")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Overexpression of PI3K p110α contributes to acquired resistance to MET inhibitor, in MET-amplified SNU-5 gastric xenografts
Authors Ji F, Liu X, Wu Y, Fang X, Huang G
Received 28 May 2015
Accepted for publication 29 June 2015
Published 19 October 2015 Volume 2015:9 Pages 5697—5704
DOI https://doi.org/10.2147/DDDT.S89410
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Wei Duan
Fujian Ji,1 Xuanwen Liu,2 Yuanyu Wu,1 Xuedong Fang,1 Guomin Huang1
1Department of General Surgery, The China–Japan Union Hospital of Jilin University, Changchun, 2Department of General Surgery, Jilin Central Hospital, Jilin, People’s Republic of China
Abstract: Gastric cancer is one of the most virulent malignant diseases and is the second leading cause of cancer mortality in the world. The receptor tyrosine kinase MET is constitutively activated in many gastric cancers and its expression is strictly required for survival of some gastric cancer cells. Targeting gastric cancers with amplified or abnormally activated MET may have therapeutic benefit based on nonclinical and emerging clinical findings. However, one of the major problems of therapies targeting tyrosine kinases is that many tumors are not responsive to treatment or eventually develop resistance to the drugs. This study aims to understand the mechanisms of MET resistance in gastric SNU-5 xenografts which developed resistance to PHA665752, a MET inhibitor, through long-period tyrosine kinase inhibitor exposure. In the current study, we found that PI3K p110α is overexpressed in PHA665752-resistant SNU-5 xenografts. These findings showed that high PI3K p110α expression contributes to tyrosine kinase inhibitor resistance. In addition, we reported the development of a carcinogen-induced gastric cancer model that recapitulates PI3K p110α expression in human disease, which will serve as a useful model to study PI3K p110α’s biology and its effectiveness as a novel biomarker and a molecular target for gastric cancer. Ultimately, PI3K p110α represents a novel target for gastric cancer.
Keywords: MET, SNU-5, gastric cancer, PI3K p110α, tyrosine kinase inhibitor resistance
Background
Despite the decreasing incidence of gastric cancer in developed countries, gastric cancer remains the second leading cause of cancer-related deaths worldwide. The greater proportion of the disease occurs within the male population in developing countries – mostly East Asia, South America, and Eastern Europe.1,2 Conventional therapy options for gastric cancer include surgery, chemotherapy, radiation therapy, and combination treatments.3 However, most patients are usually diagnosed after the cancer has progressed to an advanced stage. Moreover, even after surgical resection, tumors will recur in many patients, resulting in short survival times. The 5-year survival rate of gastric cancer patients were only 20%–25% in the Western world.4,5 Therefore, the development of novel therapies to improve the prognosis of gastric cancer patients is much needed. It is possible for us to identify molecules altered in gastric cancers and then hit them by use of specific targeted drugs.
Receptor tyrosine kinases (RTKs) are transmembrane glycoproteins that are activated by binding to their cognate ligands, resulting in the phosphorylation of tyrosine residues on the receptor and downstream signaling proteins.6 The insufficient effect of chemotherapy on advanced gastric cancer has resulted in the development of new biological therapies that modulate various targets of signal transduction pathways that are overexpressed in gastric cancer. One of the targets is MET proto-oncogene, which encodes the RTK MET. MET’s ligand has been identified as hepatocyte growth factor (HGF), which binding to MET results in tyrosine phosphorylation of the receptor and activation of downstream signaling molecules.7,8 At present, some molecules targeting MET have been evaluated in phase I or II clinical trial. Most of them are RTK inhibitors, while the others are monoclonal antibodies or biological antagonists.9–11 However, the patients first respond to targeted therapies, and that almost invariably also responding patients develop resistance during treatment just like other RTK inhibitors. Therefore, we were interested in identifying pathways whose activation could contribute to the MET inhibitor resistance.
In our work, we have developed a PHA665752-resistant gastric xenograft model by continuous dosing of PHA665752 for a long time period. And then, we discussed the underlying resistant mechanisms of this model from a perspective of molecular biology and try to find ways to circumvent these hurdles.
