WK, Chen WC, Su VY, et al Nintedanib inhibits endothelial mesenchymal transition in bleomycin-induced pulmonary fibrosis via focal adhesion kinase activity reduction. Int J Mol Sci. 2022;23(15):8193. doi:10.3390/ijms23158193
171. Campochiaro C, Lazzaroni MG, Bruni C, Zanatta E, De Luca G, Matucci-Cerinic M. Open questions on the management of targeted therapies for the treatment of systemic sclerosis-interstitial lung disease: results of a EUSTAR survey based on a systemic literature review. Ther Adv Musculoskelet Dis. 2022;14:1759720X221116408. doi:10.1177/1759720X221116408
172. Fui A, Bergantini L, Selvi E, et al Rituximab therapy in interstitial lung disease associated with rheumatoid arthritis. Intern Med J. 2020;50(3):330–336. doi:10.1111/imj.14306
173. Cragg MS, Glennie MJ. Antibody specificity controls in vivo effector mechanisms of anti-CD20 reagents. Blood. 2004;103:2738–2743. doi:10.1182/blood-2003-06-2031
174. Felberg A, Taszner M, Urban A, et al Monitoring of the complement system status in patients with b-cell malignancies treated with rituximab. Front Immunol. 2020;11:584509. doi:10.3389/fimmu.2020.584509
175. Park CS, Chung SW, Ki SY, et al Increased levels of interleukin-6 are associated with lymphocytosis in bronchoalveolar lavage fluids of idiopathic nonspecific interstitial pneumonia. Am J Respir Crit Care Med. 2000;162(3 Pt 1):1162–1168. doi:10.1164/ajrccm.162.3.9906007
176. Mozaffarian A, Brewer AW, Trueblood ES, et al Mechanisms of oncostatin M-induced pulmonary inflammation and fibrosis. J Immunol. 2008;181(10):7243–7253. doi:10.4049/jimmunol.181.10.7243
177. ClinicalTrial.govt. Available from: https://clinicaltrials.gov/ct2/show/study/NCT05181397.
178. Montero P, Milara J, Roger I, Cortijo J. Role of JAK/STAT in interstitial lung diseases; molecular and cellular mechanisms. Int J Mol Sci. 2021;22(12):6211. doi:10.3390/ijms22126211
179. Akhurst RJ, Hata A. Targeting the TGFβ signalling pathway in disease. Nat Rev Drug Discov. 2012;11(10):790–811. doi:10.1038/nrd3810
180. Janssen-Heininger Y, Reynaert NL, van der Vliet A, Anathy V. Endoplasmic reticulum stress and glutathione therapeutics in chronic lung diseases. Redox Biol. 2020;33:101516. doi:10.1016/j.redox.2020.101516
181. Pilling D, Roife D, Wang M, et al Reduction of bleomycin-induced pulmonary fibrosis by serum amyloid P. J Immunol. 2007;179(6):4035–4044. doi:10.4049/jimmunol.179.6.4035
182. Salhi S, Ribes D, Faguer S. Complement C5 inhibition reverses bleomycin-induced thrombotic microangiopathy. Clin Kidney J. 2020;14(4):1275–1276. doi:10.1093/ckj/sfaa101
183. Huizenga N, Zonozi R, Rosenthal J, Laliberte K, Niles JL, Cortazar FB. Treatment of aggressive antineutrophil cytoplasmic antibody-associated vasculitis with eculizumab. Kidney Int Rep. 2019;5(4):542–545. doi:10.1016/j.ekir.2019.11.021
184. Flohé L. Glutathione peroxidase. Basic Life Sci. 1988;49:663–668. doi:10.1007/978-1-4684-5568-7_104
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.