Materials and methods
Reagents and antibodies
A selective PI3K p110α inhibitor PI-103 and MET selective inhibitor PHA665752 were purchased from Selleck Chemicals (Houston, TX, USA). PI3K p110α, β, and γ antibodies were purchased from Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). Phospho-AKT (Ser473, p-AKT), AKT, phospho-ERK1/2 (Thr202/Tyr204, p-ERK1/2), ERK, phospho-S6 (Ser235, 236), S6, PTEN, DNA-dependent protein kinase (DNA-PK), p53 antibodies were obtained from Cell Signaling Technology (Danvers, MA, USA). The other chemicals used in this study were of analytical reagent grade. All antibodies were used as described by the manufacturer’s instructions. Tissue lysis buffer and phosphatase inhibitor cocktails were purchased from Sigma-Aldrich (St Louis, MO, USA).
Cell lines and cell culture
Human gastric cell carcinoma SNU-5 was purchased from American Type Culture Collection (ATCC) (Manassas, VA, USA), which harbor MET gene high expression and amplification.11 Cell lines were incubated at 37°C and 5% CO2 and maintained in RPMI1640 (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% heat-inactivated fetal bovine serum, 100 units/mL penicillin, 100 units/mL streptomycin, and 2 mm glutamine (Thermo Fisher Scientific). No ethics statement was required from the institutional review board for the use of these cell lines.
PHA665752-resistant SNU-5 xenograft model establishment
Six-week old female BALB/c nude mice, weighing from 18 to 20 g, were obtained from Vital River Laboratories (Beijing, People’s Republic of China). All animal experiments were performed with the approval of the Jilin University of animal use and care committee. The animals were raised under the specific pathogen-free conditions in the Experimental Animal Center of Jilin University. Mice were injected subcutaneously (sc) with 5×106 SNU-5 cells, which had been suspended in 100 μL of phosphate-buffered saline. The length and width of the tumors were measured twice weekly following the first day of SNU-5 cells’ implantation. Tumor volume was calculated using the following formula: tumor volume = length × width2 × π/6. Ten mice with tumor volume range from 100 to 150 mm3 were orally administrated with 10 mg/kg PHA665752 once daily. A continued PHA665752 treatment is needed for the development of PHA665752 resistance, marked with notable tumor growth.
cDNA library construction and sequencing
RNA isolations were performed using the RNeasyH Midi Kit (QIAGEN Inc., Valencia, CA, USA), following the protocol for isolating cytoplasmic RNA. Briefly, tumor tissues were processed and the centrifugation steps were performed at 2,850 ×g. DNA was removed using the RNase-Free DNase Set (QIAGEN Inc.) at the recommended step in the RNeasyH protocol. RNA concentration was determined using a NanoDropTM 1000 Spectrometer (Thermo Fisher Scientific Inc., Waltham, MA, USA). The sequencing library was constructed according to Illumina’s TruSeq RNA Sample Preparation Protocol.12 After normalization, the DNA sample libraries were pooled into four libraries, and the pooled libraries were sequenced on an Illumina HiSeq 2000 sequencing machine (Illumina Inc., San Diego, CA, USA).
Quantitative real-time PCR (qRT-PCR)
Total RNA above isolated was synthesized to cDNA using PrimeScript RT reagent kit with gDNA Eraser (Takara Bio Inc., Shiga, Japan) for reversed transcription PCR with mixture of oligo-dT and Random Primer. Real-time qRT-PCR was performed on CFX-96 (Bio-Rad Laboratories, Hercules, CA, USA), with endogenous control GAPDH. The reactions were performed in triplicate in a 384-well format with 10 μL reaction volume. The gene expression level was calculated relative to the expression of endogenous control GAPDH and then adjusted relative to expression in PHA665752-sensitive or-resistant tumor tissues.
Efficacy study of established PHA665752-resistant SNU-5 xenograft model in vivo
PHA665752-resistant SNU-5 tumor were isolated from mice, and cut into 3×3 mm3 pieces, then transplanted into new nude mice. When tumor volume reached 160–200 mm3, mice were randomly divided into four groups, ten mice in each group and were treated with phosphate-buffered saline, 10 mg/kg PHA665752 (once daily), 5 mg/kg PI-103, or PHA665752 plus PI-103. The tumor volume and body weight in each group were balanced. Once an appropriate tumor volume (1,500 mm3) was attained (3 weeks after treatment), tumors were excised, carefully dissected clear of surrounding skin and fat, snap-frozen in liquid nitrogen and then stored at -80°C until further processing for Western blotting analysis in vitro. All animal experiments were performed in accordance with protocols approved by the Experimental Animal Center of Jilin University Animal Care and Use Committee.
Western blotting
Proteins of tumor tissues were extracted from whole cells by lysing them in a Tris buffer (50 mmol/L, pH 8.0) containing NaCl (150 mmol/L), NP40 (1%, v/v), deoxycholic acid (0.5%, w/v), SDS (0.1%, w/v), NaF (1 mmol/L), Na3VO4 (1 mmol/L), and glycerol (10%, v/v; Sigma-Aldrich) supplemented with a protease inhibitor cocktail II (Roche Diagnostics, Indianapolis, IN, USA). The protein concentration of the supernatant was determined using the BCA Protein Assay kit (Pierce Biotechnology, Rockford, IL, USA). Equal amounts of protein were separated by sodium dodecyl sulfate/polyacrylamidegel electrophoresis (SDS/PAGE) on 12% gel, blotted on a nitrocellulose membrane, and probed with p-MET, p-ERK1/2, p-AKT, p-S6, and PI3K p110α, β, γ rabbit monoclonal antibodies and subsequently with goat anti-rabbit (horseradish peroxidase [HRP]) antibodies, and detected by chemiluminescence. Secondary antibodies were purchased from Invitrogen.
Statistical analysis of the data
All results and data were confirmed in at least three separate experiments. For comparison of metabolite levels, Student’s t-test was used with P-values ≤0.05 considered to be statistically significant. Data represent the mean ± SE.
Results
Establishment of PHA665752-resistant SNU-5 xenograft model
Nude mice-bearing SNU-5 tumors were orally dosed with 10 mg/kg PHA665752 once daily for up to 26 weeks to establish PHA665752-resistant SNU-5 xenograft model in vivo. The results indicated that treatments with 10 mg/kg PHA666762 at first significantly inhibit tumor growth PHA665752 xenografts; later, one mouse finally developed acquired resistance to PHA665752 after treatment (Figure 1A). In the end of study, the tumors were excised, carefully dissected, and weighted (Figure 1B and C).
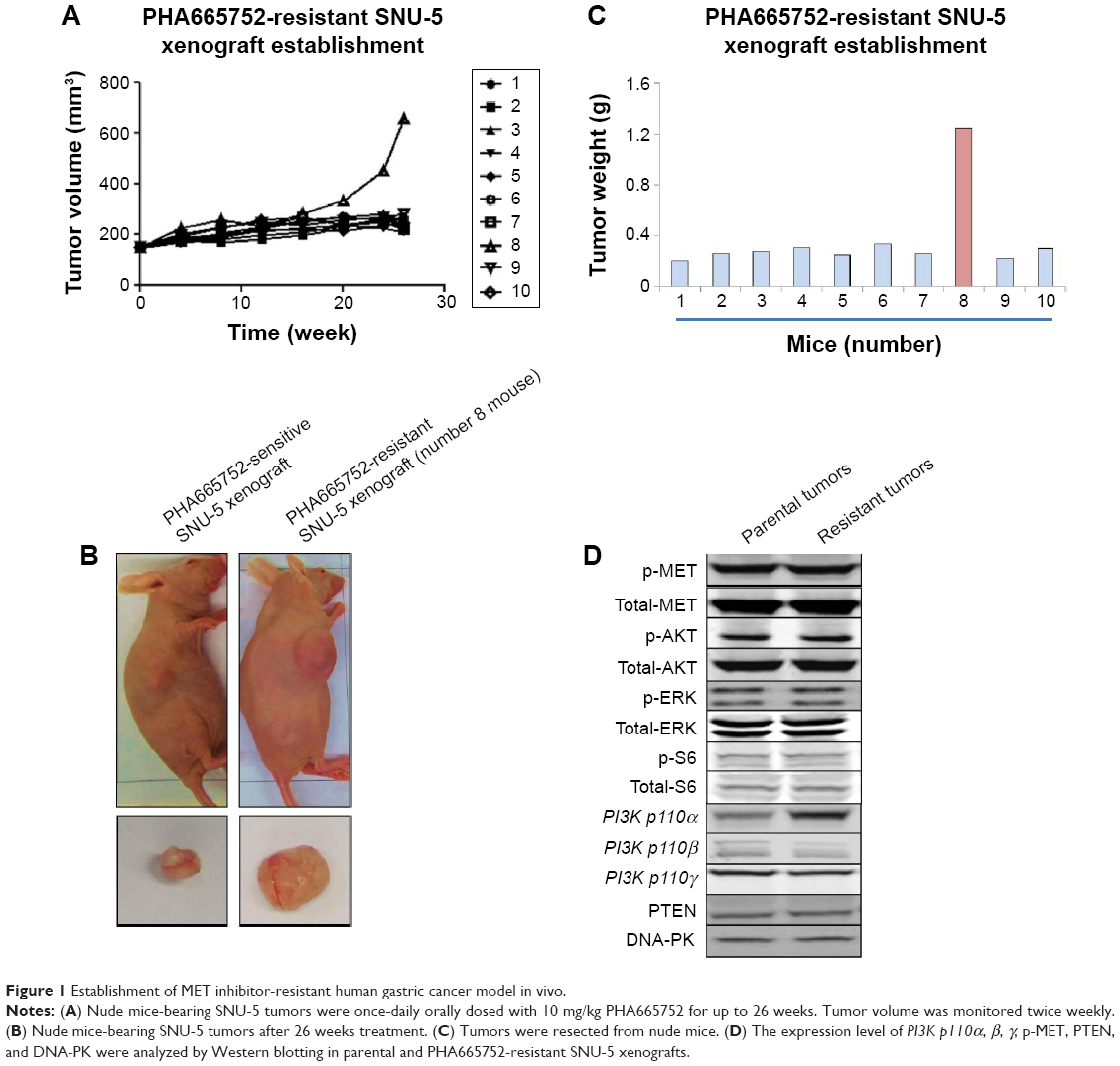 | Figure 1 Establishment of MET inhibitor-resistant human gastric cancer model in vivo. |
RNA-sequencing identifies PI3K p110α was overexpressed in the PHA665752-resistant SNU-5 xenografts
Samples of RNA had previously been isolated from parental and acquired resistant SNU-5 tumor tissues and subjected to RNA-sequencing (RNA-seq). Cuffdiff was used to determine changes common to both sensitive and resistant tumor tissues.13 As shown in Figure 1A, the tumor growth cannot be inhibited by 10 mg/kg PHA665752 after 30 weeks’ treatment and the results of RNA-seq showed that PI3K p110α was overexpressed in the PHA665752-resistant SNU-5 xenografts compared with PHA665752-sensitive tumors xenografts. In addition, we found that PI3K p110β, γ, B-raf, K-Ras, HER2, β-catenin, and PTEN expressed in a normal level (Figure 2A). According to the results, we proposed that PI3K p110α may be related to the acquired resistance to PHA665752.
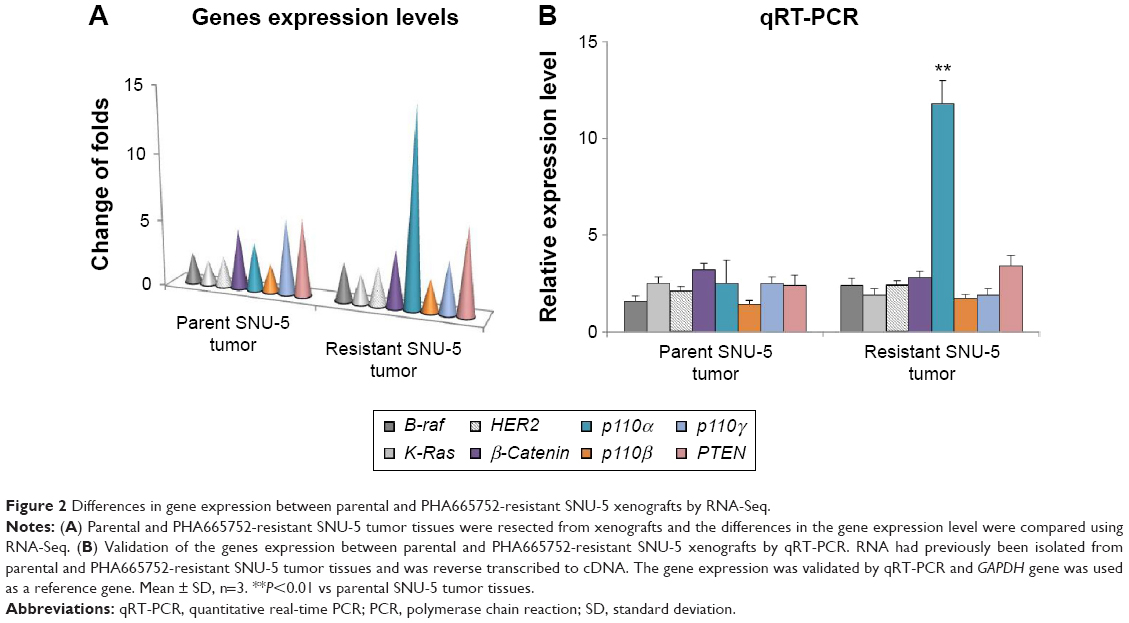 | Figure 2 Differences in gene expression between parental and PHA665752-resistant SNU-5 xenografts by RNA-Seq. |
Validation of PI3K p110α expression level by qRT-PCR
To detect the PI3K p110α expression level in PHA665752 parental and acquired resistant tumor tissues, total RNA was isolated and then synthesized to cDNA, which then subjected to qPCR. The results of qRT-PCR were highly similar to the RNA-seq results (Figure 2B). Overexpression of PI3K p110α in PHA665752-resistant tumor tissues compared with sensitive tumor tissues was observed in nude mice. Similar to the results of RNA-Seq, genes PI3K p110β, γ, B-raf, K-Ras, HER2, β-catenin, and PTEN expressed in a normal level in the resistant tumor tissue.
Combination of PHA665752 with PI-103 had a synergistic effect on PHA665752-resistant SNU-5 xenografts
We combined PHA665752 with PI-103 to treat PHA665752-resistant SNU-5 xenografts to investigate whether dual blockade of both the MET and PI3K pathways would yield inhibitory efficacy or not. The data indicated that treatment with 10 mg/kg PHA665752 could slightly inhibit tumor growth in PHA665752-resistant SNU-5 xenografts. However, 5 mg/kg PI-103 treatment has no inhibitory effects. As we expected, combination of PHA665752 with PI-103 had a synergistic effect after 3 weeks of therapy (Figure 3A–C). During the period of efficacy study, there was no significant weight loss or other toxicity regarding body weight changes (Figure 3D).
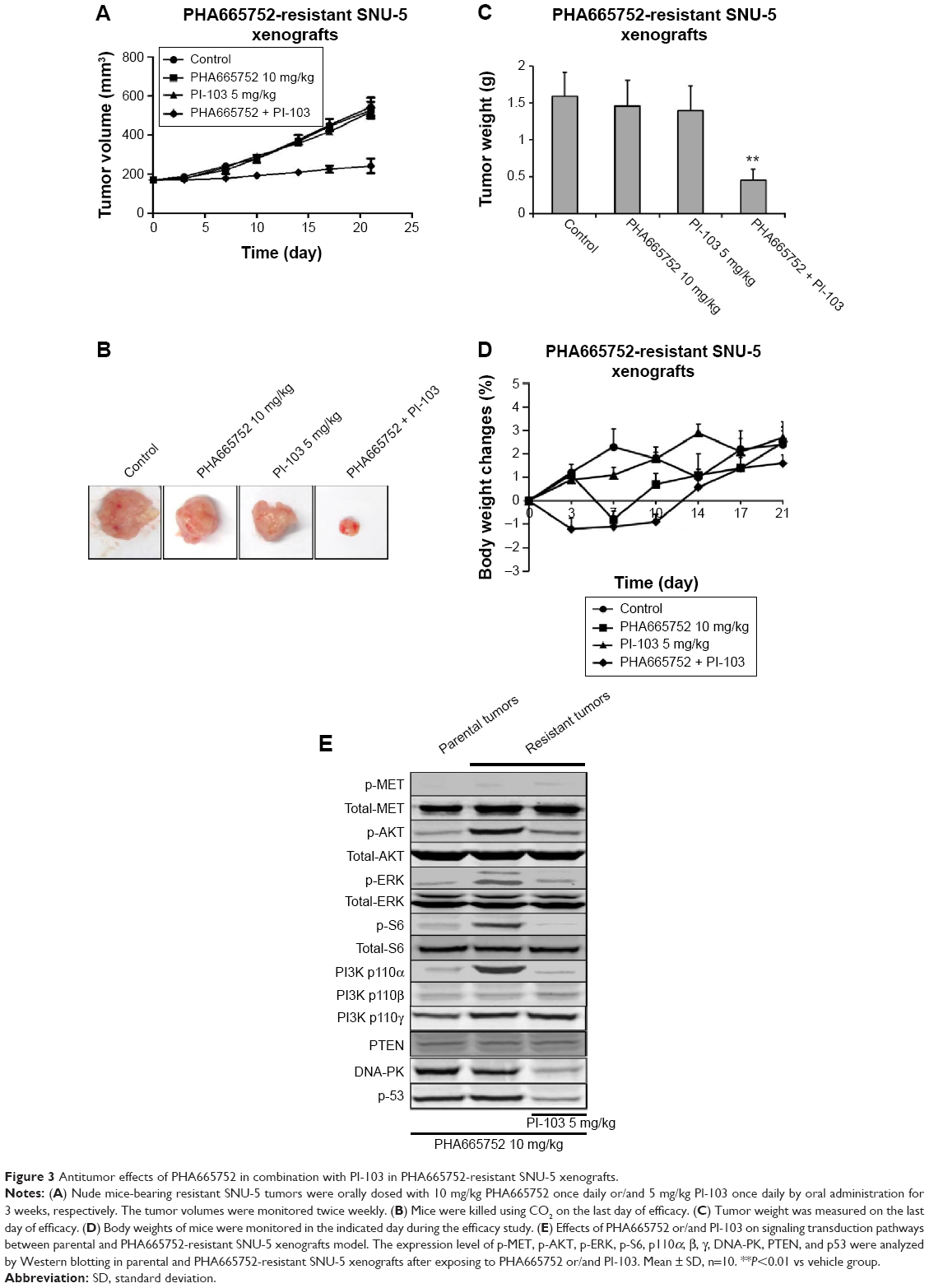 | Figure 3 Antitumor effects of PHA665752 in combination with PI-103 in PHA665752-resistant SNU-5 xenografts. |
Activation of AKT pathways is required for the resistance to MET inhibition
Since AKT and ERK are the major downstream molecules of MET pathway, Western blotting was used to observe phosphorylation status and total protein expression in PHA665752-sensitive and -resistant tumor tissues. We found that both PHA665752-sensitive and -resistant tumor tissues highly expressed p-MET and MET. The data also showed that only resistant tumor tissues highly expressed PI3K p110α. As for PI3K p110β, γ, and PTEN protein levels, there was no difference between these two models (Figure 1D). In addition, the expressions of p-MET and p-ERK protein levels were completely inhibited when both models were exposed to PHA665752. Moreover, the expressions of p-AKT and p-S6 were only observed in PHA665752-resistant tumor tissues even after treatment with PHA665752 (Figure 3E). Furthermore, combination treatment with PHA665752 and PI-103 showed a greater inhibition of PI3K p110α, p-AKT, and p-S6 in PHA665752-resistant xenografts (Figure 3E). We then test the expression of DNA-PK, which could be significantly inhibited by PI-103. The results showed that there was no difference between parental and PHA665752-resistant SNU-5 xenografts with regard to DNA-PK expression (Figure 1D). Moreover, PI-103 could potently inhibit DNA-PK as well as its downstream molecular p53 expressions (Figure 3E).
Discussion
Gastric cancer is a malignant cancer and counts for a major cause of cancer-related death worldwide. Even if traditional therapies have improved a lot in recent years, patients with advanced disease have a poor prognosis.5 Aberrant RTK activity provides growth and survival signals crucial for the development and progression of many cancers. The MET gene has recently become a popular target for molecular therapies. However, little is known regarding the possible mechanisms of resistance to treatment with MET inhibitors. In the current study, we generated an in vivo PHA665752-resistant xenograft model by using a protocol similar to Yi et al’s study.14 Gene profiles of PHA665752-sensitive and -resistant tumor tissues were then compared with RNA-seq. The results showed that overexpression of gene PI3K p110α would be found in the resistant tumor tissues but not in the sensitive ones. In addition, the highly expression level of PI3K p110α in resistant tissues was confirmed by the Western blotting. Further study showed that inhibition of the PI3K pathway by PI-103 could recover the sensitivity to PHA665752 in the resistant model. Importantly, aberrant AKT activity is required for the resistance development.
MET addiction signaling pathways and biological responses have primarily been studied, when MET activation by HGF. Lee et al reported that fusion-activated BRAF can bypass upstream MET inhibition by over-activating the mitogen-activated protein kinases pathway in an MET-independent manner and the resistance can be overcome by treating with MEK inhibitor, or a combination of MET inhibitor and RAF inhibitor.15 However, in the current resistant model, we did not find aberrant BRAF expression according to the results of RNA-seq. At present, there are few reports regarding MET inhibitor resistance and PI3K p110α in gastric cancer. However, other studies reported that inhibition of MET activity leads to the reduced activation of the PI3K/AKT/mTOR pathway in transformed cells.16–18 Our findings are consistent with these findings, since the expressions of p-AKT and p-S6 were significantly inhibited in PHA665752-sensitive tumor tissues after treatment with PHA665752. More interestingly, we found that the expressions of p-AKT and p-S6 were only observed in PHA665752-resistant tumor tissues even after exposure to MET inhibitor, and which would be reduced by the combination of PHA665752 with PI-103. According to these results, we proposed that PI3K p110α may be related to the acquired resistance to PHA665752.
In previous studies, Deven et al demonstrated that the mechanisms of MET inhibitor resistance may act through mTOR and Wnt pathways in non-small-cell lung cancer cells, since HGF could activate Wnt signaling through accumulation and nuclear translocation of β-catenin, both in vitro and in vivo.19–22 Moreover, several experimental evidences indicate the existence of biochemical relationship between the members of the HER family and MET. Simona et al reported that activation of HER family members in gastric carcinoma cells mediates resistance to MET inhibition.23–27 Nevertheless, we found that PI3K p110β, γ, B-raf, K-Ras, HER2, β-catenin, and PTEN expressed at a normal level in the current resistant in vivo gastric model. High basal p-AKT levels in PHA665752-resistant tumor tissues suggest the dependence upon PI3K signaling. Additionally, combination treatment with PHA665752 and PI-103 showed a greater inhibition of PI3K p110α, p-AKT, and p-S6, further suggesting that AKT pathways activation is required for the resistance to MET inhibition. Thus, our results imply that the expression status of PI3K p110α may be a good marker for predicting the therapeutic efficacy of MET inhibitors. However, the detailed mechanism whereby the PI3K/AKT pathway contributes to MET inhibitor resistance is currently unknown. Our studies suggest a novel mechanism of resistance in gastric cancer and further investigation is needed.
Conclusion
In summary, the MET addiction SNU-5 xenograft model developed resistance to MET inhibitor as a result of PI3K p110α gene overexpression. Combination of PHA665752 and PI-103 significantly exerted a synergistic antitumor effect on the PHA665752-resistant xenografts in vivo. Then, the antitumor effect may be induced by the inhibition of PI3K/AKT pathway. To our knowledge, this is the first study showing a relationship between the PI3K p110α and acquired MET inhibitor resistance. We suggest a novel treatment modality to overcome the acquired resistance seen in gastric cancer.
Disclosure
The authors report no conflicts of interest in this work.
References
Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2008. CA Cancer J Clin. 2008;58(2):71–96. | ||
Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55(2):74–108. | ||
Yamashita K, Sakuramoto S, Nemoto M, et al. Trend in gastric cancer: 35 years of surgical experience in Japan. World J Gastroenterol. 2011;17(29):3390–3397. | ||
Saragoni L, Morgagni P, Gardini A, et al. Early gastric cancer: diagnosis, staging, and clinical impact. Evaluation of 530 patients. New elements for an updated definition and classification. Gastric Cancer. 2013;16:549–554. | ||
Nieminen A, Kokkola A, Ylä-Liedenpohja J, Louhimo J, Mustonen H, Puolakkainen P. Early gastric cancer: clinical characteristics and results of surgery. Dig Surg. 2009;26:378–383. | ||
Birchmeier C, Birchmeier W, Gherardi E, Vande Woude GF. Met, metastasis, motility and more. Nat Rev Mol Cell Biol. 2003;4:915–925. | ||
Inoue T, Kataoka H, Goto K, et al. Activation of c-Met (hepatocyte growth factor receptor) in human gastric cancer tissue. Cancer Sci. 2004;95:803–808. | ||
Huang TJ, Wang JY, Lin SR, Lian ST, Hsieh JS. Overexpression of the c-met protooncogene in human gastric carcinoma-correlation to clinical features. Acta Oncol. 2001;40:638–643. | ||
Comoglio PM, Giordano S, Trusolino L. Drug development of MET inhibitors: targeting oncogene addiction and expedience. Nat Rev Drug Discov. 2008;7:504–516. | ||
Eder JP, Vande Woude GF, Boerner SA, LoRusso PM. Novel therapeutic inhibitors of the c-Met signaling pathway in cancer. Clin Cancer Res. 2009;15:2207–2214. | ||
Park BH, Jung KH, Yun SM, et al. KRC-327, a selective novel inhibitor of c-Met receptor tyrosine kinase with anticancer activity. Cancer Lett. 2013;331:158–166. | ||
Rathe SK, Moriarity BS, Stoltenberg CB, et al. Using RNA-seq and targeted nucleases to identify mechanisms of drug resistance in acute myeloid leukemia. Sci Rep. 2014;4:6048. | ||
Roberts A, Pimentel H, Trapnell C, Pachter L. Identification of novel transcripts in annotated genomes using RNA-Seq. Bioinformatics. 2011;27:2325–2329. | ||
Zhang Y, Pan T, Zhong X, Cheng C. Resistance to cetuximab in EGFR-overexpressing esophageal squamous cell carcinoma xenografts due to FGFR2 amplification and overexpression. J Pharmacol Sci. 2014;126:77–83. | ||
Lee NV, Lira ME, Pavlicek A, et al. A novel SND1–BRAF fusion confers resistance to c-Met inhibitor PF-04217903 in GTL16 cells through MAPK activation. PLoS One. 2012;7:e39653. | ||
Ma PC, Schaefer E, Christensen JG, Salgia R. A selective small molecule c-MET inhibitor, PHA665752, cooperates with rapamycin. Clin Cancer Res. 2005;11:2312–2319. | ||
Spix JK, Chay EY, Block ER, Klarlund JK. Hepatocyte growth factor induces epithelial cell motility through transactivation of the epidermal growth factor receptor. Exp Cell Res. 2007;313:3319–3325. | ||
Sierra JR, Cepero V, Giordano S. Molecular mechanisms of acquired resistance to tyrosine kinase targeted therapy. Mol Cancer. 2010;9:75. | ||
Fong JT, Jacobs RJ, Moravec DN, et al. Alternative signaling pathways as potential therapeutic targets for overcoming EGFR and c-Met inhibitor resistance in non-small cell lung cancer. PLoS One. 2013;8:e78398. | ||
Apte U, Zeng G, Muller P, et al. Activation of Wnt/beta-catenin pathway during hepatocyte growth factor-induced hepatomegaly in mice. Hepatology. 2006;44:992–1002. | ||
Liou G, Matragoon S, Samuel S, et al. MAP kinase and beta-catenin signaling in HGF induced RPE migration. Mol Vis. 2002;8:483–493. | ||
Ji H, Wang J, Nika H, et al. EGF-induced ERK activation promotes CK2-mediated disassociation of alpha-catenin from beta-catenin and transactivation of beta-Catenin. Mol Cell. 2009;36:547–559. | ||
Corso S, Ghiso E, Cepero V, et al. Activation of HER family members in gastric carcinoma cells mediates resistance to MET inhibition. Mol Cancer. 2010;9:121. | ||
Engelman JA, Zejnullahu K, Mitsudomi T, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–1043. | ||
Tanner M, Hollmen M, Junttila TT, et al. Amplification of HER-2 in gastric carcinoma: association with Topoisomerase II alpha gene amplification, intestinal type, poor prognosis and sensitivity to trastuzumab. Ann Oncol. 2005;16:273–278. | ||
Muller WJ, Arteaga CL, Muthuswamy SK, et al. Synergistic interaction of the Neu proto-oncogene product and transforming growth factor alpha in the mammary epithelium of transgenic mice. Mol Cell Biol. 1996;16:5726–5736. | ||
Hynes NE, MacDonald G. ErbB receptors and signaling pathways in cancer. Curr Opin Cell Biol. 2009;21:177–184. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.